 María González-Tablas1,2,3†
María González-Tablas1,2,3† Carlos Prieto1,4†
Carlos Prieto1,4† Daniel Arandia1,5
Daniel Arandia1,5 María Jara-Acevedo1,6
María Jara-Acevedo1,6 Álvaro Otero1,5Daniel Pascual1,5
Álvaro Otero1,5Daniel Pascual1,5 Laura Ruíz1,5
Laura Ruíz1,5 Iván Álvarez-Twose7,8
Iván Álvarez-Twose7,8 Andrés Celestino García-Montero1,2,8,9
Andrés Celestino García-Montero1,2,8,9 Alberto Orfao1,2,3,9‡
Alberto Orfao1,2,3,9‡ María Dolores Tabernero1,2,3,10*‡
María Dolores Tabernero1,2,3,10*‡- 1Instituto de Investigación Biomédica de Salamanca (IBSAL), University Hospital of Salamanca, Salamanca, Spain
- 2Centre for Cancer Research (Centro de Investigación del Cáncer de Salamanca (CIC)-Instituto de Biología Molecular y Celular del Cáncer (IBMCC), Centro Superior de Investigaciones Científicas (CSIC)/Universidad de Salamanca (USAL), IBSAL) and Department of Medicine, University of Salamanca, Salamanca, Spain
- 3Biomedical Research Networking Centre on Cancer– Centro de Investigación Biomédica en Red de Cáncer (CIBER-ONC) (CB16/12/00400), Instituto de Salud Carlos III, Madrid, Spain
- 4Bioinformatics Service Servicio de Apoyo a la Investigación de la Universidad de Salamanca (NUNCLEUS), University of Salamanca, Salamanca, Spain
- 5Neurosurgery Service, University Hospital of Salamanca, Salamanca, Spain
- 6Sequencing Service Servicio de Apoyo a la Investigación de la Universidad de Salamanca (NUNCLEUS), University of Salamanca, Salamanca, Spain
- 7Instituto de Estudios de Mastocitosis de Castilla La Mancha, Virgen del Valle Hospital, Toledo, Spain
- 8Spanish Network on Mastocytosis Red Española de Mastocitosis (REMA), Salamanca, Spain
- 9Spanish National DNA Bank Carlos III, University of Salamanca, Salamanca, Spain
- 10Instituto de Estudios de Ciencias de la Salud de Castilla y León (IECSCYL-IBSAL), Salamanca, Spain
Human WHO grade 1 meningiomas are generally considered benign tumors; despite this, they account for ≈50% of all recurrent meningiomas. Currently, limited data exist about the mutational profiles of grade 1 meningiomas and patient outcome. We investigated the genetic variants present in 32 WHO grade 1 meningiomas using whole exome sequencing, and correlated gene mutational profiles with tumor cytogenetics and patient outcome. Overall, WHO grade 1 meningiomas harbored numerous and heterogeneous genetic variants, which most frequently affected the NF2 (47%) gene and to a less extent the PNMA6A (22%), TIGD1 (16%), SMO (13%), PTEN (13%), CREG2 (9%), EEF1A1 (6%), POLR2A (6%), ARID1B (3%), and FAIM3 (3%) genes. Notably, non-synonymous genetic variants of SMO and POLR2A were restricted to diploid meningiomas, whereas NF2 mutations were only found among tumors that showed -22/22q─ (with or without a complex karyotype). Based on NF2 mutations and tumor cytogenetics, four genetic profiles were defined with an impact on patient recurrence-free survival (RFS). These included (1) two good-prognosis tumor subgroups—diploid meningiomas (n=9) and isolated -22/22q─ associated with NF2 mutation (n=7)—with RFS rates at 10 y of 100%; and (2) two subgroups of poor-prognosis meningiomas—isolated -22/22q─ without NF2 mutation (n=3) and tumors with complex karyotypes (n=11)—with a RFS rate at 10 y of 48% (p=0.003). Our results point out the existence of recurrent but heterogeneous mutational profiles in WHO grade 1 meningiomas which have an impact on patient outcome.
Introduction
Sporadic meningioma is the most common primary brain tumor, among which WHO (World Health Organization) grade 1 (i.e., low grade/benign) meningiomas represent the great majority (≈85%) of cases (1). Compared to WHO grade 2 (atypical) and grade 3 (anaplastic) meningiomas, WHO grade 1 tumors typically show benign histopathological features associated with a better outcome (2). However, grade 1 meningiomas still show a heterogeneous clinical behavior (3). Thus, a smaller fraction of WHO grade 1 meningioma patients (earlier or later) show tumor recurrence, associated with a poorer outcome, such recurrent WHO grade 1 tumors accounting for ≈50% of all recurrent meningiomas (2). Despite the cytogenetic profile of grade 1 meningiomas has been extensively investigated in the past, and some chromosomal alterations (e.g., monosomy of chromosome 22) and cytogenetic profiles—e.g., del(1p) and monosomy 14—have been associated with the outcome of WHO grade 1 meningiomas (3), at present there are still limited data about the type and frequency of other genetic alterations (i.e., point mutations) in these tumors (4–6) and their potential impact on patient outcome (7).
Neurofibromin 2 (NF2) was the first gene to be associated with an increased risk to develop meningioma (8). NF2 is a tumor suppressor gene located at chromosome 22q12.2, which encodes for the merlin protein, that is inactivated in a significant fraction of meningiomas by either gene mutation and/or deletion of the 22q chromosomal region—i.e., del(22q -) or monosomy 22 (9). Despite this, the potential involvement of NF2 in the development of sporadic meningioma still remains controversial (10) because it is not a hallmark of these tumors. In contrast, while early clinical studies showed no clear association in meningiomas between NF2 gene mutation and the WHO tumor grade (4, 11), recent reports showed a frequency of NF2 mutation that varies in WHO grade 1 tumors between 20–35% vs 50–72% in grade 2/3 meningiomas (6, 12), and NF2-mutated meningiomas have been reported to display a poorer outcome to that of NF2 wild-type tumors (12, 13). In line with these later findings, in vitro studies also indicate that NF2-mutated meningiomas are associated with a slower growth than NF2 wild-type tumors (9).
Other numerous genetic alterations, including extensive chromosome losses and gains (14) together with mutations in genes other than NF2, have been recurrently reported in meningiomas (5, 11, 12, 15) (Supplementary Table 1). Thus, meningiomas frequently show chromosomal alterations that, apart from del(22q)/monosomy 22 (12 vs 42%), include del(1p), del(7p), del(19p), monosomy 10, and/or monosomy 14 (16). Likewise, mutations involving the AKT1 (v-Akt murine thymoma viral oncogene homolog-1), KLF4 (Kruppel-like factor 4), PIK3CA (phosphatidylinositol 3-kinase), SMARCA4 (actin-dependent regulator of chromatin, subfamily A, member 4), and SMO (Smoothened) genes have also been recurrently reported at variable frequencies in sporadic meningioma (12, 14, 15), together with altered ARID1B, BAP1, PTEN, TP53, and TRAF7 gene profiles, as revealed via gene panel (15) and a limited number of whole exome sequencing (WES) plus whole genome sequencing (WGS) (4, 11, 12, 15) studies. In these later studies, WES/WGS of relatively limited numbers of mostly grade 2/3 meningiomas have been investigated (n=5, 5, 11 and 66 tumors) (12, 17), in addition to a few grade 1 tumors (n=17, 20 and 39, respectively) (11, 12, 15).
