 Zhonghua Tao1,2†
Zhonghua Tao1,2† Jianxia Liu3†Ting Li1,2†Hong Xu4Kai Chen4
Jianxia Liu3†Ting Li1,2†Hong Xu4Kai Chen4 Jian Zhang1,2Hao Zhou3
Jian Zhang1,2Hao Zhou3 Jie Sun3
Jie Sun3 Jinming Han5Zhaoji Guo3Hua Yang6
Jinming Han5Zhaoji Guo3Hua Yang6 Wen-Ming Cao7*
Wen-Ming Cao7* Xichun Hu1,2*
Xichun Hu1,2*- 1Department of Medical Oncology, Fudan University Shanghai Cancer Center, Shanghai, China
- 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China
- 3Department of General Surgery, The First Affiliated Hospital of Soochow University, Suzhou, China
- 4Department of Oncology, The First Affiliated Hospital of Soochow University, Suzhou, China
- 5Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 6Department of Medical Oncology, Affiliated Hospital of Hebei University, Baoding, China
- 7Department of Breast Medical Oncology, Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou, China
Background: Receptor tyrosine kinases (RTKs) are a class of tyrosine kinases that regulate cell-to-cell communication and control a variety of complex biological functions. Dysregulation of RTK signaling partly due to chromosomal rearrangements leads to novel tyrosine kinase fusion oncoproteins that are possibly driver alterations to cancers. Targeting some RTK fusions with specific tyrosine kinases inhibitors (TKIs) is an effective therapeutic strategy across a spectrum of RTK fusion-related cancers. However, there is still a paucity of extensive RTK fusion investigations in breast cancer. This study aims to characterize RTK fusions in Chinese breast cancer patients.
Methods: An in-house DNA sequencing database of 1440 Chinese breast cancer patients with a capture-based panel (520 gene or 108 gene-panel) was thoroughly reviewed. A total of 2,229 samples including 1,045 tissues and 1,184 plasmas were analyzed. RTK fusion was defined as an in-frame fusion with the tyrosine kinase domain of the RTK completely retained. Concomitant mutations were also analyzed and tumor mutational burden (TMB) was calculated. Patients’ clinical characteristics were retrieved from case records.
Results: A total of 30 RTK fusion events were identified from 27 breast cancer patients with a prevalence of 1.875%%. FGFR2 fusions were seen the most commonly (n=7), followed by RET (n=5), ROS1 (n=3), NTRK3 (n=3), BRAF (n=2), and NTRK1 (n=2). Other RTK fusions including ALK, EGFR, FGFR1, FGFR3, MET, and NTRK2 were identified in one patient each. A total of 27 unique resultant fusion proteins (22 with a novel partner) were discovered including 19 intrachromosomal rearrangements and 8 interchromosomal ones. Twenty-one fusions had the tyrosine kinase domain in-frame fused with a partner gene and six were juxtaposed with an intergenic space. Among the 27 fusions, FGFR2-WDR11 (E17: intergenic) (n=3) and ETV6-NTRK3 (E5:E15) (n=2) occurred recurrently. Of note, the normalized abundance of RTK fusion (fusion AF/max AF) correlated negatively with TMB (r=-0.48, P=0.017). Patients with TMB < 8 (Mutations/Mb) displayed a higher fusion abundance than those with TMB ≥ 8 (Mutations/Mb) (P=0.025). Moreover, CREBBP mutation only co-occurred with FGFR2 fusion (P=0.012), while NTRK3 fusion and TP53 mutation were mutually exclusive (P=0.019).
Conclusion: This is the first study comprehensively delineating the prevalence and spectrum of RTK fusions in Chinese breast cancers. Further study is ongoing to identify the enriched subpopulation who may benefit from RTK fusion inhibitors.
Introduction
Receptor tyrosine kinases (RTKs) are a subclass of tyrosine kinases that share a similar protein structure comprised of an extracellular ligand-binding domain, a transmembrane helix, and a tyrosine kinase domain (TKD)-included intracellular region (1). Approximately 58 RTK genes grouped into 20 subfamilies have been found in the human genome (2). RTKs regulate cell-to-cell communication and control a variety of complex biological functions, such as cell growth, differentiation, and metabolism (3). The RTK activity is tightly regulated under normal physiologic conditions. Dysregulation of RTK signaling leads to a number of human diseases, most notably, cancers. Constitutive activation of RTK can be caused by gain-of-function mutations, genomic amplification, or chromosomal rearrangements (4). It may confer oncogenic properties on normal cells therefore trigger RTK-induced tumorigenesis.
As one of the mechanisms mediating abnormal RTK activation in cancers, chromosomal rearrangements can result in the formation of novel tyrosine kinase fusion oncoproteins that are often therapeutically targetable with small molecule inhibitors. It has been proven that inhibiting RTK fusions with specific tyrosine kinases inhibitors (TKIs) is an effective therapeutic strategy across a spectrum of RTK fusion-driven cancers. These targetable RTK rearrangements consist of ALK fusion in lung cancer (5) and anaplastic large cell lymphoma (6), ROS1 fusion in lung cancer (7) and glioblastoma (8), RET fusion in lung (9) and thyroid cancer (10), FGFR fusion in bile duct (11) and urothelial carcinoma (12), as well as NTRK fusion in pan-cancer (13).
As a highly heterogeneous disease, breast cancer comprises distinct molecular subtypes with varied clinical outcomes (14). Patients with advanced breast cancers that are negative for both estrogen-receptor and human epidermal growth factor receptor 2 (ER-/HER2-) have very limited therapeutic options. On the other hand, although endocrine therapy and HER2-targeted therapy have achieved great success in treating ER+ or HER2+ breast cancers, approximately 50% of the advanced cases develop resistance to these treatments (15–17). Therefore, exploring RTK fusions in breast cancer may drive the discovery of novel therapy that will bring these refractory patients more treatment opportunities. Despite the research in RTK fusions has driven the approval of relevant targeted therapies in a variety of cancer types, similar investigations remain limited in breast cancer. ETV6-NTRK3 fusion, initially described in congenital fibrosarcoma and mesoblastic nephroma, was identified as a primary oncogenic event in human secretory breast carcinoma by RT-PCR and fluorescence in situ hybridization (FISH) decades ago (18, 19). Wu et al. focused on FGFR gene fusions in diverse cancers and identified FGFR2-AFF3, FGFR2-CASP7, FGFR2-CCDC6 and ERLIN2-FGFR1 in breast cancer (20). Paratala and colleagues profiled RET fusions in breast cancer and identified CCDC6-RET, NCOA4-RET and RASGEF1A-RET (21). Of note, there is still a paucity of data comprehensively characterizing RTK gene fusions in this disease so far, especially in the Eastern Asian population.
