 Nikola Curik1,2†
Nikola Curik1,2† Vaclava Polivkova1†Pavel Burda1,2Jitka Koblihova1
Vaclava Polivkova1†Pavel Burda1,2Jitka Koblihova1 Adam Laznicka1,3
Adam Laznicka1,3 Tomas Kalina4
Tomas Kalina4 Veronika Kanderova4
Veronika Kanderova4 Jana Brezinova1Sarka Ransdorfova1Dominika Karasova1Katerina Rejlova4
Jana Brezinova1Sarka Ransdorfova1Dominika Karasova1Katerina Rejlova4 Marina Bakardjieva4Daniela Kuzilkova4David Kundrat1
Marina Bakardjieva4Daniela Kuzilkova4David Kundrat1 Jana Linhartova1Hana Klamova1Cyril Salek1Pavel Klener2,5
Jana Linhartova1Hana Klamova1Cyril Salek1Pavel Klener2,5 Ondrej Hrusak4
Ondrej Hrusak4 Katerina Machova Polakova1,2*
Katerina Machova Polakova1,2*- 1Institute of Hematology and Blood Transfusion, Prague, Czechia
- 2Institute of Pathological Physiology, First Faculty of Medicine, Charles University, Prague, Czechia
- 3Second Faculty of Medicine, Charles University, Prague, Czechia
- 4CLIP-Childhood Leukaemia Investigation Prague, Department of Paediatric Haematology and Oncology, Second Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czechia
- 5First Department of Internal Medicine-Department of Hematology, Charles University General Hospital in Prague, Prague, Czechia
Somatic mutations are a common molecular mechanism through which chronic myeloid leukemia (CML) cells acquire resistance to tyrosine kinase inhibitors (TKIs) therapy. While most of the mutations in the kinase domain of BCR-ABL1 can be successfully managed, the recurrent somatic mutations in other genes may be therapeutically challenging. Despite the major clinical relevance of mutation-associated resistance in CML, the mechanisms underlying mutation acquisition in TKI-treated leukemic cells are not well understood. This work demonstrated de novo acquisition of mutations on isolated single-cell sorted CML clones growing in the presence of imatinib. The acquisition of mutations was associated with the significantly increased expression of the LIG1 and PARP1 genes involved in the error-prone alternative nonhomologous end-joining pathway, leading to genomic instability, and increased expression of the UNG, FEN and POLD3 genes involved in the base-excision repair (long patch) pathway, allowing point mutagenesis. This work showed in vitro and in vivo that de novo acquisition of resistance-associated mutations in oncogenes is the prevalent method of somatic mutation development in CML under TKIs treatment.
Introduction
Tyrosine kinase inhibitors (TKIs) are effective for treating chronic myeloid leukemia (CML); however, 10-15% of patients develop resistance to first line imatinib treatment.1 The 1st generation TKI imatinib remains the most prescribed drug recommended for frontline therapy of chronic phase (CP) CML. While 2nd-generation TKI frontline therapy has shown faster cytogenetic and molecular responses, no differences in overall survival were found between 2nd generation TKIs and imatinib, which may be the preferred choice for older patients with comorbidities and for most adult patients with low- and intermediate risk CML-CP (1, 2).
Mutations in the kinase domain (KD) of BCR-ABL1 represent a common molecular mechanism of imatinib therapy resistance and are responsible for 30% of acquired imatinib resistance cases (3, 4). Furthermore, CML relapses and progression to advanced phases of the disease are often associated with recurrent mutations in oncogenes, which are frequently mutated in other hematological malignancies (5, 6). Despite the major clinical relevance of mutation-associated TKI resistance in CML, the mechanisms underlying mutation acquisition in oncogenes of drug-resistant CML cells are not yet well understood.
It was postulated, that BCR-ABL1 KD mutations could be detected in newly diagnosed patients in accelerated or blast phases (7). This led to the general assumption that a preexisting mutated subpopulation of CML cells carrying a resistance phenotype is selected and expanded during TKI treatment. However, some studies observed the mechanism of de novo BCR-ABL1 mutation acquisition during imatinib treatment in CML cells. It was suggested that BCR-ABL1 expression, but not kinase activity, is required for the acquisition of KD mutations (8). The NAD+-dependent histone deacetylase SIRT1 was shown to promote acquisition of BCR-ABL1 KD mutations in CML cells (9). Others proposed the activity of the alternative nonhomologous end-joining (alt-NHEJ) pathway in resistant CML cells driven by BCR-ABL1 and/or MYC transcriptional activity (10, 11).
This study was based on the hypothesis that the appearance of somatic mutations during imatinib treatment is not just a passive process of selection and expansion of preexisting mutated clones; rather, it is based on de novo mutation acquisition involving deregulation of the DNA damage response and DNA repair pathways.
Materials and Methods
The detailed methods are provided as Supplementary Methods.
CML Cell Lines
CML cell lines KCL-22 (ACC 519) and CML-T1 (ACC 7) were obtained from a publicly accessible biological resource center (Leibniz Institute - Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH/DSMZ, Braunschweig, Germany).
Isolation of CML Clones
KCL-22 and CML-T1 clones were prepared by single-cell FACS sorting (BD FACS Aria III; BD Biosciences, San Jose, CA, USA) according to CD38 expression ([HB-7] anti-CD38 APC; Sony Biotechnology, San Jose, CA, USA) in 96-well plates containing optimal growing medium either imatinib-free (sensitive; S clones) or with a 0.004 µM imatinib that was subsequently increased up to 4 µM (resistant; R clones) (Supplementary Figure S1).
Patients
CML patients (n=32) were diagnosed and treated at the Institute of Hematology and Blood Transfusion in Prague (UHKT), Czech Republic (Tables 1, 2). All samples were collected after written informed consent was obtained according to the principles of the Declaration of Helsinki and approval by the UHKT Ethics Committee.
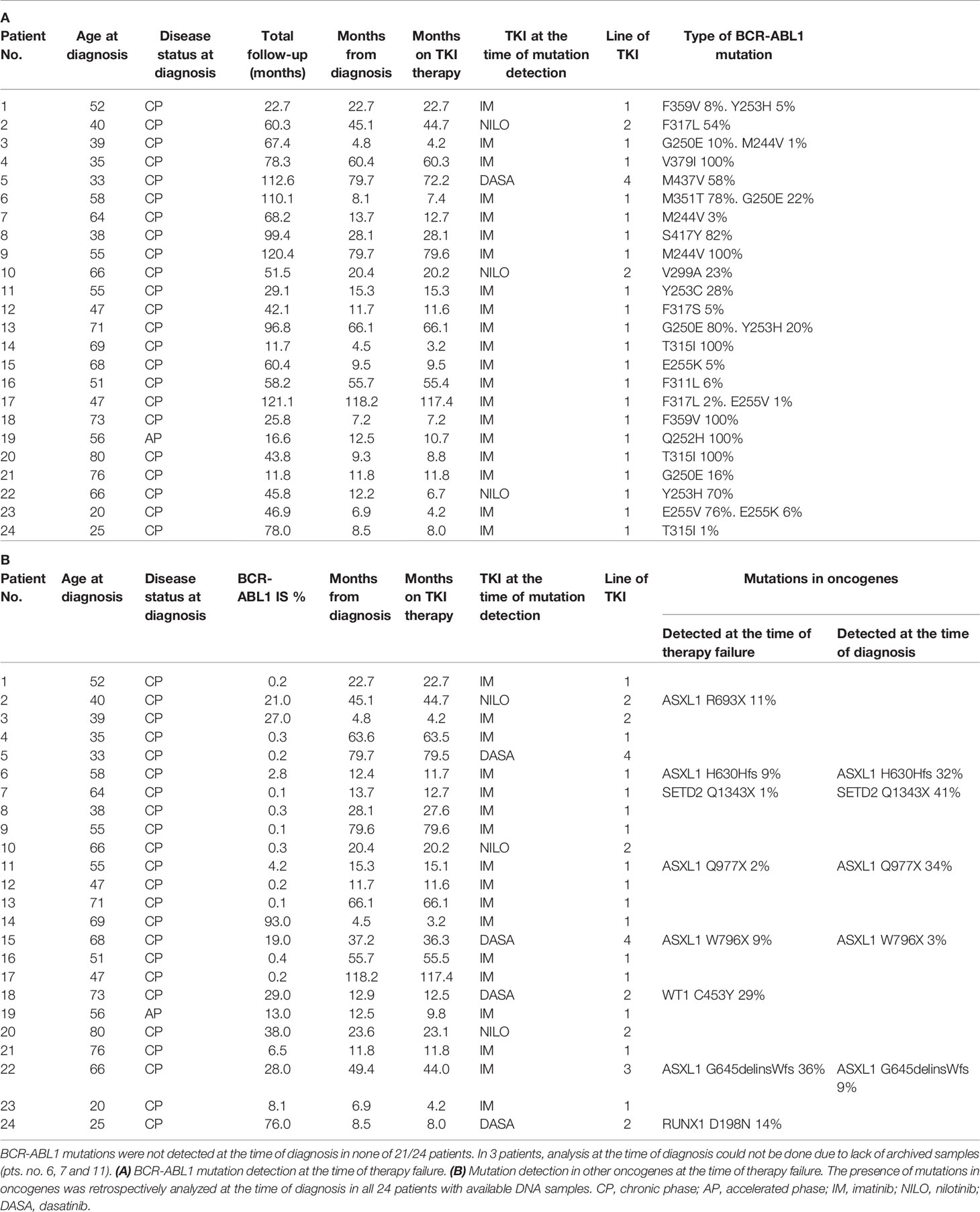
Table 1 CML patients characteristics who failed to TKI with detected BCR-ABL1 mutations (A) BCR-ABL1 mutation detection at the time of therapy failure.