Material and Methods
Patients and Samples
A total of 32 human WHO grade 1 meningioma samples from 31 patients—21 females and 10 males; median age of 66 years, range: 24 to 83 years—diagnosed with sporadic meningioma at the University Hospital of Salamanca (Spain) were retrospectively studied. At the moment of closing this study, nine patients had shown tumor recurrence after a median (range) follow-up of 7 years (2–21). Tumor samples corresponding to tissue not required for diagnosis (n=32) and paired blood samples from 3/32 patients were obtained after each donor had given his/her informed consent according to the Declaration of Helsinki and after its approval by the ethical committees. DNA was subsequently extracted from the tumor tissue and blood nucleated cells, immediately after the samples had been obtained, and stored at −80°C at the Spanish National DNA Bank Carlos III (University of Salamanca, Salamanca, Spain), until analyzed. The study was approved by the local ethics committee of the University Hospital of Salamanca (Salamanca, Spain).
Two WES datasets were additionally used in this study for the definition of the incidence of genetic variants in the general population: (i) one dataset included sequencing data from the Genome Aggregation Database (gnomAD; large-scale sequencing project from 125,748 unrelated individuals publicly available for the scientific community), and (ii) the second dataset included data on 129 unrelated Spanish subjects with similar gender (63F:66M) and age distribution (median 47 y; range 20–76 y) to the meningioma patient cohort—kindly provided by the Spanish Network on Mastocytosis (REMA)—to exclude variants that are commonly observed in the Spanish population.
Next-Generation Sequencing of Meningioma DNA Samples
Genomic DNA was extracted from tumor tissues of 32 sporadic meningiomas, and blood DNA from three of the former patients, as previously described (16). DNA quantity and quality were assessed using the NanoDrop-1000 technology (Nano-Drop Technologies, Wilmington, DE, USA), and 100 ng of gDNA was used per sample to prepare individual sequencing libraries via the Kapa HyperPlus kit (Roche Diagnostics, Penzberg, Germany). For each library, DNA hybridization and capture were performed using the SeqCap EZ MedExome kit (Roche Diagnostics) with a global coverage of 47 Mb, including the coded regions identified in (i) the CCDS 17 RefSeq CDS (version from August 2014) available at www.ncbi.nlm.nih.gov/projects/CCDS, (ii) the Ensembl 76 CDS (filtered by biotype) available at www.ensembl.org/Homo_sapiens, (iii) the VEGA 56 CDS, (iv) the GENCODE 20 CDS (www.gencodegenes.org/human/release_20.html), and (v) the miRBase 21 (www.mirbase.org) publicly available databases. Sequencing was performed on an Illumina NovaSeq genome sequencer (Illumina, San Diego, CA, USA) with an average coverage of >130× per exome and a read length of 2×150 pb. FASTQ files containing WES data were obtained for each individually sequenced tumor and blood sample.
Identification of Genetic Variants in Meningioma Samples
The Fastp software was used to check for the quality of sequencing data and to remove the remaining adaptamer sequences (18). Then, the resulting FASTQ files (with read pairs) were aligned to the GRCh38 human reference genome using the BWA (Burrows-Wheeler Aligner) software (http://bio-bwa.sourceforge.net/version 0.7.12). BWA-MEM algorithm was run with default parameters. Aligned files were then analyzed using the GATK software (version 3.5), following the best practices manual for variant calling (19). The Picard software (https://broadinstitute.github.io/picard/) was subsequently used to sort data, and duplicated reads were marked via the samtools (20). Quality of variants was recalibrated and insertions/deletions realigned using the GATK software. The UnifiedGenotyper method (GATK software) was used for variant calling. Additional filters described below were subsequently applied to discriminate between both acquired genetic variants (i.e., somatic mutations) and germline variants vs genetic variants present in the Spanish population—i.e., single nucleotide polymorphisms (SNP). The same data analysis protocol was applied for the tumor and blood DNA sequences from our series and the control (publicly available) datasets. Resulting variant calling files (VCF) were processed with SNPEffect (21) and Annovar (22), which append variant annotation information, functional effect prediction, and alternative allele frequencies (AAF) in the ExAC (23), 1000 Genomes (24), and gnomAD (25) project databases. The Linux commands to reproduce and replicate the analyses performed in this work are available upon request to the authors.
Identification of genetic variants was first focused on genes previously found to be mutated in meningiomas: AKT1, ARID1B, BAP1, CDKN2A, CDKN2C, GNA11, KLF4, NF2, NRAS, PIK3CA, POLR2A, PTEN, SF3B1, SMARCA4, SMARCB1, SMO, TERT, TP53, and TRAF7, and subsequently, in other genes that contained more mutations than found in the control dataset of the Spanish population. Cutoff values to define genetic variants were based on previous standard criteria: only those non-synonymous coding genetic variants were initially selected, and the sequences obtained were subsequently compared with our Spanish control and gnomAD population databases to discriminate between somatic variants and single-nucleotide polymorphisms. From all variants identified, only exonic variants with AAF ≥0.20 in the genomic regions sequenced which had not been previously reported in control Spanish, African, or European (non-Finnish) populations were considered in order to avoid complex genomic regions or low-quality variants. Moreover, only genes showing variants in >2 meningioma samples were further studied to avoid small size or rarely mutated genes. Filter values were adjusted based on NF2 mutations, which were used as the gold standard positive. Finally, genes with at least five times more mutations than observed in the control dataset were manually curated to obtain a list of highly mutated genes.
For the identification of somatic genetic variants, somatic genomic mutation analyses were run up after all synonymous variants had been removed; variants with <40 reads were not considered. Six paired blood (n=3) and meningioma tumor samples (n=3) were used to confirm somatic mutations in the tumor of a subset of the patients that were missing in blood DNA. Variants detected in meningiomas at allele frequencies <0.01, match with the definition of rare variants and at least 10 reads validate the mutation, were considered to correspond to somatic mutations, while above this cutoff they could correspond to germinal mutations.
Identification of Copy Number Alterations
Frozen tumor samples obtained after surgery were also assessed in parallel by interphase fluorescence in situ hybridization (iFISH) and/or single-nucleotide polymorphism (SNP) arrays, to identify numerical (copy number) alterations for 12 chromosomal regions scattered throughout the genome, which are frequently altered in meningiomas and for the (whole) 24 distinct human chromosomes, respectively. For iFISH analyses, a panel of probes directed against DNA sequences of 12 different chromosome regions frequently altered in meningiomas (1p36/1q25, 7, 9p34, 10, 14q32.3, 15q22, 17q21, 18q21, 22q11.2, X, and Y) obtained from Vysis Inc. (Abbott Laboratories, Santa Clara, CA, USA) were used in double stainings, as previously described (3). In turn, the high-density 500K GeneChip Mapping and the Genome-Wide Human SNP 6.0 Arrays (Affymetrix, Thermo-Fisher Scientific, Waltham, MA, USA) were used to determine the (cytogenetic) CNA profile of each individual meningioma, following the manufacturer’s instructions, as described elsewhere (16). SNP data are available at the GEO public database (access code: GSE42624).
Statistical Analyses
For all statistical analyses, the SPSS software (SPSS 25.0, IBM SPSS, Armonk, NY, USA) was used. The Chi-square test and the Mann-Whitney U test were used to compare different groups of patients for categorical and continuous variables, respectively. Survival curves were plotted according to the Kaplan and Meier method, and the (two-sided) log-rank test was used to assess the statistical significance of differences in recurrence-free survival (RFS; defined from tumor diagnostic surgery to disease recurrence, by imaging techniques) between distinct groups of patients. RFS data were available in 30 meningioma patients with a median follow-up of 8 years, range 1 to 21 years, after first surgery.