In this study, we aim to delineate potentially targetable RTK fusions in Chinese breast cancer patients and to explore their associations with clinical and other genetic characteristics.
Materials and Methods
Patients’ Information and Study Design
An in-house DNA-based next-generation sequencing (NGS) database of 1440 Chinese breast cancer patients was retrospectively reviewed for RTK gene fusions. The median age of the 1440 patients was 50 years. 11% of them had metastatic disease, 32.2% were at early stage, and 56.8% had clinical stage unavailable. All recruited patients had their tissue or plasma samples somatic mutation profiled for genetic testing and treatment selection from 2016 to 2020 by capture-based sequencing using a 520 gene-panel (n=1,014) or a 108 gene-panel (n=426) in a Clinical Laboratory Improvement Amendments (CLIA)/CAP-certified laboratory (Burning Rock Biotech, Guangzhou, China). A total of 684 patients had multiple samples sequenced and 2,229 samples including 1,045 tissues and 1,184 plasmas were analyzed.The sequencing depth was >1000X for tissues and >10000X for plasmas. The 20% mean depth coverage was >95%. RTK genes analyzed in this study included ALK, BRAF, EGFR, FGFR1, FGFR2, FGFR3, MET, NTRK1, NTRK2, NTRK3, RET and ROS1, which are commonly involved in cancer genome rearrangements. RTK fusions with potential functionality were identified, defined as an in-frame fusion with the intact tyrosine kinase domain of the RTK gene retained. Concomitant genomic alterations and tumor mutational burden (TMB) were also analyzed and calculated if applicable. Patients’ clinical characteristics were retrieved from case records. The RTK fusion prevalence was also compared with MSKCC (22) and TCGA (23) databases. The study was approved by the institutional review board (IRB) of Fudan University Shanghai Cancer Center. All patients had completed written informed consents before they received the genetic testing, giving the permission to use their archived samples and relative information for scientific research in the further. Due to the retrospective nature of the study, the requirement for informed consent for this study was exempted by the IRB.
NGS Data Analyses
Sequencing data were analyzed as previously described (3). Briefly, by using the BWA aligner 0.7, data in FASTQ format were aligned to the reference human genome (hg19). Local alignment optimization, duplication marking, and variant calling were conducted using the Genome Analysis Tool Kit v.3.2 (24) and VarScan v.2.4.3 (25). Low quality variants with depth <50× or mutated allele reads <8× were filtered out. Variants with a frequency >0.1% in the databases (ExAC, 1,000 Genomes, dbSNP, or ESP6500SI-V2) were excluded from further analysis. The remaining variants were annotated with ANNOVAR (2016-02-01 release) (26) and SnpEff v.3.6 (27). Stuctural variation was analyzed using an in-house script markSV (CN112349346A). The algorithm was based on two structure variation signalings: soft clipped reads and paired end reads. The copy number variation (CNV) was estimated with an in-house algorithm based on the sequencing depth as described previously (28).TMB per patient was calculated as the ratio between the total number of nonsynonymous mutations detected with the coding region size of the panel. The relative RTK fusion allele frequency (RTK. RAF) was calculated as the ratio of fusion allele frequency by the maximum allele frequency of a given sample (fusionAF/max AF).
Statistical Analyses
Statistical analyses were performed using R version 3.3.3 software. Differences in the groups were calculated and presented using Fisher’s exact test, paired two-tailed Student’s t-test, or analysis of variance as appropriate. Pearson correlation was performed to study the correlation between TMB and the RTK. RAF. P-values <0.05 were considered statistically significant.
Results
Patients’ Characteristics
A total of 27 patients with breast cancer were identified with putatively functional RTK fusions. The median age of this RTK fusion-positive cohort was 52 years (Table 1). Triple-negative breast cancer subtype (TNBC) comprised 37% of the cohort, while HR+/HER2, HR+/HER2+, and HR-/HER2+ accounting for 22.2%, 14.8% and 7.4%, respectively. Five patients (18.5%) had no histopathological information. Of the 27 patients, the majority (77.8%) had a stage IV disease and 22.2% were at stage I-III. Ten patients (51.9%) were treatment-naïve and fourteen (52.9%) were previously treated. Twenty-four patients were sequenced with the OncoScreen panel (Burning Rock, Guangzhou, China) and 3 with a 108 breast-cancer related gene panel (PurePlasma, Burning Rock); 11 and 15 patients had tissue and plasma samples sequenced, respectively, and 1 patient had both sample types. TMB was only calculated for patients sequenced with the OncoScreen panel and showed a median value of 3.98 mutations/Mb. The median relative RTK fusion allele frequency (RTK. RAF) was 42.15% in this cohort.

Table 1 Clinicopathological and molecular characteristics of patients.
Prevalence and Spectrum of RTK Fusions in Breast Cancer
A total of 30 RTK fusion events were identified from 27 breast cancer patients with a prevalence of 1.875% (27/1440). Three patients harbored double fusions. Among the 30 evens, FGFR2 fusions occurred most commonly (n=9), followed by RET (n=5), ROS1 (n=3), NTRK3 (n=3), BRAF (n=2), and NTRK1 (n=2). Other RTK gene fusions including ALK, EGFR, FGFR1, FGFR3, MET, and NTRK2 only occurred once (Figure 1A). The overall RTK fusion prevalence as well as fusion frequencies in different genes were comparable among different clinical stages and sample types (Table S1).