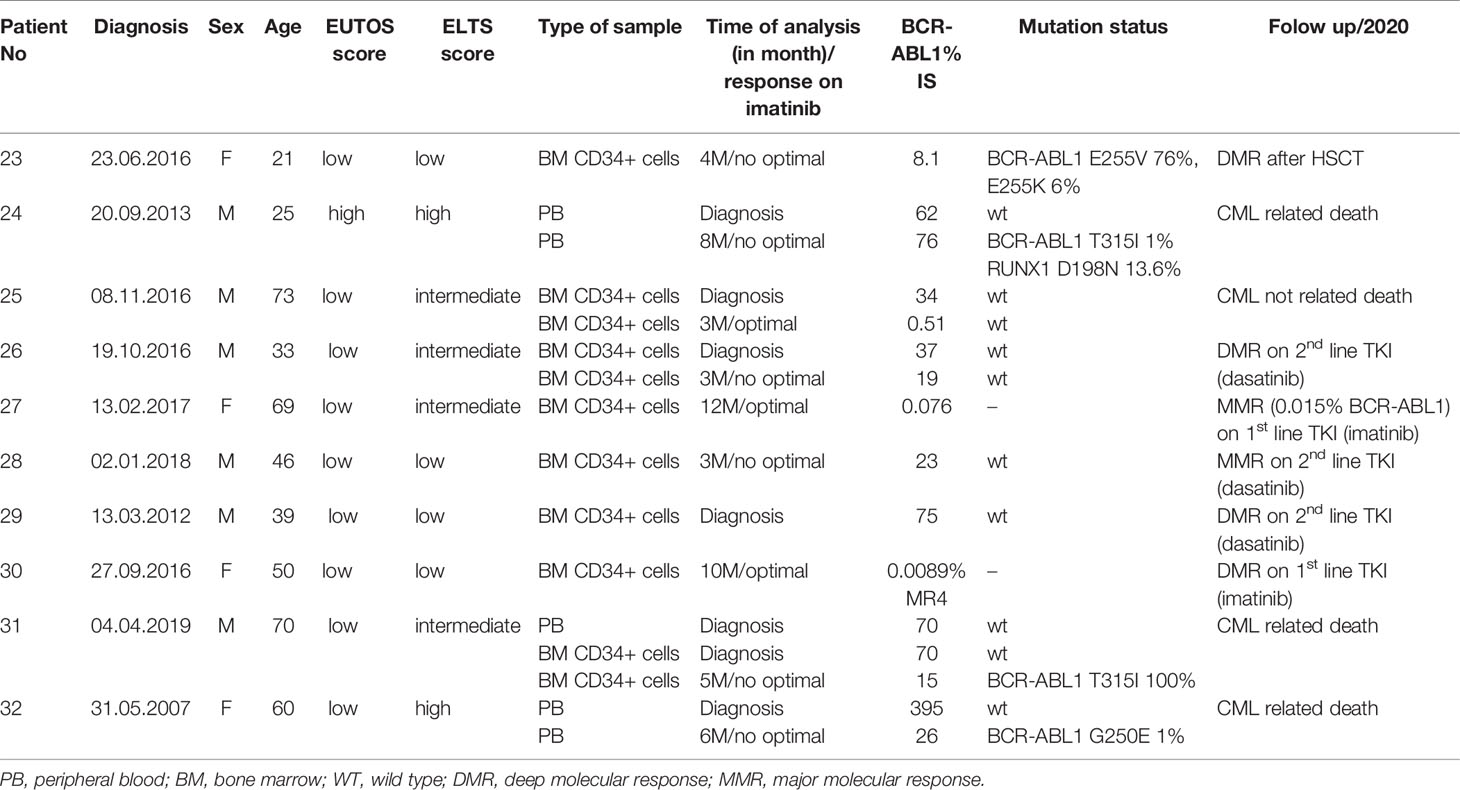
Table 2 Characteristics of patients and samples used for the evaluation of gene expression analysis of DNA repair and response to DNA damage genes.
In Vivo Models
Experiments were performed on NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG)-immunodeficient female mice (n=16) aged 8-10 weeks. Mice were subcutaneously inoculated with 1x 106 KCL-22R imatinib-resistant clones. Treated mice were orally administered imatinib (100 mg/kg/mouse) once per day, with the initiation of treatment 3 days after inoculation. The design of all experiments was approved by the institutional Animal Care and the Use Committee.
Droplet Digital PCR
Mutations were analyzed on DNA level using commercially available allele-specific assays for droplet digital PCR (ASO-ddPCR) (QX200 Droplet Digital PCR system and Auto Droplet generator, Bio-Rad, Hercules, CA, USA).
Gene Expression PCR Arrays
Human DNA Damage Signaling Pathway RT2 Profiler™ PCR Array (PAHS-029Z; Qiagen, Valencia, CA, USA) and Human DNA Repair RT2 Profiler™ PCR Array (PAHS-042Z; Qiagen) were performed on the StepOnePlus system (Thermo Fisher Scientific, Waltham, MA, USA). The complete list of 128 measured genes is provided (Supplementary Table S1). The differentially expressed genes were determined following stringent criteria: 1) the mean 2ΔC value for R clones was ≥ 2.5 or ≤ 0.4-fold of the mean 2-(Cts-Ctc) value for S clones and 2) the difference of 2ΔC values for R clones and S clones reached the level of significance p=0.05.
Next-Generation Sequencing
BCR-ABL1 KD amplicon libraries were prepared using the Nextera XT DNA Library Prep Kit (cat. number FC-131-1096, Illumina, San Diego, CA, USA).
The DNA custom-designed NGS panel of 33 genes often mutated in myeloid malignancies (Supplementary Table S2) was analyzed using SeqCap EZ HyperCap Workflow (Roche, San Diego, CA, USA).
Sequence analysis and mutation identification were performed using NextGENe software (SoftGenetics, State College, PA, USA).
Mass Cytometry
The protein expression in leukemic cells and clones was determined by staining with CyTOF metal-conjugated antibodies against proteins of interest (Supplementary Table S3) and analyzed by a CyTOF2 mass cytometer (Fluidigm, South San Francisco, CA, USA) as described in detail previously (12).
Statistical Analysis
The statistical analyses were performed using Student´s t-test in MS Excel (Microsoft Corporation, Redmond, WA, USA). Fish plots were generated in the program Fishplot package for R (13).
Results
TKI-Resistant Mutation Acquisition in CML Patients
From 253 CML patients diagnosed within the years 2010-2018 and treated with TKIs as frontline therapy 87 responded to therapy as warning or failure according to the ELN recommendation.1 This group of non-optimally responded patients were regularly evaluated for the presence of BCR-ABL1 KD mutations. BCR-ABL1 mutations were detected in 24/87 patients (28%) at a median time of 12.2 months since TKI start (range 3.2-117.4; Table 1A). Presence of BCR-ABL1 mutations was retrospectively analyzed by NGS at the time of diagnosis in 21/24 patients with available RNA samples. Mutations were not detected in any of the 21 patients at the time of diagnosis with the sensitivity range of 1-3.7% (Supplementary Methods). Thus, it is highly probable, that these mutations were acquired de novo under TKI treatment. DNA NGS myeloid panel (Supplementary Table S2) was used to reveal co-occurrence of somatic mutations in oncogenes in the samples of non-optimal responders with BCR-ABL1 mutations. The oncogenic mutations were detected in 8/24 patients (33%) (Table 1B). Presence of oncogenic mutations was retrospectively analyzed by NGS myeloid panel at the time of diagnosis in all 24 patients. The analysis revealed mutations in 5/24 patients at the time of diagnosis, while 3 mutations were likely acquired de novo under TKI treatment.
Resistance-Conferring Mutations Are Acquired De Novo After Exposure to Imatinib
The hypothesis of de novo mutation acquisition in CML cells resistant to TKI was tested on FACS sorted single cells of KCL-22 exposed to imatinib. To address whether the putative BCR-ABL1 mutation acquisition is related to CD38 (non)expression as suggested previously (14), cells were sorted according to the expression of CD38. Out of the 120 sorted single cells, 33 growing clones emerged. No BCR-ABL1 KD mutations were detected by NGS in KCL-22 clones in day 21 post-sorting. Finally, 6 KCL-22R clones resistant to 4 µM imatinib were established from both CD38- (n=4) and CD38+ (n=2) cells of origin (Supplementary figure S1A).
The following three of four CD38- KCL-22R clones carried a mutation in the BCR-ABL1 KD: Clone 1, T315I (50%); Clone 2, Y253H (30%); and Clone 4, E255K (50%) (Figure 1A). DNA analyses using ASO-ddPCR confirmed the same level of mutated genomic BCR-ABL1 in the resistant clones, suggesting that clones with 50% mutated BCR-ABL1 carried mutations on only 1 Ph chromosome. The presence of an additional cryptic unmutated BCR-ABL1 fusion was confirmed in Clone 2 with 30% mutated BCR-ABL1 by FISH analysis (Figure 1B).