Results
Genetic Profile of WHO Grade 1 Meningiomas
NF2 (15/32 tumors, 47%) gene was the most frequently showed non-synonymous and/or synonymous genetic variants among WHO grade 1 meningiomas (Table 1 and Supplementary Table 1). Other genes that recurrently carried non-synonymous genetic variants at lower frequencies included the PNMA6A (7/32 tumors, 22%), TIGD1 (5/32, 16%), SMO (4/32, 13%), PTEN (4/32, 13%), CREG2 (3/32, 9%), EEF1A1 (2/32, 6%), and POLR2A (2/32, 6%) genes; in turn, the ARID1B and FAIM3 genes were found to be mutated in a single tumor each (1/32, 3%) (Table 1). From these later nine genes, TIGD1 was the only gene for which synonymous genetic variants were also detected (in addition to non-synonymous variants) in 5/32 tumors (16%). Out of all recurrent non-synonymous genetic variants identified in our patients, only a subset of them involving the ARID1B, NF2, POLR2A, PTEN, SMARCA4, and SMO genes had been previously reported in sporadic meningiomas (Supplementary Table 2).
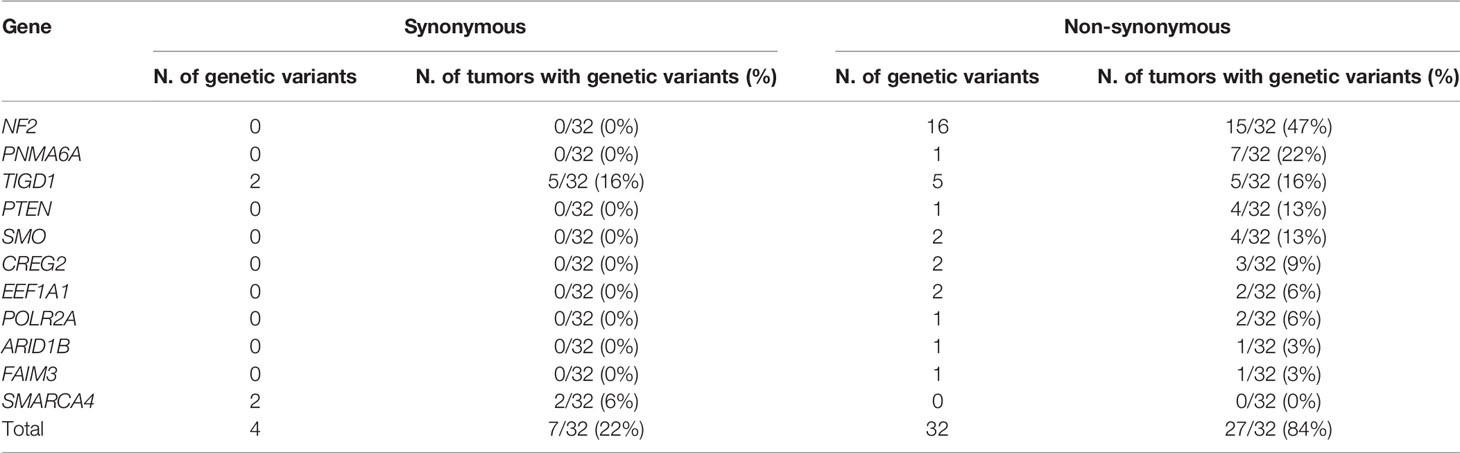
Table 1 Synonymous and non-synonymous genetic variants detected in WHO grade 1 meningiomas (n = 32) ordered by the frequency of tumors with non-synonymous genetic variants identified.
In more detail, a total of 16 different non-synonymous variants of the NF2 gene for which no synonymous variants were found, were identified in 15/32 tumors (47%), one of which displayed two different variants (Table 1 and Figure 1). Of note, all 16 genetic variants of the NF2 gene identified affected the FERM domain of the NF2-coded merlin protein with potential (predicted) impact at the functional protein level (Supplementary Figure 1).
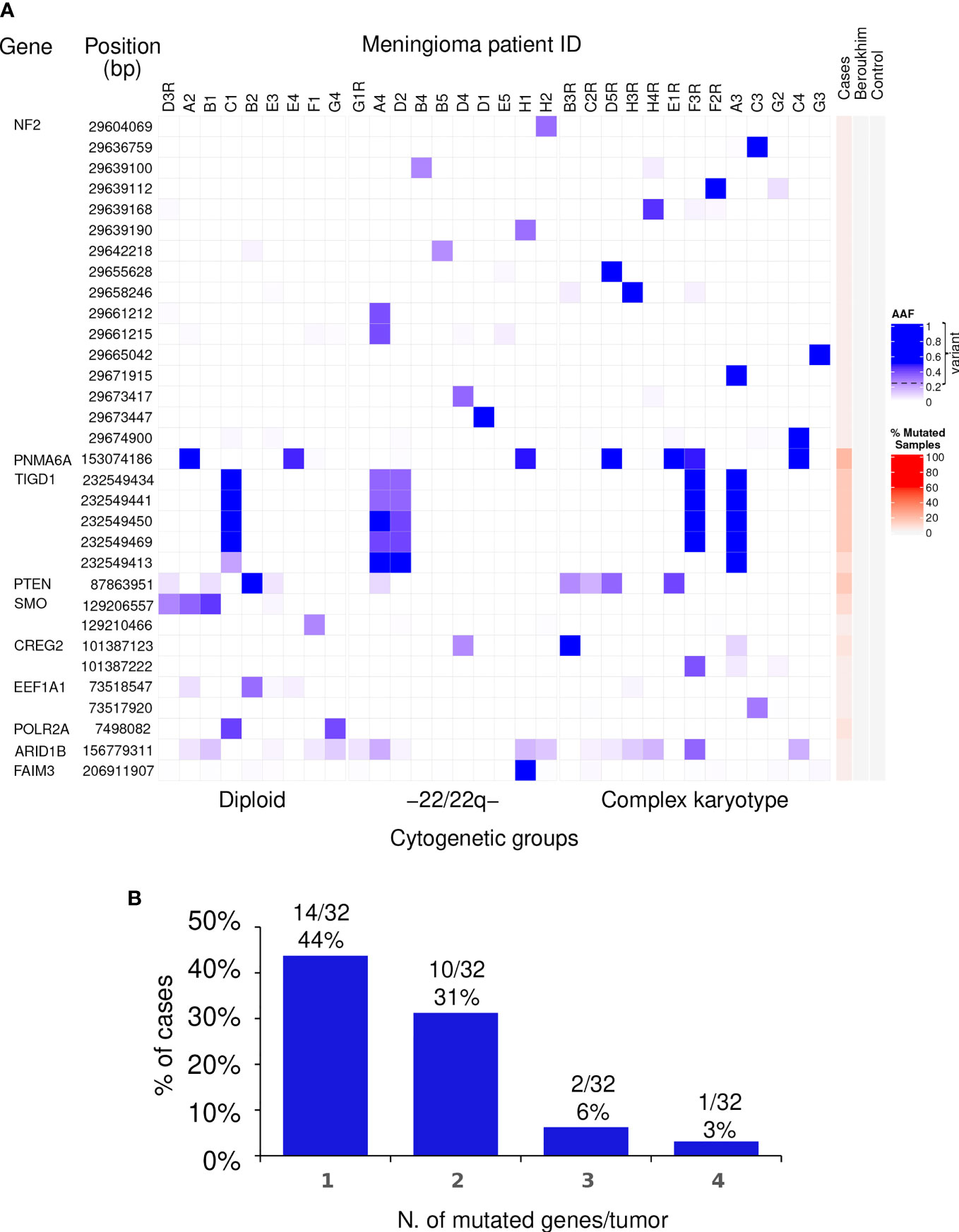
Figure 1 Distribution of non-synonymous genetic variants found altered in WHO grade 1 meningiomas. (A) Heat map of the non-synonymous genetic variants identified (rows) in individual WHO grade 1 meningiomas (columns) classified according to their chromosomal copy number cytogenetic profile. For each genetic variant, alternative allele frequencies (AAF) are color coded in blue from the lowest (light blue) to the highest (dark blue) percent values using the 0.25 AAF value as the cutoff for variant detection. Patients that showed tumor recurrence are identified on top of each column by the patient ID code followed or not by the letter “R” for the recurrent vs non-recurrent tumors. The red color scale represents the overall percentage of mutated samples among the WHO grade 1 meningiomas studied in our cohort as well in the controls. (B) The number and frequency of tumors carrying non-synonymous genetic variants in different (numbers of) genes are displayed.