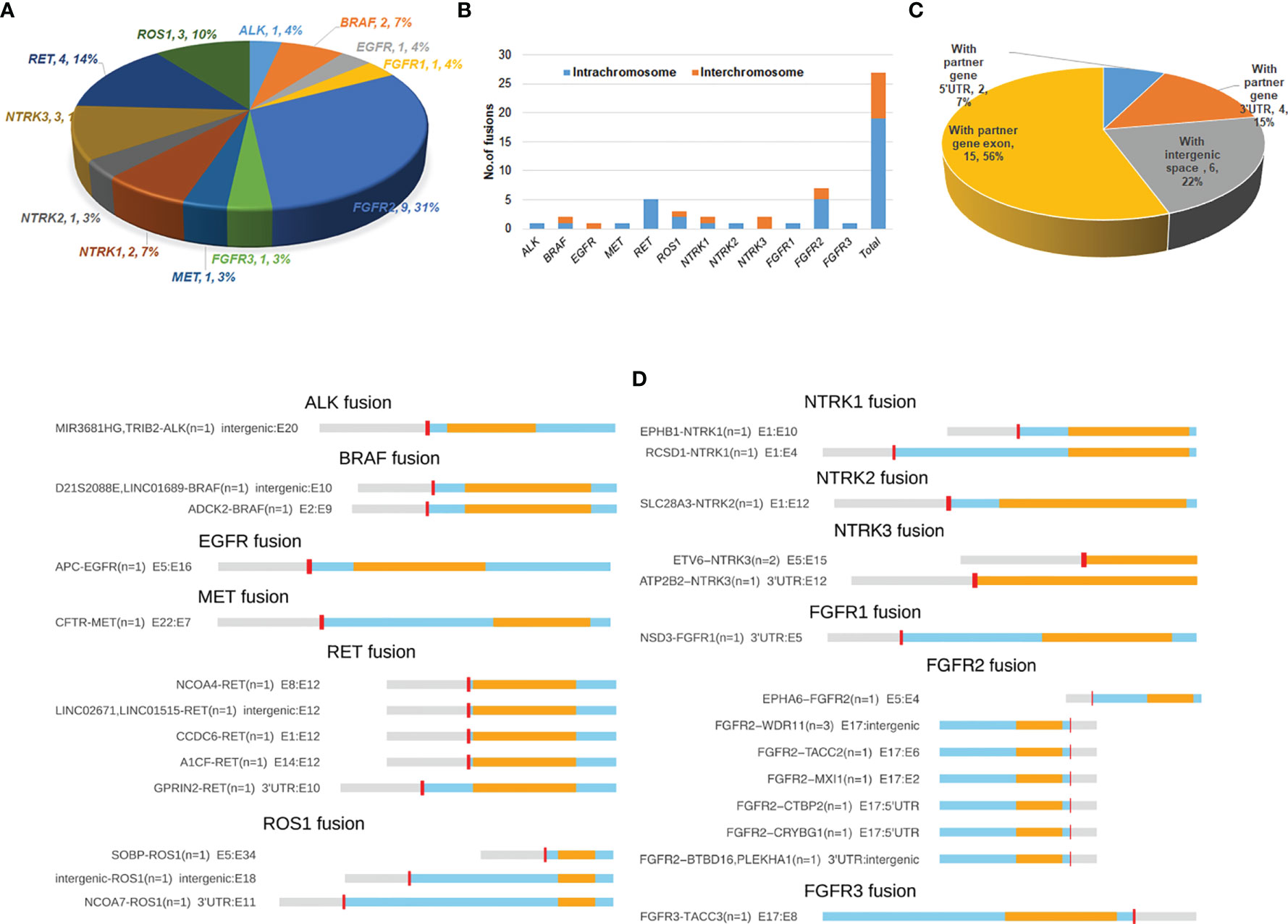
Figure 1 Distribution and spectrum of RTK fusions. (A) The distribution of RTK fusion events in different genes (n=29). (B) Distrubution of fusion variants by RTK genes and different chromosomal rearrangements (n=27); (C) RTK fusion variants classified by different types of fusion partner (n=27); (D) Spectrum of RTK fusions.
A total of 27 unique resultant fusions were discovered. The majority (n=19, 70.3%) of them were resulted from intrachromosomal translocation involving all 12 RTK genes except for EGFR and NTRK3, while interchromosomal fusions (n=8, 29.7%) only happened for BRAF, ROS1, NTRK1, NTRK3 and FGFR2 genes (Figure 1B). Twenty-one resultant fusions had the tyrosine kinase domain in-frame fused with a partner gene either at the 5’-end (n=16) or 3’-end (n=5), including 15 juxtaposed with an exon of the partner gene, 4 with the 3’-UTR (GPRIN2-RET, NCOA7-ROS1, ATP2B2-NTRK3, and ESD3-FGFR1), and 2 with the 5’-UTR of a partner gene (FGFR2-CTBP2, FGFR2-CRYBG1) (Figures 1C, D). We also observed 6 fusions of which the kinase domains were juxtaposed with an intergenic space (Figures 1C, D). Among the 27 fusions, FGFR2-WDR11 (E17: intergenic) (n=3) and ETV6-NTRK3 (E5:E15) (n=2) were recurrent (Figure 1D). The remaining fusions were only seen in one patient each. Of note, 1 patient harbored both A1CF-RET (E14:E12) and GPRIN2-RET (3’UTR: E10) fusions, and 2 out of the 3 patients identified with FGFR2-WDR11 (E17: intergenic) harbored an additional FGFR2 fusion: one with FGFR2-BTBD16 (3’UTR: intergenic) and the other with FGFR2-TACC2 (E17:E6). Of note, the vast majority of the 27 fusions we identified were rearranged with novel partners, with only 4 previously reported in breast cancer, including ETV6-NTRK3, CCDC6-RET, NCOA4-RET and FGFR3-TACC3 (Table 2).