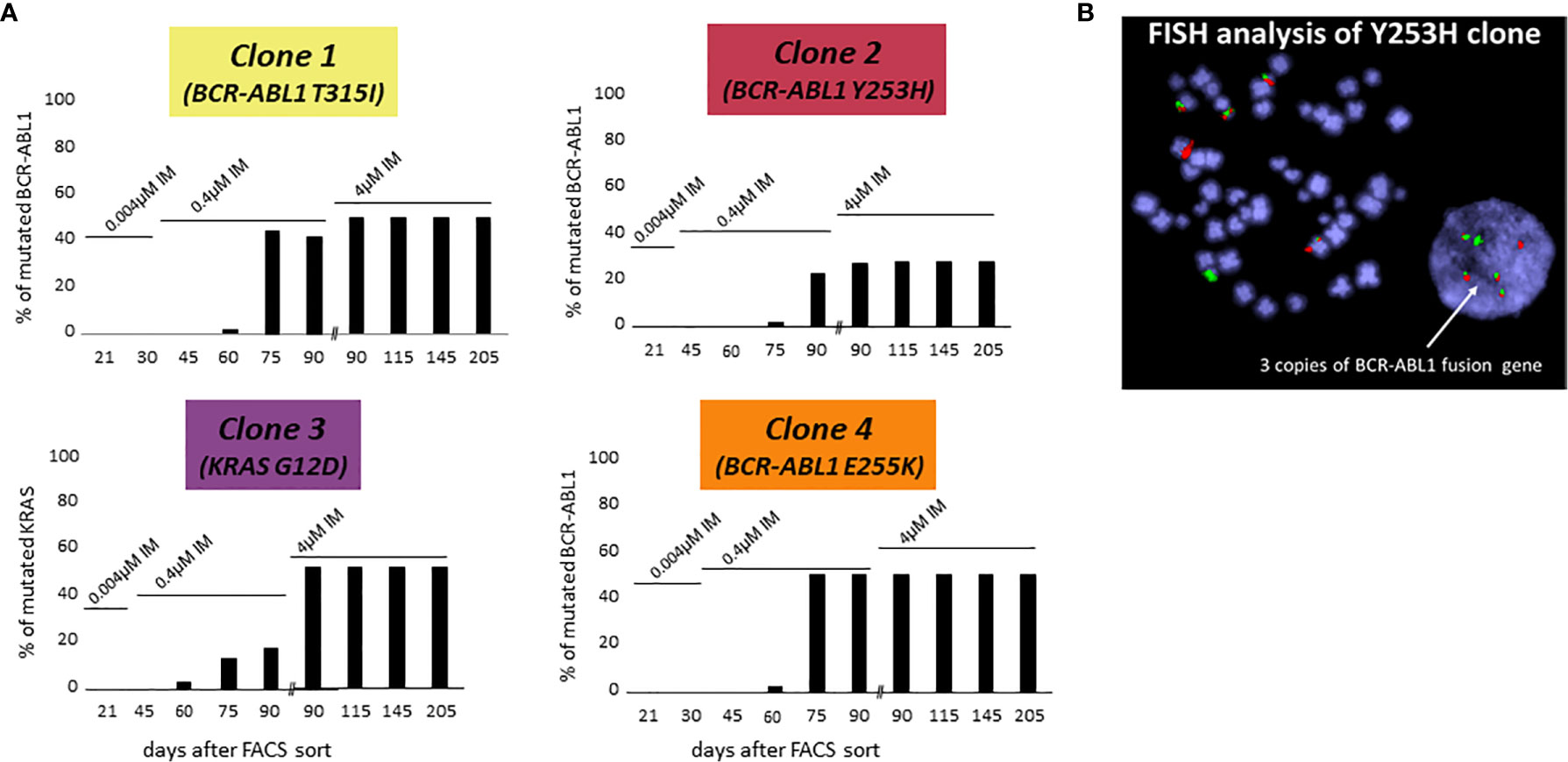
Figure 1 De novo acquisition of somatic mutations during the development of KCL-22 clones of CD38- cell of origin resistant to 4 µM imatinib. KCL-22R clones were prepared by single cell FACS sorting into medium with 0.004 µM imatinib. The imatinib concentration was increased to 0.4 µM in day 31 and further increased to 4 µM in day 75 after sort. (A) The presence of mutated mRNA for BCR-ABL1 or DNA for KRAS during the development of resistant KCL-22R clones up to day 205 post single-cell FACS sorting: Clone 1, Clone 2, Clone 3 and Clone 4. No BCR-ABL1/KRAS mutations were detected in KCL-22R clones until days 60-75 postscoring after the escalation of the imatinib concentration up to 0.4 µM. The growing concentration of imatinib is indicated above the columns. The presence of mutation in day 90 was determined both in the culture with 4 µM imatinib and in the precedent culture with 0.4 µM imatinib growing simultaneously. (B) FISH analysis XL BCR/ABL DF of Clone 2 (Y253H) revealed the presence of 3 BCR-ABL1 fusions.
In Clone 3, BCR-ABL1 was unmutated during the whole follow-up. The NGS myeloid panel was applied to all four KCL-22R clones growing in 4 µM imatinib (Supplementary Table S4). The analysis revealed that Clone 3 was bearing mutations in the ATRX (K1933Kfs), RUNX1 (G74R) and KRAS (G12D) genes. The ATRX and RUNX1 mutations were first detected 21 days post-sorting. The KRAS mutation, confirmed by Sanger sequencing in day 205 in 4 µM imatinib, was first detected 60 days post-sorting on the 0.4 µM imatinib level, similar to the acquisition of BCR-ABL1 mutations in other KCL-22R clones (Figure 1A). Y253H-bearing Clone 2 acquired a de novo mutation in BCOR (R1454Q). CD38+ KCL-22R Clone 5 and Clone 6 acquired BCR-ABL1 mutations Y235H and T315I, respectively (Supplementary Figure S2).
To support data suggesting de novo acquisition of mutations conferring imatinib resistance, cells of the original isolated KCL-22 Clones 1-4 stored at day 20 after the sort to 0.004µM IM were used to repeat experiments of resistance development in biological triplicates. Interestingly, the newly isolated resistant clones (KCL-22R´) developed a different spectrum of BCR-ABL1 and KRAS mutations (Figure 2).
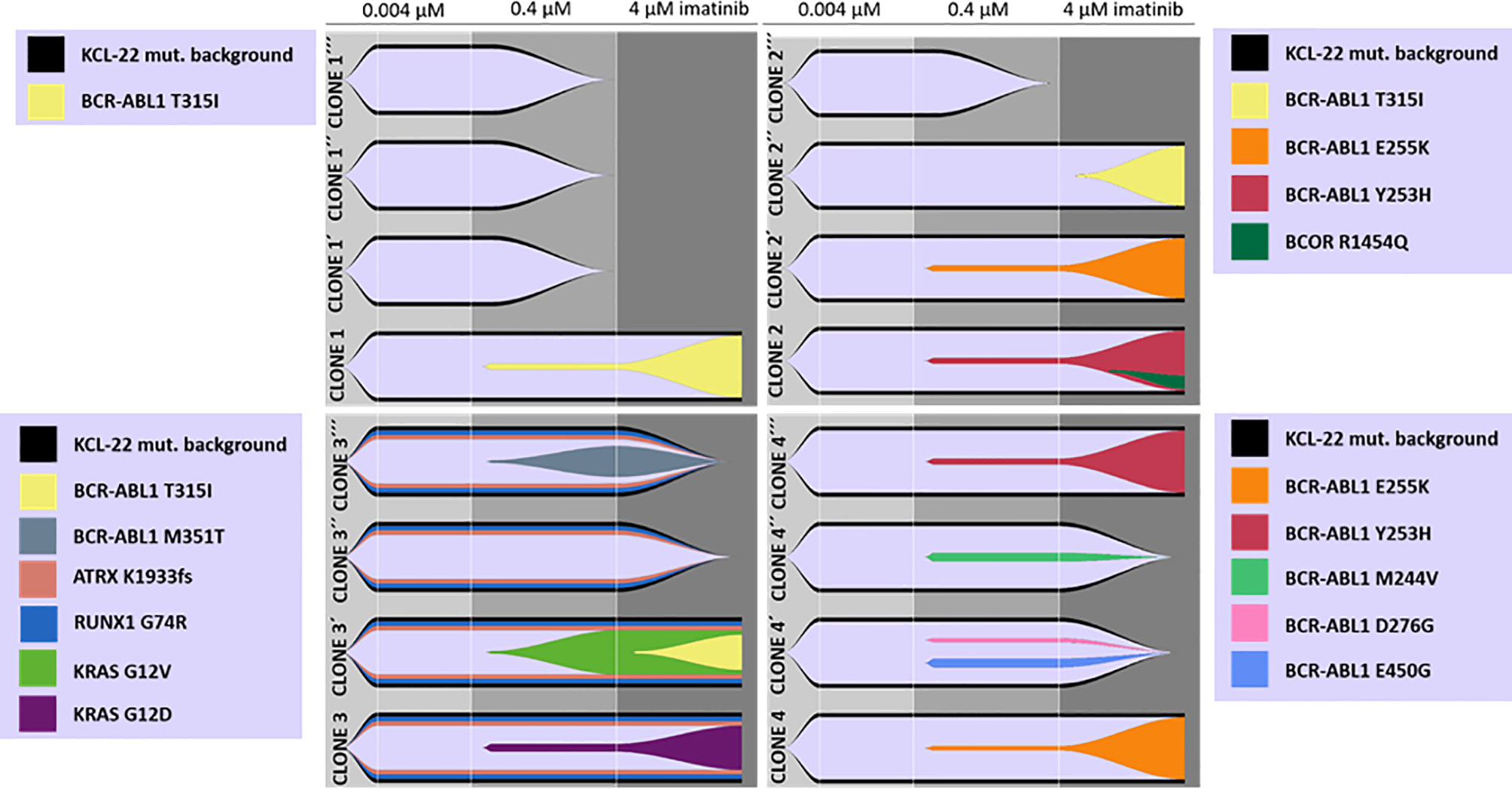
Figure 2 De novo acquisition and evolution of somatic mutations during the development of KCL-22 clones resistant to 4 µM imatinib. Originally, 4 single-cell FACS-resistant CD38- clones, Clone 1, Clone 2, Clone 3 and Clone 4, represented by the lower fish plots in each quadrant, were isolated by a stepwise increase in imatinib concentration and analyzed for mutations by a DNA NGS custom panel. The cells of each clone (using cryopreserved samples from day 21 after sorting) were used to repeat resistance development in biological triplicates, represented by ´, ´´ and ´´´ fish plots in each quadrat. The imatinib concentration was gradually increased up to 4 µM during repeated resistance development, performed exactly as before. DNA NGS custom panel analysis revealed newly obtained resistant clones that exhibit different spectra of acquired mutations, while some clones failed to repeatedly gain a resistant phenotype. The Fishplot package for R was used for data visualization. KCL-22 mutation background is shown elsewhere in detail (Supplementary Table S4).
The de novo acquisition of mutations was also confirmed in the experiment with CML-T1 cell line by similarly designed experiment (Supplementary Results; Supplementary Figure S3). KCL-22 and CML-T1 cells have capability of mutation acquisition irrespectively to the expression of CD38.