In contrast to NF2, recurrent non-synonymous genetic variants were observed for a total of six other genes, i.e., PNMA6A, TIGD1, PTEN, SMO, CREG2, and POLR2A, with the following distribution (Figure 1A): (i) a single recurrent non-synonymous genetic variant of the PNMA6A gene at 153074186bp was identified in 7/32 (22%) meningiomas; (ii) the same four TIGD1 variants were identified in 5/32 (16%) meningiomas at 232549434bp, 232549441bp, 232549450bp, and 232549469bp, four of these five tumors showing an additional genetic variant of this gene located at 232549413bp, all five non-synonymous genetic variants of the TIGD1 gene being located along a relatively short region of 36bp, from the 232549413bp to the 232549469bp position (Figure 1A); (iii) a single non-synonymous variant of PTEN at the 87863951bp position was identified in 4/32 tumors (13%); (iv) one non-synonymous genetic variant of the SMO gene at 129206557bp was detected in 3/32 (9%) patients; (v) one CREG2 variant at 101387123bp was identified in another 2/32 tumors (6%); and finally, (vi) one POLR2A variant at 7498082bp was detected in 2/32 (6%) meningiomas (Figure 1A). Overall, non-synonymous genetic variants in ≥1 gene were found in 27/32 WHO grade 1 meningiomas, the great majority (24/32; 75%) of them showing one or two altered genes, three in two tumors (6%), and ≥4 genes were found to carry non-synonymous genetic variants in one patient (3%) (Figure 1B).
In a subset of 3/32 patients, mutational analyses were performed in paired blood and primary tumor samples. One genetic variant involving the PNMA6A gene was present in 1/3 meningiomas investigated. In turn, non-synonymous variants of the NF2, POLR2A, and PTEN genes were identified in tumor samples from 2/3, 1/3, and 1/3 patients investigated, respectively, while they were systematically absent in blood DNA (Supplementary Table 3), supporting the specific acquisition of these genetic variants in the tumor cells of these patients.
Frequency of Non-Synonymous Genetic Variants in Different Cytogenetic Subgroups of WHO Grade 1 Meningiomas
From the cytogenetic point of view, our 32 WHO grade 1 meningiomas were divided into three groups based on their chromosome copy number status by iFISH and/or SNP-arrays: (i) diploid tumors (meningiomas that showed no chromosome losses and/or gains), 9 cases (28%); (ii) meningiomas with an isolated loss of chromosome 22/del(22q), 10 tumors (31%); and, (iii) meningiomas with complex karyotypes consisting of losses and/or gains involving ≥2 chromosomes/chromosome regions, 13 cases (41%) (Table 2). Overall, the mean number of altered genes and/or the mean ( ± 1SD) number of non-synonymous genetic variants per tumor slightly increased (p>0.05) from diploid tumors (1.2 ± 0.7 altered genes/tumor and 1.7 ± 1.5 variants/tumor) and meningiomas carrying isolated monosomy 22/22q─ (1.2 ± 0.9 altered genes/tumor and 2.1 ± 2.3 genetic variants/tumor), to cases with complex karyotypes (1.6 ± 1.0 altered genes/tumor and 2.1 ± 1.9 genetic variants/tumor) (Table 2). In turn, the frequency of non-synonymous genetic variants that affected individual genes in these three cytogenetic subgroups of meningiomas revealed different mutational profiles among them for the NF2, SMO, and POLR2A genes (Table 2). Thus, NF2 mutations were restricted to the majority of tumors displaying monosomy 22/22q─ (70%) and complex karyotypes (62%)—all such cases with complex karyotypes plus NF2 mutation including monosomy 22/22q─ —while absent (0%) in diploid meningiomas (p= 0.001) (Table 2); in contrast, SMO and POLR2A genetic variants were restricted to diploid meningiomas—44 and 22% cases vs 0% among tumors with isolated monosomy 22/22q─ and complex karyotypes (p=0.001 and p=0.02, respectively)—and PTEN variants were exclusively found in meningiomas that had complex and diploid karyotypes (23 and 11%, respectively), while absent in tumors with an isolated monosomy 22/22q─ (p=0.15) (Table 2).

Table 2 Frequency of non-synonymous genetic variants in different cytogenetic subtypes of WHO grade 1 meningiomas as defined by their chromosome copy number profile.
Non-Synonymous Genetic Variants in Recurrent vs Non-Recurrent WHO Grade 1 Meningiomas
At the moment of closing this study, 10/32 WHO grade 1 tumors had shown recurrence of the disease. These included 1/9 (11%) diploid meningiomas, 1/10 (10%) tumors carrying an isolated monosomy 22/22q─, and 8/13 (62%) meningiomas with complex karyotypes (p=0.009). Overall, a high frequency of tumors carrying non-synonymous genetic variants of the PTEN gene (all located at the 87863951pb chromosomal region) were found in recurrent vs non-recurrent tumors (30 vs 5%, p=0.04) (Table 3) in association with a poorer outcome (p=0.002)—recurrence-free survival (RFS) rate at 10 years of 85% (95% confidence interval: 68–100%) for PTEN mutated patients vs not reached for PTEN wild-type tumors—(Figure 2A). In contrast, no significantly different (p>0.05) frequencies of genetic variants involving the other altered NF2, PNMA6A, CREG2, ARID1B, NF2, TIGD1, SMO, EEF1A1, POLR2A, and FAIM3 genes were observed in recurrent vs non-recurrent tumors (Table 3). In addition to the PTEN gene status, the overall cytogenetic profile of WHO grade 1 meningiomas also showed an impact (p=0.018) on RFS (Figure 2B). Based on the cytogenetic profile (diploid vs isolated monosomy 22/22q─ vs complex karyotype) and the presence vs absence of NF2 gene mutations, four distinct genetic subgroups were defined, which could be pooled in two groups with significantly different RFS rates at 10 years (p=0.003): (i) a good-prognosis group (n=16) with median ( ± 95% confidence interval) RFS at 10 y of 100% (100%) consisting of diploid tumors (n=9) and meningiomas with isolated monosomy 22/22q─ associated with NF2 mutation (n=7), and (ii) poor-prognosis meningiomas (n=14) with isolated monosomy 22/22q─ in the absence of NF2 mutation (n=3) and complex karyotypes (n=11), with median ±95% confidence interval RFS at 10 y of 48% (19–77%) (Figure 2C).