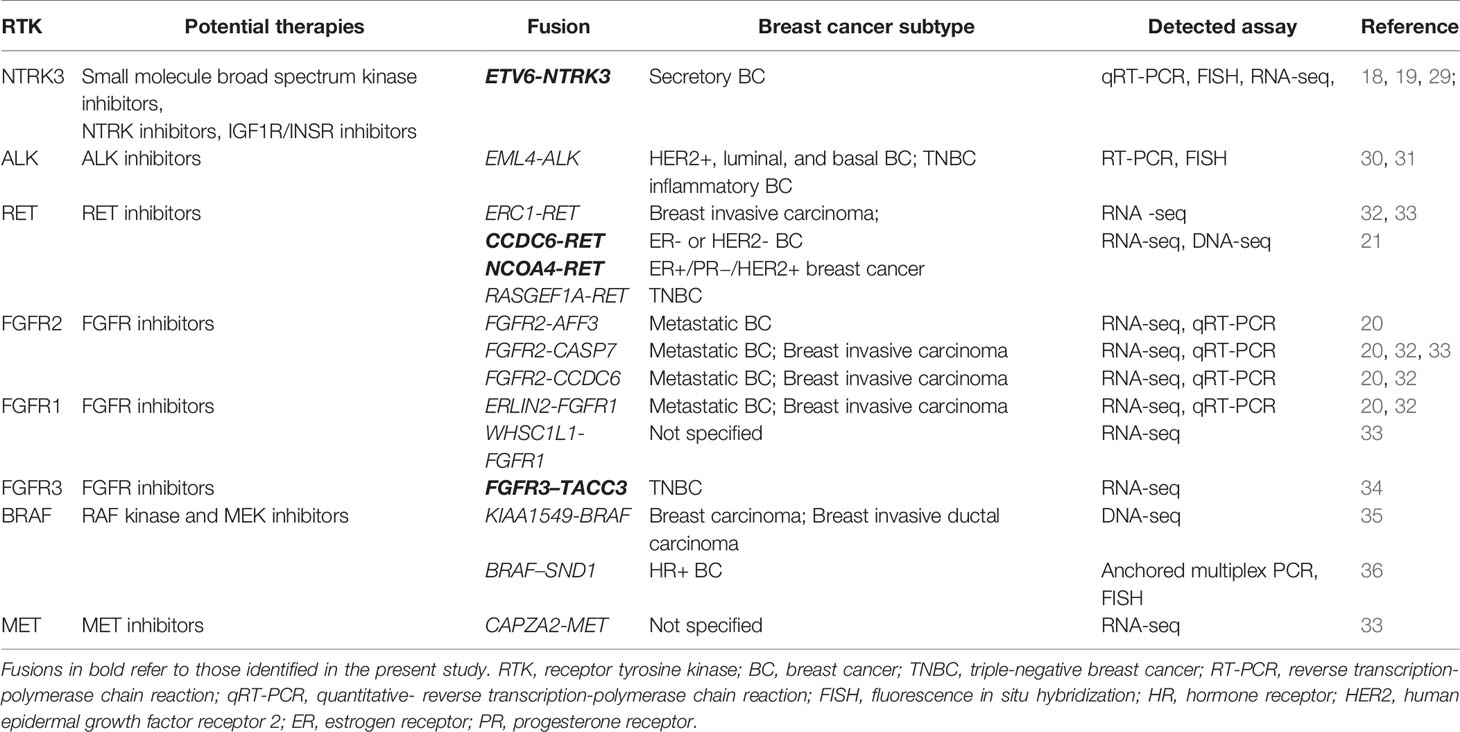
Table 2 List of RTK gene fusions previously reported in breast cancers.
Next, we also compared the RTK fusion frequency among different databases. As shown in Figure 2, our cohort displayed higher overall RTK fusion (1.875% vs. 0.6%, P<0.001) and RET fusion (0.3% vs. 0%, P=0.021) frequencies than MSKCC (22). TCGA (0.6%) (23) revealed significantly more frequent FGFR2 fusions than MSKCC (0.1%, P=0.017) as well as our cohort (0.1%, P=0.027). Other RTK genes did not show significant differences in fusion frequencies among databases.
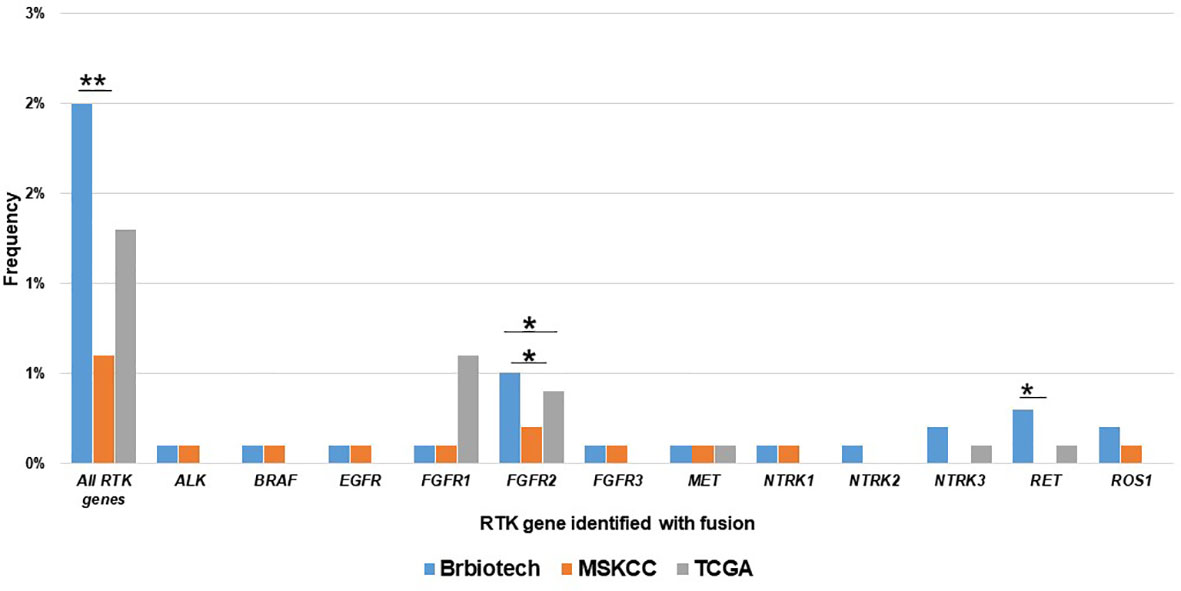
Figure 2 Comparison of the prevalence RTK fusions among different databases. Brbiotech (n=1440), MSKCC (n=1756), TCGA (n=996) *P-value <0.05; **P-value <0.01.
RTK Fusion Abundance Correlated With TMB
We evaluated the association between the RTK fusion abundance (defined as RTK. RAF) and TMB value and found a negative linear correlation (Figure 3A, r=-0.48, P=0.017). We defined the cut-off as the second tertile of TMB in the given cohort. Patients with TMB < 8 (Mutations/Mb) displayed a higher fusion abundance than those with TMB ≥ 8 (Mutations/Mb) (50.3%vs 19.0%, P=0.025, Figure 3B). Besides, in the eight TMB-high (>8 mutations/Mb) patients, four had received platinum-based chemotherapy; while only two out of the sixteen TMB-low (<8 mutations/Mb) patients had received platinum-based chemotherapy. In the subset of patients with plasma sample sequenced, using the cutoff of 9 (Mutations/Mb), patients with higher blood TMB (bTMB) also possessed lower fusion abundance than those with lower bTMB (5.0% vs 53.5%, P=0.037, Figure 3C). Similarly, patients with higher fusion abundance showed both significantly lower TMB (P=0.042, Figure 3D) and bTMB (P=0.025, Figure 3E). The phenomenon suggested a higher likelihood of subclonal nature for RTK fusions in TMB-high patients.