De Novo Acquisition of Mutations in CML Cells Is Associated With Transcriptional Activation of the Base-Excision Repair and Alt-NHEJ Repair Pathways In Vitro and In Vivo
Based on the observed results, the de novo acquisition of mutations during imatinib treatment seems to be the result of the interplay between stochastic processes and impaired DNA damage signaling and repair. To test this hypothesis, gene expression PCR arrays carrying DNA damage response and DNA repair genes were applied to reveal the distinct expression profiles of KCL-22R and KCL-22S (sensitive) clones, respectively (Supplementary Results; Supplementary Figure S4A) and to identify the genes with significantly different expression in KCL-22R clones at the time of mutation acquisition. For this and following analysis, KCL-22R clones originating from CD38- cells were used. Two distal clusters were formed from KCL-22R clones and KCL-22S clones, respectively. Eleven significantly differentially expressed genes were identified: decreased CDKN1A and increased BRCA2, BRIP1, CDC25A, EXO1, FEN1, H2AFX, LIG1, MLH1, RAD51 and UNG. Differentially expressed genes were analyzed with KEGG Pathways (https://www.genome.jp/kegg/pathway.html) and Reactome Pathways (https://reactome.org/) databases to delineate putative dysregulated molecular mechanisms/pathways. Mismatch repair and BER pathways were proposed as the most relevant by KEGG (p<.001 and p=0.0001, respectively), and homology repair and mismatch repair were proposed by the Reactome. However, K-means clustering supported the transcriptional activity of BER (Supplementary Figures S4B, C and Supplementary Results).
To decipher the putative pathway involved in mutation acquisition, the expression of genes associated with mismatch repair (RPM1, RFC1, POLD3, PCNA, LIG1, EXO1, MLH1, MSH6, MLH3, MSH3 and PMS2) was determined in KCL-22R clones and compared to that in KCL-22S clones. The increased expression of MLH1 and genes with more general functions in DNA repair (e.g., PCNA, POLD3, LIG1) was found, while the expression of genes involved in the direct recognition of mismatched DNA (MSH6, MLH3, MSH3, PMS2) was not significantly altered, making mismatch repair a questionable candidate to participate in mutation acquisition in KCL-22R clones (Supplementary Figure S5A). Next, the expression of genes participating in BER was determined. The expression of genes involved in short-patch BER was not altered, while significant upregulation of genes participating in long-patch BER was found (Figure 3A). In addition to its roles in BER and mismatch repair, LIG1 is also associated with the error-prone alt-NHEJ pathway of DNA double-strand break (DSB) repair. To investigate the possible role of DNA DSB repair pathways, the expression of DNA ligases and their functional partners was determined. The expression of LIG1 and PARP1 was found to be significantly increased, while the expression of LIG3, LIG4 and their respective partners was not altered (Figure 3B). Increased expression of BER and alt-NHEJ genes was also confirmed at the time of mutation acquisition in the imatinib-resistant KCL-22R´ and CML-T1R clones (Supplementary Figure S5B, C). Moreover, regulatory regions of the LIG1 and PARP1 genes were found to be epigenetically active during and after mutation acquisition and, in the case of LIG1, occupied by the MYC transcription factor, while the DNA methylation was found in KCL-22 at regulatory region of LIG4 (Supplementary Results; Supplementary Figures S6–S8). PARP1 inhibitor NU-1025 treatment prevented or delayed BCR-ABL1 mutation acquisition in CML-T1 and KCL-22 cells after the exposure to imatinib (Supplementary Results; Supplementary Figure S9).
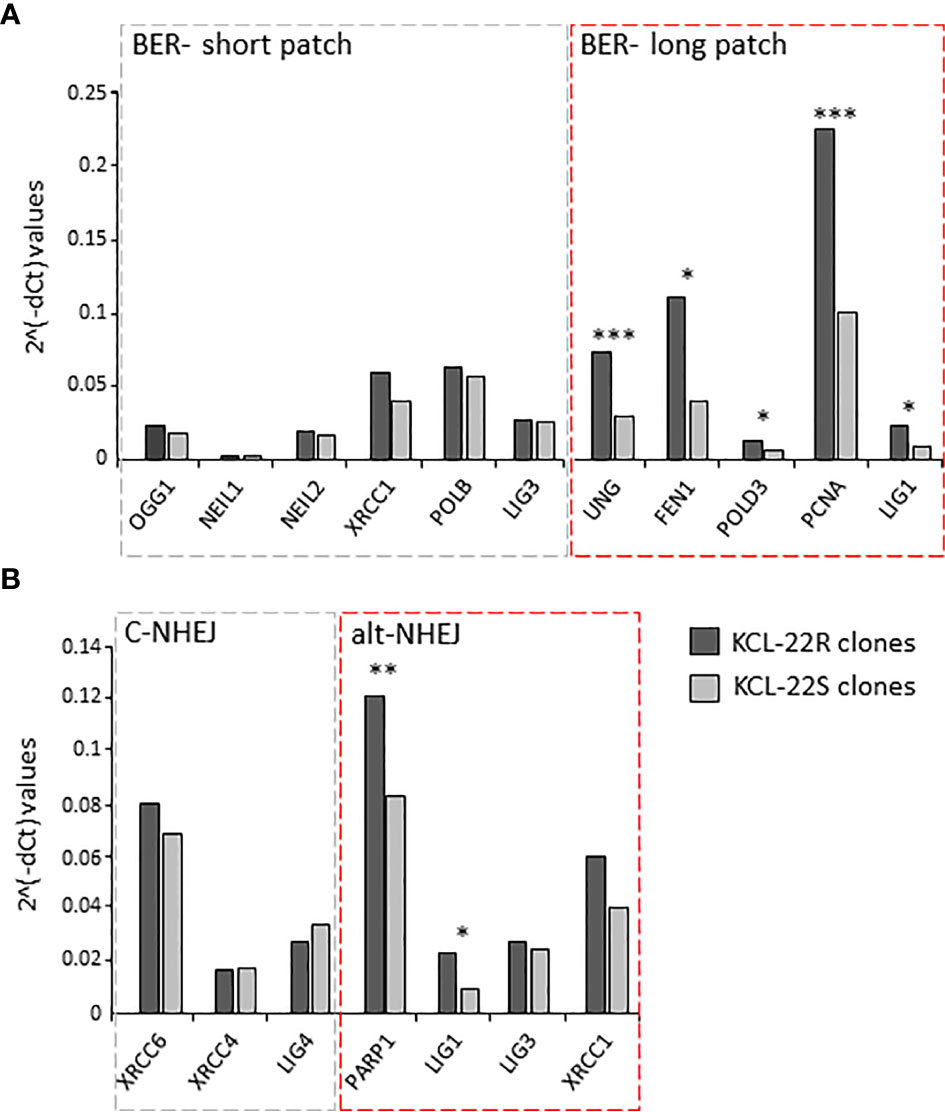
Figure 3 The expression of DNA damage response and DNA repair genes putatively associated with mutagenesis in KCL-22 clones. The expression data are shown for the genes of short- and long-patch BER (A) and for DNA ligases and their functional partners - LIG1 and PARP1; LIG3 and XRCC1; LIG4, XRCC4 and XRCC6, respectively, involved in classical/c-NHEJ and alternative/alt-NHEJ (B) pathways. The expression was measured at the days of mutation acquisitions; 60-75 days postsorting. Dark gray columns represent the average gene expression for KCL-22R imatinib-resistant clones. Light gray columns represent the average gene expression in control, imatinib-naïve KCL-22S clones. The gene expression was normalized to mean Ct values for 4 control genes (B2M, GAPDH, RPLP0, HPRT1). The level of significance is indicated: *P < 0.05, **P < 0.01 and ***P < 0.001.
The expression of DNA ligases and their functional partners was examined in CD34+ primary cells of CML-CP patients (Table 2). The expression of LIG1 and PARP1, representing alt-NHEJ pathway, was found to be relatively augmented to that of LIG4 and XRCC6, representing classical NHEJ (c-NHEJ) pathway, at the time of diagnosis and resistance to imatinib (therapy failure) in comparison with prevalently non-leukemic CD34+ cells at the time of response to TKIs (Figure 4A). Gene expression was also examined in the total leukocytes from peripheral blood (PB) of CML patients at diagnosis (n=3), who on TKI therapy subsequently acquired mutations, compared with the pooled samples of PB leukocytes of healthy donors (n=5) (Figure 4B). No difference in the expression of DNA ligases was found, while the increased expression of FEN1, PCNA and, to a lesser degree, UNG was confirmed in CML-CP. Suggested molecular mechanisms were studied during mutation acquisition in CML patients resistant to TKIs. The expression of LIG1, FEN1, UNG and, to a lesser degree, POLD3 was augmented during mutation acquisition (Figure 4B).