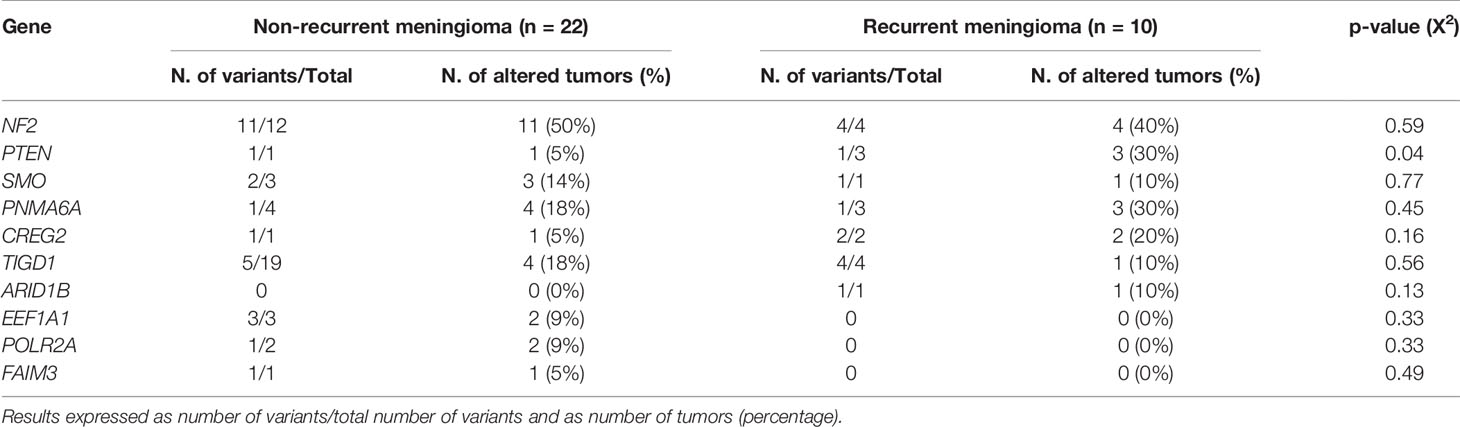
Table 3 Distribution of the non-synonymous genetic variants identified in non-recurrent vs recurrent WHO grade 1 meningiomas.
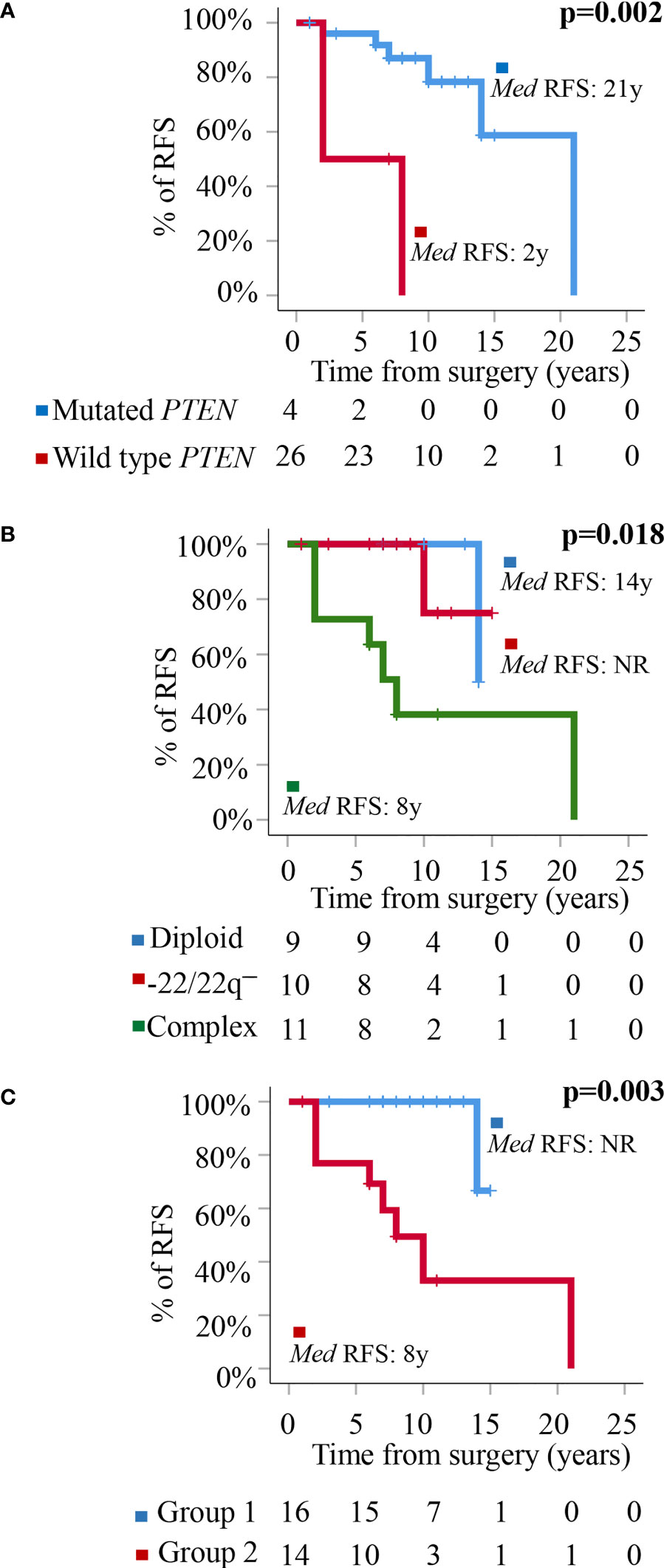
Figure 2 Prognostic impact of PTEN mutation and the cytogenetic and molecular tumor profiles in the outcome. (A) Prognostic impact of the presence of genetic variants of PTEN on patient relapse-free survival (RFS). (B) Prognostic impact of the cytogenetic profile of WHO grade 1 meningiomas on patient RFS. (C) Prognostic impact of the overall cytogenetic and mutational profile on RFS of WHO grade 1 meningiomas distributed into a good-prognosis group (Group 1: meningiomas with a diploid karyotype and isolated monosomy 22/22q─ associated with NF2 mutation) (n = 16) and a poor-prognosis group (Group 2: meningiomas with isolated monosomy 22/22q─ in the absence of NF2 mutations and tumors with a complex karyotype associated or not with mutation of the NF2 gene) (n = 14).
Discussion
Previous studies have shown multiple genes and chromosomes to be recurrently altered in meningiomas (26). Among all altered genes, NF2 is by far the most frequently mutated (0–46%) in the literature (4, 6, 8, 9, 12, 15, 27–29), followed by the TRAF7 (2–38%) (4, 12, 13, 30), AKT1 (8–31%) (6, 7, 12, 13, 15, 31, 32), KLF4 (9–15%; 100% of secretory meningiomas) (12, 13, 30, 32), SMO (6–32%) (7, 10, 12, 15), TERT (2–14%) (10, 33–38), and PIK3CA (0–7%) (10, 12, 39) genes, and other less studied genes, such as the ARID1B, POLR2A, and PTEN genes found to be mutated in 22, 6, and 1% of meningiomas, respectively (4, 5, 40). Typically, all above genes are recurrently altered in meningiomas across all WHO grades, despite controversial results have been reported about a greater frequency of the NF2, KLF4, and SMO gene variants in WHO grade 2/3 vs WHO grade 1 meningiomas (12, 15).
Despite all the above, so far, only a few studies (4, 11, 12) have applied NGS to investigate the presence of genetic variants across the whole exome/genome of meningiomas, usually in a series of patients that also included other CNS tumors (e.g., gliomas) (41) and/or that were preferentially focused on WHO grade 2/3 meningiomas (11). Thus, only a limited number of studies specifically included WES/WGS data on WHO grade 1 meningiomas for a total of 76 WHO grade 1 tumors with available data reported in the literature (n=17, 20, and 39) (11, 12, 15). Here we used WES to search for those genes more frequently altered in WHO grade 1 meningiomas and investigate the potential association between their alterations and both tumor cytogenetics and patient outcome.