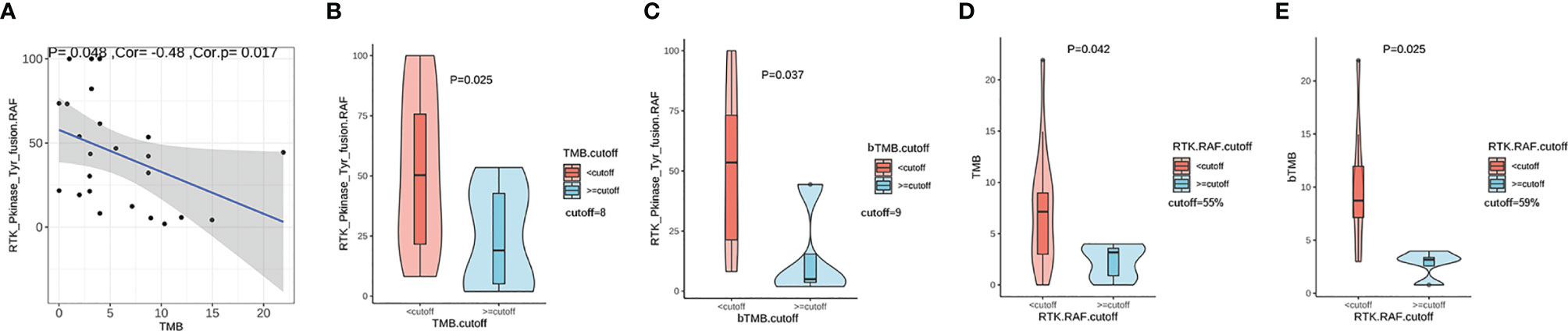
Figure 3 The correlation between TMB and relative RTK fusion allele frequency (RTK. RAF). (A) Pearson correlation between TMB and RTK. RAF (fusion AF/max AF) (n=24); (B) Comparison of RTK. RAF in TMB-high vs. TMB-low groups (n=24). (C) Comparison of RTK. RAF in blood TMB-high vs. blood TMB-low groups (n=13). (D) Comparison of the TMB in RTK. RAF -high vs. RTK. RAF -low groups (n=24). (E) Comparison of the blood TMB in RTK. RAF -high vs. RTK. RAF -low groups (n=13).
Genomic Alterations Co-Occurring With RTK Fusions
We next characterized the concomitant alterations in the 27 RTK fusion-positive breast cancers (Figure 4A). Fusions in different RTK genes were mutually exclusive except for one BRAF fusion-positive patient who also harbored a rearrangement of FGFR1 fused with intergenic space. Of note, this fusion lacked the intact FGFR1 kinase domain therefore had been excluded from our analyses. Moreover, 4 out of the 7 FGFR2 fusion-positive patients harbored FGFR2 amplifications: three harbored FGFR2-WDR11 (E17: intergenic) concomitant with another FGFR2 fusion (FGFR2-BTBD16 (3’UTR: intergenic), n=1; FGFR2-TACC2 (E17:E6), n=1) or alone (n=1) and one had FGFR2-CTBP2 (E17: 5’UTR). In addition, amplified FGFR1 and NTRK1 were also observed from the patient with ESD3-FGFR1 (3’UTR: E5) and the one with EPHB1-NTRK1 (E1:E10), respectively.
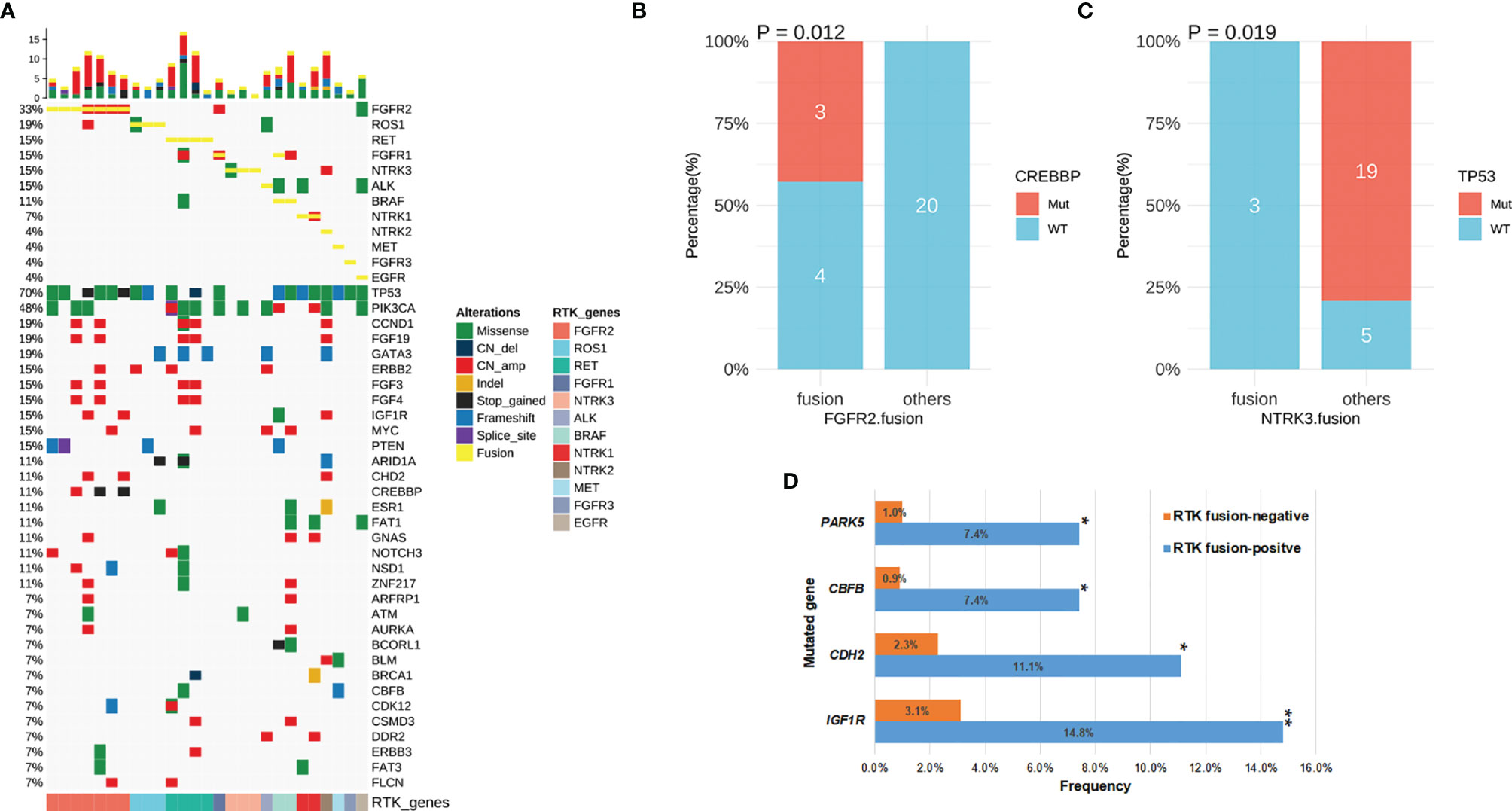
Figure 4 The concomitant mutations in RTK fusion-positive patients. (A) The oncoprint of 27 breast cancer patients with RTK fusion. (B) CREBBP mutation frequency in patients with FGFR2 fusion vs. those with other RTK fusion. (C) TP53 mutation frequency in patients with NTRK3 fusion vs. those with other RTK fusion. (D) Comparison of mutation frequency in RTK fusion-positive vs. negative patients. *P-value <0.05; **P-value <0.01.