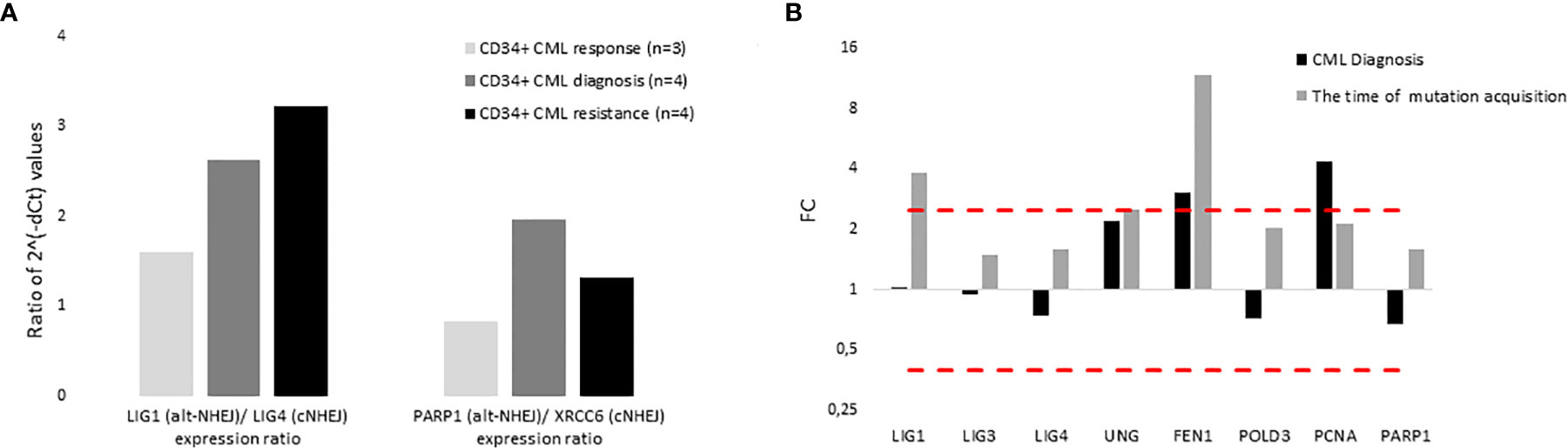
Figure 4 The expression of DNA ligases, their functional partners and BER – long patch genes in primary cells of CML patients. (A) The columns represent the ratios of 2-(Cts-Ctc) values of gene expression for indicated pairs of genes (involved either in altNHEJ or cNHEJ pathway) in CD34+ BM cells of CML patients at the time of optimal response to imatinib (n = 3; Nr. 25, Nr. 27, Nr. 30; light gray), at the CML diagnosis (n = 4; Nr. 25, Nr. 26, Nr. 29, Nr. 31; dark gray) and at the time of resistance to imatinib (n = 4; Nr. 23, Nr. 26, Nr. 28, Nr. 31; black). (B) The black columns represent the fold change (FC; relative expression) of average gene expression in total PB leukocytes of CML patients at the time of diagnosis (n = 3; Nr. 24, Nr. 31 and Nr. 32) compared to gene expression in healthy donors (n = 5; pooled). The gray columns represent the FC of average gene expression in total PB leukocytes of CML patients (n = 2; Nr. 24 and Nr. 32) at the time of first detection of somatic mutations at low levels (RUNX1 D198N 14% + T315I 1% and BCR-ABL1 G250E 1%, respectively) compared to expression in healthy donors (n = 5; pooled). Red dashed lines mark cut-offs indicating strongly differentially expressed genes with FC ≤ 0.4 or ≥ 2.5, similar to inclusion criteria applied for KCL-22R clones.
De Novo TKI-Resistant Mutation Acquisition and the Evolution of Resistant CML Clones Depends on the Culture Conditions, Time, and Dose of Imatinib
The processes of natural mutation appearance and mutated cell expansion was studied in detail in KCL-22 cells. Invariably, no BCR-ABL1 KD mutations were detected in imatinib-naïve cells by NGS. However, BCR-ABL1 mutations were repeatedly detected by NGS after the exposure of cells to 0.4 µM imatinib. The competition of mutated clones was driven not solely by the type of mutations but also by their frequency and by the environmental conditions (Supplementary Results; Supplementary Figure S10). In progressed KCL-22 cells (Supplementary Methods), BCR-ABL1 mutations did not appear after the exposure of cells to 0.4 µM imatinib. Compared to de novo KCL-22 cells, the progressed cells revealed decreased sensitivity to imatinib and putative involvement of BCR-ABL1-independent resistance mechanisms (Supplementary Results; Supplementary Figure S11).
To address a clonal evolution upon the constant imatinib dose, equal numbers of cells from the 4 KCL-22R clones growing in 4 µM imatinib were mixed, split, and cultured in 4 µM, 10 µM imatinib and imatinib-free medium. The T315I-bearing clone outgrew other 3 clones in both 4 µM and 10 µM imatinib (Figures 5A, B). The superior proliferation of the T315I-bearing clone in 4 µM imatinib was also confirmed using a mixture of cell tracking dyes (Supplementary Figure S12A) and by cell cycle analysis (Supplementary Figures S12B, C). In the mixture of clones growing without imatinib, the Y253H-bearing clone showed a proliferative advantage over the other 3 clones (Figure 5C). Clonal evolution was followed for the artificially mixed 3 CML-T1R clones, where Y253H-mutated cells outgrown in all conditions (Supplementary Figure S13).
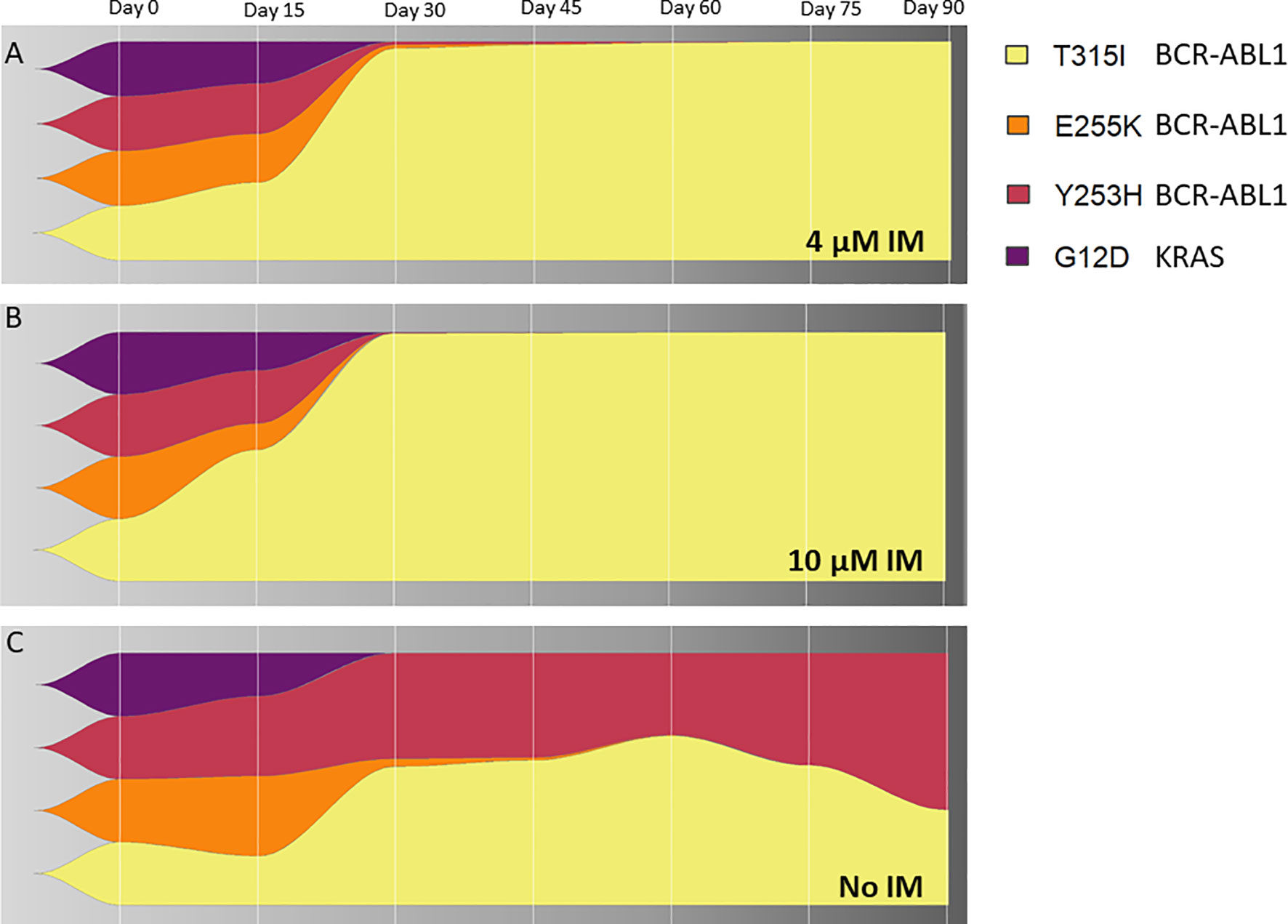
Figure 5 The clonal evolution of KCL-22R resistant clones. Starting from an equal number of cells (1x106) for each clone (Clone 1, Clone 2, Clone 3 and Clone 4 of KCL-22) growing in 4 µM imatinib, subsequent clonal evolution was followed by the monitoring of mutated BCR-ABL1 transcripts and mutated KRAS at selected time points indicated above the charts. Cells were cultivated for 90 days in stable conditions of (A) 4 µM imatinib, (B) 10 µM imatinib or (C) without imatinib. The superior growing fitness of Y253H-bearing Clone 2 in imatinib-free conditions is likely due to the presence of additional unmutated BCR-ABL1 fusion in this clone, as revealed by detected copy numbers by ddPCR and confirmed by cytogenetic analysis. The presence of BCR-ABL1 KD and KRAS mutations is shown as a percentage of total BCR-ABL1 or KRAS in the cells, respectively, determined by ASO-ddPCR.
The signaling properties of leukemic cells and resistant clones were studied at the protein level using a CyTOF custom-designed panel. BCR-ABL1-dependent pathways via STAT5, ERK1/2 and AKT were inhibited after the imatinib exposure and remained diminished in mutated resistant clones with slightly distinct pathway preferences dependent on the type of mutation and/or imatinib dose. Imatinib-resistant clones KCL-22R displayed persistently high expression of MYC, while overexpression of BCL-2 was additionally found in CML-T1R clones (Supplementary Results; Supplementary Figures S14–S17).
All 4 KCL-22R clones were able to engraft in the mice without any apparent difference between the imatinib-treated and untreated groups. Individual clones exhibited different engraftment abilities (Figure 6A). T315I-mutated Clone 1 exhibited the fastest growth in the imatinib-treated cohort, similar to in vitro conditions (Figure 6B). Similar data were observed in the imatinib-untreated cohort.
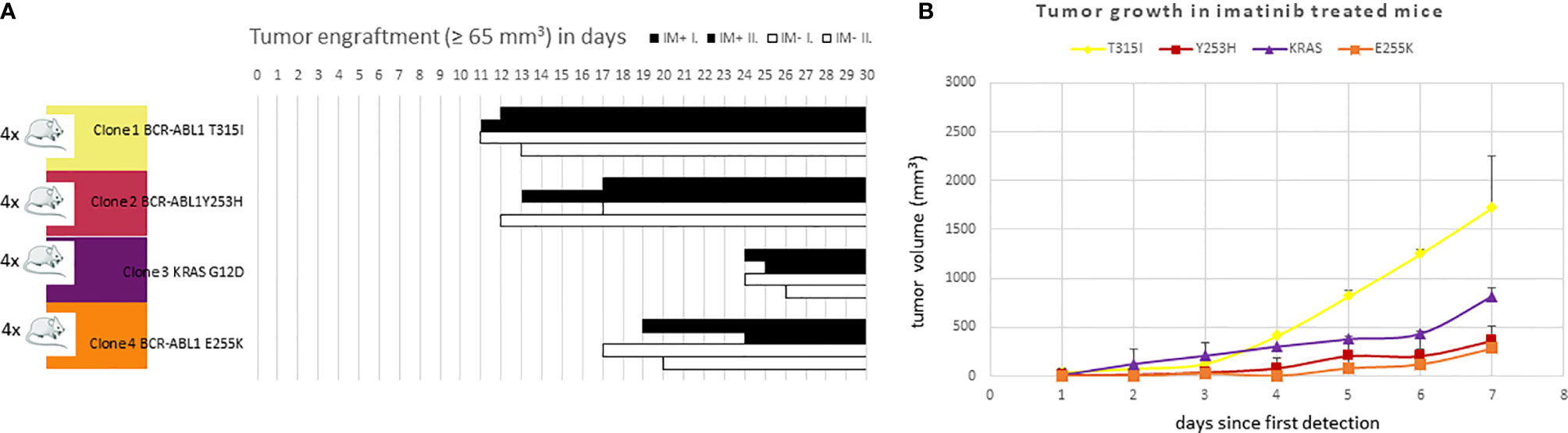
Figure 6 The efficacy of (A) engraftment and (B) tumor growth of imatinib-resistant KCL-22 clones in NSG mice. (A) A total of 1x106 cells of individual resistant clones were subcutaneously inoculated into mice (n = 4 per clone). After three days, mice were randomly distributed into imatinib-treated (n = 2 per clone; 100 mg/kg in PBS 1 x per day by oral gavage; IM+; black columns) and untreated/control (n = 2 per clone; 200 µl PBS 1 x per day by oral gavage; IM-; white columns) groups and monitored for tumor detection. The X-axis represents days since transplantation. Bars represent detected and growing tumors. (B) Tumor growth of imatinib-resistant KCL-22R clones in NSG mice treated with imatinib. The X-axis represents days since the first tumor detection. The Y-axis represents tumor volume. Error bars represent standard deviations.
Discussion
Both the selection of preexisting BCR-ABL1 mutated clones (15) and de novo acquisition during TKIs therapy (8, 9) were proposed to explain the emergence of TKI-resistant mutations. This study worked with the hypothesis of de novo acquisition of mutations conferring resistance to imatinib as the prevalent mechanism in CML cells.
The established in vitro model of CML KCL-22 cells has the ability to relapse after exposure to imatinib upon acquiring the T315I mutation (8). Despite differences in the procedure of establishing imatinib-resistant clones using single-cell FACS sorting, de novo acquisition of BCR-ABL1 KD and other somatic mutations was confirmed in this work by sensitive NGS and ASO-ddPCR. The same clonal population identically exposed to imatinib in parallel experiments was able to either develop different mutations or fail to acquire any mutation conferring resistance, thus proving the de novo acquisition process and supporting the hypothesis that the existence of the specific pool of pre-mutated or mutationally destined cells is unlikely for KCL-22 cells. De novo acquisition of BCR-ABL1 mutations was also confirmed in the CML-T1 model.
Recently, it was established that the genetic landscape of CML is significantly more diverse than assumed in the past, particularly in recurrent disease and advanced phases (6). Indeed, several mutated genes in addition to BCR-ABL1 were identified by the DNA NGS myeloid panel in resistant KCL-22R clones, namely, BCOR, ATRX, RUNX1 and KRAS. All these genes were previously found to be mutated in CML, although their clinical importance for CML patients remains unclear (5, 16, 17). RUNX1 was found to be commonly mutated during both CML-CP diagnosis and blast phase, often in cooccurrence with other recurrent mutations (17, 18). RUNX1 mutations are associated with the block of cell differentiation and with aggressive AML (19, 20). Notably, KCL-22 carries isochromosome 21 [i(21)(q10)], possessing 3 alleles of RUNX1, and mutations in RUNX1 were found to be associated with Chr21 trisomy (21). In the case of KRAS, de novo mutation acquisition was proven in the clone bearing mutations in ATRX and RUNX1. While KRAS mutations in CML seem to be particularly rare events (22), it should be noted that the pattern of acquisition of a KRAS mutation following RUNX1 genetic lesions was described in AML (23). The association of the BCOR and ATRX mutations with prognosis remains contradictory in CML (5, 24). In contrast to KCL-22R clones, no somatic mutations beside BCR-ABL1 KD were identified in CML-T1R clones. However, it must be mentioned that some of the genes frequently found to be mutated in the lymphoid blast phase (e.g., IKZF1) (17, 25) were omitted in the NGS myeloid gene panel.
The exact molecular mechanism of mutagenesis of BCR-ABL1 KD and other oncogenes remains unclear. It was proposed that BCR-ABL1-induced reactive oxygen species (ROS) are responsible for genomic instability and BCR-ABL1 mutation acquisition in CML cells (26–28). While ROS may definitely increase the genomic instability of CML cells and contribute to deregulation of DNA repair (29), the proposed pathways of ROS induction via STAT5 and PI3K/Akt were found to be inhibited in KCL-22 and CML-T1 cells treated with imatinib and remained diminished even after mutation acquisition, making their contribution to de novo mutagenesis questionable.
Therefore, the DNA damage response and DNA repair genes with notably changed expression at the time of mutation acquisition were identified and subsequently annotated to the functions in the mismatch repair and BER pathways. Only a few studies are examining the role of mismatch repair in CML. Rather, an inhibitory effect of BCR-ABL1 on the expression of mismatch repair genes was found in CML cells (30). The data from expression arrays in this work revealed that the expression of most of the mismatch repair-specific genes was unchanged during mutagenesis. By contrast, the expression of genes involved in the long-patch variant of the BER pathway was significantly upregulated during mutation acquisition. FEN1 has been observed overexpressed in a broad spectrum of cancers and was previously associated with PCNA-dependent genetic instability and DNA damage (31, 32). The increased expression of POLD3 was also observed during mutation acquisition. The POLD3 subunit of DNA polymerase δ was found to induce mutagenesis by promoting translesion synthesis. POLD3 is required for inducing C-to-T mutagenesis in an abasic site (33). Notably, C-to-T base exchange is behind the T315I BCR-ABL1 mutation, which is frequently acquired in KCL-22R clones and KCL-22 cells and is one of the most common mutations in advanced CML.
In addition to their role in BER (34, 35), UNG and LIG1 are also associated with the error-prone alt-NHEJ repair pathway of DSB (36). Alt-NHEJ was originally depicted as a backup pathway in cells with defective c-NHEJ (37), while some recent studies suggest that inactivation of c-NHEJ is not required for alt-NHEJ activity (38). The group of Chen et al. (8, 9) proposed that KCL-22 cells utilize NAD+-dependent c-NHEJ to induce point mutations (14). Overexpression of CD38 NAD+ hydrolase leads to abrogation of BCR-ABL1 mutation acquisition (14). The data of this work do not provide strong support for c-NHEJ involvement in de novo mutation acquisition. First, in contrast to Wang et al., CD38+ resistant KCL-22R clones with BCR-ABL1 KD mutations were also isolated. This discrepancy may be caused by the different origins of CD38+ KCL-22 cells tested in both studies, which include naturally occurring CD38+ cell subpopulation vs. cells with artificial CD38 overexpression, which may lose or diminish the mutagenesis ability. Second, the expression of LIG4, XRCC4 and XRCC6 genes was not changed in the KCL-22 (and CML-T1) model, and DNA methylation associated with repressive chromatin marks was found in the regulatory regions of LIG4. However, it should be noted, that upregulation of LIG4 expression, along with upregulation of alt-NHEJ genes, was observed during mutation acquisition in PB leukocytes of CML patients. Notwithstanding, data in general are in agreement with the studies proposing that the alt-NHEJ pathway is activated in TKI-resistant CML cells (10, 39). Increased expression of LIG1, UNG and PARP1 was found in cells undergoing mutagenesis. The expression of alt-NHEJ factors in CML was previously found to be augmented by the oncogenic transcription factor MYC (11). Accordingly, MYC is strongly expressed in both KCL-22R and CML-T1R clones and CML-resistant primary cells, as described previously (40), and its binding was enriched in the LIG1 transcription start site (TSS) at the time of mutation acquisition.
The growth abilities of individual KCL-22R and CML-T1R clones were assessed in different environmental conditions in vitro and in vivo with the conclusion that clonal evolution is driven by both mutation-type specific oncogenic properties and additional genetic/cytogenetic lesions in concert with environmental selection pressures. At high imatinib concentrations, T315I-bearing Clone 1 showed superior growth over other KCL-22R clones, in agreement with the superior resistance associated with the T315I mutation (41). The growing abilities of individual KCL-22R clones only partially reflect kinase activity revealed by CyTOF, a fact already described by others (42, 43). Conversely, the finding that the activity of the main BCR-ABL1-associated signaling pathways in KCL-22R clones is generally lower than that in imatinib-naïve KCL-22 cells is in accordance with the fact that BCR-ABL1 mutations do not confer a growth advantage in the absence of imatinib (44).
Taken together, this study provides evidence for the responsibility of de novo mutation acquisition for acquired imatinib resistance in CML-CP. Imatinib has been the most used frontline TKI to combat CML-CP, and it likely will remain in this position in the future owing to the benefits of low toxicity and cost-effectiveness even after emerging therapy goal of treatment-free remission has been considered (45, 46). For CML-CP patients, no robust evidence has conclusively shown that BCR-ABL1 KD mutations conferring resistance to imatinib may already be detectable at diagnosis, which was also confirmed in this work (47, 48). Somatic mutations acquired de novo in the follow-up constitute a bulwark of lesions associated with refractory and progressed CML with poor outcomes (5).
The data support the considered role of the alt-NHEJ repair pathway (11) and provide the first hint that BER, particularly the long-patch variant, may be involved in the acquisition of somatic point mutations in oncogenes in CML. Contrary to some other natural in vitro models of BCR-ABL1 mutagenesis (49), BCR-ABL1 KD mutations acquired in resistant KCL-22R and CML-T1R clones belong among the mutations most commonly detected in both CML-CP and advanced CML (50). Moreover, the same mechanism of acquisition may be undergoing in the case of somatic mutations in other cancer genes, thus underlying the clinical relevance of the presented findings.
In conclusion, somatic mutations associated with resistance to imatinib are acquired in CML-CP de novo, thus early mutation detection is highly required to prevent progression because several therapeutic options exist for cases with mutated KD of BCR-ABL1. However, even when mutations in other cancer genes can be uncovered early by currently feasible NGS approaches, the resistance associated with other somatic mutations or in combination with mutated BCR-ABL1 remains therapeutically challenging, which will require highly individualized treatment management in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject PRJNA750307.
Ethics Statement
The studies involving human participants were reviewed and approved by UHKT Ethics Committee. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Animal Care and the Use Committee, First Faculty of Medicine, Charles University, Prague.
Author Contributions
NC wrote the manuscript, performed experiments, evaluated data, and contributed to the interpretation of the results. VP performed the majority of the experiments, evaluated data, and contributed to the interpretation of the results. PB performed experiments, analyzed data, and revised the paper. JK performed NGS analysis and evaluated data. AL performed in vivo experiments. TK and DKuz designed and interpreted mass cytometry data. JB and SR performed cytogenetic analysis and interpreted data. DKar performed expression arrays. KR prepared samples for mass cytometry analysis and interpreted data. VK and MB performed cell tracking and cell cycle analysis. DKun evaluated clonal development. JL performed resistant clone establishment. HK and CS provided patient samples and clinical data. PK supervised the in vivo experiments. OH supervised mass cytometry and flow cytometry analysis, interpreted data and critically revised the manuscript. KMP designed experiments, coordinated the research, interpreted the results, and revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Czech Science Foundation (GACR) project no. 18-18407 and the Czech Ministry of Health project no. 00023736 for the conceptual development of a research institute.
Conflict of Interest
HK was a consultant for and received honoraria from Novartis. KMP was a consultant for and received honoraria from Incyte and Angelini.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
A murine cell line-derived xenograft (CDX) model of BCR-ABL1 mutation acquisition and evolution was kindly provided by the Center for Experimental Biomodels, First Faculty of Medicine, Charles University. We would like to acknowledge Eva Pokorna (Institute of Pathological Physiology, First Faculty of Medicine, Charles University) for the excellent technical support for in vivo mice experiments. We would like to acknowledge Pavel Semerak (CLIP laboratories, Faculty Hospital Motol and Second Faculty of Medicine, Charles University) for the excellent technical support for single-cell sorting and CyTOF analysis. CyTOF2 instrument was financed through Operational Program Prague Competitiveness (CZ.2.16/3.1.00/21540).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.744373/full#supplementary-material
Abbreviations
Alt-NHEJ, alternative nonhomologous end-joining; BER, base-excision repair; BM, bone marrow; CML, chronic myeloid leukemia; c-NHEJ, classical nonhomologous end-joining; CP, chronic phase; DMR, deep molecular response; DSB, double strand break; ELN, European Leukemia Net; FC, fold change; IdU, 5-iodo-2′-deoxyuridine; KD, kinase domain; MMR, major molecular response; NGS, Next Generation Sequencing; PB, peripheral blood; TKIs, tyrosine kinase inhibitors; TSS, transcription start site; WT, wild type.
References
1. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia (2020) 34(4):966–84. doi: 10.1038/s41375-020-0776-2
2. Radich JP, Deininger M, Abboud CN, Altman JK, Berman E, Bhatia R, et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw (2018) 16(9):1108–35. doi: 10.6004/jnccn.2018.0071
3. Quintás-Cardama A, Kantarjian HM, Cortes JE. Mechanisms of Primary and Secondary Resistance to Imatinib in Chronic Myeloid Leukemia. Cancer Control (2009) 16(2):122–31. doi: 10.1177/107327480901600204
4. Jabbour E, Kantarjian H, Jones D, Talpaz M, Bekele N, O’Brien S, et al. Frequency and Clinical Significance of BCR-ABL Mutations in Patients With Chronic Myeloid Leukemia Treated With Imatinib Mesylate. Leukemia (2006) 20(10):1767–73. doi: 10.1038/sj.leu.2404318
5. Kim T, Tyndel MS, Kim HJ, Ahn J, Choi SH, Park HJ, et al. Spectrum of Somatic Mutation Dynamics in Chronic Myeloid Leukemia Following Tyrosine Kinase Inhibitor Therapy. Blood (2017) 129(1):38–47. doi: 10.1182/blood-2016-04-708560
6. Branford S, Kim DDH, Apperley JF, Eide CA, Mustjoki S, Ong ST, et al. Laying the Foundation for Genomically-Based Risk Assessment in Chronic Myeloid Leukemia. Leukemia (2019) 33(8):1835–50. doi: 10.1038/s41375-019-0512-y
7. Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, et al. High-Sensitivity Detection of BCR-ABL Kinase Domain Mutations in Imatinib-Naive Patients: Correlation With Clonal Cytogenetic Evolution But Not Response to Therapy. Blood (2005) 106(6):2128–37. doi: 10.1182/blood-2005-03-1036
8. Yuan H, Wang Z, Gao C, Chen W, Huang Q, Yee J, et al. BCR-ABL Gene Expression is Required for its Mutations in a Novel KCL-22 Cell Culture Model for Acquired Resistance of Chronic Myelogenous Leukemia. J Biol Chem (2010) 285(7):5085–96. doi: 10.1074/jbc.M109.039206
9. Wang Z, Yuan H, Roth M, Stark JM, Bhatia R, Chen WY. SIRT1 Deacetylase Promotes Acquisition of Genetic Mutations for Drug Resistance in CML Cells. Oncogene (2013) 32(5):589–98. doi: 10.1038/onc.2012.83
10. Tobin LA, Robert C, Rapoport AP, Gojo I, Baer MR, Tomkinson AE, et al. Targeting Abnormal DNA Double-Strand Break Repair in Tyrosine Kinase Inhibitor-Resistant Chronic Myeloid Leukemias. Oncogene (2013) 32(14):1784–93. doi: 10.1038/onc.2012.203
11. Muvarak N, Kelley S, Robert C, Baer MR, Perrotti D, Gambacorti-Passerini C, et al. C-MYC Generates Repair Errors via Increased Transcription of Alternative-NHEJ Factors, LIG3 and PARP1, in Tyrosine Kinase-Activated Leukemias. Mol Cancer Res (2015) 13(4):699–712. doi: 10.1158/1541-7786.MCR-14-0422
12. Pelák O, Kužílková D, Thürner D, Kiene M, Stanar K, Stuchlý J, et al. Lymphocyte Enrichment Using CD81-Targeted Immunoaffinity Matrix. Cytometry A (2017) 91(1):62–72. doi: 10.1002/cyto.a.22918
13. Miller CA, McMichael J, Dang HX, Maher CA, Ding L, Ley TJ, et al. Visualizing Tumor Evolution With the Fishplot Package for R. BMC Genomics (2016) 17(1):880. doi: 10.1186/s12864-016-3195-z
14. Wang Z, Liu Z, Wu X, Chu S, Wang J, Yuan H, et al. ATRA-Induced Cellular Differentiation and CD38 Expression Inhibits Acquisition of BCR-ABL Mutations for CML Acquired Resistance. PloS Genet (2014) 10(6):e1004414. doi: 10.1371/journal.pgen.1004414
15. Carella AM, Garuti A, Cirmena G, Catania G, Rocco I, Palermo C, et al. Kinase Domain Mutations of BCR-ABL Identified at Diagnosis Before Imatinib-Based Therapy are Associated With Progression in Patients With High Sokal Risk Chronic Phase Chronic Myeloid Leukemia. Leuk Lymphoma (2010) 51(2):275–8. doi: 10.3109/10428190903503446
16. Soverini S, de Benedittis C, Mancini M, Martinelli G. Mutations in the BCR-ABL1 Kinase Domain and Elsewhere in Chronic Myeloid Leukemia. Clin Lymphoma Myeloma Leuk (2015) 15 Suppl:120. doi: 10.1016/j.clml.2015.02.035
17. Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative Genomic Analysis Reveals Cancer-Associated Mutations at Diagnosis of CML in Patients With High-Risk Disease. Blood (2018) 132(9):948–61. doi: 10.1182/blood-2018-02-832253
18. Grossmann V, Kohlmann A, Klein H, Schindela S, Schnittger S, Dicker F, et al. Targeted Next-Generation Sequencing Detects Point Mutations, Insertions, Deletions and Balanced Chromosomal Rearrangements as Well as Identifies Novel Leukemia-Specific Fusion Genes in a Single Procedure. Leukemia (2011) 25(4):671–80. doi: 10.1038/leu.2010.309
19. Khan M, Cortes J, Kadia T, Naqvi K, Brandt M, Pierce S, et al. Clinical Outcomes and Co-Occurring Mutations in Patients With RUNX1-Mutated Acute Myeloid Leukemia. Int J Mol Sci (2017) 18(8):1618. doi: 10.3390/ijms18081618
20. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations From an International Expert Panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
21. Roche-Lestienne C, Deluche L, Corm S, Tigaud I, Joha S, Philippe N, et al. RUNX1 DNA-Binding Mutations and RUNX1-PRDM16 Cryptic Fusions in BCR-ABL+ Leukemias are Frequently Associated With Secondary Trisomy 21 and may Contribute to Clonal Evolution and Imatinib Resistance. Blood (2008) 111(7):3735–41. doi: 10.1182/blood-2007-07-102533
22. Agarwal A, Eide CA, Harlow A, Corbin AS, Mauro MJ, Druker BJ, et al. An Activating KRAS Mutation in Imatinib-Resistant Chronic Myeloid Leukemia. Leukemia (2008) 22(12):2269–72. doi: 10.1038/leu.2008.124
23. Martignoles J, Delhommeau F, Hirsch P. Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. Int J Mol Sci (2018) 19(12):3850. doi: 10.3390/ijms19123850
24. Adnan Awad S, Kankainen M, Ojala T, Koskenvesa P, Eldfors S, Ghimire B, et al. Mutation Accumulation in Cancer Genes Relates to Nonoptimal Outcome in Chronic Myeloid Leukemia. Blood Adv (2020) 4(3):546–59. doi: 10.1182/bloodadvances.2019000943
25. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 Lymphoblastic Leukaemia is Characterized by the Deletion of Ikaros. Nature (2008) 453(7191):110–4. doi: 10.1038/nature06866
26. Grundschober E, Warsch W, Sexl V. The Transcription Factor STAT5 Drives Mutation and Imatinib Resistance in Chronic Myeloid Leukemia. Via ROS Prod BMC Pharmacol (2011) 11(Suppl 2):A22. doi: 10.1186/1471-2210-11-S2-A22
27. Nieborowska-Skorska M, Flis S, Skorski T. AKT-Induced Reactive Oxygen Species Generate Imatinib-Resistant Clones Emerging From Chronic Myeloid Leukemia Progenitor Cells. Leukemia (2014) 28(12):2416–8. doi: 10.1038/leu.2014.249
28. Cheng Y, Hao Y, Zhang A, Hu C, Jiang X, Wu Q, et al. Persistent STAT5-Mediated ROS Production and Involvement of Aberrant P53 Apoptotic Signaling in the Resistance of Chronic Myeloid Leukemia to Imatinib. Int J Mol Med (2018) 41(1):455–63. doi: 10.3892/ijmm.2017.3205
29. Pawlowska E, Blasiak J. DNA Repair–A Double-Edged Sword in the Genomic Stability of Cancer Cells–The Case of Chronic Myeloid Leukemia. Int J Mol Sci (2015) 16(11):27535–49. doi: 10.3390/ijms161126049
30. Stoklosa T, Poplawski T, Koptyra M, Nieborowska-Skorska M, Basak G, Slupianek A, et al. BCR/ABL Inhibits Mismatch Repair to Protect From Apoptosis and Induce Point Mutations. Cancer Res (2008) 68(8):2576–80. doi: 10.1158/0008-5472.CAN-07-6858
31. Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, et al. Overexpression and Hypomethylation of Flap Endonuclease 1 Gene in Breast and Other Cancers. Mol Cancer Res (2008) 6(11):1710–7. doi: 10.1158/1541-7786.MCR-08-0269
32. Becker JR, Gallo D, Leung W, Croissant T, Thu YM, Nguyen HD, et al. Flap Endonuclease Overexpression Drives Genome Instability and DNA Damage Hypersensitivity in a PCNA-Dependent Manner. Nucleic Acids Res (2018) 46(11):5634–50. doi: 10.1093/nar/gky313
33. Hirota K, Yoshikiyo K, Guilbaud G, Tsurimoto T, Murai J, Tsuda M, et al. The POLD3 Subunit of DNA Polymerase δ can Promote Translesion Synthesis Independently of DNA Polymerase ζ. Nucleic Acids Res (2015) 43(3):1671–83. doi: 10.1093/nar/gkv023
34. Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base Excision Repair Initiation Revealed by Crystal Structures and Binding Kinetics of Human Uracil-DNA Glycosylase With DNA. EMBO J (1998) 17(17):5214–26. doi: 10.1093/emboj/17.17.5214
35. DeMott MS, Zigman S, Bambara RA. Replication Protein A Stimulates Long Patch DNA Base Excision Repair. J Biol Chem (1998) 273(42):27492–8. doi: 10.1074/jbc.273.42.27492
36. Limpose KL, Corbett AH, Doetsch PW. BERing the Burden of Damage: Pathway Crosstalk and Posttranslational Modification of Base Excision Repair Proteins Regulate DNA Damage Management. DNA Repair (Amst) (2017) 56:51–64. doi: 10.1016/j.dnarep.2017.06.007
37. Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, et al. Mechanisms of DNA Double Strand Break Repair and Chromosome Aberration Formation. Cytogenet Genome Res (2004) 104(1-4):14–20. doi: 10.1159/000077461
38. Xiong X, Du Z, Wang Y, Feng Z, Fan P, Yan C, et al. 53BP1 Promotes Microhomology-Mediated End-Joining in G1-Phase Cells. Nucleic Acids Res (2015) 43(3):1659–70. doi: 10.1093/nar/gku1406
39. Sallmyr A, Tomkinson AE, Rassool FV. Up-Regulation of WRN and DNA Ligase IIIalpha in Chronic Myeloid Leukemia: Consequences for the Repair of DNA Double-Strand Breaks. Blood (2008) 112(4):1413–23. doi: 10.1182/blood-2007-07-104257
40. Srutova K, Curik N, Burda P, Savvulidi F, Silvestri G, Trotta R, et al. BCR-ABL1 Mediated miR-150 Downregulation Through MYC Contributed to Myeloid Differentiation Block and Drug Resistance in Chronic Myeloid Leukemia. Haematologica (2018) 103(12):2016–25. doi: 10.3324/haematol.2018.193086
41. O’Hare T, Eide CA, Deininger MWN. Bcr-Abl Kinase Domain Mutations, Drug Resistance, and the Road to a Cure for Chronic Myeloid Leukemia. Blood (2007) 110(7):2242–9. doi: 10.1182/blood-2007-03-066936
42. Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, et al. Phosphorylation of the ATP-Binding Loop Directs Oncogenicity of Drug-Resistant BCR-ABL Mutants. Proc Natl Acad Sci U S A (2006) 103(51):19466–71. doi: 10.1073/pnas.0609239103
43. Griswold IJ, MacPartlin M, Bumm T, Goss VL, O’Hare T, Lee KA, et al. Kinase Domain Mutants of Bcr-Abl Exhibit Altered Transformation Potency, Kinase Activity, and Substrate Utilization, Irrespective of Sensitivity to Imatinib. Mol Cell Biol (2006) 26(16):6082–93. doi: 10.1128/MCB.02202-05
44. Miething C, Feihl S, Mugler C, Grundler R, von Bubnoff N, Lordick F, et al. The Bcr-Abl Mutations T315I and Y253H do Not Confer a Growth Advantage in the Absence of Imatinib. Leukemia (2006) 20(4):650–7. doi: 10.1038/sj.leu.2404151
45. Hantel A, Larson RA. Imatinib is Still Recommended for Frontline Therapy for CML. Blood Adv (2018) 2(24):3648–52. doi: 10.1182/bloodadvances.2018018614
46. Shih YT, Cortes JE, Kantarjian HM. Treatment Value of Second-Generation BCR-ABL1 Tyrosine Kinase Inhibitors Compared With Imatinib to Achieve Treatment-Free Remission in Patients With Chronic Myeloid Leukaemia: A Modelling Study. Lancet Haematol (2019) 6(8):e398–408. doi: 10.1016/S2352-3026(19)30087-0
47. Schmitt MW, Pritchard JR, Leighow SM, Aminov BI, Beppu L, Kim DS, et al. Single-Molecule Sequencing Reveals Patterns of Preexisting Drug Resistance That Suggest Treatment Strategies in Philadelphia-Positive Leukemias. Clin Cancer Res (2018) 24(21):5321–34. doi: 10.1158/1078-0432.CCR-18-0167
48. Machova Polakova K, Kulvait V, Benesova A, Linhartova J, Klamova H, Jaruskova M, et al. Next-Generation Deep Sequencing Improves Detection of BCR-ABL1 Kinase Domain Mutations Emerging Under Tyrosine Kinase Inhibitor Treatment of Chronic Myeloid Leukemia Patients in Chronic Phase. J Cancer Res Clin Oncol (2015) 141(5):887–99. doi: 10.1007/s00432-014-1845-6
49. Dong Y, Gao X, Zhao Y, Wei M, Xu L, Yang G, et al. Semi-random Mutagenesis Profile of BCR-ABL During Imatinib Resistance Acquirement in K562 Cells. Mol Med Rep (2017) 16(6):9409–14. doi: 10.3892/mmr.2017.7835
Keywords: chronic myelogenous leukemia (CML), base excision repair (BER), alt-nonhomologous end joining (alt-NHEJ), TKI resistance, oncogene
Citation: Curik N, Polivkova V, Burda P, Koblihova J, Laznicka A, Kalina T, Kanderova V, Brezinova J, Ransdorfova S, Karasova D, Rejlova K, Bakardjieva M, Kuzilkova D, Kundrat D, Linhartova J, Klamova H, Salek C, Klener P, Hrusak O and Machova Polakova K (2021) Somatic Mutations in Oncogenes Are in Chronic Myeloid Leukemia Acquired De Novo via Deregulated Base-Excision Repair and Alternative Non-Homologous End Joining. Front. Oncol. 11:744373. doi: 10.3389/fonc.2021.744373
Received: 20 July 2021; Accepted: 02 September 2021;
Published: 20 September 2021.
Edited by:
Swami P. Iyer, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Tej K. Pandita, Baylor College of Medicine, United StatesVijaya Charaka, Houston Methodist Research Institute, United States
Copyright © 2021 Curik, Polivkova, Burda, Koblihova, Laznicka, Kalina, Kanderova, Brezinova, Ransdorfova, Karasova, Rejlova, Bakardjieva, Kuzilkova, Kundrat, Linhartova, Klamova, Salek, Klener, Hrusak and Machova Polakova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katerina Machova Polakova, a2F0ZXJpbmEubWFjaG92YUB1aGt0LmN6
†These authors have contributed equally to this work