Overall, WES revealed recurrent but heterogeneous mutational profiles in WHO grade 1 meningiomas with up to 10 genes showing non-synonymous genetic variants in at least one tumor, some of which were closely associated with the cytogenetic (i.e., chromosomal) tumor profiles. Thus, from those 9/10 genes found to be altered in ≥2 grade 1 meningiomas, the NF2 gene was the most frequently carried non-synonymous genetic variants, in combination or not with other synonymous genetic variants, respectively. Another four genes (PNMA6A, TIGD1, PTEN, and SMO) were found to be altered in >10% of all WHO grade 1 meningiomas investigated, while the remaining CREG2, EEF1A1, and POLR2A genes were recurrently altered in a minority (<10%) of cases. Of note, paired analysis of blood and meningioma tissue samples: in a subset of three patients, both samples were available, revealing the presence of high-alternative allele frequencies (range: 96 to 98%) in both tissues for the PNMA6A non-synonymous gene variants identified, suggesting they might correspond to germinal variants of this gene. In contrast, all genetic variants of the NF2, POLR2A, and PTEN genes found in these three patients in which paired tumor and blood DNA were studied, and were absent in blood DNA and restricted to the tumor cell DNA where they were detected at frequencies ≤40%—except for NF2 tumors with -22/del(22q), suggesting these later variants would correspond to tumor-specific non-synonymous genetic variants, e.g., somatic mutations. However, further studies in which paired blood and tumor samples from larger series of meningioma patients are studied are necessary to confirm these preliminary observations. Overall, these results confirm the high frequency of genetic variants of the NF2 gene and to a less extent also of the PTEN, SMO, and POLR2A genes, in WHO grade 1 meningiomas. At the same time we report here for the first time a relatively high frequency of genetic variants of the PNMA6A and TIGD1 genes, which were absent in other series of healthy subjects and patients investigated in our laboratory (42–44). In contrast, we could not find in our WHO grade 1 tumors and control populations, non-synonymous genetic variants, for other genes previously reported to be recurrently mutated in meningiomas such as the TRAF7, AKT1, KLF4, TERT, and PIK3CA genes (10, 12, 39). Further investigations are thereby required to better understand the reason for these differences and the potential role of these later genes in meningioma.
Up to 80 different NF2 mutations have been previously reported in meningiomas (4, 6, 8, 9, 11, 12, 15, 26–29). In line with these findings, 16 different non-recurrent non-synonymous variants of the NF2 gene coded at chromosome 22q12.2 were observed in almost half of our WHO grade 1 meningiomas, in close association with monosomy 22/22q─, either as an isolated chromosomal alteration or in the context of a complex karyotype. Of note, only a quarter of all NF2 variants found (genomic position and bp change: 29604069, c.71_72insA; 29639100, c.A251C; 29642218, c.T380G; 29661212, c.683delG) had been previously identified in meningiomas (5, 12, 27), while the other 12 are reported here for the first time. Lack of recurrent NF2 mutations among our WHO grade 1 meningiomas, as well as in most series reported in the literature, suggests that sporadic meningioma cells frequently acquire NF2 mutations randomly (11), in both WHO grade 1 and higher-grade tumors (9, 12). Despite such high variability on the specific NF2 mutations found among our WHO grade1 meningiomas, in silico analysis revealed a common effect at the merlin protein level. Thus, all 16 genetic variants identified would target the FERM functional domain of the NF2-coded merlin protein. These results support the notion that despite NF2 mutations are heterogenous, they all lead to inactivation of the merlin protein in the absence of wild-type NF2, and support a relevant role for the NF2 gene in the oncogenesis of a significant fraction (≈50%) of WHO grade 1 meningiomas (11). In line with this hypothesis and previous literature data (9, 11), we found a close association between NF2 mutation and monosomy 22/22q─, as all NF2-mutated tumors also carried monosomy 22/22q -, in association or not with a complex karyotype. In contrast with previous studies, mutations in genes other than NF2, which are coded in the vicinity of the NF2 gene at chromosome 22q (e.g., the SMARCB1 gene), were not detected in our patients, suggesting that mutations in these genes (12) are absent or rare (≤5%) in WHO grade 1 meningiomas, and thereby, they would play a limited role in the pathogenesis of WHO grade 1 meningiomas, in contrast with NF2 gene mutations.
Other genes that were recurrently mutated in our series included the PNMA6A, TIGD1, SMO, PTEN, CREG2, EEF1A1, and POLR2A genes. Interestingly, some of these genes (SMO and POLR2A) were restricted to NF2-wild-type diploid tumors, or they (PTEN) predominated in NF2-wild-type cases. In turn, recurrent mutations in the TIGD1 gene were found across all cytogenetic subgroups of meningiomas including both NF2-mutated and wild-type tumors. These results suggest that some of these later altered genes (e.g., SMO, POLR2A) might act as driver genes in a significant fraction of NF2-wild-type WHO grade 1 meningiomas (12, 15, 39), further studies being required to confirm this hypothesis.
When individually considered, only PTEN mutations together with the cytogenetic tumor profile showed a significant impact on patient outcome (i.e., RFS). These results confirm previous data from our group (3) indicating that complex karyotypes within WHO grade 1 meningiomas are associated with an increased risk of relapse. In turn, here we show for the first time an adverse prognostic impact for PTEN mutation, although most PTEN-mutated cases also carried a complex karyotype. In line with these findings, some authors have recently reported that the PTEN gene might play a critical role in regulating chromosome segregation to prevent gross genomic instability via regulation of the mitotic arrest deficient 2 (MAD2) gene (45); these finding might contribute to explain the association here reported between mutated PTEN and both a complex karyotype and a poorer outcome. Of note, this is the first study in which an association between PTEN gene mutations and a poor outcome is reported among WHO grade 1 meningiomas, similarly to what has been reported in tumor types other than meningioma, including glioma (46). However, these findings are in contrast with previous studies that found a similar frequency of PTEN mutations in meningiomas to the one here reported, but no association with RFS (47). Most interestingly, when we simultaneously considered the tumor cytogenetic patterns and the NF2 mutation profile, four distinct genetic profiles were identified, which could be grouped into two good-prognosis genetic profiles (diploid karyotype and isolated monosomy 22/22q─ associated with NF2 mutation), and two poor-prognosis genetic patterns (isolated monosomy 22/22q─ in the absence of NF2 mutation and complex karyotypes) with significantly different RFS rates. Altogether, these results suggest that presence of NF2 mutation in the context of an isolated monosomy 22/22q─ confer a low risk of recurrence for WHO grade 1 meningiomas, while complex karyotypes and isolated loss of chromosome 22 in the absence of NF2 mutation would confer a poorer prognosis. So far, few studies have combined cytogenetic and molecular data for risk-stratification of WHO grade 1 and also grade 2/3 meningiomas. Therefore, our preliminary results on the prognostic impact of the above described molecular/genetic profiles still need to be confirmed in larger prospective series of WHO grade 1 meningiomas followed for long periods of time (11).
In summary, our results based on WES of WHO grade 1 meningiomas reveal recurrent but heterogeneous mutational and cytogenetic profiles, which might reflect different oncogenic pathways associated with distinct patterns of chromosomal instability and patient outcome. Further studies in larger series of both WHO grade 1 and grade 2/3 meningiomas are required to confirm these observations.
Data Availability Statement
The datasets presented in this study can be found in online repositories. Genomic data analyzed in this study is available at EBI ENA under the PRJEB43143 project identifier.