Concomitant genomic alterations in other genes were also comprehensively assessed in RTK fusion-positive cancers. TP53 remained the most frequently mutated gene (70%) with the majority being missense mutations (12/19) (Figure 4A). PICK3CA alterations co-occurred the second most commonly (n=13, 48%). Other common concomitant alterations included amplifications in CCND1and FGF19 (19%), as well as frameshift mutations in GATA3 (19%).
In the RTK fusion-positive cohort, CREBBP mutation only co-occurred with FGFR2 fusion (P=0.012, Figure 4B), while NTRK3 fusion and TP53 mutation were mutually exclusive (P=0.019, Figure 4C). By comparing the mutation frequency in RTK fusion-positive versus RTK fusion-negative breast cancer patients, we observed that IGF1R (14.8% vs. 3.1%, P=0.004), CHD2 (11.1% vs. 2.3%, P=0.022), CBFB (7.4% vs. 0.9%, P=0.018) and PAK5 (7.4% vs. 1.0%, P=0.024) mutated more commonly in the fusion-positive cohort (Figure 4D).
Discussion
Our study comprehensively characterized the RTK gene fusions in Chinese breast cancer patients and identified 27 unique fusions that are potential oncogenic drivers. Among them, ETV6-NTRK3, CCDC6-RET, NCOA4-RET and FGFR3-TACC3 have been reported in breast cancer previously. ETV6-NTRK3 has been described as a primary oncogenic event in a rare subset of breast cancer secretory breast carcinoma (18, 19, 29). Clinical trials are currently ongoing that test the efficacy of entrectinib (a broad-spectrum kinase inhibitor for NTRKs, ROS, and ALK) in NTRK-rearranged solid tumors including breast cancer (NCT02568267, CT02097810). CCDC6-RET and NCOA4-RET have been previously characterized as oncogenic and occur recurrently in papillary thyroid and non-small cell lung cancers (37, 38). Recently, Paratala et al. identified CCDC6-RET (n=6) and NCOA4-RET (n=1) out of 9693 breast cancers. They also observed a rapid response to the RET inhibitor cabozantinib in a case with NCOA4-RET-positive breast cancer (21). Shaver et al. discovered FGFR3-TACC3, a canonical fusion across multiple solid tumors, in 1/80 TNBC tumors and in vitro studies indicate this fusion protein is a targetable driver in TNBC (34). FGFR2-TACC2 that has been described in glioblastoma (39), NSCLC (40) and cervical cancer (41), was first identified in breast cancer in our study. Table 2 also summarizes other previously reported RTK gene fusions in breast cancer that were not detected in our cohort. The expression of the canonical NSCLC EML4-ALK fusion was detected in 2.4% of breast cancers (30). Robertson et al. also identified EML4-ALK in 1/25 inflammatory breast cancers (31). Several studies profiling the landscape of kinase fusions across diverse cancers discovered ERC1-RET, CAPZA2-MET, and various FGFR fusions in breast cancer (20, 32, 33), of which ERC1-RET, FGFR2-CASP7, FGFR2-CCDC6 and ERLIN2-FGFR1 were recurrent. Besides, KIAA1549-BRAF was described in 2 breast cancers (35), and BRAF-SND1 was identified in 2 hormone receptor-positive breast cancers (36).
Of note, we also discovered a variety of novel fusions including 16 with an unreported partner gene and 6 juxtaposed with an intergenic space (Figure 1D). Among them, FGFR2-WDR11 (E17: intergenic), FGFR2-BTBD16 (3’UTR: intergenic), FGFR2-CTBP2 (E17: 5’UTR), ESD3-FGFR1 (3’UTR: E5) and EPHB1-NTRK1 (E1:E10) co-occurred with the amplification of the corresponding RTK gene. Although retaining the intact kinase domain, these amplicon-associated RTK fusions might represent the by-products of chromosomal amplifications known as passenger aberrations instead of oncogenic fusions (42). Therefore, their oncogenic significance merits further validation.
Intriguingly, we observed a negative correlation between relative RTK fusion abundance and TMB, suggesting that RTK fusions in TMB-low tumors are more likely to function as oncogenic drivers while fusion in TMB-high tumors are prone to be passenger alterations. Of note, in the eight TMB-high (>8 mutations/Mb) patients, four had received platinum-based chemotherapy; while only two out of the sixteen TMB-low (<8 mutations/Mb) patients had received platinum-based chemotherapy. The observation suggests the high mutation load is more likely to be caused by DNA damaging agent. Similarly in lung cancer, most driver mutations are found in non-smoking TMB-low NSCLC patients and high-TMB is associated with smoking history (43, 44). This can be explained by that the presence of an oncogenic driver is sufficient for the tumorigenesis in non-smokers while in patients with smoking history, tobacco carcinogens cause direct DNA damage and confer a high somatic mutation load that eventually increase the cancer risk (45, 46).
Our study has several limitations. Due to the retrospective nature of the study, we recuirted patients sequenced with un-uniform panels and diverse sample types. Enrolled patient were also with diverse clinical scenarios and a portion of them missed the clinical information. The heterogeneity may diminish the strength of the findings of our study. Our cohort was selected from patients who had underwent NGS, which tends to enroll more patients with advance disease, because patients with metastatic settings are more likely to seek for therapeutic option. Targeted DNA-based sequencing was used to detect RTK fusion in this study. Compared with RNA-based sequencing, this approach has certain technical limitations on detecting gene fusions. For instance, fusions with the breakpoint region insufficiently covered by the panel or those with breakpoint spanning repetitive sequence may not be identified (47). Therefore, this technique is likely to attenuate the capability of identifying unknown fusions and underestimate the prevalence of RTK fusions. Besides, DNA-based sequencing fails to provide direct evidence for the expression of resultant fusions at the mRNA level, so further evaluation of their transcripts is warranted to determine their significance. Moreover, the therapeutic information and clinical outcomes of patients were not provided, thus the therapeutic relevance of these potentially targetable RTK fusions remains unrevealed in breast cancer and merits further elucidation.
In conclusion, this is the first study comprehensively delineating the prevalence and spectrum of potentially targetable RTK fusions in Chinese breast cancers. Further study is ongoing to identify the enriched subpopulation who may benefit from RTK fusion inhibitors.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Fudan University Shanghai Cancer Center. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
W-MC and XH contributed to the conception or design of the work. HX, KC, JZ, HZ, JS, JH, ZG, and HY contributed to the acquisition of data. ZT, JL, and TL contributed to the analysis and interpretation of data. ZT, JL, and TL drafted the MS. All authors contributed to the article and approved the submitted version.
Funding
The authors are grateful for the financial grant from the National Science and Technology Major Project (2020ZX09201-013).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Dr. Jianxing Xiang, Dr. Lin Shao, Dr. Bing Li and Yuchen Yang from Burning Rock Biotech for their assistance in data analysis and manuscript writing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.741142/full#supplementary-material
References
1. Hubbard SR. Structural Analysis of Receptor Tyrosine Kinases. Prog Biophys Mol Biol (1999) 71(3-4):343–58. doi: 10.1016/S0079-6107(98)00047-9
2. Robinson DR, Wu YM, Lin SF. The Protein Tyrosine Kinase Family of the Human Genome. Oncogene (2000) 19(49):5548–57. doi: 10.1038/sj.onc.1203957
3. Mao X, Zhang Z, Zheng X, Xie F, Duan F, Jiang L, et al. Capture-Based Targeted Ultradeep Sequencing in Paired Tissue and Plasma Samples Demonstrates Differential Subclonal CtDNA-Releasing Capability in Advanced Lung Cancer. J Thorac Oncol (2017) 12(4):663–72. doi: 10.1016/j.jtho.2016.11.2235
4. Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell (2010) 141(7):1117–34. doi: 10.1016/j.cell.2010.06.011
5. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic Lymphoma Kinase Inhibition in Non-Small-Cell Lung Cancer. N Engl J Med (2010) 363(18):1693–703. doi: 10.1056/NEJMoa1006448
6. Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, et al. Novel ALK Inhibitors in Clinical Use and Development. J Hematol Oncol (2015) 8:17. doi: 10.1186/s13045-015-0122-8
7. Arnaoutakis K. Crizotinib in ROS1-Rearranged non-Small-Cell Lung Cancer. N Engl J Med (2015) 372(7):683. doi: 10.1056/NEJMc1415359
8. Kiehna EN, Arnush MR, Tamrazi B, Cotter JA, Hawes D, Robison NJ, et al. Novel GOPC(FIG)-ROS1 Fusion in a Pediatric High-Grade Glioma Survivor. J Neurosurg Pediatr (2017) 20(1):51–5. doi: 10.3171/2017.2.PEDS16679
9. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in Patients With Advanced RET-Rearranged non-Small-Cell Lung Cancer: An Open-Label, Single-Centre, Phase 2, Single-Arm Trial. Lancet Oncol (2016) 17(12):1653–60. doi: 10.1016/S1470-2045(16)30562-9
10. Kloos RT, Ringel MD, Knopp MV, Hall NC, King M, Stevens R, et al. Phase II Trial of Sorafenib in Metastatic Thyroid Cancer. J Clin Oncol (2009) 27(10):1675–84. doi: 10.1200/JCO.2008.18.2717
11. Borad MJ, Gores GJ, Roberts LR. Fibroblast Growth Factor Receptor 2 Fusions as a Target for Treating Cholangiocarcinoma. Curr Opin Gastroenterol (2015) 31(3):264–8. doi: 10.1097/MOG.0000000000000171
12. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med (2019) 381(4):338–48. doi: 10.1056/NEJMoa1817323
13. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med (2018) 378(8):731–9. doi: 10.1056/NEJMoa1714448
14. Malhotra GK, Zhao X, Band H, Band V. Histological, Molecular and Functional Subtypes of Breast Cancers. Cancer Biol Ther (2010) 10(10):955–60. doi: 10.4161/cbt.10.10.13879
15. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of Chemotherapy Plus a Monoclonal Antibody Against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N Engl J Med (2001) 344(11):783–92. doi: 10.1056/NEJM200103153441101
16. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. J Clin Oncol (2002) 20(3):719–26. doi: 10.1200/JCO.2002.20.3.719
17. Osborne CK, Schiff R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu Rev Med (2011) 62:233–47. doi: 10.1146/annurev-med-070909-182917
18. Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, et al. Expression of the ETV6-NTRK3 Gene Fusion as a Primary Event in Human Secretory Breast Carcinoma. Cancer Cell (2002) 2(5):367–76. doi: 10.1016/S1535-6108(02)00180-0
19. Makretsov N, He M, Hayes M, Chia S, Horsman DE, Sorensen PH, et al. A Fluorescence In Situ Hybridization Study of ETV6-NTRK3 Fusion Gene in Secretory Breast Carcinoma. Genes Chromosomes Cancer (2004) 40(2):152–7. doi: 10.1002/gcc.20028
20. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov (2013) 3(6):636–47. doi: 10.1158/2159-8290.CD-13-0050
21. Paratala BS, Chung JH, Williams CB, Yilmazel B, Petrosky W, Williams K, et al. RET Rearrangements are Actionable Alterations in Breast Cancer. Nat Commun (2018) 9(1):4821. doi: 10.1038/s41467-018-07341-4
22. Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell (2018) 34(3):427–38.e6. doi: 10.1016/j.ccell.2018.08.008
23. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors From 33 Types of Cancer. Cell (2018) 173(2):291–304.e6. doi: 10.1016/j.cell.2018.03.022
24. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A Mapreduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res (2010) 20(9):1297–303. doi: 10.1101/gr.107524.110
25. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. Varscan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res (2012) 22(3):568–76. doi: 10.1101/gr.129684.111
26. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38(16):e164. doi: 10.1093/nar/gkq603
27. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, Snpeff: Snps in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly (Austin) (2012) 6(2):80–92. doi: 10.4161/fly.19695
28. Yang L, Ye F, Bao L, Zhou X, Wang Z, Hu P, et al. Somatic Alterations of TP53, ERBB2, PIK3CA and CCND1 Are Associated With Chemosensitivity for Breast Cancers. Cancer Sci (2019) 110(4):1389–400. doi: 10.1111/cas.13976
29. Kim J, Kim S, Ko S, In YH, Moon HG, Ahn SK, et al. Recurrent Fusion Transcripts Detected by Whole-Transcriptome Sequencing of 120 Primary Breast Cancer Samples. Genes Chromosomes Cancer (2015) 54(11):681–91. doi: 10.1002/gcc.22279
30. Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, et al. Exon Array Profiling Detects EML4-ALK Fusion in Breast, Colorectal, and Non-Small Cell Lung Cancers. Mol Cancer Res (2009) 7(9):1466–76. doi: 10.1158/1541-7786.MCR-08-0522
31. Robertson FM, Petricoin Iii EF, Van Laere SJ, Bertucci F, Chu K, Fernandez SV, et al. Presence of Anaplastic Lymphoma Kinase in Inflammatory Breast Cancer. Springerplus (2013) 2:497. doi: 10.1186/2193-1801-2-497
32. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The Landscape of Kinase Fusions in Cancer. Nat Commun (2014) 5:4846. doi: 10.1038/ncomms5846
33. Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, et al. The Landscape and Therapeutic Relevance of Cancer-Associated Transcript Fusions. Oncogene (2015) 34(37):4845–54. doi: 10.1038/onc.2014.406
34. Shaver TM, Lehmann BD, Beeler JS, Li CI, Li Z, Jin H, et al. Diverse, Biologically Relevant, and Targetable Gene Rearrangements in Triple-Negative Breast Cancer and Other Malignancies. Cancer Res (2016) 76(16):4850–60. doi: 10.1158/0008-5472.CAN-16-0058
35. Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The Distribution of BRAF Gene Fusions in Solid Tumors and Response to Targeted Therapy. Int J Cancer (2016) 138(4):881–90. doi: 10.1002/ijc.29825
36. Matissek KJ, Onozato ML, Sun S, Zheng Z, Schultz A, Lee J, et al. Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor-Positive Breast Cancer. Cancer Discov (2018) 8(3):336–53. doi: 10.1158/2159-8290.CD-17-0535
37. Santoro M, Moccia M, Federico G, Carlomagno F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes (Basel) (2020) 11(4). doi: 10.3390/genes11040424
38. Gainor JF, Shaw AT. The New Kid on the Block: RET in Lung Cancer. Cancer Discov (2013) 3(6):604–6. doi: 10.1158/2159-8290.CD-13-0174
39. Georgescu MM, Islam MZ, Li Y, Traylor J, Nanda A. Novel Targetable FGFR2 and FGFR3 Alterations in Glioblastoma Associate With Aggressive Phenotype and Distinct Gene Expression Programs. Acta Neuropathol Commun (2021) 9(1):69. doi: 10.1038/onc.2014.406
40. Cai X, Xu C, Wang WX, Zhang Q, Chen Y, Fang Y, et al. Incidence of FGFR-TACC Gene Fusions in Chinese Non-Small Cell Lung Cancer (NSCLC): A Multicenter Study. Int J Cancer (2019) 37(15_suppl):e13001–e. doi: 10.1200/JCO.2019.37.15_suppl.e13001
41. Gill CM, Orfanelli T, Yoxtheimer L, Roy-McMahon C, Suhner J, Tomita S, et al. Histology-Specific FGFR2 Alterations and FGFR2-TACC2 Fusion in Mixed Adenoid Cystic and Neuroendocrine Small Cell Carcinoma of the Uterine Cervix. Gynecol Oncol Rep (2020) 34:100668. doi: 10.1016/j.gore.2020.100668
42. Kalyana-Sundaram S, Shankar S, Deroo S, Iyer MK, Palanisamy N, Chinnaiyan AM, et al. Gene Fusions Associated With Recurrent Amplicons Represent a Class of Passenger Aberrations in Breast Cancer. Neoplasia (2012) 14(8):702–8. doi: 10.1593/neo.12914
43. Spigel DR, Schrock AB, Fabrizio D, Frampton GM, Sun J, He J, et al. Total Mutation Burden (TMB) in Lung Cancer (LC) and Relationship With Response to PD-1/PD-L1 Targeted Therapies. J Clin Oncol (2016) 34: (15_suppl):9017–. doi: 10.1200/JCO.2016.34.15_suppl.9017
44. Nagahashi M, Sato S, Yuza K, Shimada Y, Ichikawa H, Watanabe S, et al. Common Driver Mutations and Smoking History Affect Tumor Mutation Burden in Lung Adenocarcinoma. J Surg Res (2018) 230:181–5. doi: 10.1016/j.jss.2018.07.007
45. Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational Signatures Associated With Tobacco Smoking in Human Cancer. Science (2016) 354(6312):618–22. doi: 10.1126/science.aag0299
46. Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco Smoke Carcinogens, DNA Damage and P53 Mutations in Smoking-Associated Cancers. Oncogene (2002) 21(48):7435–51. doi: 10.1038/sj.onc.1205803
47. Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas With No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res (2019) 25(15):4712–22. doi: 10.1158/1078-0432.CCR-19-0225
Keywords: receptor tyrosine kinase, gene fusion, genomic rearrangments, breast cancer, next-generation sequencing
Citation: Tao Z, Liu J, Li T, Xu H, Chen K, Zhang J, Zhou H, Sun J, Han J, Guo Z, Yang H, Cao W-M and Hu X (2021) Profiling Receptor Tyrosine Kinase Fusions in Chinese Breast Cancers. Front. Oncol. 11:741142. doi: 10.3389/fonc.2021.741142
Received: 14 July 2021; Accepted: 10 September 2021;
Published: 28 September 2021.
Edited by:
Claudio Sette, Catholic University of the Sacred Heart, ItalyReviewed by:
Chi-Cheng Huang, Taipei Veterans General Hospital, TaiwanReza Ghasemi, Washington University School of Medicine in St. Louis, United States
Copyright © 2021 Tao, Liu, Li, Xu, Chen, Zhang, Zhou, Sun, Han, Guo, Yang, Cao and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xichun Hu, aHV4aWNodW4yMDE3QDE2My5jb20=; Wen-Ming Cao, Y2Fvd21AempjYy5vcmcuY24=
†These authors have contributed equally to this work