Ethics Statement
The studies involving human participants were reviewed and approved by the local ethics committee of the University Hospital of Salamanca (Salamanca, Spain). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All authors have contributed significantly to this article: AOr and MT contributed to the study design. DA, AOt, DP, LR, IÁ-T, and AG-M contributed to the collection of samples and clinical data. M-GT, CP, MJ-A, and MT developed the methodology and analysis. Finally, MG-T, CP, AOr, and MT contributed to the original draft preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants: IBY 17/00002 from IBSAL (Salamanca, Spain), GRS2132/A2020 from Junta de Castilla y León (Spain), and CB16/12/00400 from CIBER-ONC and FONDOS FEDER (Instituto de Salud Carlos III, Ministerio de Economía y Competitividad, Madrid, Spain).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We want to acknowledge the REMA (Spanish Network on Mastocytosis), the Spanish National DNA Bank Carlos III (University of Salamanca and Plataforma de Biobancos, Instituto de Salud Carlos III, Madrid, Spain) for their collaboration in this study, and both the Sequencing and Bioinformatics Service of NUCLEUS (University of Salamanca, Salamanca, Spain) for their support in WES and data analysis, respectively.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.740782/full#supplementary-material
Abbreviations
AAF, alternative allele frequencies; Bp, pair base; CAN, copy number alterations; Del, deletion; FERM, Four-point-one, Ezrin, Radixin, Moesin; iFISH, fluorescence in situ hybridization; gnomAD, Genome Aggregation Database; NGS, next-generation sequencing; NRAS, NRAS Proto-Oncogene; GTPase; REMA, Spanish Network on Mastocytosis; SNP, nucleotide polymorphism; RFS, recurrence-free survival; WES, whole exome sequencing; WGS, whole genome sequencing; WHO, World Health Organization.
References
1. Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol (2019) 21:v1–v100. doi: 10.1093/neuonc/noz150
2. Linsler S, Kraemer D, Driess C, Oertel J, Kammers K, Rahnenfuhrer J, et al. Molecular Biological Determinations of Meningioma Progression and Recurrence. PloS One (2014) 9:e94987. doi: 10.1371/journal.pone.0094987
3. Domingues PH, Sousa P, Otero Á, Gonçalves JM, Ruiz L, de Oliveira C, et al. Proposal for a New Risk Stratification Classification for Meningioma Based on Patient Age, WHO Tumor Grade, Size, Localization, and Karyotype. Neuro Oncol (2014) 16:735–47. doi: 10.1093/neuonc/not325
4. Zhang X, Jia H, Lu Y, Dong C, Hou J, Wang Z, et al. Exome Sequencing on Malignant Meningiomas Identified Mutations in Neurofibromatosis Type 2 (NF2) and Meningioma 1 (MN1) Genes. Discovery Med (2014) 18:301–11.
5. Clark VE, Harmancl AS, Bai H, Youngblood MW, Lee TI, Baranoski JF, et al. Recurrent Somatic Mutations in POLR2A Define a Distinct Subset of Meningiomas. Nat Genet (2016) 48:1253–9. doi: 10.1038/ng.3651
6. Shankar GM, Abedalthagafi M, Vaubel RA, Merrill PH, Nayyar N, Gill CM, et al. Germline and Somatic BAP1 Mutations in High-Grade Rhabdoid Meningiomas. Neuro Oncol (2017) 19:535–45. doi: 10.1093/neuonc/now235
7. Boetto J, Bielle F, Sanson M, Peyre M, Kalamarides M. SMO Mutation Status Defines a Distinct and Frequent Molecular Subgroup in Olfactory Groove Meningiomas. Neuro Oncol (2017) 19:345–51. doi: 10.1093/neuonc/now276
8. Szijan I, Rochefort D, Bruder C, Surace E, Machiavelli G, Dalamon V, et al. NF2 Tumor Suppressor Gene: A Comprehensive and Efficient Detection of Somatic Mutations by Denaturing HPLC and Microarray-CGH. Neuromolecular Med (2003) 3:41–52. doi: 10.1385/NMM:3:1:41
9. Tabernero MD, Jara-Acevedo M, Nieto AB, Caballero AR, Otero A, Sousa P, et al. Association Between Mutation of the NF2 Gene and Monosomy 22 in Menopausal Women With Sporadic Meningiomas. BMC Med Genet (2013) 14:114. doi: 10.1186/1471-2350-14-114
10. Jungwirth G, Warta R, Beynon C, Sahm F, von Deimling A, Unterberg A, et al. Intraventricular Meningiomas Frequently Harbor NF2 Mutations But Lack Common Genetic Alterations in TRAF7, AKT1, SMO, KLF4, PIK3CA, and TERT. Acta Neuropathol Commun (2019) 7:140. doi: 10.1186/s40478-019-0793-4
11. Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK, et al. Genomic Landscape of High-Grade Meningiomas. NPJ Genomic Med (2017) 2:1–14. doi: 10.1038/s41525-017-0014-7
12. Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic Analysis of Non-NF2 Meningiomas Reveals Mutations in TRAF7, KLF4, AKT1, and SMO. Science (2013) 339:1077–80. doi: 10.1126/science.1233009
13. Hao S, Huang G, Feng J, Li D, Wang K, Wang L, et al. Non-NF2 Mutations Have a Key Effect on Inhibitory Immune Checkpoints and Tumor Pathogenesis in Skull Base Meningiomas. J Neurooncol (2019) 144:11–20. doi: 10.1007/s11060-019-03198-9
14. Bi WL, Abedalthagafi M, Horowitz P, Agarwalla PK, Mei Y, Aizer AA, et al. Genomic Landscape of Intracranial Meningiomas. J Neurosurg (2016) 125:525–35. doi: 10.3171/2015.6.JNS15591
15. Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, et al. Genomic Sequencing of Meningiomas Identifies Oncogenic SMO and AKT1 Mutations. Nat Genet (2013) 45:285–9. doi: 10.1038/ng.2526
16. Tabernero MD, Maíllo A, Nieto AB, Diez-Tascón C, Lara M, Sousa P, et al. Delineation of Commonly Deleted Chromosomal Regions in Meningiomas by High-Density Single Nucleotide Polymorphism Genotyping Arrays. Genes Chromosomes Cancer (2012) 51:606–17. doi: 10.1002/gcc.21948
17. Chen F, Xiang CX, Zhou Y, Ao XS, Zhou DQ, Peng P, et al. Gene Expression Profile for Predicting Survival of Patients With Meningioma. Int J Oncol (2015) 46:791–7. doi: 10.3892/ijo.2014.2779
18. Chen S, Zhou Y, Chen Y, Gu J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
19. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr Protoc Bioinforma (2013) 43:11 10 1–33. doi: 10.1002/0471250953.bi1110s43
20. Li H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation From Sequencing Data. Bioinformatics (2011) 27:2987–93. doi: 10.1093/bioinformatics/btr509
21. De Baets G, Van Durme J, Reumers J, Maurer-Stroh S, Vanhee P, Dopazo J, et al. SNPeffect 4.0: on-Line Prediction of Molecular and Structural Effects of Protein-Coding Variants. Nucleic Acids Res (2012) 40:D935–9. doi: 10.1093/nar/gkr996
22. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38:e164. doi: 10.1093/nar/gkq603
23. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
24. 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A Global Reference for Human Genetic Variation. Nature (2015) 526:68–74. doi: 10.1038/nature15393
25. Karczewski KJ, Francioli LC, Tiao G, Cummings JA, Wang Q, Collins RL, et al. The Mutational Constraint Spectrum quantified from variation in 141,456 humans. Nature (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
26. Domingues P, González-Tablas M, Otero Á, Pascual D, Ruiz L, Miranda D, et al. Genetic/molecular Alterations of Meningiomas and the Signaling Pathways Targeted. Oncotarget (2015) 6:10671–88. doi: 10.18632/oncotarget.3870
27. Hansson CM, Buckley PG, Grigelioniene G, Piotrowski A, Hellstrom AR, Mantripragada K, et al. Comprehensive Genetic and Epigenetic Analysis of Sporadic Meningioma for Macro-Mutations on 22q and Micro-Mutations Within the NF2 Locus. BMC Genomics (2007) 8:16. doi: 10.1186/1471-2164-8-16
28. Kim JH, Kim IS, Kwon SY, Jang BC, Il SS, DH S, et al. Mutational Analysis of the NF2 Gene in Sporadic Meningiomas by Denaturing High-Performance Liquid Chromatography. Int J Mol Med (2006) 18:27–32. doi: 10.3892/ijmm.18.1.27
29. Hartmann C, Sieberns J, Gehlhaar C, Simon M, Paulus W, von Deimling A. NF2 Mutations in Secretory and Other Rare Variants of Meningiomas. Brain Pathol (2006) 16:15–9. doi: 10.1111/j.1750-3639.2006.tb00557.x
30. Reuss DE, Piro RM, Jones DT, Simon M, Ketter R, Kool M, et al. Secretory Meningiomas Are Defined by Combined KLF4 K409Q and TRAF7 Mutations. Acta Neuropathol (2013) 125:351–8. doi: 10.1007/s00401-013-1093-x
31. Sahm F, Bissel J, Koelsche C, Schweizer L, Capper D, Reuss D, et al. AKT1E17K Mutations Cluster With Meningothelial and Transitional Meningiomas and can be Detected by SFRP1 Immunohistochemistry. Acta Neuropathol (2013) 126:757–62. doi: 10.1007/s00401-013-1187-5
32. Yesiloz U, Kirches E, Hartmann C, Scholz J, Kropf S, Sahm F, et al. Frequent AKT1E17K Mutations in Skull Base Meningiomas Are Associated With mTOR and ERK1/2 Activation and Reduced Time to Tumor Recurrence. Neuro Oncol (2017) 19:1088–96. doi: 10.1093/neuonc/nox018
33. Goutagny S, Nault JC, Mallet M, Henin D, Rossi JZ, Kalamarides M. High Incidence of Activating TERT Promoter Mutations in Meningiomas Undergoing Malignant Progression. Brain Pathol (2014) 24:184–9. doi: 10.1111/bpa.12110
34. Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J Natl Cancer Inst (2016) 108:1–4. doi: 10.1093/jnci/djv377
35. Peyre M, Gauchotte G, Giry M, Froehlich S, Pallud J, Graillon T, et al. De Novo and Secondary Anaplastic Meningiomas: A Study of Clinical and Histomolecular Prognostic Factors. Neuro Oncol (2018) 20:1113–21. doi: 10.1093/neuonc/nox231
36. Biczok A, Kraus T, Suchorska B, Terpolilli NA, Thorsteinsdottir J, Giese A, et al. TERT Promoter Mutation is Associated With Worse Prognosis in WHO Grade II and III Meningiomas. J Neurooncol (2018) 139:671–8. doi: 10.1007/s11060-018-2912-7
37. Spiegl-Kreinecker S, Lotsch D, Neumayer K, Kastler L, Gojo J, Pirker C, et al. TERT Promoter Mutations Are Associated With Poor Prognosis and Cell Immortalization in Meningioma. Neuro Oncol (2018) 20:1584–93. doi: 10.1093/neuonc/noy104
38. Lu VM, Goyal A, Lee A, Jentoft M, Quinones-Hinojosa A, Chaichana KL. The Prognostic Significance of TERT Promoter Mutations in Meningioma: A Systematic Review and Meta-Analysis. J Neurooncol (2019) 142:1–10. doi: 10.1007/s11060-018-03067-x
39. Abedalthagafi M, Bi WL, Aizer AA, Merrill PH, Brewster R, Agarwalla PK, et al. Oncogenic PI3K Mutations Are as Common as AKT1 and SMO Mutations in Meningioma. Neuro Oncol (2016) 18:649–55. doi: 10.1093/neuonc/nov316
40. Peters N, Wellenreuther R, Rollbrocker B, Hayashi Y, Meyer-Puttlitz B, Duerr EM, et al. Analysis of the PTEN Gene in Human Meningiomas. Neuropathol Appl Neurobiol (1998) 24:3–8. doi: 10.1046/j.1365-2990.1998.00093.x
41. Lorenz J, Rothhammer-Hampl T, Zoubaa S, Bumes E, Pukrop T, Kölbl O, et al. A Comprehensive DNA Panel Next Generation Sequencing Approach Supporting Diagnostics and Therapy Prediction in Neurooncology. Acta Neuropathol Commun (2020) 8:124. doi: 10.1186/s40478-020-01000-w
42. Muñoz-González JI, Álvarez-Twose I, Jara-Acevedo M, Henriques A, Viñas E, Prieto C, et al. Frequency and Prognostic Impact of KIT and Other Genetic Variants in Indolent Systemic Mastocytosis. Blood (2019) 134:456–68. doi: 10.1182/blood.2018886507
43. Muñoz-González JI, Jara-Acevedo M, Alvarez-Twose I, Merker JD, Teodosio C, Hou Y, et al. Impact of Somatic and Germline Mutations on the Outcome of Systemic Mastocytosis. Blood Adv (2018) 2:2814–28. doi: 10.1182/bloodadvances.2018020628
44. Díaz-Casado E, Gómez-Nieto R, de Pereda JM, Muñoz LJ, Jara-Acevedo M, López DE. Analysis of Gene Variants in the GASH/Sal Model of Epilepsy. PloS One (2020) 15:e0229953. doi: 10.1371/journal.pone.0229953
45. Sun Z, Lu J, Wu M, Li M, Bai L, Shi Z, et al. Deficiency of PTEN Leads to Aberrant Chromosome Segregation Through Downregulation of MAD2. Mol Med Rep (2019) 20(5):4235–43. doi: 10.3892/mmr.2019.10668
46. Paganin M, Grillo MF, Silvestri D, Scapinello G, Buldini B, Cazzaniga G, et al. The Presence of Mutated and Deleted PTEN Is Associated With an Increased Risk of Relapse in Childhood T Cell Acute Lymphoblastic Leukaemia Treated With AIEOP-BFM ALL Protocols. Br J Haematol (2018) 182:705–11. doi: 10.1111/bjh.15449
Keywords: mutational profiles, whole exome sequencing (WES), NF2, PTEN, cytogenetics, WHO grade 1 meningioma
Citation: González-Tablas M, Prieto C, Arandia D, Jara-Acevedo M, Otero Á, Pascual D, Ruíz L, Álvarez-Twose I, García-Montero AC, Orfao A and Tabernero MD (2021) Whole-Exome Sequencing Reveals Recurrent but Heterogeneous Mutational Profiles in Sporadic WHO Grade 1 Meningiomas. Front. Oncol. 11:740782. doi: 10.3389/fonc.2021.740782
Received: 13 July 2021; Accepted: 27 October 2021;
Published: 17 November 2021.
Edited by:
Rengyun Liu, The First Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Hidetoshi Kasuya, Tokyo Women’s Medical University Medical Center East, JapanAnne Barlier, Aix Marseille Université, France
Copyright © 2021 González-Tablas, Prieto, Arandia, Jara-Acevedo, Otero, Pascual, Ruíz, Álvarez-Twose, García-Montero, Orfao and Tabernero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Dolores Tabernero, dGFiZXJuZXJAdXNhbC5lcw==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship