Abstract
Non-small cell lung carcinoma (NSCLC) comprises 80%–85% of lung cancer cases. EGFR is involved in several cancer developments, including NSCLC. The EGFR pathway regulates the Bax/Bcl-2 cascade in NSCLC. Increasing understanding of the molecular mechanisms of fundamental tumor progression has guided the development of numerous antitumor drugs. The development and improvement of rationally planned inhibitors and agents targeting particular cellular and biological pathways in cancer have been signified as a most important paradigm shift in the strategy to treat and manage lung cancer. Newer approaches and novel chemotherapeutic agents are required to accompany present cancer therapies for improving efficiency. Using natural products as a drug with an effective delivery system may benefit therapeutics. Naturally originated compounds such as phytochemicals provide crucial sources for novel agents/drugs and resources for tumor therapy. Applying the small-molecule inhibitors (SMIs)/phytochemicals has led to potent preclinical discoveries in various human tumor preclinical models, including lung cancer. In this review, we summarize recent information on the molecular mechanisms of the Bax/Bcl-2 cascade and EGFR pathway in NSCLC and target them for therapeutic implications. We further described the therapeutic potential of Bax/Bcl-2/EGFR SMIs, mainly those with more potent and selectivity, including gefitinib, EGCG, ABT-737, thymoquinone, quercetin, and venetoclax. In addition, we explained the targeting EGFR pathway and ongoing in vitro and in vivo and clinical investigations in NSCLC. Exploration of such inhibitors facilitates the future treatment and management of NSCLC.
1 Introduction
Among all cancer deaths, lung cancer mortality is very common, estimated up to 1.7 million per year worldwide (1). There are two histological subtypes: non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC). NSCLC comprises around 80% to 85% of all lung cancer cases. Tobacco smoking is the root cause of NSCLC which comprises around 80% of cases in the United States and other countries where smoking is common (2). Although the most common etiology behind NSCLC and SCLC is smoking, lung adenocarcinoma (LUAD) is mostly associated with non-smokers. Non-smoker LUAD is commonly found in East Asian women with environmental exposure and genetic reasons. According to the recent document, the standard 5-year survival rate for NSCLC is barely 16% (3). Over half of lung tumor cases are diagnosed following metastasis, for which the mean survival time is about 8 months. Among lung tumor bone metastasis, the majority common target organ is the vertebral column, which causes more severe effects on patients’ recovery rate and life worth (4, 5).
EGFR overexpression has been involved in cancers, including NSCLC (6). EGFR is one of the most generally mutated genes in NSCLC (7). EGFR is associated with several human malignancies (8). Around 10%–30% of NSCLC patients have activating mutations in EGFR (9, 10). Increased EGFR protein and mRNA expressions are linked with poor prognosis, tumor growth, metastasis, and resistance to chemotherapy (11). EGFR activation is linked with proliferation, metastasis, apoptosis inhibition, and radio-chemotherapy resistance in cancer (12). Aberrant activation of the EGFR pathway axis has been found to play a major role in cancer (13). A study reported MAPK1 amplification in an erlotinib-resistant EGFR-mutant NSCLC (14). The histological transformation to SCLC in EGFR mutant-NSCLC patients with acquired EGFR TKI resistance. Similarly, aberrant induction of the EGFR–STAT-3–Bcl-xL signaling axis has been observed to fuel cancer progression (13, 15, 16). Bcl-2 and Bcl-xL are Akt/EGFR downstream pathway proteins which were reported to be conscientious to drug resistance in several tumors, such as SCLC (17, 18).
EGFR has been considered an important target for NSCLC therapeutics (19). Numerous studies have documented the mechanisms engaged in the progression of AR to EGFR TKI, which may be potential therapeutic strategies (20, 21). The inhibition of EGFR could be a potential clinical strategy for inhibiting and overcoming EMT-linked acquired drug resistance that affords inspiration for the clinical trial of combined EGFR and FGFR blockage in EGFR-mutated NSCLCs (21, 22). Targeted therapy opens new dynamics in lung cancer management by identifying altered target genes. Targeting EGFR in the patients with stimulating mutations showed initial and considerable success in the clinic (23, 24). However, the inhibition of EGFR leads to upregulation of pro-apoptotic proteins and, lastly, results in apoptosis by activating the intrinsic apoptotic pathway (25, 26). Earlier reports (27, 28) demonstrated the important function of Bcl-2 in the resistance of NSCLC for EGFR TKIs. It validated that growth inhibition is induced by the treatment activation of caspase-3 and Bax, supposedly by EGFR, ERKs, and MMP-2 downregulation (29). To develop effective therapies against lung cancer, it is very important to understand its biology at the molecular level (30). In the current study, we presented recent information on the molecular mechanisms of the Bax/Bcl-2 cascade-mediated EGFR pathway in NSCLC and its therapeutic implications along with clinical investigations that facilitate the treatment and management of several cancers, including NSCLC.
2 Regulation of Bax in NSCLC
Bax is a pro-apoptotic protein that plays a pivotal role in controlling apoptosis. It is generally present in the cytoplasm, which is heterodimerized with anti-apoptotic proteins. When a cell is exposed to an apoptotic stimulus, Bax protein is translocated to the mitochondria (31). The increased expression of Bax mediates in early apoptosis (32). Bcl-2 family members share at least one of four types of homology, namely, BH1, BH2, BH3, and BH4 (33). Their structure can be homodimers or heterodimers having nine α helices and a hydrophobic α-helix embedded in the core with a transmembrane terminal C attached at the mitochondrial lining (33–35). The activation of Bax can be initiated by various abiotic factors such as heat, pH change, and stress conditions (36, 37). p53 upregulates Bax in the stress environment as a stimulus response, further activating downstream target genes like Bax (38). The Bax gene was first reported as one of the important pro-apoptotic Bcl-2 family proteins (39). The tertiary structure of Bax is exhibited in Figure 1A.
Figure 1
The human Bax gene is present in chromosome 19q and consists of six exons and four variants (41). Mutations and alterations in the coding regions and promoters of the Bax gene have been detected for affecting the protein expression and function in a variety of malignancies (42, 43). Mutations in the Bax gene are very frequent, resulting in loss of the tumor-suppressor function and resistance to apoptosis and chemotherapy (44, 45). The single-nucleotide polymorphism (SNP) of the Bax gene, i.e., at -125 nucleotides, a G to A transition, from the beginning of transcription, and at -248 nucleotides from the translation initiation, has been recognized in cancer. This SNP was linked with altered mRNA/protein expression associated with cancer development and chemoresistance (46, 47). An elevated risk of HNSCC in patients having the AA genotype of G(-248)A SNP was reported (42). Hence, Bax is an important gene in oral cancer development. The effects of promoter methylation/SNP, mutations of the exons, and reduced expression of Bax were associated with oral cancer development (48).
A low expression of Bax has been drastically linked with NSCLC patients (49) and poor prognosis in NSCLC patients (50). A decreased expression of Bax and p53 was comparatively resistant to cisplatin and decreased apoptosis in lung cancer cells (51). Radio-resistant NSCLC exhibits little box/Bak activation compared with radiosensitive NSCLCs (52). A low Bax protein expression was demonstrated for contributing to oral cancer progression (53, 54). A low expression of Bax is associated with decreased apoptosis, advanced-stage neoplasms, poor prognosis, cancer progression, and resistance to chemotherapy in colorectal cancer (55, 56). The regulation of Bax-induced apoptosis in NSCLC is incompletely understood.
3 Regulation of Bcl-2 in NSCLC
B cell lymphoma 2 (Bcl-2) is an anti-apoptotic protein encoded by the Bcl-2 gene in the human genome specified as an oncogene (57). It was recognized for its involvement in t(14;18) chromosomal translocations detected in non-Hodgkin’s lymphomas (57). Bcl-2 is ~250 kb in length and made up of three (03) exons and two (02) promoters (58), exon 1 and exon 2 encoding all four BH domains, whereas exon 3 encodes the TM domain that connects the protein to intracellular membranes (59, 60). It was the primary protein to be recognized among Bcl-2 family proteins. There are two isoforms of Bcl-2: Bcl-2α and Bcl-2β. Hence, Bcl-2α is anti-apoptotic (61). The Bcl-2 protein has a common genetic region in the BH domain; it includes up to four conserved BH domains (62, 63). Bcl-2 (239 amino acids) contains four domains, namely, BH1, BH2, BH3, and BH4 (64, 65). Additionally, these domains compose BH4 domain (10–30) residues, BH3 domain (93–107) residues, BH1 domain (136–155) residues, and BH2 domain (187–202) residues (64). Bcl-2 explains a tertiary structure (Figure 1B) enclosing two hydrophobic α-helices (Hα5 and Hα6) surrounded by amphipathic α-helices (40, 66). The Bcl-2 gene activates via a chromosomal translocation mechanism in many human tumors (67, 68). Bcl-2 inhibits apoptosis via inhibiting the liberation of cyt-c, thus blocking the activation of caspases that stimulate apoptosis (69). Bcl-2 binds with BH3 domains of Bax and inhibits their functioning (70, 71).
Bcl-2 promotes cell survival and viability and regulates mitochondrial dynamics like fusion and fission. An increased Bcl-2 expression has been observed in NSCLC (72). The expression of Bcl-2 and mutated p53 might be highly resistant to cisplatin and have low susceptibility for apoptosis in lung cancer cells (51). Bcl-2 has induced cancer growth and resistance to chemotherapeutics in xenograft models of NSCLC (73, 74). Bcl-2 is considered a good prognostic marker in NSCLC (75, 76) and a constructive prognostic biomarker in LUSC (77). The evaluation of Bcl-2 expression by tumors could provide predictive data on the clinical manners of NSCLC (78). An excessive expression of Bcl-2 and a suppressed expression of Bax lead to homeostatic disbalance of cells, subsequently causing cancer. However, according to a recent study, 76% of SCLC is caused by the overexpression of Bcl-2 (79, 80). Bcl-2 is a critical player in imparting resistance to cancer cells (81, 82). Increased Bcl-2 expression is linked with advanced-stage neoplasms and poor differentiation (83) and is found to resist chemotherapy in many cancers (56). A high expression of Bcl-2 protein was found in many drug-resistant cancer cells (53, 84). However, Bcl-2 transcript cleavage induces cell death and impairs cell survival (85, 86). The upregulation of Bcl-2 defends drug-mediated cells from apoptosis (87). Bcl-2 participates in a fundamental function in cancer growth, angiogenesis, and tumor vascular density (88). Therefore, Bcl-2 is involved in NSCLC.
4 Regulation of EGFR in NSCLC
EGFR contains an extracellular EGF-attaching domain, a transmembrane domain, and a cytoplasmic domain (89). EGFR is a transmembrane cell-surface receptor. The tyrosine kinase (TK) receptor is commonly activated in epithelial tumors (90). The corresponding mRNA is encoded from 28 exons spanning approximately 190,000 (nucleotides) on chromosome 7p12. It belongs to the ErbB family of receptor tyrosine kinases that also includes ErbB2 (HER-2 or Neu), ErbB3 (HER-3), and ErbB4 (HER-4) (91). The activated EGFR causes the activation of several pathways, including ERK and Stat-3 pathways (92). ERK and Stat-3 are the important signaling molecules under EGFR (93). EGFR activates the PI3K-Akt, STAT, and MAPK pathways, eventually leading to enhanced cell proliferation, survival, and migration (26, 94–96) (Figure 2). EGFR overexpression has been involved in multiple cancers, including NSCLC (6). EGFR is one of the most generally mutated genes in NSCLC (7). Around 10%–30% of NSCLC patients have activating mutations in EGFR (9, 10). Major scientific and clinical studies have proved that alleles of patients with NSCLC revealed mutations in KRAS and EGFR genes, which demonstrate their function in tumor development and progression (100, 101). KRAS and EGFR mutations occur mutually and have symbiotic relations where KRAS mutations may bestow resistance to EGFR inhibitors (102, 103).
Figure 2
Several studies have explained mutations in the EGFR gene (6, 104). EGFR mutations are frequently oncogenic; specifically, they trigger the EGFR pathway in the lack of ligand and endorse cell survival and anti-apoptotic signals (6, 105). EGFR, TK type I receptors, and its gene are situated at the short arm of human chromosome 7 (106). In EGFR, 28 exons form a protein, which is dispensed on the cell membrane of several epithelial cells, wherever it attaches to EGF or heparin-binding EGF and controls cell growth (107). By comparison, exon 20 insertions and exon 18-point alterations/mutations are less general than exon 19 deletions and exon 21 L858R substitutions in EGFR mutations in NSCLC (108, 109). The regulation and activation of EGFR and downstream genes initiate apoptosis, angiogenesis, and proliferation (110). Hence, the most frequent are first short in-frame deletions about the LREA motif of exon 19 (~45%–50% mutations) and 2nd point mutations (CTG to CGG) in exon 21, which affects the substitution of leucine via arginine on codon 858, L858R (~45%–50% mutations) (111, 112). These alterations/mutations are more common in NSCLC (111–113). The histological transformation to SCLC in EGFR mutant-NSCLC patients with acquired EGFR TKI resistance. Numerous studies have documented the mechanisms engaged in the progression of AR to EGFR TKI, which may be potential therapeutic strategies (20, 21).
More EGFR protein and mRNA expressions are linked with poor prognosis, more tumor growth, metastasis, and resistance to chemotherapy (11). EGFR plays a major role in various human malignancies (8). EGFR activation is linked with the malignant phenotype, blockage of apoptosis, increased proliferation, metastasis, and resistance to radio-chemotherapy (12). Aberrant activation of the EGFR pathway axis was found to play a major role in HNSCC (13). EGFR is associated with oral cancer development and chemoresistance (93, 114). EGFR activation has been detected in oral cancer (8, 115). EGFR is linked with inhibition of apoptosis and resistance to chemotherapy in various tumors (116, 117). Numerous signaling molecules lie downstream of EGFR involved in cancer development (93).
5 Targeting the EGFR Pathway in NSCLC
Targeted therapy opens new dynamics in lung cancer management by identifying altered target genes. According to the biological function of diverse forms of EGFR in NSCLC, EGFR-targeted therapy is divided into two parts: “EGFR mutant targeted therapy” and “wt EGFR targeted therapy.” Among NSCLC patients, ~10%–30% have lung cancers with EGFR mutations (118). EGFR mutant targeted therapy targets the main oncogene linked with tumorigenesis and is important for cancer maintenance. However, this therapy displays a remarkable comeback in clinical TKI treatment through induction of apoptosis. Similarly, above 70% of NSCLC patients by wt EGFR treated with TKIs are getting “wt EGFR targeted therapy.” Therefore, it targets a protein not openly connected to cancer initiation but more liable to cell growth. This therapy is far less efficient in the clinical phase. It is normally linked with growth arrest and stable disease (SD) (118, 119).
The EGFR mutational summary of NSCLCs is a good predictor of reaction for therapy with the extremely efficient TKIs (120, 121). TKIs have been considered proficient drugs in NSCLC and have provided brilliantly targeted drugs (122). TKIs targeting EGFR were checked in clinical trials approved by the FDA (121, 123). Multiple agents/drugs targeting EGFR have appeared, including gefitinib, erlotinib, panitumumab, and cetuximab (124–126). Among the approved EGFR-TKIs, gefitinib, lapatinib, erlotinib, and icotinib are categorized as first-generation and afatinib, neratinib, and dacomitinib are categorized as second-generation EGFR inhibitors. The third-generation EGFR inhibitors include olmutinib, almonertinib, and osimertinib (127). In addition, vandetanib, brigatinib, and pyrotinib are categorized as multi-kinase inhibitors, because of their inhibitory actions against kinases, excluding the EGFR (128).
Targeting EGFR signaling represents a new strategy for personalized medicine in NSCLC. Targeting EGFR in the patients with stimulating mutations showed initial and considerable success in the clinic (23, 24). Some were developed to target EGFR, including TKIs and BRAF inhibitors (129, 130). Inhibiting EGFR-mediated activation of the downstream pathway, EGFR TKIs can influence the cellular level of apoptotic-linked proteins, primarily the pro-apoptotic consequence of EGFR targeting (131, 132). EGFR may be a potential clinical strategy for inhibiting EMT-linked acquired drug resistance and EGFR blockage in EGFR-mutated NSCLCs (21, 22). However, the inhibition of EGFR leads to upregulation of pro-apoptotic proteins and, lastly, results in apoptosis by activating the intrinsic apoptotic pathway (25, 26) (Figure 3). Earlier reports (27, 28) demonstrated the important function of Bcl-2 in the resistance of NSCLC for EGFR TKIs. It validated that growth inhibition is induced by the treatment activation of caspase-3 and Bax, supposedly by EGFR, ERK, and MMP-2 downregulation (29, 134).
Figure 3
6 Therapeutic Target of Bax/Bcl-2 Cascade and EGFR-Mediated NSCLC by Phytochemicals/Small-Molecule Inhibitors
Scientific data exhibit that phytochemicals have considerable anticancer potential. Roughly 50% of approved antitumor drugs originated from natural produces or derived from that place (135). However, these phytochemicals were tested for antitumor efficiency at in vitro and in vivo levels. They possess balancing and overlapping mechanisms for slowing down the carcinogenic procedure via scavenging free radicals (136), repressing growth and proliferation (137), and reducing angiogenesis and invasiveness of cancer cells (138). They exert an extensive and complex range of acts on several molecular targets as well as signal transduction pathways such as membrane receptors (139), kinases (140), downstream tumor-activator or -suppressor proteins (141), and transcriptional factors (142). Several phytochemicals/SMIs have been exhibited as potential therapeutics for NSCLC. The inhibitors of Bcl-2 are used for the therapeutic targeting of several tumors (Table 1). Some of the incredible antitumor phytochemicals/SMIs in this regard are explained in the present study. Here, we have discussed selective phytochemicals/SMIs including gefitinib, EGCG, ABT-737, thymoquinone, quercetin, and venetoclax that inhibit and target EGFR pathways in NSCLC (Figure 3).
Table 1
| Agents | Structure | IC50 for Bcl-2 (μM) | Clinical status | References |
|---|---|---|---|---|
| Gefitinib | 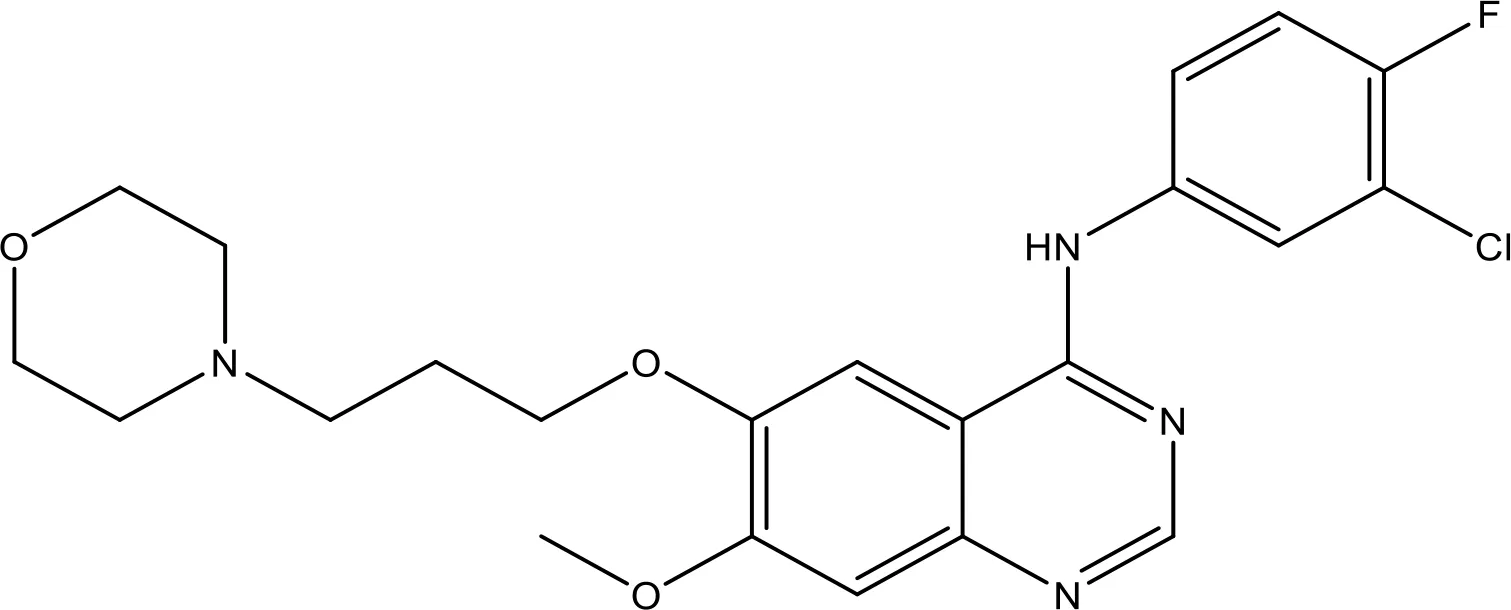 | 17.12 | Phase 2 | (143) |
| Thymoquinone | 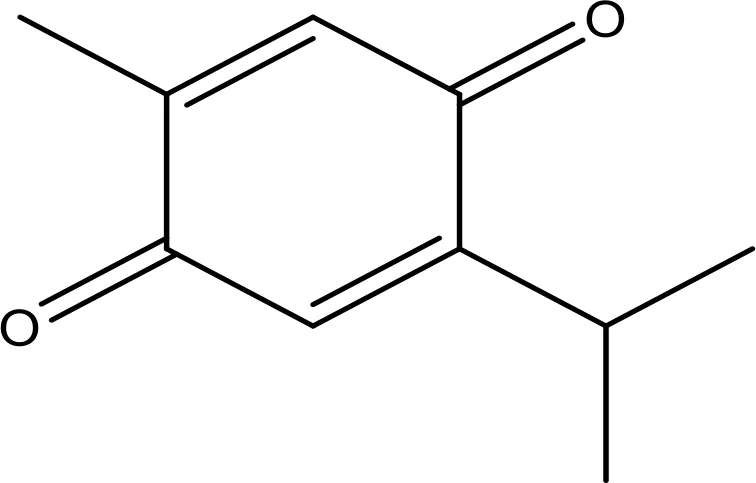 | 45.78 | Phase 2 | (144) |
| Quercetin | 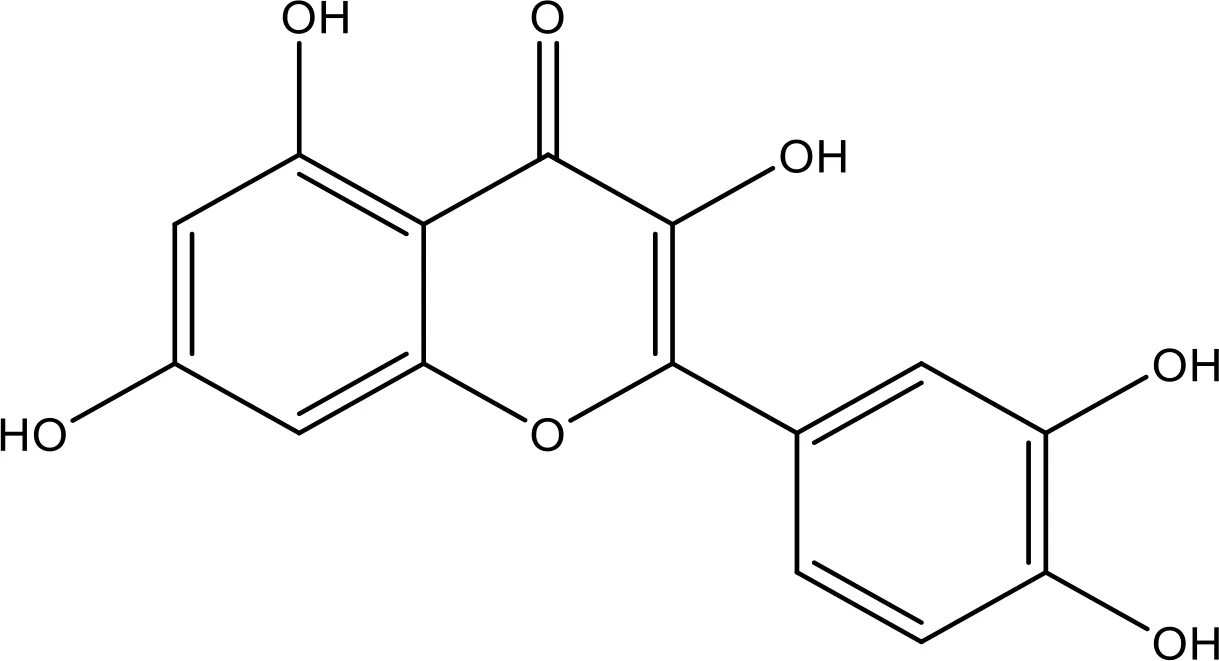 | 35.69 | Phase 2 | (144) |
| EGCG | 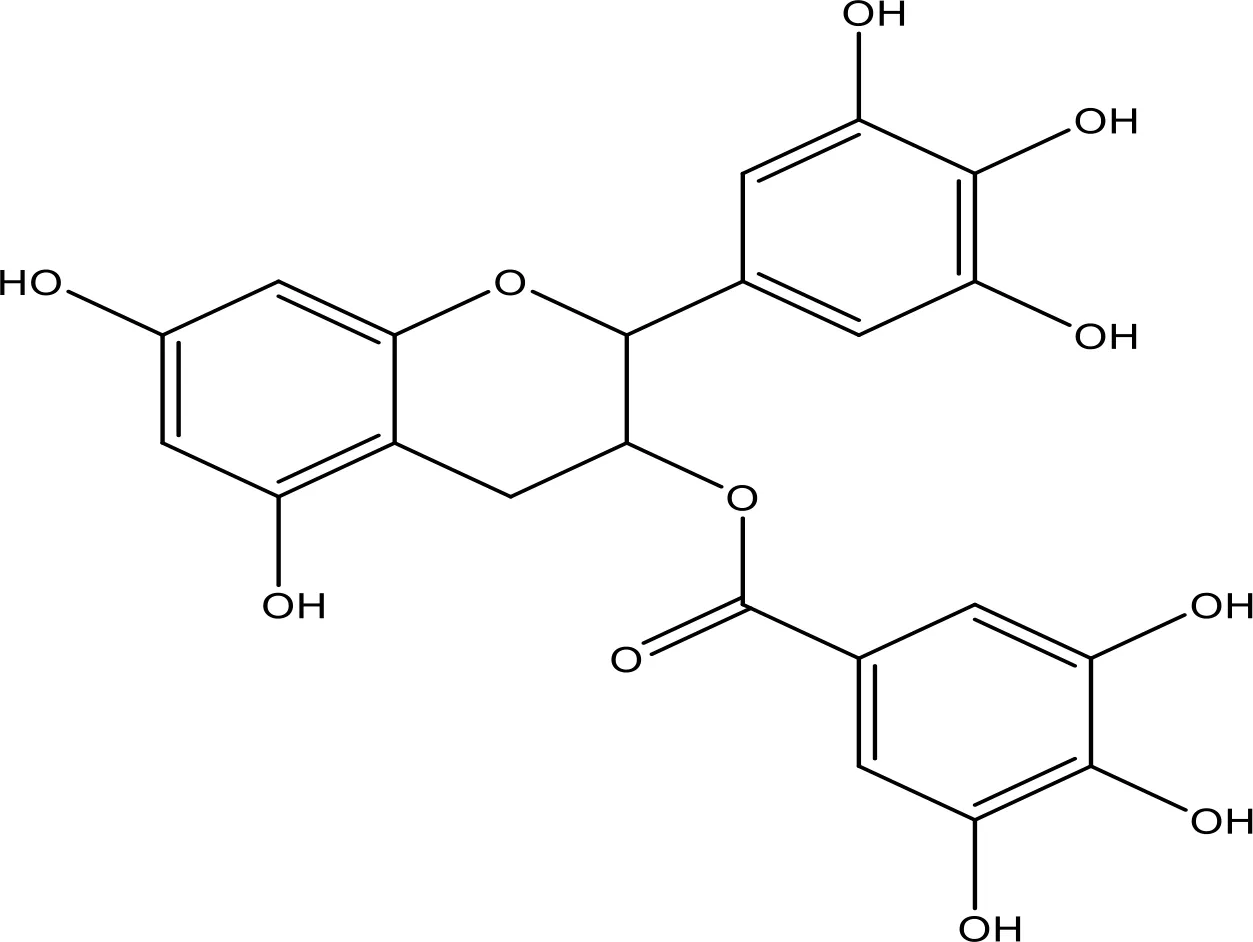 | 0.45 | Phase 1/2 | (145, 146) |
| ABT-737 | 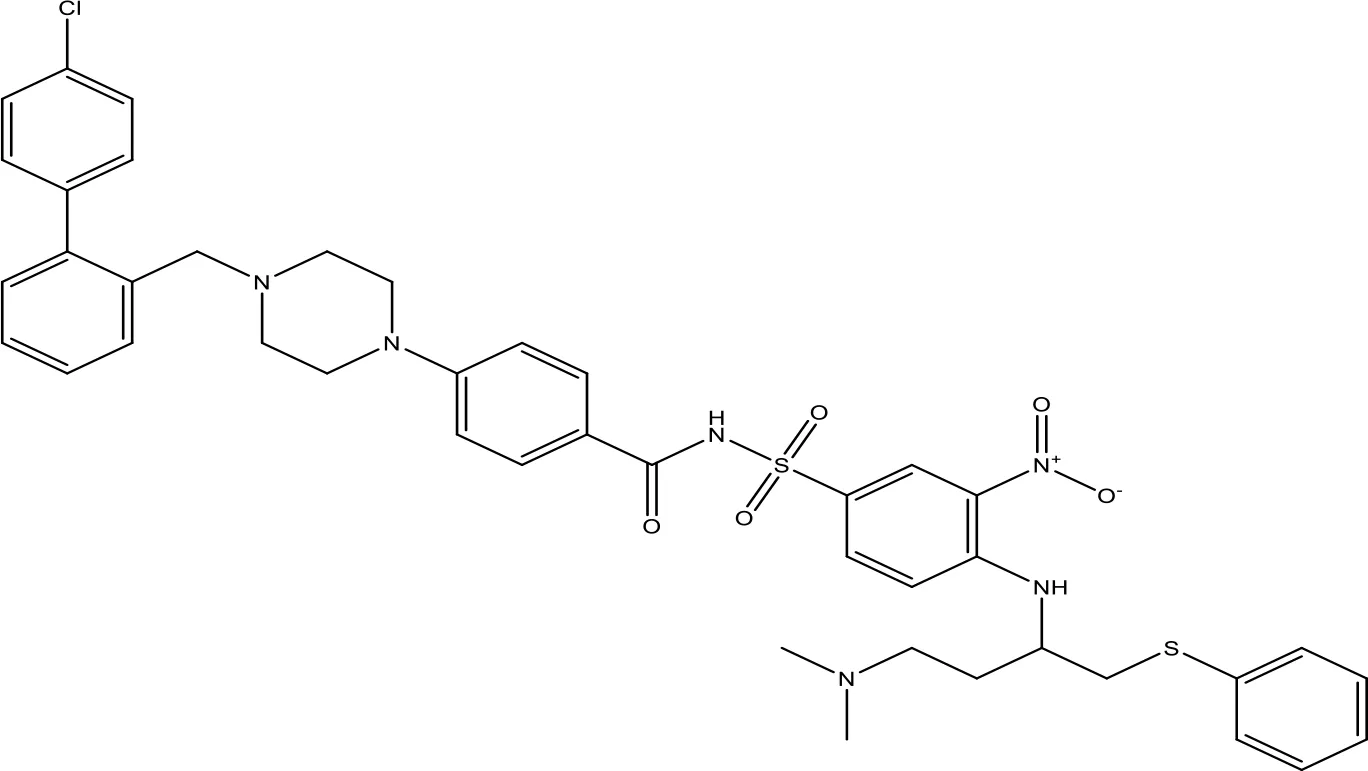 | 0.12 | Phase 1/2 | (147, 148) |
| ABT-263 | 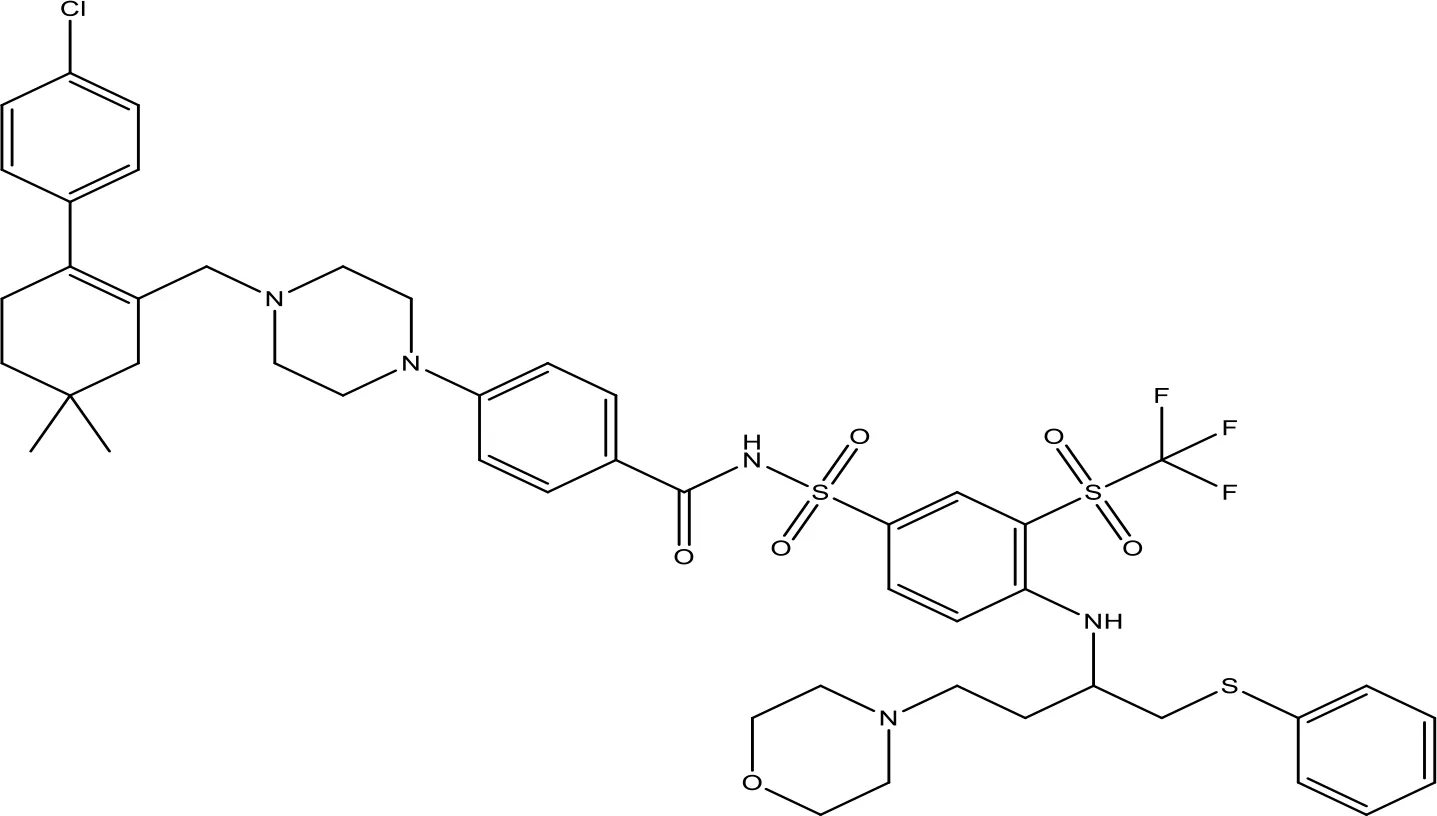 | NA | Phase 1/2 | (149, 150) |
| ABT-199 | 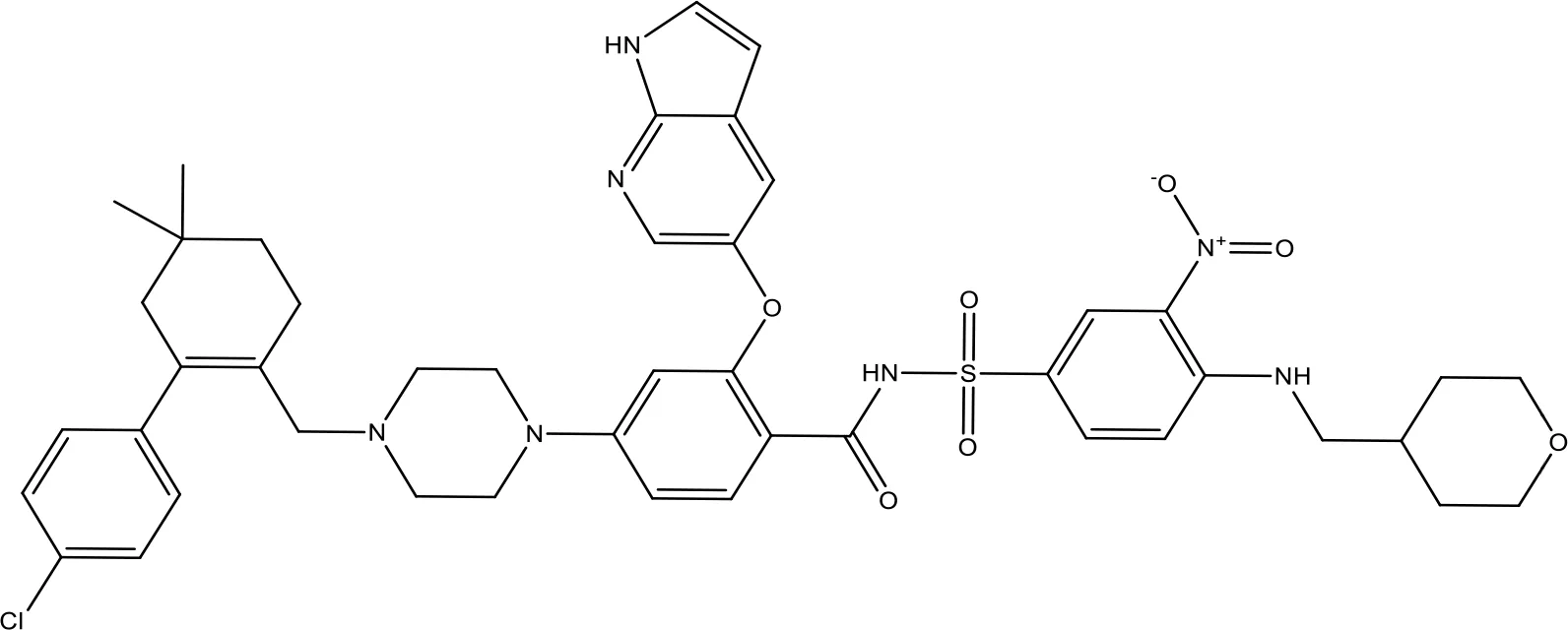 | 0.1 | Approved for use in CLL | (151) |
| TW-37 | 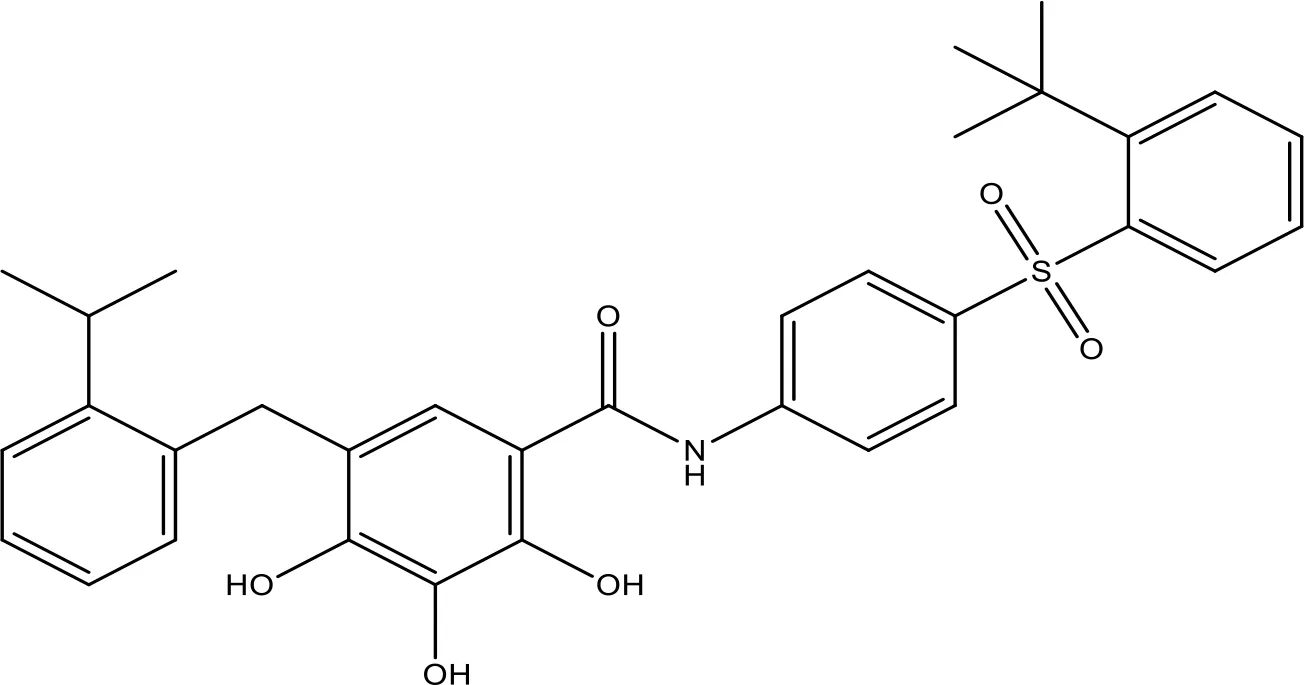 | NA | Phase 1/2 | (152, 153) |
| Gossypol | 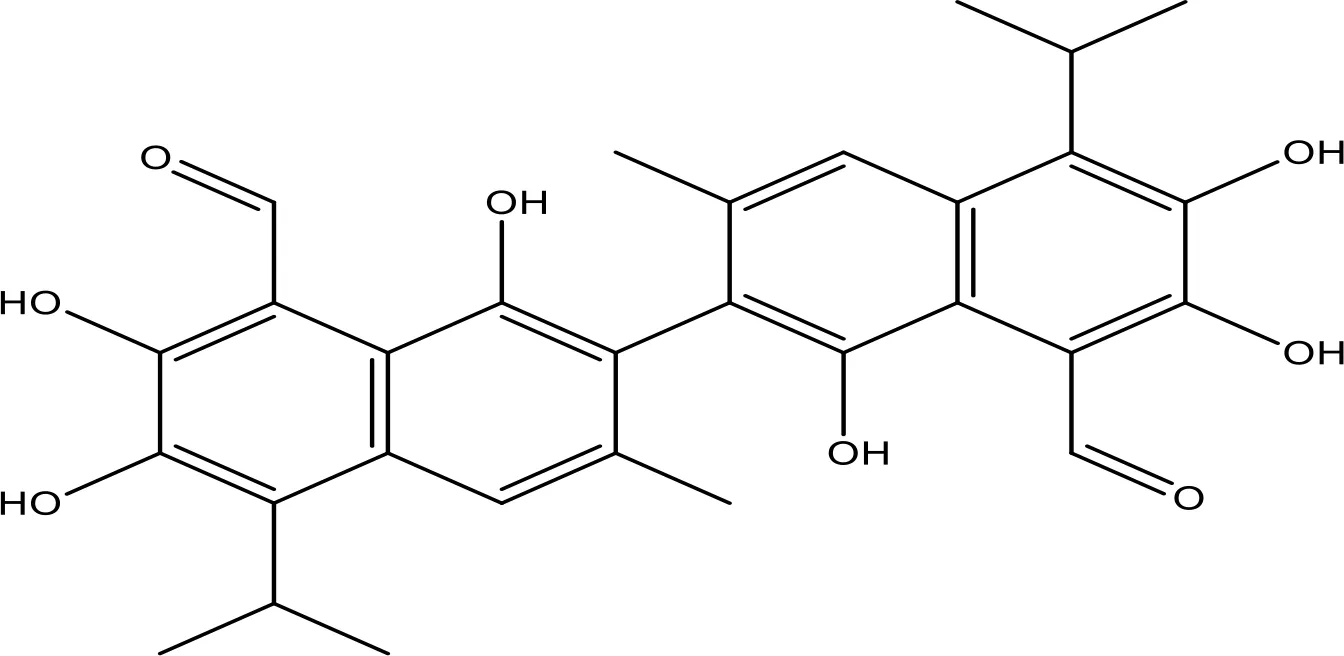 | 0.28-10 | Phase 1/2 | (154) |
| GX15-070 (obatoclax) | 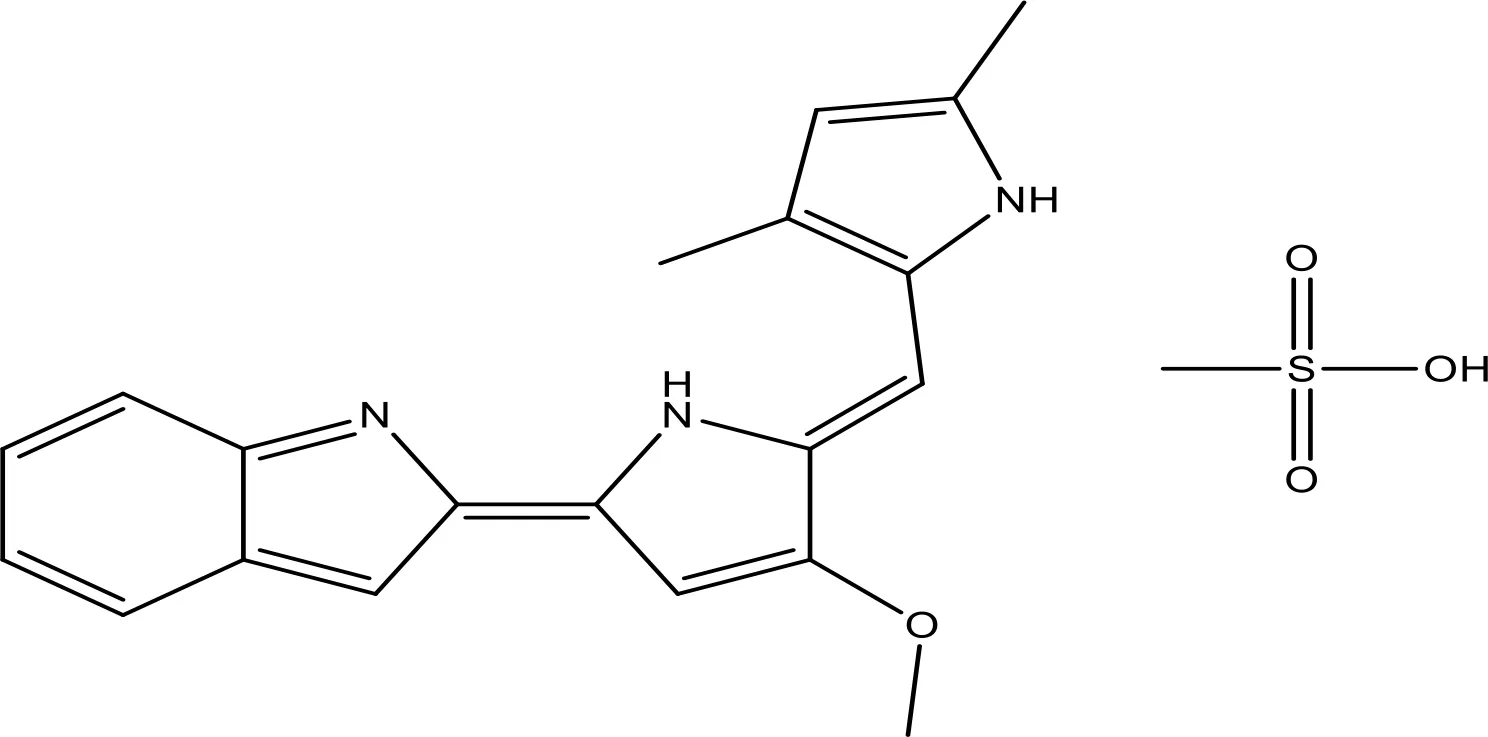 | NA | Phase 1 | (155, 156) |
| HA14-1 | 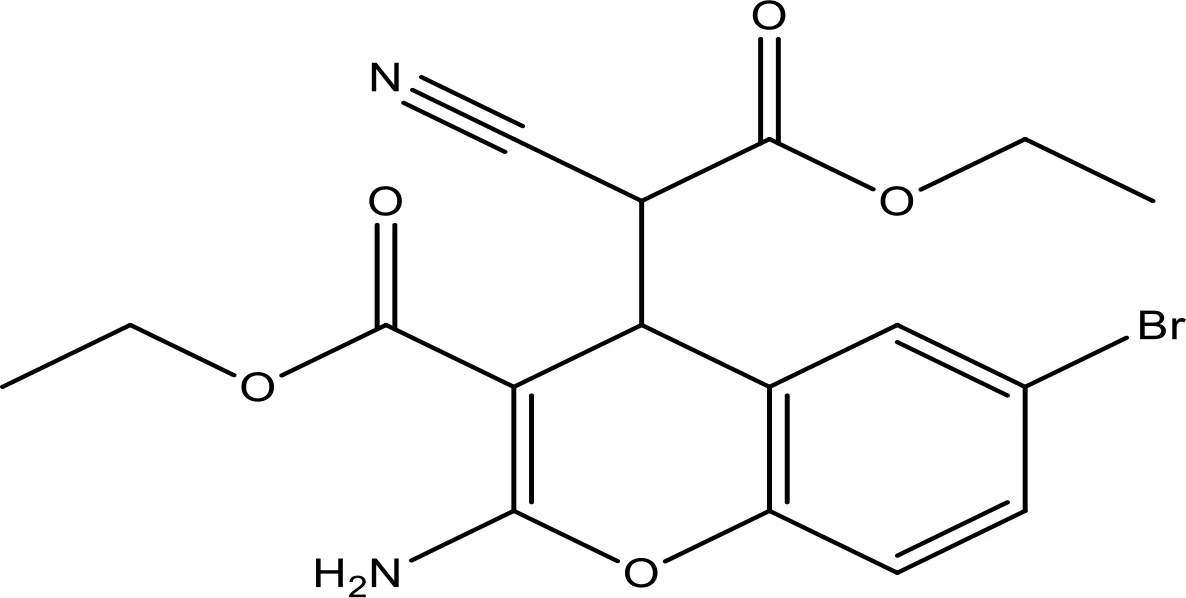 | ~9 | Preclinical | (155, 157) |
| Chelerythrine | 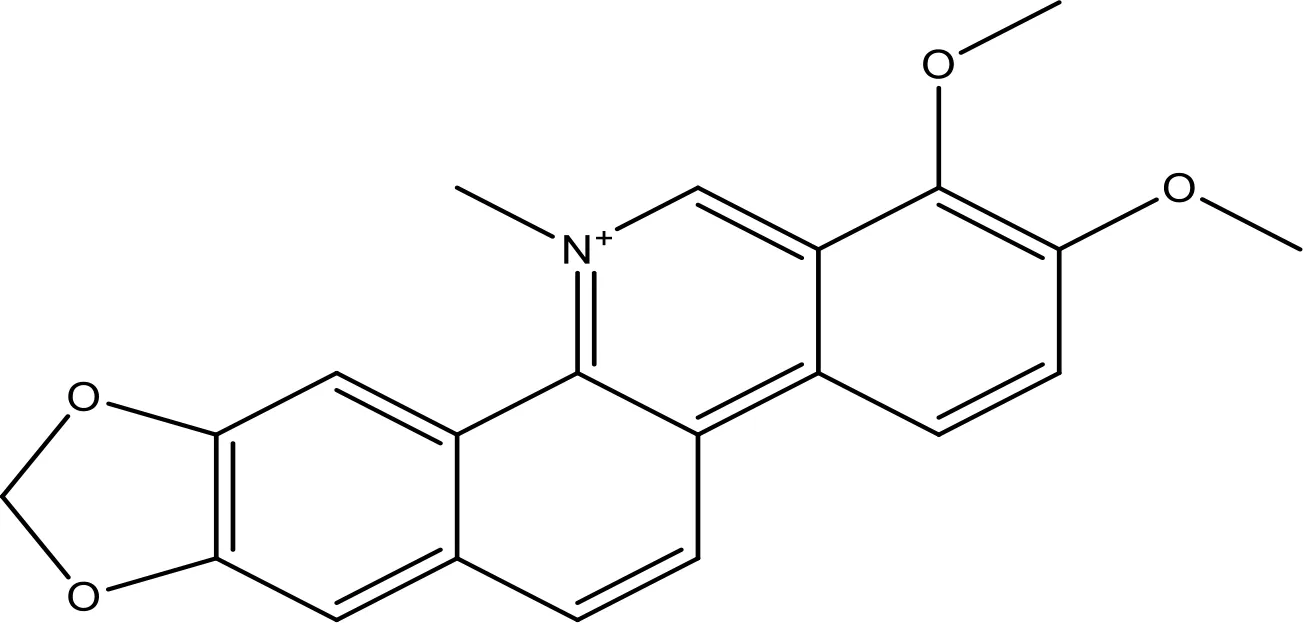 | ~10 | (158) | |
| S55746 |  | NA | Phase 1 | (159) |
The inhibitors of Bcl-2 are used for the therapeutic targeting of several tumors.
6.1 Mechanism of Gefitinib in NSCLC
Gefitinib showed potent activity in NSCLC (160). IDEAL 1 and 2 trials have been designed to further examine the efficiency and safety of two diverse gefitinib doses in patients with pretreated NSCLC (161, 162). This test validated that gefitinib is dynamic in greatly pretreated NSCLC patients, with a reaction rate of 11.8% and symptom enhancement in 43% of patients in the 250-mg arm (162). A randomized phase II trial compared gefitinib with docetaxel for advanced NSCLC patients (163, 164). Six phase III trials estimated the force on survival of erlotinib/gefitinib alone or in combination with therapy in metastatic or advanced NSCLC patients (165–167). Hence, the in vitro actions of gefitinib against susceptible and resistant cancer cells have been evaluated in numerous reports (168, 169). A study (170) examined the result of gefitinib on the cell proliferation of NSCLC cell lines utilizing the MTS test and colony formation tests. However, the results exhibited IC50 values of 4–42 μM.
6.1.1 Effect of Gefitinib on Bax/Bcl-2 Cascade
Gefitinib-mediated apoptosis is increased via accumulation of the BH3 mimetic ABT-737 (171). It stimulates apoptosis by activation of Baxin cancer cells. However, it stimulates G1 arrest and apoptosis via regulating p21 and p27 and the activation of Bax in GBC cells (172). The less regulation of Bcl-2 via RNAi in gefitinib-resistant H1975 cells with T790M mutation increased the results of gefitinib and can offer a new therapeutic approach for NSCLC treatment (28). Gefitinib could be most efficient in NSCLC patients (173). It repressed the expression of Bcl-2 and Bcl-xL, rendering HCC prone to cell death (174). Gefitinib combined with cinobufotalin obstructs viability and assists the apoptosis of A549 cells, showing that the combined therapy may be a potential novel treatment for lung cancer patients resistant to gefitinib (143). This is a new therapy for the treatment of NSCLC.
6.1.2 Effect of Gefitinib on the EGFR-Mediated Pathway
Gefitinib has been the initial SMI of EGFR identified for clinical application (175). Gefitinib is a TKI that treats NSCLC patients whose cancers have particular EGFR mutations (176). It was approved for cancer treatment in May 2003 (176) and approved for metastatic EGFR mutation-positive NSCLC in July 2015 (177). Gefitinib blocks EGF-mediated EGFR autophosphorylation in many EGFR-expressing cancer cells (178). The kinase inhibitory action of gefitinib, along with eight of the approved kinase inhibitors, has been assessed (179, 180). The kinase inhibitory actions have been evaluated against 310 kinases utilizing an activity-based kinase test, and it has been observed that gefitinib especially prevents EGFR and its mutants. In addition to the inhibition of EGFR, the results exhibited gefitinib’s capability to inhibit the serine/threonine kinases at IC50 = 50 and 90 nmol/l, respectively (RICK and GAK). However, gefitinib might have cellular option modes of activity. The cellular IC50 of gefitinib against several EGFR mutants has been established (181). A study reported the relationship of growth inhibition on the ERK1/2 and Akt activation and in response to the EGFR pathway (170). Hence, a study displays that the accumulation of a BH3 mimetic drastically increases the killing of NSCLC cells via EGFR TKI gefitinib (171).
6.2 Mechanism of EGCG in NSCLC
EGCG is the most plentiful polyphenol in green tea (182, 183). The therapeutic results of EGCG were identified against various tumors (184–186). EGCG was examined in many tumor cells and a few clinical trials with minimum information on its efficiency in lung cancer. EGCG damages growth in SCLC cells. However, a variable result on the limited number of NSCLC cells was checked (187, 188). EGCG is a promising antioxidant with various beneficial results in oxidative stress-mediated disorders (189). EGCG contributes to blocking NO and H2O2 production in human skin (190). EGCG might powerfully inhibit oxidative stress-induced protein tyrosine nitration via oxidative stress in blood platelets (191), and antioxidants may progress the function of mitochondria (192). However, EGCG is a well-recognized antioxidant and quenches ROS, supporting oxidative DNA break, cancer endorsement, and mutagenesis that lead to anticancer effects (193).
6.2.1 Effect of EGCG on the Bax/Bcl-2 Cascade
EGCG induced apoptosis by enhancing the Bax and cleaved caspase-3 expression and dropping the Bcl-xL expression in cancer cells (194). EGCG diminished the regulation of Bcl-2 and Bcl-xL (40, 195). EGCG mediated apoptosis via an intrinsic pathway by caspase-9 activation in PC3 cells (196) and MCF-7 cells (197). However, EGCG-mediated cell death of tumor cells has been correlated with the decline in the expression of Bcl-2. EGCG has displayed the induction of apoptosis by enhancing caspase-3, caspase-9, BAD, cyt-c, PTEN, SMAC, and Fas and repressed Bcl-2, Bcl-xL, and c-Myc in cancer cells (198, 199). Green tea improved the apoptotic efficiency in cancer (200). It has anticancer effects, which enhanced Bax, Bak, and PUMA and reduced Bcl-xL and Bcl-2 that activate caspases-9, inducing apoptosis in cancers (97, 201, 202). Recently, the interaction of EGCG with p53 disrupts p53 with its regulatory E3 ligase MDM2 and reduces the ubiquitination of p53 through MDM2. Since EGCG interrupts the binding of p53 for its regulator MDM2, p53 is stabilized through blocking p53 ubiquitination as well as degradation (203). EGCG was recognized from a library of about 2,295 phytochemicals like an inhibitor of p53 with MDM2 interaction (204).
6.2.2 Effect of EGCG on the EGFR-Mediated Pathway
EGCG prevents several signal transduction pathways in tumor cells. Hence, EGCG blocked proliferation in various NSCLC cells (205). EGCG induces apoptosis by the mitochondrial pathway and inhibits EGFR, ERK, and STAT3 signaling in HNSCC (145, 146). EGCG prevented STAT3 activation and downregulation of the target genes, including Bcl-2, Bcl-xL, Mcl-1, cyclin D1, and VEGF (206). However, EGCG blocked NF-κB, ERK1/2, and Akt-induced pathways, thereby modifying the Bcl-2 family protein ratio that activates caspases in tumor cells (207–209). Inhibition of c-Jun N-terminal kinase via EGCG induced apoptosis in OSCC cells (201, 210). EGCG might inhibit p-Akt/p-mTOR expression through PTEN to control the PI3K/Akt pathway (211, 212). EGCG exposure noticeably reduced EGF-mediated EGFR, ERK1/2, and Akt activation. However, long-term EGCG treatment prevented the total and membranous expression of EGFR and noticeably attenuated EGFR nuclear localization and cyclin D1 expression, showing that EGCG treatment repressed EGFR transactivation. However, inhibition of the EGFR pathway could partially contribute to the antitumor action of EGCG in lung cancer (213).
The wnt/β-catenin pathway was encouraged in lung tumor stem cells. Hence, EGCG decreased lung tumor stem cells’ action by reducing lung tumor stem cell markers, blocking tumorsphere formation, reducing cell proliferation, and promoting cell death (214). EGCG was observed for blocking angiogenesis and diminishing xenograft cancer growth via inhibiting IGF-1 by repressing HIF-1a and VEGF in A549 cells (215–217). EGCG blocked HGF-induced cell growth and invasion via repression of HGF/c-Met signaling in SCC VII/SF cells, whereas it blocked xenograft cancer survival in vivo by increasing cell death (218). Numerous in vitro studies explained the anticancer effect and potential mechanisms of EGCG on tumor cells. The combination treatment blocked the EGFR pathway and reduced the p-EGFR, p-ERK, and p-Akt expression in vitro and in vivo. EGCG and cDDP have exhibited a potential therapeutic effect in NSCLC patients (219). Nano-EGCG can prevent lung cancer cell proliferation, invasion, and migration via the activation of AMPK pathways. However, this mechanism of nano-EGCG recommends its application in lung cancer treatment and prevention (220).
6.3 Mechanism of ABT-737 in NSCLC
ABT-737 is an SMI designed to especially block anti-apoptotic Bcl-2 proteins (221, 222). This molecule, a BH3 mimetic, attaches with more affinity to Bcl-2, and Bcl-xL stimulates apoptosis (221). ABT-737 may improve the radiosensitivity of a variety of solid cancers. However, the radiosensitizing effect of ABT-737 has been examined in NSCLC (223). Despite its potential results in in-vitro studies and tests on animal models, ABT-737 was approved for clinical trials due to unfavorable pharmacological features such as thrombocytopenia (222, 224–227). The clinical significance of Bax is largely reported in several studies. Several Bax-targeted anticancer drugs are approved for medical use, for example, ABT-737 (228). This class of drugs has proved their potential in reversing the resistance effect. Therefore, there is the bigger necessity to develop cost-effective gene-targeted combinatorial drugs from natural products (66, 228).
6.3.1 Effect of ABT-737 on Bax/Bcl-2 Cascade
Abbott developed ABT-737, a novel inhibitor of Bc-l-2, in the last decade which was expected to target Bcl-2, Bcl-2-X, and Bcl-2-w to show promising results at the research stage. These were developed as BH3-targetable small-molecule inhibitors (SMI). They enhance the apoptotic effects in SCLC (229). ABT-737 is a potent inhibitor of Bcl-2, Bcl-w, and Bcl-xL (222). It is a promising SMI of anti-apoptotic proteins such as Bcl-2 in HNSCC (147). ABT-737 activates caspase-3, which leads to apoptosis. ABT-737 upregulates the Noxa expression. Noxa by small interfering RNA attenuates cell death (147). It attaches with a very high affinity with Bcl-2. Bcl-2 is hindered because of ABT-737 into its hydrophobic groove, and this binding dislocates any bound BH3-enclosing proteins (148). ABT-737 induced the caspase-3 activation and cleavage of PARP that stimulated apoptosis. Glioblastoma cells’ large quantities of Bax protein are prebound with Bcl-2, which are acutely liberated via ABT-737 treatment. However, the highly “addicted” cells become Bax-neutralizing Bcl-2 proteins (230). ABT-737 holds huge promise, as it passionately attaches the pro-survival proteins similar to Bcl-2 and stimulates Bax/Bak-dependent destruction. BT-737 regulated Bax/Bak-induced apoptosis (231).
6.3.2 Effect of ABT-737 on EGFR-Mediated Pathway
Several studies exhibited the potential effect of ABT-737 on signaling molecules. BIM polymorphism is strongly linked to a poor clinical reaction for EGFR TKIs in EGFR-mutant NSCLC patients; hence, BH3-mimetic ABT-737 returns BIM functionality EGFR-TKI sensitivity (232). ABT-737 drastically increases erlotinib-mediated cell death, and more strong responses for EGFR inhibitors in lung tumor patients harbor EGFR kinase domain mutations (233). Bcl-2 antagonist ABT-737 slights the apoptotic threshold to chemotherapeutic drugs via the PI3K/Akt signaling inhibition in cancer cells. Inhibition of Bcl-2 and Bcl-xL increases Akt/PI3K inhibition-induced apoptosis in cancer cells (234, 235). The p53 and Akt pathways were examined to be associated with the effect of ABT-737 and naringenin in gastric cells (236). The PI3K/Akt inhibitor BEZ235 with ABT-737 regulates ovarian cancer cell apoptosis (237). However, targeting the Akt/PI3K/mTOR and/or ERK/MAPK pathways may disturb the imbalance between anti-apoptotic and pro-apoptotic partners that might constitute an important approach for sensitizing cancer cells for ABT-737 (238). Inactivation of ERK1/2 with ABT-737 enhanced the BIM expression that induced apoptosis in oral cancer cells. Targeting the ERK1/2-bim pathway via BH3-mimetic ABT-737 is an optional therapeutic approach for oral cancer (239). The phosphorylation of Bcl-2 on Ser-70 through JNK and paclitaxel synergizes by ABT-737 and reinstates paclitaxel sensitivity in breast cancer cells (240). ABT-737 recovered the radiation sensitivity of HeLa cells, thereby stimulating cell death, and showed that ABT-737 reduced HeLa cell proliferation and activated JNK (c-Jun), which resulted in more regulation of BIM (241). The sensitizing results were detected when ABT-737 was combined with sorafenib that effectively repressed levels of STAT3. They suggested that targeting STAT3 in combination with inducers of the apoptosis pathway may be a potential novel strategy for treating tumor cells (242).
6.4 Mechanism of Thymoquinone in NSCLC
There are numerous bioactive ingredients extracted from Nigella sativa (black seeds), which show anticancer activities by modulation of cell-cycle pathways, but thymoquinone (TQ) is regarded as the most potent anticancer bioactive compound found in black seeds (243). Others compounds are dithymoquinone (DTQ), thymohydroquinone (THQ), and thymol (THY) (244, 245). N. sativa has numerous valuable constituents that effectively treat various diseases (246). TQ has been identified to exert antioxidative, anti-inflammatory, and anticancer effects (247, 248). TQ plays an effective role in cancer treatment by inducing apoptosis or suppressing the expression of carcinogens (249–251). TQ displays inhibitory results on numerous processes of NSCLC, including apoptosis proliferation, migration, and angiogenesis (252). TQ blocked the growth and decreased expression of cyclin D1 in A549 cells (NSCLC) (253, 254). However, TQ synergistically augments conventional medicine prevention of NCI-H460 cells (252). It displays a therapeutic role in lung cancer (255–257). TQ blocks cell proliferation, stimulates apoptosis, and obstructs xenograft cancers’ in vivo growth of numerous tumor cells, including lungs (247, 252).
TQ is a powerful anti-carcinogenic and anti-mutagenic mediator (258, 259). Aqueous and alcohol extracts of N. sativa were effectual in inactivating MCF-7 breast cancer cells (258, 260). In-silico screening is an excellent approach used to screen potential anticancer compounds. It also helps our body soak up the medicine quickly when taken in petite dosages. Therefore, it indicates a potential aspect of combinatorial therapy (261, 262). Although there are several constituents of N. sativa which played a beneficial role in disease management, its chief constituent TQ has proved its active role in cancer prevention. TQ imparts antioxidant effects in animal models (263–265). Cancer as a disease relies on multiple factors like modification in genetic pathways (266, 267). Black seed preparation also helps to reduce the toxicity and side effects of anticancer synthetic drugs (268, 269). TQ showed a protective role in oxidative stress conditions when administered orally by inducing free radical generation (270–272). Few researchers suggested that oral administration of TQ alleviates quinine reductase and glutathione transferase (273, 274). Therefore, TQ can be used as a drug to counter the toxicity of liver carcinogens (275, 276). This property of TQ attributes to its protective role in balancing the toxicity of chemotherapy-based treatments (277). TQ has therapeutic implications in health and cancer management by genetic cascade modulations. It functions through the activation of the cancer suppression gene (278).
6.4.1 Effect of Thymoquinone on Bax/Bcl-2 Cascade
Alterations in the normal process of cell death (apoptosis) increase the chances of cell survival and thus lead to cancer development and progression. Bax/Bcl-2 cascade is critical to inducing apoptosis, as already reviewed. Still, there is scope to determine its role in other important cancer-related pathways like modulation of Bax-Bcl-2 cascade (279). TQ phytosomes stimulated apoptosis at 4.31 ± 2.21 µM by caspase-3 activation and generation of ROS, besides gathering cells on G2-M and pre-G1 in A549 cells (280). TQ increased apoptosis via enhancing the Bax/Bcl-2 ratio and more regulating the expression of p53 in A549 cells (281). TQ stimulates the tumoricidal action of NK against lung tumor cells via more regulating pro-apoptotic genes and less anti-apoptotic genes (282, 283). TQ considerably diminished the viability of HCT116 cells in a concentration- and time-dependent way. However, treatment of cells with TQ-mediated cell death has been linked with the more regulation of Bax and downregulation of Bcl-2 and Bcl-xL (284). TQ stimulated caspase-9,-7, and -3 and activated PARP. TQ modulates the action of the Bax/Bcl-2 cascade. Here, TQ was found to downregulate the expression of Bcl-2, thus inducing apoptosis (285). This study also showed that TQ elevates ROS expression, which leads to a decrease in MMP, also known as DNA laddering, and the subsequent release of cyt-c (285). TQ upregulates the Bax/Bcl-2 ratio, thus inhibiting downstream caspases in the hepatic ischemia–reperfusion injury (I/R) model (286). TQ also increases the Bax/Bcl-2 ratio while upregulating the expression of Bax. In one of the studies, TQ was found to elevate the expression of Bax in Hl-60 cells, thus activating upstream caspase 8 results in the release of cytochrome c (287). These observations regarding TQ inspire the scientific community to develop gene target-based combinatorial therapies, where TQ can play a significant role in blocking the expression of Bcl-2 and counter toxicity of chemotherapy drugs.
6.4.2 Effect of Thymoquinone on EGFR-Mediated Pathway
TQ is associated with multiple pathways; treatment with TQ reduced the phosphorylation of JAK2, Src kinase, and EGFR. TQ stimulated apoptosis in HCT116 cells via inhibiting the STAT3 pathway by inhibition of JAK2- and Src-induced phosphorylation of EGFR-TK (253, 284, 288). TQ attenuated the STAT3 expression target gene products, including c-Myc, survivin, and cyclin-D1 and -D2, and increased p27 and p21 (284). TQ might target multiple kinases, such as PI3K, MAPK, JAK/STAT, PLK1, and tyrosine kinase, in diverse cancer cells as well as animal models. However, inhibiting the action of kinases or repressing their expression may be among the mechanisms of TQ antitumor action. Targeting kinases with TQ, which is a molecularapproach for tumor therapeutics. (288). It illustrated the capability for suppressing the ERK1/2 pathway, which blocked the invasion and migration of A549 cells (254), and the therapeutic promise of TQ as an anti-metastatic drug in lung cancer treatment. Hence, subcutaneous doses of the TQ-I3M combination repressed the lung tumor metastasis and decreased tumor growth by the inhibition of the NF-κB/Akt/mTOR pathway in the xenograft model (253). It shows significant anticancer activities via upregulation of PTEN during transcription. It is well-known that PTEN played a role in inducing p53 expression and inhibits the Akt pathway (267). Apart from this, TQ also modulates various genetic pathways. It also inhibits NF-ĸB activation, which results in the downregulation of inflammatory genes. It upregulates miR34a and downregulates Rac1 expression (278).
6.5 Mechanism of Quercetin in NSCLC
Quercetin (Qu) is a flavonoid found abundantly in fruits (apple) and vegetables (onions), citrus foods, and tea. It has excellent antioxidant properties (289). It shows anticancer properties like growth factor suppression, apoptosis induction, and anti-proliferative actions (290). When treating A549 and H1975 cells for 24 h using the vehicle of Qu, there was no sign of altered viability compared to control (291). Qu suppresses the wound closure and invasive and migratory abilities of NSCLC cells at low concentrations (10–50 µM), indicating its anti-proliferative properties (292). Qu notably stimulated the apoptosis of cancer cells. It demonstrates antitumor roles both in vivo and in vitro (293). Apart from good results both in vitro and in vivo, some clinical studies have also evaluated the antitumor therapeutic results of Qu in ovarian cancer (294). In both in vitro and in vivo cancer studies, Qu was said to have a beneficial effect against prostate cancer (295).
This antioxidant effect of Qu can be attributed to its protective nature against the toxicity of drugs (296, 297). Studies were conducted to access its apoptosis induction potential and chemosensitivity, and the results were found to be appreciable and show the tumor inhibitory actions of Qu (298–300). Due to its antioxidant properties, it can also be used as a nutritional supplement in human health management. Several studies proved that it protects from the harmful effects of free radicals caused by smoking (296). Qu has excellent modulation properties toward inflammatory agents. It inhibits the core inflammatory enzyme COX (301–305). In one such study conducted by Cruz-Correa et al., combination doses of curcumin (480 mg) and Qu (20 mg) were orally administered to familial adenomatous polyposis (FAP) patients for 6 months three times a day. The result shows that this combination treatment reduces the size of rectal adenomas with no side effects (306). Qu inhibits hexavalent chromium (Cr[VI]) and shows a chemical carcinogen-induced cell transformation such as cell visibility loss, ROS generation, and microRNA-21 (miR-21) elevation in human colon cancer Caco-2-cells (307, 308).
6.5.1 Effect of Quercetin on Bax/Bcl-2 Cascade
Qu inhibits cell proliferation, stimulates apoptosis, and functions as an antioxidant. It may modulate apoptosis by Bcl-2 family proteins that downregulate Bcl-xL and Bcl-2 and upregulate Bax and Bad (309, 310). However, Qu modulates the expression of the Bax/Bcl-2 cascade and thus mediates apoptosis (311–313). It stimulates apoptosis in caspase-3-dependent signaling via blocking Cox-2 expression and controls Bcl-2 and Bax expression. It might be a promising and potent agent that can be safely utilized in leukemia therapy (314). The results recommended that NSCLC H-520 cells with Qu enhanced the cisplatin-mediated apoptosis. However, this has been accompanied by downregulation of Bcl-2 and Bcl-xL and upregulation of Bax. Qu acts as an efficient chemo-sensitizer in the chemotherapy of lung tumor through controlling the expression of several apoptosis-linked genes (315). TQ and Qu drastically decrease the expression of Bcl-2 and induce Bax, indicative of sensitizing NSCLC cells stimulating apoptosis (144). Bcl-2 and Bcl-xL protein expressions were significantly decreased, and Bax and caspase-3 were increased treated by Qu (312).
6.5.2 Effect of Quercetin on the EGFR-Mediated Pathway
The role of Qu-mediated molecular regulation in repressing NSCLC metastasis recommends that it has promising therapeutic functions for metastatic NSCLC (292). Qu is an effectual inhibitor for managing NSCLC harboring the EGFR C797S mutation. However, Qu displayed promising cytotoxic results on NSCLCs harboring the EGFR C797S mutation via preventing AXL and stimulating cell death (316). It mimics the interfaces of ATP in the active location of RTKs (EGFR, FGFR1, and c-Met) that lead to the prevention of C RTK overexpression (317). Qu and its permethylated form blocked migration and cell viability, downregulated VEGFR-2, and decreased Akt, JNK, and ERK levels on human primary endothelial cells (318). Qu altered the Akt/mTOR/AMPK/signaling (319). Qu used the anti-NSCLC effect by blocking Src-mediated Fn14/NF-κB signaling in vitro and in vivo (320). The antitumor roles of Qu generally occur via the modulation of VEGF, apoptosis, P13K/Akt/mTOR, MAPK/ERK1/2, and Wnt/β catenin pathways (321). Qu blocks the proteasome action via modulation of pathways including Akt/PI3K and ERK (322, 323). Qu was reported to suppress the melanoma and breast cancer cells by inhibiting MMP3 expression (324). Qu significantly shows anti-migratory effects (292).
6.6 Mechanism of Venetoclax in NSCLC
Researchers developed a successful Bcl-2 inhibitor called venetoclax (325). It inhibited Bcl-2 only and developed the same BH3 mimetic SMI (326). After showing successful results in clinical trials while treating chronic lymphocytic leukemia (CCL), venetoclax was approved by FDA as a second-line drug to treat CCL (327). It is a selective inhibitor of Bcl-2 family proteins that stimulate apoptosis in several cancers, including lung cancer. Venetoclax (ABT-199) has revealed clinical efficiency in numerous hematological tumors (328)
6.6.1 Effect of Venetoclax on Bax/Bcl-2 Cascade
Venetoclax is a selective and potent inhibitor of Bcl-2. It has revealed clinical efficiency in numerous hematological tumors (328). This inhibitor continues to attach to Bcl-2 (329). Venetoclax stimulated BIM-dependent cell death in vitro, inhibited cancer growth, and stimulated tumor failures in mice bearing more Bcl-2–expressing SCLC cancers in vivo. However, venetoclax is a potential therapy for more Bcl-2-expressing SCLCs (330). SCLC displays elevated Bcl-2 expression and may be accountable to single-drug treatment via venetoclax. Increased Bcl-xL venetoclax explained preclinical trial action in breast tumor cells (151). Hence, venetoclax is a promising Bcl-2 inhibitor. Bcl-2 is a target in specific subtypes of human T-ALL that can be utilized by venetoclax (331). The new combination of decitabine with venetoclax was proficient and well tolerated in old AML patients (332). However, venetoclax presents the first-in-class Bcl-2 inhibitor careful platelets (333).
6.6.2 Effect of Venetoclax on EGFR-Mediated Pathway
Venetoclax can work by diverse signaling pathways for accomplishing synergistic cytotoxicity with AZD9291 in NSCLC (H1975AR). Hence, this inhibitor might provide an effective choice in combination therapy with EGFR-TKIs for treating NSCLC with EGFR-TKI resistance (334). However, combining radiation for EGFR and Bcl-2 obstruction may be a new plan for targeting cancer stem cells (335). A study supports a preventive, therapeutic targeting of bioenergetics and mitochondrial primarily for impacting early drug-escape appearance using the EGFR accuracy inhibitor combined with a wide BH3 mimetic for interrupting Bcl-xL/Bcl-2 together (336). NF-κB was drastically less regulated in AZD9291+ABT-199 treatment groups than AZD9291 or ABT-199 treatment only (334). The combination of ABT-199 + irradiation + cetuximab enhanced the blockage of the 2D and 3D cell proliferation, migration, and resistance to cell death. Additionally, in a nude model with a heterotopic tumor xenograft, a treatment combining ABT-199 with fractional cetuximab irradiation delayed the cancer growth and enhanced in vivo lifespan without side effects (335). ABT-199 has been checked in combinations with TKIs such as imatinib, nilotinib, and dasatinib in cells with blast-crisis CML. This study revealed in a CML mouse model that ABT-199 alone or in combination is better than nilotinib in removing CML stem cells in vivo. However, to study the dual inhibition of Bcl-2 and Mcl-1, HHT and ABT-199 were combined and examined in seven diffuse large B-cell lymphoma cells (337).
7 Combined Therapeutic Strategy for EGFR-Mediated NSCLC
EGFR belongs to the tyrosine kinase family. EGFR dimerization is responsible for cell proliferation, survival, and invasion (26, 338). Inhibiting pathways by EGFR presents an excellent strategy for therapeutic interference. Gefitinib and erlotinib are selective EGFR TKIs explaining anticancer action either singly or combined with radiation therapy and chemotherapy in human cancer xenografts (339–341). Effective drugs used in EGFR-embattled therapies are erlotinib and gefitinib (121, 342), also known as EGFR TKIs, which face resistance during the treatment of advanced-stage NSCLC (95, 343, 344). This resistance is due to a mutation caused by exon 19 deletions and missense mutation on exon 21 (120, 345, 346). Until the current utilization of TKIs, the standard first-line management and treatment for patients with unresectable NSCLC and excellent presentation status have engaged the employment of combined chemotherapy with regimens (347).
Vemurafenib and dabrafenib are used in single-agent target therapy against NSCLC patients with BRAF mutation (348–351). This mutation is caused due to a single transversion at exon 15, where valine is replaced through glutamate (residue 600) (352). Hence, other potential gene targets in the case of NSCLC are HER2, NTRK, Bax, and Bcl-2 (353, 354). Cetuximab-induced targeting of EGFR stimulated tumor cell death (355, 356). The TGFα-EGFR pathway in both cancer-associated endothelial cells and cancer cells themselves is essential in the development of colon cancer. However, repealing the pathway activation via a double tyrosine kinase inhibitor in combined therapy may significantly reduce cancer cell proliferation and stimulate apoptosis in both cells. However, targeting the VEGFR and EGFR pathway in cancer vasculature with anti-neovascular therapy offers a new plan for colon cancer treatment. Cetuximab, an anti-EGFR antibody, is moderately effective in EGFR-expressing cells (357). The T790M mutation is a promising target for NSCLC patients (358). However, new therapies are required to conquer resistance to the drug. Crizotinib (MET inhibitor) might enhance the gefitinib susceptibility in NSCLC (359).
Numerous in vitro studies explained the anticancer effect and potential mechanisms of EGCG on tumor cells. The combination treatment blocked the EGFR pathway and reduced the p-EGFR, p-ERK, and p-Akt expression. EGCG and cDDP have exhibited a potential therapeutic effect in NSCLC patients (219). BIM polymorphism is strongly linked to a poor clinical reaction for EGFR TKIs in EGFR-mutant NSCLC patients; hence, BH3-mimetic ABT-737 returns BIM functionality EGFR-TKI sensitivity (232). ABT-737 drastically increases erlotinib-mediated cell death, and more strong responses for EGFR inhibitors in lung tumor patients harbor EGFR kinase domain mutations (233). TQ is associated with many pathways and stimulated apoptosis in tumor cells via inhibiting the STAT3 pathway by inhibiting JAK2- and Src-induced phosphorylation of EGFR-TK (253, 284, 288). However, Qu displayed potent cytotoxic results on NSCLC cells harboring the EGFR C797S mutation via preventing AXL and stimulating cell death (316). Venetoclax may work by synergistic cytotoxicity with AZD9291 in NSCLC (H1975AR). Hence, this inhibitor might provide a productive choice in combination therapy with EGFR-TKIs for treating NSCLC with EGFR-TKI resistance (334). Therefore, several therapeutic strategies to target the EGFR pathway demonstrated various efficiencies that overcome drug resistance and cancer development.
8 Conclusions and Future Prospects
The EGFR pathway is associated with several cancer progressions, including NSCLC. The EGFR pathway regulates Bax/Bcl-2 cascade in NSCLC. Inhibition of EGFR leads to upregulation of pro-apoptotic proteins and stimulates apoptosis by activating the intrinsic apoptotic pathway. Targeted therapy might finally alter the treatment model for lung cancer and provide an expectation for patients with inadequate treatment opportunities. New targeted therapies offer a novel hope for cancer patients, including NSCLC, a rare disease for standard treatments. In the last decades, the improvement in cellular, molecular, and cancer biology research could be distinct by some foundational pillars—one of the most significant ones being the beginning of SMIs/phytochemicals. Targeting the EGFR with SMIs is a suitable validated strategy in tumor therapy. EGFR SMIs have been approved worldwide for the treatment of multiple cancers. However, these drugs explained high efficiency in cancer therapy.
Several clinical trials for the SMIs/agents of targeted cancer therapy are ongoing and have illustrated potent and promising effects to date. Hence, these trials assist in describing the function of targeted cancer therapy in the management and treatment of tumors, including NSCLC. Therefore, the dispute for the clinical improvement and utilization in cancer therapy of anti-EGFR agents alone and/or in combination with other SMIs/phytochemicals treatments would be the suitable assortment of potentially responding NSCLC patients. In the future, combined therapies with molecular mechanisms might lead to the eventual therapeutic option. However, targeting EGFR-mediated Bax/Bcl-2 cascade would be a potential therapy for NSCLC. In prospect studies, this study should significantly assist in the approach of new inhibitors for the EGFR-mediated Bax/Bcl-2 cascade that facilitate the treatment and management of NSCLC. Additionally, a close collaboration between molecular biologists, clinicians, and pathologists is critical for developing target therapy for NSCLC.
Funding
This work is supported and funded through the Indian Council of Medical Research (Grant No. 45/6/2020-DDI/BMS).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
MaA: conceptualization, writing—original draft preparation, data curation, investigation, methodology. SA: writing—original draft preparation, data analysis, validation, visualization. MoA: formal analysis, writing—review and editing, investigation, and validation. AME: data curation validation, writing—review and editing. WA: methodology, writing—review and editing. MAl: investigation, validation, writing—review and editing. YH: investigation, validation, writing—review and editing. AT: data curation, writing—review and editing. AS: data curation, writing—review and editing. VIP: conceptualization, data analysis, validation, project administration, writing—review and editing. MIH: conceptualization, investigation, writing—original draft preparation, review and editing.
Acknowledgments
MaA expresses thanks to the Indian Council of Medical Research for financial support (Grant No. 45/6/2020-DDI/BMS). The authors express gratitude to India’s Department of Science and Technology Government for the FIST support (FIST program no. SR/FST/LSI-541/2012).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Bcl-2, B cell lymphoma 2; OSCC, oral squamous cell carcinoma; MMP, mitochondrial membrane permeabilization; MMP mitochondrial membrane potential; CLL, chronic lymphocytic leukemia; BH, Bcl-2 homology; NSCLC, non-small cell lung carcinoma/cancer; LUSC, lung squamous cell carcinoma; PKB, Akt/protein kinase B; NFκB, nuclear factor κB; JAK2, Janus-activated kinase-2; EGFR, epidermal growth factor receptor; TNFR, tumor necrosis factor receptor; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase1/2; JNK, c-Jun N-terminal kinase; AP-1, activator protein-1; PARP, poly-(ADP-ribose) polymerase; TQ-I3M, TQ–indirubin-3-monoxime; IDEAL, Iressa Dose Evaluation in Advanced Lung Cancer; TKIs, tyrosine kinase inhibitors; NTRK, neurotrophic tyrosine receptor kinase; STAT3, signal transducer and activator of transcription-3; EGCG, epigallocatechin-3-gallate.
References
1
TorreLABrayFSiegelRLFerlayJLortet-TieulentJJemalA. Global Cancer Statistics 2012. CA: Cancer J Clin (2015) 65:87–108. doi: 10.3322/caac.21262
2
AlbergAJBrockMVFordJGSametJMSpivackSD. Epidemiology of Lung Cancer: Diagnosis and Management of Lung Cancer: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest (2013) 143:e1S–e29S. doi: 10.1378/chest.12-2345
3
SinkeviciusKWKriegelCBellariaKJLeeJLauANLeemanKTet al. Neurotrophin Receptor TrkB Promotes Lung Adenocarcinoma Metastasis. Proc Natl Acad Sci (2014) 111:10299–304. doi: 10.1073/pnas.1404399111
4
AbrahmJLBanffyMBHarrisMB. Spinal Cord Compression in Patients With Advanced Metastatic Cancer:”All I Care About is Walking and Living My Life”. Jama (2008) 299:937–46. doi: 10.1001/jama.299.8.937
5
SunHNemecekAN. Optimal Management of Malignant Epidural Spinal Cord Compression. Emergency Med Clinics North Am (2009) 27:195–208. doi: 10.1016/j.emc.2009.02.001
6
ZhangZStieglerALBoggonTJKobayashiSHalmosB. EGFR-Mutated Lung Cancer: A Paradigm of Molecular Oncology. Oncotarget (2010) 1:497. doi: 10.18632/oncotarget.186
7
ImielinskiMBergerAHHammermanPSHernandezBPughTJHodisEet al. Mapping the Hallmarks of Lung Adenocarcinoma With Massively Parallel Sequencing. Cell (2012) 150:1107–20. doi: 10.1016/j.cell.2012.08.029
8
RabinowitsGHaddadRI. Overcoming Resistance to EGFR Inhibitor in Head and Neck Cancer: A Review of the Literature. Oral Oncol (2012) 48:1085–9. doi: 10.1016/j.oraloncology.2012.06.016
9
ChenY-M. Update of Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in non-Small-Cell Lung Cancer. J Chin Med Assoc (2013) 76:249–57. doi: 10.1016/j.jcma.2013.01.010
10
KrisMJohnsonBKwiatkowskiDIafrateAWistubaIAronsonSet al. Identification of Driver Mutations in Tumor Specimens From 1,000 Patients With Lung Adenocarcinoma: The Nci’s Lung Cancer Mutation Consortium (LCMC). J Clin Oncol (2011) 29:CRA7506–CRA7506.
11
NormannoNDe LucaABiancoCStrizziLMancinoMMaielloMRet al. Epidermal Growth Factor Receptor (EGFR) Signaling in Cancer. Gene (2006) 366:2–16. doi: 10.1016/j.gene.2005.10.018
12
FungCGrandisJR. Emerging Drugs to Treat Squamous Cell Carcinomas of the Head and Neck. Expert Opin Emerg Drugs (2012) 15:355–73. doi: 10.1517/14728214.2010.497754
13
JohnsonDE. Targeting Proliferation and Survival Pathways in Head and Neck Cancer for Therapeutic Benefit. Chin J Cancer (2012) 31:319–26. doi: 10.5732/cjc.011.10404
14
ErcanDXuCYanagitaMMonastCSPratilasCAMonteroJet al. Reactivation of ERK Signaling Causes Resistance to EGFR Kinase Inhibitors. Cancer Discov (2012) 2:934–47. doi: 10.1158/2159-8290.CD-12-0103
15
AlamMMishraR. Role of PI3K and EGFR in Oral Cancer Progression and Drug Resistance. Int J Res Appl Sci Biotechnol (2020) 7:85–9. doi: 10.31033/ijrasb.7.6.14
16
AlamMMishraR. Bcl-xL Expression and Regulation in the Progression, Recurrence, and Cisplatin Resistance of Oral Cancer. Life Sci (2021) 280:119705. doi: 10.1016/j.lfs.2021.119705
17
EmiMKimRTanabeKUchidaYTogeT. Targeted Therapy Against Bcl-2-Related Proteins in Breast Cancer Cells. Breast Cancer Res (2005) 7:1–13. doi: 10.1186/bcr1323
18
NakajimaWSharmaKHicksMALeNBrownRKrystalGWet al. Combination With Vorinostat Overcomes ABT-263 (Navitoclax) Resistance of Small Cell Lung Cancer. Cancer Biol Ther (2016) 17:27–35. doi: 10.1080/15384047.2015.1108485
19
ZhangGYeXJiDZhangHSunFShangCet al. Inhibition of Lung Tumor Growth by Targeting EGFR/VEGFR-Akt/NF-κb Pathways With Novel Theanine Derivatives. Oncotarget (2014) 5:8528. doi: 10.18632/oncotarget.2336
20
RemonJMoranTMajemMReguartNDalmauEMarquez-MedinaDet al. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in EGFR-Mutant non-Small Cell Lung Cancer: A New Era Begins. Cancer Treat Rev (2014) 40:93–101. doi: 10.1016/j.ctrv.2013.06.002
21
ZhongW-ZZhouQWuY-L. The Resistance Mechanisms and Treatment Strategies for EGFR-Mutant Advanced non-Small-Cell Lung Cancer. Oncotarget (2017) 8:71358. doi: 10.18632/oncotarget.20311
22
RaoofSMulfordIJFrisco-CabanosHNangiaVTimoninaDLabrotEet al. Targeting FGFR Overcomes EMT-Mediated Resistance in EGFR Mutant non-Small Cell Lung Cancer. Oncogene (2019) 38:6399–413. doi: 10.1038/s41388-019-0887-2
23
PaoWChmieleckiJ. Rational, Biologically Based Treatment of EGFR-Mutant non-Small-Cell Lung Cancer. Nat Rev Cancer (2010) 10:760–74. doi: 10.1038/nrc2947
24
SequistLVJoshiVAJännePAMuzikanskyAFidiasPMeyersonMet al. Response to Treatment and Survival of Patients With non-Small Cell Lung Cancer Undergoing Somatic EGFR Mutation Testing. Oncol (2007) 12:90–8. doi: 10.1634/theoncologist.12-1-90
25
CostaDBHalmosBKumarASchumerSTHubermanMSBoggonTJet al. BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers With Oncogenic EGFR Mutations. PloS Med (2007) 4:e315. doi: 10.1371/journal.pmed.0040315
26
SharmaSVBellDWSettlemanJHaberDA. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat Rev Cancer (2007) 7:169–81. doi: 10.1038/nrc2088
27
LuMLiuBXiongHWuFHuCLiuP. Trans-3, 5, 4´-Trimethoxystilbene Reduced Gefitinib Resistance in NSCLC s via Suppressing MAPK/Akt/Bcl-2 Pathway by Upregulation of miR-345 and miR-498. J Cell Mol Med (2019) 23:2431–41. doi: 10.1111/jcmm.14086
28
ZouMXiaSZhuangLHanNChuQChaoTet al. Knockdown of the Bcl-2 Gene Increases Sensitivity to EGFR Tyrosine Kinase Inhibitors in the H1975 Lung Cancer Cell Line Harboring T790M Mutation. Int J Oncol (2013) 42:2094–102. doi: 10.3892/ijo.2013.1895
29
YangFYangY-HZengW-H. The Inhibition of Cell Growth Through the EGFR/ERK/MMP-2 Pathway Induced by Ampelopsin in the Human Malignant Melanoma A375 Cell Line. Natural Product Commun (2020) 15:1934578X20912864. doi: 10.1177/1934578X20912864
30
GuptaPTaiyabAHussainAAlajmiMFIslamAHassanMI. Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy. Cancers (2021) 13:1898. doi: 10.3390/cancers13081898
31
WolterKGHsuYTSmithCLNechushtanAXiXGYouleRJ. Movement of Bax From the Cytosol to Mitochondria During Apoptosis. J Cell Biol (1997) 139:1281–92. doi: 10.1083/jcb.139.5.1281
32
KiddVJ. Proteolytic Activities That Mediate Apoptosis. Annu Rev Physiol (1998) 60:533–73. doi: 10.1146/annurev.physiol.60.1.533
33
CosulichSCWorrallVHedgePJGreenSClarkePR. Regulation of Apoptosis by BH3 Domains in a Cell-Free System. Curr Biol (1997) 7:913–20. doi: 10.1016/S0960-9822(06)00410-6
34
LeibowitzBYuJ. Mitochondrial Signaling in Cell Death via the Bcl-2 Family. Cancer Biol Ther (2010) 9:417–22. doi: 10.4161/cbt.9.6.11392
35
YouleRJStrasserA. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat Rev Mol Cell Biol (2008) 9:47–59. doi: 10.1038/nrm2308
36
Pećina-ŠlausN. Wnt Signal Transduction Pathway and Apoptosis: A Review. Cancer Cell Int (2010) 10:1–5. doi: 10.1186/1475-2867-10-22
37
ZhangHHolzgreveWDe GeyterC. Bcl2-L-10, a Novel Anti-Apoptotic Member of the Bcl-2 Family, Blocks Apoptosis in the Mitochondria Death Pathway But Not in the Death Receptor Pathway. Hum Mol Genet (2001) 10:2329–39. doi: 10.1093/hmg/10.21.2329
38
SatoTHanadaMBodrugSIrieSIwamaNBoiseLHet al. Interactions Among Members of the Bcl-2 Protein Family Analyzed With a Yeast Two-Hybrid System. Proc Natl Acad Sci (1994) 91:9238–42. doi: 10.1073/pnas.91.20.9238
39
OltvaiZNMillimanCLKorsmeyerSJ. Bcl-2 Heterodimerizes In Vivo With a Conserved Homolog, Bax, That Accelerates Programmed Cell Death. Cell (1993) 74:609–19. doi: 10.1016/0092-8674(93)90509-O
40
AlamMAliSMohammadTHasanGMYadavDKHassanM. B Cell Lymphoma 2: A Potential Therapeutic Target for Cancer Therapy. Int J Mol Sci (2021) 22:10442. doi: 10.3390/ijms221910442
41
WeiMCZongWXChengEHLindstenTPanoutsakopoulouVRossAJet al. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science (2001) 292:727–30. doi: 10.1126/science.1059108
42
ChenKHuZWangLESturgisEMEl-NaggarAKZhangWet al. Single-Nucleotide Polymorphisms at the TP53-Binding or Responsive Promoter Regions of BAX and BCL2 Genes and Risk of Squamous Cell Carcinoma of the Head and Neck. Carcinogenesis (2007) 28:2008–12. doi: 10.1093/carcin/bgm172
43
MoshynskaOSankaranKSaxenaA. Molecular Detection of the G(-248)A BAX Promoter Nucleotide Change in B Cell Chronic Lymphocytic Leukaemia. Mol Pathol (2003) 56:205–9. doi: 10.1136/mp.56.4.205
44
LeBlancHLawrenceDVarfolomeevETotpalKMorlanJSchowPet al. Tumor-Cell Resistance to Death Receptor–Induced Apoptosis Through Mutational Inactivation of the Proapoptotic Bcl-2 Homolog Bax. Nat Med (2002) 8:274–81. doi: 10.1038/nm0302-274
45
MiquelCBorriniFGrandjouanSAuperinAViguierJVelascoVet al. Role of Bax Mutations in Apoptosis in Colorectal Cancers With Microsatellite Instability. Am J Clin Pathol (2005) 123:562–70. doi: 10.1309/JQ2X3RV3L8F9TGYW
46
MoshynskaOMoshynskyyIMisraVSaxenaA. G125A Single-Nucleotide Polymorphism in the Human BAX Promoter Affects Gene Expression. Oncogene (2005) 24:2042–9. doi: 10.1038/sj.onc.1208377
47
SaxenaAMoshynskaOSankaranKViswanathanSSheridanDP. Association of a Novel Single Nucleotide Polymorphism, G(-248)A, in the 5'-UTR of BAX Gene in Chronic Lymphocytic Leukemia With Disease Progression and Treatment Resistance. Cancer Lett (2002) 187:199–205. doi: 10.1016/S0304-3835(02)00378-6
48
AlamMKashyapTMishraPPandaAKNaginiSMishraR. Role and Regulation of Proapoptotic Bax in Oral Squamous Cell Carcinoma and Drug Resistance. Head Neck (2019) 41:185–97. doi: 10.1002/hed.25471
49
LimH-YChoiJ-HKangSYJungYMKimHCOhYTet al. Low Expression of Bax in non-Small Cell Lung Cancer: Predictor of Poor Disease-Free Survival in Adenocarcinoma Patients With Surgical Resection (AACR). (2006).
50
JeongSHLeeH-WHanJHKangSYChoiJ-HJungYMet al. Low Expression of Bax Predicts Poor Prognosis in Resected non-Small Cell Lung Cancer Patients With non-Squamous Histology. Jpn J Clin Oncol (2008) 38:661–9. doi: 10.1093/jjco/hyn089
51
HanJ-YChungY-JParkSWKimJSRhyuM-GKimH-Ket al. The Relationship Between Cisplatin-Induced Apoptosis and P53, Bcl-2 and Bax Expression in Human Lung Cancer Cells. Korean J Intern Med (1999) 14:42. doi: 10.3904/kjim.1999.14.1.42
52
ViktorssonKEkedahlJLindebroMCLewensohnRZhivotovskyBLinderSet al. Defective Stress Kinase and Bak Activation in Response to Ionizing Radiation But Not Cisplatin in a Non-Small Cell Lung Carcinoma Cell Line. Exp Cell Res (2003) 289:256–64. doi: 10.1016/S0014-4827(03)00264-7
53
AlamMKashyapTPramanikKKSinghAKNaginiSMishraR. The Elevated Activation of NFkappaB and AP-1 is Correlated With Differential Regulation of Bcl-2 and Associated With Oral Squamous Cell Carcinoma Progression and Resistance. Clin Oral Investig (2017) 21:2721–31. doi: 10.1007/s00784-017-2074-6
54
CamisascaDRHonoratoJBernardoVda SilvaLEda FonsecaECde FariaPAet al. Expression of Bcl-2 Family Proteins and Associated Clinicopathologic Factors Predict Survival Outcome in Patients With Oral Squamous Cell Carcinoma. Oral Oncol (2009) 45:225–33. doi: 10.1016/j.oraloncology.2008.05.021
55
ManoochehriMKarbasiABandehpourMKazemiB. Down-Regulation of BAX Gene During Carcinogenesis and Acquisition of Resistance to 5-FU in Colorectal Cancer. Pathol Oncol Res (2014) 20:301–7. doi: 10.1007/s12253-013-9695-0
56
NixPCawkwellLPatmoreHGreenmanJStaffordN. Bcl-2 Expression Predicts Radiotherapy Failure in Laryngeal Cancer. Br J Cancer (2005) 92:2185–9. doi: 10.1038/sj.bjc.6602647
57
TsujimotoYFingerLRYunisJNowellPCCroceCM. Cloning of the Chromosome Breakpoint of Neoplastic B Cells With the T(14;18) Chromosome Translocation. Science (1984) 226:1097–9. doi: 10.1126/science.6093263
58
HockenberyDNunezGMillimanCSchreiberRDKorsmeyerSJ. Bcl-2 is an Inner Mitochondrial Membrane Protein That Blocks Programmed Cell Death. Nature (1990) 348:334–6. doi: 10.1038/348334a0
59
Chen-LevyZNourseJClearyML. The Bcl-2 Candidate Proto-Oncogene Product is a 24-Kilodalton Integral-Membrane Protein Highly Expressed in Lymphoid Cell Lines and Lymphomas Carrying the T(14;18) Translocation. Mol Cell Biol (1989) 9:701–10. doi: 10.1128/mcb.9.2.701-710.1989
60
SetoMJaegerUHockettRDGraningerWBennettSGoldmanPet al. Alternative Promoters and Exons, Somatic Mutation and Deregulation of the Bcl-2-Ig Fusion Gene in Lymphoma. EMBO J (1988) 7:123–31. doi: 10.1002/j.1460-2075.1988.tb02791.x
61
TsujimotoY. Stress-Resistance Conferred by High Level of Bcl-2 Alpha Protein in Human B Lymphoblastoid Cell. Oncogene (1989) 4:1331–6.
62
DanialNNKorsmeyerSJ. Cell Death: Critical Control Points. Cell (2004) 116:205–19. doi: 10.1016/S0092-8674(04)00046-7
63
WillisSNAdamsJM. Life in the Balance: How BH3-Only Proteins Induce Apoptosis. Curr Opin Cell Biol (2005) 17:617–25. doi: 10.1016/j.ceb.2005.10.001
64
LiuZWildCDingYYeNChenHWoldEAet al. BH4 Domain of Bcl-2 as a Novel Target for Cancer Therapy. Drug Discov Today (2016) 21:989–96. doi: 10.1016/j.drudis.2015.11.008
65
ReedJC. Proapoptotic Multidomain Bcl-2/Bax-Family Proteins: Mechanisms, Physiological Roles, and Therapeutic Opportunities. Cell Death Differ (2006) 13:1378–86. doi: 10.1038/sj.cdd.4401975
66
PetrosAMMedekANettesheimDGKimDHYoonHSSwiftKet al. Solution Structure of the Antiapoptotic Protein Bcl-2. Proc Natl Acad Sci USA (2001) 98:3012–7. doi: 10.1073/pnas.041619798
67
IkegakiNKatsumataMMinnaJTsujimotoY. Expression of Bcl-2 in Small Cell Lung Carcinoma Cells. Cancer Res (1994) 54:6–8.
68
MinnAJVelezPSchendelSLLiangHMuchmoreSWFesikSWet al. Bcl-X(L) Forms an Ion Channel in Synthetic Lipid Membranes. Nature (1997) 385:353–7. doi: 10.1038/385353a0
69
YinXMOltvaiZNKorsmeyerSJ. BH1 and BH2 Domains of Bcl-2 are Required for Inhibition of Apoptosis and Heterodimerization With Bax. Nature (1994) 369:321–3. doi: 10.1038/369321a0
70
HardwickJMSoaneL. Multiple Functions of BCL-2 Family Proteins. Cold Spring Harbor Perspect Biol (2013) 5:a008722. doi: 10.1101/cshperspect.a008722
71
LucianiDSWhiteSAWidenmaierSBSaranVVTaghizadehFHuXet al. Bcl-2 and Bcl-xL Suppress Glucose Signaling in Pancreatic β-Cells. Diabetes (2013) 62:170–82. doi: 10.2337/db11-1464
72
ZhuCShihWLingCTsaoM. Immunohistochemical Markers of Prognosis in non-Small Cell Lung Cancer: A Review and Proposal for a Multiphase Approach to Marker Evaluation. J Clin Pathol (2006) 59:790–800. doi: 10.1136/jcp.2005.031351
73
ChoiJChoiKBenvenisteENHongY-SLeeJ-HKimJet al. Bcl-2 Promotes Invasion and Lung Metastasis by Inducing Matrix Metalloproteinase-2. Cancer Res (2005) 65:5554–60. doi: 10.1158/0008-5472.CAN-04-4570
74
HuYBebbGTanSNgRYanHSartorJRet al. Antitumor Efficacy of Oblimersen Bcl-2 Antisense Oligonucleotide Alone and in Combination With Vinorelbine in Xenograft Models of Human Non–Small Cell Lung Cancer. Clin Cancer Res (2004) 10:7662–70. doi: 10.1158/1078-0432.CCR-04-1036
75
MartinBPaesmansMBerghmansTBranleFGhisdalLMascauxCet al. Role of Bcl-2 as a Prognostic Factor for Survival in Lung Cancer: A Systematic Review of the Literature With Meta-Analysis. Br J Cancer (2003) 89:55–64. doi: 10.1038/sj.bjc.6601095
76
ShibataYHidakaSTagawaYNagayasuT. Bcl-2 Protein Expression Correlates With Better Prognosis in Patients With Advanced non-Small Cell Lung Cancer. Anticancer Res (2004) 24:1925–8.
77
FengCWuJYangFQiuMHuSGuoSet al. Expression of Bcl-2 is a Favorable Prognostic Biomarker in Lung Squamous Cell Carcinoma. Oncol Lett (2018) 15:6925–30. doi: 10.3892/ol.2018.8198
78
LaudanskiJChyczewskiLNiklińskaWKretowskaMFurmanMSawickiBet al. Expression of Bcl-2 Protein in non-Small Cell Lung Cancer: Correlation With Clinicopathology and Patient Survival. Neoplasma (1999) 46:25–30.
79
KaiserUSchilliMHaagUNeumannKKreipeHKoganEet al. Expression of Bcl-2—Protein in Small Cell Lung Cancer. Lung Cancer (1996) 15:31–40. doi: 10.1016/0169-5002(96)00568-5
80
VauxDLCorySAdamsJM. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates With C-Myc to Immortalize Pre-B Cells. Nature (1988) 335:440–2. doi: 10.1038/335440a0
81
Aharoni-SimonMShumiatcherRYeungAShihAZDolinskyVWDoucetteCAet al. Bcl-2 Regulates Reactive Oxygen Species Signaling and a Redox-Sensitive Mitochondrial Proton Leak in Mouse Pancreatic β-Cells. Endocrinology (2016) 157:2270–81. doi: 10.1210/en.2015-1964
82
OtakeYSoundararajanSSenguptaTKKioEASmithJCPineda-RomanMet al. Overexpression of Nucleolin in Chronic Lymphocytic Leukemia Cells Induces Stabilization of Bcl2 mRNA. Blood (2007) 109:3069–75. doi: 10.1182/blood-2006-08-043257
83
JackelMCDorudianMAMarxDBrinckUSchauerASteinerW. Spontaneous Apoptosis in Laryngeal Squamous Cell Carcinoma is Independent of Bcl-2 and Bax Protein Expression. Cancer (1999) 85:591–9. doi: 10.1002/(SICI)1097-0142(19990201)85:3<591::AID-CNCR9>3.0.CO;2-F
84
ZhangPZhangZZhouXQiuWChenFChenW. Identification of Genes Associated With Cisplatin Resistance in Human Oral Squamous Cell Carcinoma Cell Line. BMC Cancer (2006) 6:224. doi: 10.1186/1471-2407-6-224
85
GibsonSAPellenzCHutchisonREDaveyFRShillitoeEJ. Induction of Apoptosis in Oral Cancer Cells by an Anti-Bcl-2 Ribozyme Delivered by an Adenovirus Vector. Clin Cancer Res (2000) 6:213–22.
86
MeseHSasakiAAlcaldeRENakayamaSMatsumuraT. Regulation of Apoptosis Reduction in the Cisplatin-Resistant A431 Cell Line by Bcl-2 and CPP32. Chemotherapy (2000) 46:69–76. doi: 10.1159/000007258
87
RaffoAJPerlmanHChenMWDayMLStreitmanJSButtyanR. Overexpression of Bcl-2 Protects Prostate Cancer Cells From Apoptosis In Vitro and Confers Resistance to Androgen Depletion In Vivo. Cancer Res (1995) 55:4438–45.
88
NorJEChristensenJLiuJPetersMMooneyDJStrieterRMet al. Up-Regulation of Bcl-2 in Microvascular Endothelial Cells Enhances Intratumoral Angiogenesis and Accelerates Tumor Growth. Cancer Res (2001) 61:2183–8.
89
LeeNYHazlettTLKolandJG. Structure and Dynamics of the Epidermal Growth Factor Receptor C-Terminal Phosphorylation Domain. Protein Sci (2006) 15:1142–52. doi: 10.1110/ps.052045306
90
CallaghanTAntczakMFlickingerTRainesMMyersMKungHJ. A Complete Description of the EGF-Receptor Exon Structure: Implication in Oncogenic Activation and Domain Evolution. Oncogene (1993) 8:2939–48.
91
YardenYSliwkowskiMX. Untangling the ErbB Signalling Network. Nat Rev Mol Cell Biol (2001) 2:127–37. doi: 10.1038/35052073
92
GrandisJRSokJC. Signaling Through the Epidermal Growth Factor Receptor During the Development of Malignancy. Pharmacol Ther (2004) 102:37–46. doi: 10.1016/j.pharmthera.2004.01.002
93
SohJWLeeEHPrywesRWeinsteinIB. Novel Roles of Specific Isoforms of Protein Kinase C in Activation of the C-Fos Serum Response Element. Mol Cell Biol (1999) 19:1313–24. doi: 10.1128/MCB.19.2.1313
94
AlamMHasanGMHassanMI. A Review on the Role of TANK-Binding Kinase 1 Signaling in Cancer. Int J Biol Macromol (2021) 183:2364–75. doi: 10.1016/j.ijbiomac.2021.06.022
95
LynchTJBellDWSordellaRGurubhagavatulaSOkimotoRABranniganBWet al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of non–Small-Cell Lung Cancer to Gefitinib. N Engl J Med (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
96
PaoWMillerVZakowskiMDohertyJPolitiKSarkariaIet al. EGF Receptor Gene Mutations are Common in Lung Cancers From “Never Smokers” and are Associated With Sensitivity of Tumors to Gefitinib and Erlotinib. Proc Natl Acad Sci (2004) 101:13306–11. doi: 10.1073/pnas.0405220101
97
AlamMAliSAshrafGMBilgramiALYadavDKHassanMI. Epigallocatechin 3-Gallate: From Green Tea to Cancer Therapeutics. Food Chem (2022) 379:132135. doi: 10.1016/j.foodchem.2022.132135
98
AbourehabMAAlqahtaniAMYoussifBGGoudaAM. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules (2021) 26:6677.
99
AlamMAshrafGMSheikhKKhanAAliSAnsariMMet al. Potential Therapeutic Implications of Caffeic Acid in Cancer Signaling: Past, Present, and Future. Front Pharmacol (2022) 13:845871. doi: 10.3389/fphar.2022.845871
100
AliSAlamMHasanGMHassanMI. Potential Therapeutic Targets of Klebsiella Pneumoniae: A Multi-Omics Review Perspective. Briefings Funct Genomics (2021). doi: 10.1093/bfgp/elab038
101
PaoWWangTYRielyGJMillerVAPanQLadanyiMet al. KRAS Mutations and Primary Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib. PloS Med (2005) 2:e17. doi: 10.1371/journal.pmed.0020017
102
DingLGetzGWheelerDAMardisERMcLellanMDCibulskisKet al. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature (2008) 455:1069–75. doi: 10.1038/nature07423
103
ReckMRabeKF. Precision Diagnosis and Treatment for Advanced non–Small-Cell Lung Cancer. N Engl J Med (2017) 377:849–61. doi: 10.1056/NEJMra1703413
104
MitsudomiT. Molecular Epidemiology of Lung Cancer and Geographic Variations With Special Reference to EGFR Mutations. Trans Lung Cancer Res (2014) 3:205. doi: 10.3978/j.issn.2218-6751.2014.08.04
105
McCubreyJASteelmanLSAbramsSLLeeJTChangFBertrandFEet al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT Pathways in Malignant Transformation and Drug Resistance. Adv Enzyme Regul (2006) 46:249–79. doi: 10.1016/j.advenzreg.2006.01.004
106
ZhaoJGuerreroAKelnarKPeltierHJBaderAG. Synergy Between Next Generation EGFR Tyrosine Kinase Inhibitors and miR-34a in the Inhibition of non-Small Cell Lung Cancer. Lung Cancer (2017) 108:96–102. doi: 10.1016/j.lungcan.2017.02.020
107
ZhongMMaXSunCChenL. MicroRNAs Reduce Tumor Growth and Contribute to Enhance Cytotoxicity Induced by Gefitinib in non-Small Cell Lung Cancer. Chemico-biol Interact (2010) 184:431–8. doi: 10.1016/j.cbi.2010.01.025
108
DownwardJ. Targeting RAS Signalling Pathways in Cancer Therapy. Nat Rev Cancer (2003) 3:11–22. doi: 10.1038/nrc969
109
ZhouJ-YChenXZhaoJBaoZChenXZhangPet al. MicroRNA-34a Overcomes HGF-Mediated Gefitinib Resistance in EGFR Mutant Lung Cancer Cells Partly by Targeting MET. Cancer Lett (2014) 351:265–71. doi: 10.1016/j.canlet.2014.06.010
110
RicciutiBLeonardiGCMetroGGrignaniFPaglialungaLBellezzaGet al. Targeting the KRAS Variant for Treatment of non-Small Cell Lung Cancer: Potential Therapeutic Applications. Expert Rev Respir Med (2016) 10:53–68. doi: 10.1586/17476348.2016.1115349
111
LadanyiMPaoW. Lung Adenocarcinoma: Guiding EGFR-Targeted Therapy and Beyond. Modern Pathol (2008) 21:S16–22. doi: 10.1038/modpathol.3801018
112
SequistLVBellDWLynchTJHaberDA. Molecular Predictors of Response to Epidermal Growth Factor Receptor Antagonists in Non–Small-Cell Lung Cancer. J Clin Oncol (2007) 25:587–95. doi: 10.1200/JCO.2006.07.3585
113
ShigematsuHLinLTakahashiTNomuraMSuzukiMWistubaIIet al. Clinical and Biological Features Associated With Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. J Natl Cancer Inst (2005) 97:339–46. doi: 10.1093/jnci/dji055
114
PramanikKKSinghAKAlamMKashyapTMishraPPandaAKet al. Reversion-Inducing Cysteine-Rich Protein With Kazal Motifs and its Regulation by Glycogen Synthase Kinase 3 Signaling in Oral Cancer. Tumor Biol (2016) 37:15253–64. doi: 10.1007/s13277-016-5362-x
115
LaimerKSpizzoGGastlGObristPBrunhuberTFongDet al. High EGFR Expression Predicts Poor Prognosis in Patients With Squamous Cell Carcinoma of the Oral Cavity and Oropharynx: A TMA-Based Immunohistochemical Analysis. Oral Oncol (2007) 43:193–8. doi: 10.1016/j.oraloncology.2006.02.009
116
ModjtahediHEssapenS. Epidermal Growth Factor Receptor Inhibitors in Cancer Treatment: Advances, Challenges and Opportunities. Anticancer Drugs (2009) 20:851–5. doi: 10.1097/CAD.0b013e3283330590
117
ShaibWKonoSSabaN. Antiepidermal Growth Factor Receptor Therapy in Squamous Cell Carcinoma of the Head and Neck. J Oncol (2012) 2012:521215. doi: 10.1155/2012/521215
118
JiHSharplessNEWongK-K. EGFR Targeted Therapy: View From Biological Standpoint. Cell Cycle (2006) 5:2072–6. doi: 10.4161/cc.5.18.3277
119
WargoJAReubenACooperZAOhKSSullivanRJ. Immune Effects of Chemotherapy, Radiation, and Targeted Therapy and Opportunities for Combination With Immunotherapy. Paper Presented At: Semin Oncol (Elsevier) (2015) 42:601–16. doi: 10.1053/j.seminoncol.2015.05.007
120
MaemondoMInoueAKobayashiKSugawaraSOizumiSIsobeHet al. Gefitinib or Chemotherapy for non–Small-Cell Lung Cancer With Mutated EGFR. N Engl J Med (2010) 362:2380–8. doi: 10.1056/NEJMoa0909530
121
MokTSWuY-LThongprasertSYangC-HChuD-TSaijoNet al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N Engl J Med (2009) 361:947–57. doi: 10.1056/NEJMoa0810699
122
BlumenscheinGRSaintignyPLiuSKimESTsaoASHerbstRSet al. Comprehensive Biomarker Analysis and Final Efficacy Results of Sorafenib in the BATTLE Trial. Clin Cancer Res (2013) 19:6967–75. doi: 10.1158/1078-0432.CCR-12-1818
123
InoueAKobayashiKUsuiKMaemondoMOkinagaSMikamiIet al. First-Line Gefitinib for Patients With Advanced non-Small-Cell Lung Cancer Harboring Epidermal Growth Factor Receptor Mutations Without Indication for Chemotherapy. J Clin Oncol (2009) 27:1394–400. doi: 10.1200/JCO.2008.18.7658
124
BlumenscheinGJrSmitEPlanchardDKimD-WCadranelJDe PasTet al. A Randomized Phase II Study of the MEK1/MEK2 Inhibitor Trametinib (GSK1120212) Compared With Docetaxel in KRAS-Mutant Advanced non-Small-Cell Lung Cancer (NSCLC). Ann Oncol (2015) 26:894–901. doi: 10.1093/annonc/mdv072
125
FerrerIZugazagoitiaJHerbertzSJohnWPaz-AresLSchmid-BindertG. KRAS-Mutant non-Small Cell Lung Cancer: From Biology to Therapy. Lung Cancer (2018) 124:53–64. doi: 10.1016/j.lungcan.2018.07.013
126
PapadimitrakopoulouVLeeJJWistubaIITsaoASFossellaFVKalhorNet al. The BATTLE-2 Study: A Biomarker-Integrated Targeted Therapy Study in Previously Treated Patients With Advanced non–Small-Cell Lung Cancer. J Clin Oncol (2016) 34:3638. doi: 10.1200/JCO.2015.66.0084
127
NagasakaMZhuVWLimSMGrecoMWuFOuS-HI. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors for Advanced EGFR+ NSCLC. J Thorac Oncol (2021) 16:740–63. doi: 10.1016/j.jtho.2020.11.028
128
ZhangCLeighlNBWuY-LZhongW-Z. Emerging Therapies for non-Small Cell Lung Cancer. J Hematol Oncol (2019) 12:1–24. doi: 10.1186/s13045-019-0731-8
129
MassarelliEVarella-GarciaMTangXXavierACOzburnNCLiuDDet al. KRAS Mutation is an Important Predictor of Resistance to Therapy With Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in non–Small-Cell Lung Cancer. Clin Cancer Res (2007) 13:2890–6. doi: 10.1158/1078-0432.CCR-06-3043
130
WilhelmSMCarterCTangLWilkieDMcNabolaARongHet al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res (2004) 64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443
131
BaselgaJ. The EGFR as a Target for Anticancer Therapy—Focus on Cetuximab. Eur J Cancer (2001) 37:16–22. doi: 10.1016/S0959-8049(01)00233-7
132
CiardielloFBiancoRDamianoVFontaniniGCaputoRPomaticoGet al. Antiangiogenic and Antitumor Activity of Anti-Epidermal Growth Factor Receptor C225 Monoclonal Antibody in Combination With Vascular Endothelial Growth Factor Antisense Oligonucleotide in Human GEO Colon Cancer Cells. Clin Cancer Res (2000) 6:3739–47.
133
AlamMAhmedSElasbaliAMAdnanMAlamSHassanMIet al. Therapeutic Implications of Caffeic Acid in Cancer and Neurological Diseases. Front Oncol (2022) 12:860508. doi: 10.3389/fonc.2022.860508
134
PramanikKKNaginiSSinghAKMishraPKashyapTNathNet al. Glycogen Synthase Kinase-3β Mediated Regulation of Matrix Metalloproteinase-9 and its Involvement in Oral Squamous Cell Carcinoma Progression and Invasion. Cell Oncol (2018) 41:47–60. doi: 10.1007/s13402-017-0358-0
135
NewmanDJCraggGM. Natural Products as Sources of New Drugs From 1981 to 2014. J Natural Products (2016) 79:629–61. doi: 10.1021/acs.jnatprod.5b01055
136
LeeW-LHuangJ-YShyurL-F. Phytoagents for Cancer Management: Regulation of Nucleic Acid Oxidation, ROS, and Related Mechanisms. Oxid Med Cell Longevity (2013) 2013. doi: 10.1155/2013/925804
137
YanX-BXieTWangS-DWangZLiH-YYeZ-M. Apigenin Inhibits Proliferation of Human Chondrosarcoma Cells via Cell Cycle Arrest and Mitochondrial Apoptosis Induced by ROS Generation-an In Vitro and In Vivo Study. Int J Clin Exp Med (2018) 11:1615–31.
138
LuLZhaoZLiuLGongWDongJ. Combination of Baicalein and Docetaxel Additively Inhibits the Growth of non-Small Cell Lung Cancer In Vivo. Tradit Med Modern Med (2018) 1:213–8. doi: 10.1142/S2575900018500131
139
DengQ-PWangM-JZengXChenGGHuangR-Y. Effects of Glycyrrhizin in a Mouse Model of Lung Adenocarcinoma. Cell Physiol Biochem (2017) 41:1383–92. doi: 10.1159/000467897
140
DouJWangZMaLPengBMaoKLiCet al. Baicalein and Baicalin Inhibit Colon Cancer Using Two Distinct Fashions of Apoptosis and Senescence. Oncotarget (2018) 9:20089. doi: 10.18632/oncotarget.24015
141
AdamsLSPhungSYeeNSeeramNPLiLChenS. Blueberry Phytochemicals Inhibit Growth and Metastatic Potential of MDA-MB-231 Breast Cancer Cells Through Modulation of the Phosphatidylinositol 3-Kinase Pathway. Cancer Res (2010) 70:3594–605. doi: 10.1158/0008-5472.CAN-09-3565
142
ZhangWSuJXuHYuSLiuYZhangYet al. Dicumarol Inhibits PDK1 and Targets Multiple Malignant Behaviors of Ovarian Cancer Cells. PloS One (2017) 12:e0179672. doi: 10.1371/journal.pone.0179672
143
HanYMaRCaoGLiuHHeLTangLet al. Combined Treatment of Cinobufotalin and Gefitinib Exhibits Potent Efficacy Against Lung Cancer. Evid-Based Complement Altern Med (2021) 2021:6612365. doi: 10.1155/2021/6612365
144
AlamSMohammadTPadderRAHassanMIHusainM. Thymoquinone and Quercetin Induce Enhanced Apoptosis in non-Small Cell Lung Cancer in Combination Through the Bax/Bcl2 Cascade. J Cell Biochem (2022) 123:259–74. doi: 10.1002/jcb.30162
145
LeoneMZhaiDSarethSKitadaSReedJCPellecchiaM. Cancer Prevention by Tea Polyphenols is Linked to Their Direct Inhibition of Antiapoptotic Bcl-2-Family Proteins. Cancer Res (2003) 63:8118–21.
146
MasudaMSuzuiMWeinsteinIB. Effects of Epigallocatechin-3-Gallate on Growth, Epidermal Growth Factor Receptor Signaling Pathways, Gene Expression, and Chemosensitivity in Human Head and Neck Squamous Cell Carcinoma Cell Lines. Clin Cancer Res (2001) 7:4220–9.
147
LiRZangYLiCPatelNSGrandisJRJohnsonDE. ABT-737 Synergizes With Chemotherapy to Kill Head and Neck Squamous Cell Carcinoma Cells via a Noxa-Mediated Pathway. Mol Pharmacol (2009) 75:1231–9. doi: 10.1124/mol.108.052969
148
VoglerM. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv Med (2014) 2014:943648. doi: 10.1155/2014/943648
149
RobertsAWSeymourJFBrownJRWierdaWGKippsTJKhawSLet al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J Clin Oncol (2012) 30:488–96. doi: 10.1200/JCO.2011.34.7898
150
TseCShoemakerARAdickesJAndersonMGChenJJinSet al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res (2008) 68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836
151
VaillantFMerinoDLeeLBreslinKPalBRitchieMEet al. Targeting BCL-2 With the BH3 Mimetic ABT-199 in Estrogen Receptor-Positive Breast Cancer. Cancer Cell (2013) 24:120–9. doi: 10.1016/j.ccr.2013.06.002
152
AshimoriNZeitlinBDZhangZWarnerKTurkieniczIMSpaldingACet al. TW-37, a Small-Molecule Inhibitor of Bcl-2, Mediates S-Phase Cell Cycle Arrest and Suppresses Head and Neck Tumor Angiogenesis. Mol Cancer Ther (2009) 8:893–903. doi: 10.1158/1535-7163.MCT-08-1078
153
WangZSongWAboukameelAMohammadMWangGBanerjeeSet al. TW-37, a Small-Molecule Inhibitor of Bcl-2, Inhibits Cell Growth and Invasion in Pancreatic Cancer. Int J Cancer (2008) 123:958–66. doi: 10.1002/ijc.23610
154
OliverCLBauerJAWolterKGUbellMLNarayanAO'ConnellKMet al. In Vitro Effects of the BH3 Mimetic, (-)-Gossypol, on Head and Neck Squamous Cell Carcinoma Cells. Clin Cancer Res (2004) 10:7757–63. doi: 10.1158/1078-0432.CCR-04-0551
155
ChenJFreemanALiuJDaiQLeeRM. The Apoptotic Effect of HA14-1, a Bcl-2-Interacting Small Molecular Compound, Requires Bax Translocation and is Enhanced by PK11195. Mol Cancer Ther (2002) 1:961–7.
156
HuangSOkumuraKSinicropeFA. BH3 Mimetic Obatoclax Enhances TRAIL-Mediated Apoptosis in Human Pancreatic Cancer Cells. Clin Cancer Res (2009) 15:150–9. doi: 10.1158/1078-0432.CCR-08-1575
157
SinicropeFAPeningtonRCTangXM. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis is Inhibited by Bcl-2 But Restored by the Small Molecule Bcl-2 Inhibitor, HA 14-1, in Human Colon Cancer Cells. Clin Cancer Res (2004) 10:8284–92. doi: 10.1158/1078-0432.CCR-04-1289
158
HoffmannTKLeenenKHafnerDBalzVGerharzCDGrundAet al. Antitumor Activity of Protein Kinase C Inhibitors and Cisplatin in Human Head and Neck Squamous Cell Carcinoma Lines. Anticancer Drugs (2002) 13:93–100. doi: 10.1097/00001813-200201000-00011
159
CasaraPDavidsonJClaperonALe Toumelin-BraizatGVoglerMBrunoAet al. S55746 is a Novel Orally Active BCL-2 Selective and Potent Inhibitor That Impairs Hematological Tumor Growth. Oncotarget (2018) 9:20075–88. doi: 10.18632/oncotarget.24744
160
RansonMHammondLAFerryDKrisMTulloAMurrayPIet al. ZD1839, a Selective Oral Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor, is Well Tolerated and Active in Patients With Solid, Malignant Tumors: Results of a Phase I Trial. J Clin Oncol (2002) 20:2240–50. doi: 10.1200/JCO.2002.10.112
161
FukuokaMYanoSGiacconeGTamuraTNakagawaKDouillardJ-Yet al. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients With Advanced non–Small-Cell Lung Cancer. J Clin Oncol (2003) 21:2237–46. doi: 10.1200/JCO.2003.10.038
162
KrisMGNataleRBHerbstRSLynchTJJrPragerDBelaniCPet al. Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients With non–Small Cell Lung Cancer: A Randomized Trial. Jama (2003) 290:2149–58. doi: 10.1001/jama.290.16.2149
163
CuferTVrdoljakE. Results From a Phase II, Open-Label, Randomized Study (SIGN) Comparing Gefitinib With Docetaxel as Second-Line Therapy in Patients With Advanced (Stage IIIb or IV) Non-Small-Cell Lung Cancer. J Clin Oncol (2005) 23:7035–5. doi: 10.1200/jco.2005.23.16_suppl.7035
164
CuferTVrdoljakEGaafarRErensoyIPembertonKGroupSS. Phase II, Open-Label, Randomized Study (SIGN) of Single-Agent Gefitinib (IRESSA) or Docetaxel as Second-Line Therapy in Patients With Advanced (Stage IIIb or IV) non-Small-Cell Lung Cancer. Anti-cancer Drugs (2006) 17:401–9. doi: 10.1097/01.cad.0000203381.99490.ab
165
GiacconeGHerbstRManegoldCScagliottiGVRosellRMillerVet al. Gefitinib in Combination With Gemcitabine and Cisplatin in Advanced non-Small-Cell Lung Cancer: A Phase III Trial–INTACT 1. J Clin Oncol (2004) 22:2004–784. doi: 10.1200/JCO.2004.08.001
166
HerbstRGiacconeGSchillerJNataleRMillerVManegoldCet al. Gefitinib in Combination With Paclitaxel and Carboplatin in Advanced Non-Small-Cell Lung Cancer: A Phase III Trial–INTACT 2. J Clin Oncol (2004) 22:785–94. doi: 10.1200/JCO.2004.07.215
167
ShepherdFARodrigues PereiraJCiuleanuTTanEHHirshVThongprasertSet al. Erlotinib in Previously Treated non–Small-Cell Lung Cancer. N Engl J Med (2005) 353:123–32. doi: 10.1056/NEJMoa050753
168
FestucciaCMuziPMillimaggiDBiordiLGravinaGSpecaSet al. Molecular Aspects of Gefitinib Antiproliferative and Pro-Apoptotic Effects in PTEN-Positive and PTEN-Negative Prostate Cancer Cell Lines. Endoc-Rel Cancer (2005) 12:983–98. doi: 10.1677/erc.1.00986
169
WangMChangAY-C. Molecular Mechanism of Action and Potential Biomarkers of Growth Inhibition of Synergistic Combination of Afatinib and Dasatinib Against Gefitinib-Resistant non-Small Cell Lung Cancer Cells. Oncotarget (2018) 9:16533. doi: 10.18632/oncotarget.24814
170
OnoMHirataAKometaniTMiyagawaMUedaS-IKinoshitaHet al. Sensitivity to Gefitinib (Iressa, ZD1839) in non-Small Cell Lung Cancer Cell Lines Correlates With Dependence on the Epidermal Growth Factor (EGF) Receptor/Extracellular Signal-Regulated Kinase 1/2 and EGF Receptor/Akt Pathway for Proliferation. Mol Cancer Ther (2004) 3:465–72.
171
CraggMSKurodaJPuthalakathHHuangDCSStrasserA. Gefitinib-Induced Killing of NSCLC Cell Lines Expressing Mutant EGFR Requires BIM and can be Enhanced by BH3 Mimetics. PloS Med (2007) 4:e316. doi: 10.1371/journal.pmed.0040316
172
AriyamaHQinBBabaETanakaRMitsugiKHaradaMet al. Gefitinib, a Selective EGFR Tyrosine Kinase Inhibitor, Induces Apoptosis Through Activation of Bax in Human Gallbladder Adenocarcinoma Cells. J Cell Biochem (2006) 97:724–34. doi: 10.1002/jcb.20678
173
CappuzzoFMagriniECeresoliGLBartoliniSRossiELudoviniVet al. Akt Phosphorylation and Gefitinib Efficacy in Patients With Advanced non–Small-Cell Lung Cancer. J Natl Cancer Inst (2004) 96:1133–41. doi: 10.1093/jnci/djh217
174
HöpfnerMSutterAPHuetherASchuppanDZeitzMScherüblH. Targeting the Epidermal Growth Factor Receptor by Gefitinib for Treatment of Hepatocellular Carcinoma. J Hepatol (2004) 41:1008–16. doi: 10.1016/j.jhep.2004.08.024
175
WakelingAEGuySPWoodburnJRAshtonSECurryBJBarkerAJet al. ZD1839 (Iressa): An Orally Active Inhibitor of Epidermal Growth Factor Signaling With Potential for Cancer Therapy. Cancer Res (2002) 62:5749–54.
176
CohenMHWilliamsGASridharaRChenGPazdurR. FDA Drug Approval Summary: Gefitinib (ZD1839)(Iressa®) Tablets. Oncol (2003) 8:303–6. doi: 10.1634/theoncologist.8-4-303
177
KazandjianDBlumenthalGMYuanWHeKKeeganPPazdurR. FDA Approval of Gefitinib for the Treatment of Patients With Metastatic EGFR Mutation–Positive non–Small Cell Lung Cancer. Clin Cancer Res (2016) 22:1307–12. doi: 10.1158/1078-0432.CCR-15-2266
178
HirataAOgawaS-IKometaniTKuwanoTNaitoSKuwanoMet al. ZD1839 (Iressa) Induces Antiangiogenic Effects Through Inhibition of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res (2002) 62:2554–60.
179
BrehmerDGreffZGodlKBlenckeSKurtenbachAWeberMet al. Cellular Targets of Gefitinib. Cancer Res (2005) 65:379–82.
180
KitagawaDYokotaKGoudaMNarumiYOhmotoHNishiwakiEet al. Activity-Based Kinase Profiling of Approved Tyrosine Kinase Inhibitors. Genes Cells (2013) 18:110–22. doi: 10.1111/gtc.12022
181
KanchaRKvon BubnoffNPeschelCDuysterJ. Functional Analysis of Epidermal Growth Factor Receptor (EGFR) Mutations and Potential Implications for EGFR Targeted Therapy. Clin Cancer Res (2009) 15:460–7. doi: 10.1158/1078-0432.CCR-08-1757
182
LambertJDYangCS. Cancer Chemopreventive Activity and Bioavailability of Tea and Tea Polyphenols. Mutat Res (2003) 523-524:201–8. doi: 10.1016/S0027-5107(02)00336-6
183
YangCSLambertJDJuJLuGSangS. Tea and Cancer Prevention: Molecular Mechanisms and Human Relevance. Toxicol Appl Pharmacol (2007) 224:265–73. doi: 10.1016/j.taap.2006.11.024
184
MukhtarHAhmadN. Tea Polyphenols: Prevention of Cancer and Optimizing Health. Am J Clin Nutr (2000) 71:1698S–1702S; discussion 1703S-1694S. doi: 10.1093/ajcn/71.6.1698S
185
TangSNSinghCNallDMeekerDShankarSSrivastavaRK. The Dietary Bioflavonoid Quercetin Synergizes With Epigallocathechin Gallate (EGCG) to Inhibit Prostate Cancer Stem Cell Characteristics, Invasion, Migration and Epithelial-Mesenchymal Transition. J Mol Signal (2010) 5:14. doi: 10.1186/1750-2187-5-14
186
YangCSMaliakalPMengX. Inhibition of Carcinogenesis by Tea. Annu Rev Pharmacol Toxicol (2002) 42:25–54. doi: 10.1146/annurev.pharmtox.42.082101.154309
187
SadavaDWhitlockEKaneSE. The Green Tea Polyphenol, Epigallocatechin-3-Gallate Inhibits Telomerase and Induces Apoptosis in Drug-Resistant Lung Cancer Cells. Biochem Biophys Res Commun (2007) 360:233–7. doi: 10.1016/j.bbrc.2007.06.030
188
SuganumaMKurusuMSuzukiKTasakiEFujikiH. Green Tea Polyphenol Stimulates Cancer Preventive Effects of Celecoxib in Human Lung Cancer Cells by Upregulation of GADD153 Gene. Int J Cancer (2006) 119:33–40. doi: 10.1002/ijc.21809
189
El-MowafyASalemHAl-GayyarMEl-MeseryMEl-AzabM. Evaluation of Renal Protective Effects of the Green-Tea (EGCG) and Red Grape Resveratrol: Role of Oxidative Stress and Inflammatory Cytokines. Natural Product Res (2011) 25:850–6. doi: 10.1080/14786419.2010.533669
190
KatiyarSKAfaqFPerezAMukhtarH. Green Tea Polyphenol (–)-Epigallocatechin-3-Gallate Treatment of Human Skin Inhibits Ultraviolet Radiation-Induced Oxidative Stress. Carcinogenesis (2001) 22:287–94. doi: 10.1093/carcin/22.2.287
191
SabetkarMLowSYBradleyNJJacobsMNaseemKMRichard BruckdorferK. The Nitration of Platelet Vasodilator Stimulated Phosphoprotein Following Exposure to Low Concentrations of Hydrogen Peroxide. Platelets (2008) 19:282–92. doi: 10.1080/09537100801915142
192
MengQVelalarCNRuanR. Regulating the Age-Related Oxidative Damage, Mitochondrial Integrity, and Antioxidative Enzyme Activity in Fischer 344 Rats by Supplementation of the Antioxidant Epigallocatechin-3-Gallate. Rejuvenation Res (2008) 11:649–60. doi: 10.1089/rej.2007.0645
193
LambertJDEliasRJ. The Antioxidant and Pro-Oxidant Activities of Green Tea Polyphenols: A Role in Cancer Prevention. Arch Biochem Biophys (2010) 501:65–72. doi: 10.1016/j.abb.2010.06.013
194
LiMLiJ-JGuQ-HAnJCaoL-MYangH-Pet al. EGCG Induces Lung Cancer A549 Cell Apoptosis by Regulating Ku70 Acetylation. Oncol Rep (2016) 35:2339–47. doi: 10.3892/or.2016.4587
195
LinH-YHouS-CChenS-CKaoM-CYuC-CFunayamaSet al. (–)-Epigallocatechin Gallate Induces Fas/CD95-Mediated Apoptosis Through Inhibiting Constitutive and IL-6-Induced JAK/STAT3 Signaling in Head and Neck Squamous Cell Carcinoma Cells. J Agric Food Chem (2012) 60:2480–9. doi: 10.1021/jf204362n
196
HagenRMChedeaVSMintoffCPBowlerEMorseHRLadomeryMR. Epigallocatechin-3-Gallate Promotes Apoptosis and Expression of the Caspase 9a Splice Variant in PC3 Prostate Cancer Cells. Int J Oncol (2013) 43:194–200. doi: 10.3892/ijo.2013.1920
197
TangYZhaoDYElliottSZhaoWCurielTJBeckmanBSet al. Epigallocatechin-3 Gallate Induces Growth Inhibition and Apoptosis in Human Breast Cancer Cells Through Survivin Suppression. Int J Oncol (2007) 31:705–11. doi: 10.3892/ijo.31.4.705
198
AlamMAliSAhmedSElasbaliAMAdnanMIslamAet al. Therapeutic Potential of Ursolic Acid in Cancer and Diabetic Neuropathy Diseases. Int J Mol Sci (2021) 22:12162. doi: 10.3390/ijms222212162
199
WeiRMaoLXuPZhengXHackmanRMMackenzieGGet al. Suppressing Glucose Metabolism With Epigallocatechin-3-Gallate (EGCG) Reduces Breast Cancer Cell Growth in Preclinical Models. Food Funct (2018) 9:5682–96. doi: 10.1039/C8FO01397G
200
LiNChenXLiaoJYangGWangSJosephsonYet al. Inhibition of 7,12-Dimethylbenz[a]Anthracene (DMBA)-Induced Oral Carcinogenesis in Hamsters by Tea and Curcumin. Carcinogenesis (2002) 23:1307–13. doi: 10.1093/carcin/23.8.1307
201
ShankarSGanapathySHingoraniSRSrivastavaRK. EGCG Inhibits Growth, Invasion, Angiogenesis and Metastasis of Pancreatic Cancer. Front Biosci (2008) 13:440–52. doi: 10.2741/2691
202
ShankarSGanapathySSrivastavaRK. Green Tea Polyphenols: Biology and Therapeutic Implications in Cancer. Front Biosci (2007) 12:4881–99. doi: 10.2741/2435
203
ZhaoJBlayneyALiuXGandyLJinWYanLet al. EGCG Binds Intrinsically Disordered N-Terminal Domain of P53 and Disrupts P53-MDM2 Interaction. Nat Commun (2021) 12:986. doi: 10.1038/s41467-021-21258-5
204
RiazMAshfaqUAQasimMYasmeenEUl QamarMTAnwarF. Screening of Medicinal Plant Phytochemicals as Natural Antagonists of P53-MDM2 Interaction to Reactivate P53 Functioning. Anticancer Drugs (2017) 28:1032–8. doi: 10.1097/CAD.0000000000000548
205
MilliganSABurkePColemanDTBigelowRLSteffanJJCarrollJLet al. The Green Tea Polyphenol EGCG Potentiates the Antiproliferative Activity of C-Met and Epidermal Growth Factor Receptor Inhibitors in non–Small Cell Lung Cancer Cells. Clin Cancer Res (2009) 15:4885–94. doi: 10.1158/1078-0432.CCR-09-0109
206
LinHYHouSCChenSCKaoMCYuCCFunayamaSet al. Epigallocatechin Gallate Induces Fas/CD95-Mediated Apoptosis Through Inhibiting Constitutive and IL-6-Induced JAK/STAT3 Signaling in Head and Neck Squamous Cell Carcinoma Cells. J Agric Food Chem (2012) 60:2480–9. (-)-. doi: 10.1021/jf204362n
207
AdhamiVMAhmadNMukhtarH. Molecular Targets for Green Tea in Prostate Cancer Prevention. J Nutr (2003) 133:2417S–24S. doi: 10.1093/jn/133.7.2417S
208
AminARKhuriFRChenZGShinDM. Synergistic Growth Inhibition of Squamous Cell Carcinoma of the Head and Neck by Erlotinib and Epigallocatechin-3-Gallate: The Role of P53-Dependent Inhibition of Nuclear factor-kappaB. Cancer Prev Res (Phila) (2009) 2:538–45. doi: 10.1158/1940-6207.CAPR-09-0063
209
ChungLYCheungTCKongSKFungKPChoyYMChanZYet al. Induction of Apoptosis by Green Tea Catechins in Human Prostate Cancer DU145 Cells. Life Sci (2001) 68:1207–14. doi: 10.1016/S0024-3205(00)01020-1
210
YamamotoTDigumarthiHAranbayevaZWatahaJLewisJMesserRet al. EGCG-Targeted P57/KIP2 Reduces Tumorigenicity of Oral Carcinoma Cells: Role of C-Jun N-Terminal Kinase. Toxicol Appl Pharmacol (2007) 224:318–25. doi: 10.1016/j.taap.2006.11.013
211
AlmatroodiSAAlmatroudiAKhanAAAlhumaydhiFAAlsahliMARahmaniAH. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and its Role in the Therapy of Various Types of Cancer. Molecules (2020) 25:3146. doi: 10.3390/molecules25143146
212
LuoK-WLungW-YChun-XieX-LLHuangW-R. EGCG Inhibited Bladder Cancer T24 and 5637 Cell Proliferation and Migration via PI3K/AKT Pathway. Oncotarget (2018) 9:12261. doi: 10.18632/oncotarget.24301
213
MaY-CLiCGaoFXuYJiangZ-BLiuJ-Xet al. Epigallocatechin Gallate Inhibits the Growth of Human Lung Cancer by Directly Targeting the EGFR Signaling Pathway. Oncol Rep (2014) 31:1343–9. doi: 10.3892/or.2013.2933
214
ZhuJJiangYYangXWangSXieCLiXet al. Wnt/beta-Catenin Pathway Mediates (-)-Epigallocatechin-3-Gallate (EGCG) Inhibition of Lung Cancer Stem Cells. Biochem Biophys Res Commun (2017) 482:15–21. doi: 10.1016/j.bbrc.2016.11.038
215
HeLZhangEShiJLiXZhouKZhangQet al. (–)-Epigallocatechin-3-Gallate Inhibits Human Papillomavirus (HPV)-16 Oncoprotein-Induced Angiogenesis in non-Small Cell Lung Cancer Cells by Targeting HIF-1α. Cancer Chemother Pharmacol (2013) 71:713–25. doi: 10.1007/s00280-012-2063-z
216
LiXFengYLiuJFengXZhouKTangX. Epigallocatechin-3-Gallate Inhibits IGF-I-Stimulated Lung Cancer Angiogenesis Through Downregulation of HIF-1α and VEGF Expression. Lifestyle Genomics (2013) 6:169–78. doi: 10.1159/000354402
217
RelatJBlancafortAOliverasGCufíSHaroDMarreroPFet al. Different Fatty Acid Metabolism Effects of (–)-Epigallocatechin-3-Gallate and C75 in Adenocarcinoma Lung Cancer. BMC Cancer (2012) 12:1–8. doi: 10.1186/1471-2407-12-280
218
KohYWChoiECKangSUHwangHSLeeMHPyunJet al. Green Tea (–)-Epigallocatechin-3-Gallate Inhibits HGF-Induced Progression in Oral Cavity Cancer Through Suppression of HGF/c-Met. J Nutr Biochem (2011) 22:1074–83. doi: 10.1016/j.jnutbio.2010.09.005
219
WangJSunPWangQZhangPWangYZiCet al. (–)-Epigallocatechin-3-Gallate Derivatives Combined With Cisplatin Exhibit Synergistic Inhibitory Effects on non-Small-Cell Lung Cancer Cells. Cancer Cell Int (2019) 19:1–16. doi: 10.1186/s12935-019-0981-0
220
ChenB-HHsiehC-HTsaiS-YWangC-YWangC-C. Anticancer Effects of Epigallocatechin-3-Gallate Nanoemulsion on Lung Cancer Cells Through the Activation of AMP-Activated Protein Kinase Signaling Pathway. Sci Rep (2020) 10:1–11. doi: 10.1038/s41598-020-62136-2
221
CorySAdamsJM. Killing Cancer Cells by Flipping the Bcl-2/Bax Switch. Cancer Cell (2005) 8:5–6. doi: 10.1016/j.ccr.2005.06.012
222
OltersdorfTElmoreSWShoemakerARArmstrongRCAugeriDJBelliBAet al. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature (2005) 435:677–81. doi: 10.1038/nature03579
223
LeeJMKimHSKimAChangYSLeeJGChoJet al. ABT-737, a BH3 Mimetic, Enhances the Therapeutic Effects of Ionizing Radiation in K-Ras Mutant Non-Small Cell Lung Cancer Preclinical Model. Yonsei Med J (2022) 63:16. doi: 10.3349/ymj.2022.63.1.16
224
GandhiLCamidgeDRDe OliveiraMRBonomiPGandaraDKhairaDet al. Phase I Study of Navitoclax (ABT-263), a Novel Bcl-2 Family Inhibitor, in Patients With Small-Cell Lung Cancer and Other Solid Tumors. J Clin Oncol (2011) 29:909. doi: 10.1200/JCO.2010.31.6208
225
HauckPChaoBHLitzJKrystalGW. Alterations in the Noxa/Mcl-1 Axis Determine Sensitivity of Small Cell Lung Cancer to the BH3 Mimetic ABT-737. Mol Cancer Ther (2009) 8:883–92. doi: 10.1158/1535-7163.MCT-08-1118
226
KaeferAYangJNoertersheuserPMensingSHumerickhouseRAwniWet al. Mechanism-Based Pharmacokinetic/Pharmacodynamic Meta-Analysis of Navitoclax (ABT-263) Induced Thrombocytopenia. Cancer Chemother Pharmacol (2014) 74:593–602. doi: 10.1007/s00280-014-2530-9
227
RudinCMHannCLGaronEBDe OliveiraMRBonomiPDCamidgeDRet al. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients With Relapsed Small Cell Lung Cancer. Clin Cancer Res (2012) 18:3163–9. doi: 10.1158/1078-0432.CCR-11-3090
228
HuangZ. Bcl-2 Family Proteins as Targets for Anticancer Drug Design. Oncogene (2000) 19:6627–31. doi: 10.1038/sj.onc.1204087
229
HannCLDanielVCSugarEADobromilskayaIMurphySCCopeLet al. Therapeutic Efficacy of ABT-737, a Selective Inhibitor of BCL-2, in Small Cell Lung Cancer. Cancer Res (2008) 68:2321–8. doi: 10.1158/0008-5472.CAN-07-5031
230
TagschererKEFasslACamposBFarhadiMKraemerABöckBet al. Apoptosis-Based Treatment of Glioblastomas With ABT-737, a Novel Small Molecule Inhibitor of Bcl-2 Family Proteins. Oncogene (2008) 27:6646–56. doi: 10.1038/onc.2008.259
231
Van DelftMFWeiAHMasonKDVandenbergCJChenLCzabotarPEet al. The BH3 Mimetic ABT-737 Targets Selective Bcl-2 Proteins and Efficiently Induces Apoptosis via Bak/Bax If Mcl-1 is Neutralized. Cancer Cell (2006) 10:389–99. doi: 10.1016/j.ccr.2006.08.027
232
XiaJBaiHYanBLiRShaoMXiongLet al. Mimicking the BIM BH3 Domain Overcomes Resistance to EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant non-Small Cell Lung Cancer. Oncotarget (2017) 8:108522. doi: 10.18632/oncotarget.19411
233
GongYSomwarRPolitiKBalakMChmieleckiJJiangXet al. Induction of BIM is Essential for Apoptosis Triggered by EGFR Kinase Inhibitors in Mutant EGFR-Dependent Lung Adenocarcinomas. PloS Med (2007) 4:e294. doi: 10.1371/journal.pmed.0040294
234
PremkumarDRJaneEPDiDomenicoJDVukmerNAAgostinoNRPollackIF. ABT-737 Synergizes With Bortezomib to Induce Apoptosis, Mediated by Bid Cleavage, Bax Activation, and Mitochondrial Dysfunction in an Akt-Dependent Context in Malignant Human Glioma Cell Lines. J Pharmacol Exp Ther (2012) 341:859–72. doi: 10.1124/jpet.112.191536
235
RahmaniMAustMMAttkissonEWilliamsDCJrFerreira-GonzalezAGrantS. Dual Inhibition of Bcl-2 and Bcl-xL Strikingly Enhances PI3K Inhibition-Induced Apoptosis in Human Myeloid Leukemia Cells Through a GSK3- and Bim-Dependent Mechanism. Cancer Res (2013) 73:1340–51. doi: 10.1158/0008-5472.CAN-12-1365
236
ZhangHZhongXZhangXShangDZhouYIZhangC. Enhanced Anticancer Effect of ABT-737 in Combination With Naringenin on Gastric Cancer Cells. Exp Ther Med (2016) 11:669–73. doi: 10.3892/etm.2015.2912
237
Petigny-LechartierCDubocCJebahiALouisM-HAbeilardEDenoyelleCet al. The Mtorc1/2 Inhibitor AZD8055 Strengthens the Efficiency of the MEK Inhibitor Trametinib to Reduce the Mcl-1/[Bim and Puma] Ratio and to Sensitize Ovarian Carcinoma Cells to ABT-737. Mol Cancer Ther (2017) 16:102–15. doi: 10.1158/1535-7163.MCT-16-0342
238
JebahiAVilledieuMPétigny-LechartierCcBrotinELouisM-HAbeilardEet al. PI3K/mTOR Dual Inhibitor NVP-BEZ235 Decreases Mcl-1 Expression and Sensitizes Ovarian Carcinoma Cells to Bcl-xL-Targeting Strategies, Provided That Bim Expression is Induced. Cancer Lett (2014) 348:38–49. doi: 10.1016/j.canlet.2014.03.001
239
ShinJAKimLHLeeSJJeongJHJungJYLeeHNet al. Targeting ERK1/2-Bim Signaling Cascades by BH3-Mimetic ABT-737 as an Alternative Therapeutic Strategy for Oral Cancer. Oncotarget (2015) 6:35667–83. doi: 10.18632/oncotarget.5523
240
ShajahanANDobbinZCHickmanFEDakshanamurthySClarkeR. Tyrosine-Phosphorylated Caveolin-1 (Tyr-14) Increases Sensitivity to Paclitaxel by Inhibiting BCL2 and BCLxL Proteins via C-Jun N-Terminal Kinase (JNK). J Biol Chem (2012) 287:17682–92. doi: 10.1074/jbc.M111.304022
241
WangHYangYBShenHMGuJLiTLiXM. ABT-737 Induces Bim Expression via JNK Signaling Pathway and its Effect on the Radiation Sensitivity of HeLa Cells. PloS One (2012) 7:e52483. doi: 10.1371/journal.pone.0052483
242
KiprianovaIRemyJMiloschNMohrenzIVSeifertVAignerAet al. Sorafenib Sensitizes Glioma Cells to the BH3 Mimetic ABT-737 by Targeting MCL1 in a STAT3-Dependent Manner. Neoplasia (2015) 17:564–73. doi: 10.1016/j.neo.2015.07.003
243
HmzaAOsmanMAdnanAOmarE. Immunomodulatory Effect of Nigella Sativa Oil in the Disease Process of Type 1 Diabetic Rats. Res J Pharmaceut Biol Chem Sci (2013) 4:980–8.
244
Al-JassirMS. Chemical Composition and Microflora of Black Cumin (Nigella Sativa L.) Seeds Growing in Saudi Arabia. Food Chem (1992) 45:239–42. doi: 10.1016/0308-8146(92)90153-S
245
GhoshehOAHoudiAACrooksPA. High Performance Liquid Chromatographic Analysis of the Pharmacologically Active Quinones and Related Compounds in the Oil of the Black Seed (Nigella Sativa L.). J Pharm Biomed Anal (1999) 19:757–62. doi: 10.1016/S0731-7085(98)00300-8
246
NajmiANasiruddinMKhanRAHaqueSF. Effect of Nigella Sativa Oil on Various Clinical and Biochemical Parameters of Insulin Resistance Syndrome. Int J Diabetes Develop Cntr (2008) 28:11. doi: 10.4103/0973-3930.41980
247
BanerjeeSPadhyeSAzmiAWangZPhilipPAKucukOet al. Review on Molecular and Therapeutic Potential of Thymoquinone in Cancer. Nutr Cancer (2010) 62:938–46. doi: 10.1080/01635581.2010.509832
248
KunduJKimD-HKunduJKChunK-S. Thymoquinone Induces Heme Oxygenase-1 Expression in HaCaT Cells via Nrf2/ARE Activation: Akt and Ampkα as Upstream Targets. Food Chem Toxicol (2014) 65:18–26. doi: 10.1016/j.fct.2013.12.015
249
KaeferCMMilnerJA. The Role of Herbs and Spices in Cancer Prevention. J Nutr Biochem (2008) 19:347–61. doi: 10.1016/j.jnutbio.2007.11.003
250
MathurMLGaurJSharmaRHaldiyaKR. Antidiabetic Properties of a Spice Plant Nigella Sativa. J Endocrinol Metab (2011) 1:1–8. doi: 10.4021/jem12e
251
SteinmetzKAPotterJD. Vegetables, Fruit, and Cancer Prevention: A Review. J Am Dietetic Assoc (1996) 96:1027–39. doi: 10.1016/S0002-8223(96)00273-8
252
JafriSHGlassJShiRZhangSPrinceMKleiner-HancockH. Thymoquinone and Cisplatin as a Therapeutic Combination in Lung Cancer: In Vitro and In Vivo. J Exp Clin Cancer Res (2010) 29:1–11. doi: 10.1186/1756-9966-29-87
253
AlmajaliBAl-JamalHANTaibWRWIsmailIJohanMFDoolaaneaAAet al. Thymoquinone, as a Novel Therapeutic Candidate of Cancers. Pharmaceuticals (2021) 14:369. doi: 10.3390/ph14040369
254
YangJKuangX-rLvP-tYanX-x. Thymoquinone Inhibits Proliferation and Invasion of Human Nonsmall-Cell Lung Cancer Cells via ERK Pathway. Tumor Biol (2015) 36:259–69. doi: 10.1007/s13277-014-2628-z
255
BanerjeeSKasebAOWangZKongDMohammadMPadhyeSet al. Antitumor Activity of Gemcitabine and Oxaliplatin is Augmented by Thymoquinone in Pancreatic Cancer. Cancer Res (2009) 69:5575–83. doi: 10.1158/0008-5472.CAN-08-4235
256
El-AzizMAAHassanHAMohamedMHMekiARMAbdel-GhaffarSKHusseinMR. The Biochemical and Morphological Alterations Following Administration of Melatonin, Retinoic Acid and Nigella Sativa in Mammary Carcinoma: An Animal Model. Int J Exp Pathol (2005) 86:383–96. doi: 10.1111/j.0959-9673.2005.00448.x
257
SalimEIFukushimaS. Chemopreventive Potential of Volatile Oil From Black Cumin (Nigella Sativa L.) Seeds Against Rat Colon Carcinogenesis. Nutr Cancer (2003) 45:195–202. doi: 10.1207/S15327914NC4502_09
258
BourgouSKsouriRBellilaASkandraniIFallehHMarzoukB. Phenolic Composition and Biological Activities of Tunisian Nigella Sativa L. Shoots and Roots. Comptes Rendus Biol (2008) 331:48–55. doi: 10.1016/j.crvi.2007.11.001
259
KhaderMBresgenNEcklP. Antimutagenic Effects of Ethanolic Extracts From Selected Palestinian Medicinal Plants. J Ethnopharmacol (2010) 127:319–24. doi: 10.1016/j.jep.2009.11.001
260
FarahIOBegumRA. Effect of Nigella Sativa (N. Sativa L.) and Oxidative Stress on the Survival Pattern of MCF-7 Breast Cancer Cells. Biomed Sci Instrumentation (2003) 39:359–64.
261
PadhyeSBanerjeeSAhmadAMohammadRSarkarFH. From Here to Eternity-the Secret of Pharaohs: Therapeutic Potential of Black Cumin Seeds and Beyond. Cancer Ther (2008) 6:495.
262
RandhawaMA. Black Seed, Nigella Sativa, Deserves More Attention. J Ayub Med Coll Abbottabad (2008) 20:1–2.
263
Ait MbarekLAit MouseHElabbadiNBensalahMGamouhAAboufatimaRet al. Anti-Tumor Properties of Blackseed (Nigella Sativa L.) Extracts. Braz J Med Biol Res (2007) 40:839–47. doi: 10.1590/S0100-879X2006005000108
264
BadaryOAAl-ShabanahONagiMAl-RikabiAElmazarMM. Inhibition of Benzo (a) Pyrene-Induced Forestomach Carcinogenesis in Mice by Thymoquinone. Eur J Cancer Prevent: Off J Eur Cancer Prev Organisation (ECP) (1999) 8:435–40. doi: 10.1097/00008469-199910000-00009
265
El-AbharHAbdallahDSalehS. Gastroprotective Activity of Nigella Sativa Oil and its Constituent, Thymoquinone, Against Gastric Mucosal Injury Induced by Ischaemia/Reperfusion in Rats. J Ethnopharmacol (2003) 84:251–8. doi: 10.1016/S0378-8741(02)00324-0
266
AldebasiYHRahmaniAHKhanAAAlySM. The Effect of Vascular Endothelial Growth Factor in the Progression of Bladder Cancer and Diabetic Retinopathy. Int J Clin Exp Med (2013) 6:239.
267
ArafaE-SAZhuQShahZIWaniGBarakatBMRacomaIet al. Thymoquinone Up-Regulates PTEN Expression and Induces Apoptosis in Doxorubicin-Resistant Human Breast Cancer Cells. Mutat Research/Fundamental Mol Mech Mutagene (2011) 706:28–35. doi: 10.1016/j.mrfmmm.2010.10.007
268
SalomiMNairSCPanikkarK. Inhibitory Effects of Nigella Sativa and Saffron (Crocus Sativus) on Chemical Carcinogenesis in Mice. (1991). doi: 10.1080/01635589109514142
269
SalomiNNairSJayawardhananKVargheseCPanikkarK. Antitumour Principles From Nigella Sativa Seeds. Cancer Lett (1992) 63:41–6. doi: 10.1016/0304-3835(92)90087-C
270
BadaryOATahaRAGamal El-DinAMAbdel-WahabMH. Thymoquinone is a Potent Superoxide Anion Scavenger. Drug Chem Toxicol (2003) 26:87–98. doi: 10.1081/DCT-120020404
271
NagiMNAlmakkiHASayed-AhmedMMAl-BekairiAM. Thymoquinone Supplementation Reverses Acetaminophen-Induced Oxidative Stress, Nitric Oxide Production and Energy Decline in Mice Liver. Food Chem Toxicol (2010) 48:2361–5. doi: 10.1016/j.fct.2010.05.072
272
SamaliANordgrenHZhivotovskyBPetersonEOrreniusS. A Comparative Study of Apoptosis and Necrosis in HepG2 Cells: Oxidant-Induced Caspase Inactivation Leads to Necrosis. Biochem Biophys Res Commun (1999) 255:6–11. doi: 10.1006/bbrc.1998.0139
273
HuatBTKSwamySMK. Intracellular Glutathione Depletion and Reactive Oxygen Species Generation are Important in α-Hederin-Induced Apoptosis of P388 Cells. Mol Cell Biochem (2003) 245:127–39. doi: 10.1023/A:1022807207948
274
NagiMNAlmakkiHA. Thymoquinone Supplementation Induces Quinone Reductase and Glutathione Transferase in Mice Liver: Possible Role in Protection Against Chemical Carcinogenesis and Toxicity. Phytother Res (2009) 23:1295–8. doi: 10.1002/ptr.2766
275
AbdelmeguidNEFakhouryRKamalSMAl WafaiRJ. Effects of Nigella Sativa and Thymoquinone on Biochemical and Subcellular Changes in Pancreatic β-Cells of Streptozotocin-Induced Diabetic Rats. J Diabetes (2010) 2:256–66. doi: 10.1111/j.1753-0407.2010.00091.x
276
FathyMNikaidoT. In Vivo Modulation of iNOS Pathway in Hepatocellular Carcinoma by Nigella Sativa. Environ Health Prev Med (2013) 18:377–85. doi: 10.1007/s12199-013-0336-8
277
FoudaAMMDabaMHYDahabGMSharaf el-DinOA. Thymoquinone Ameliorates Renal Oxidative Damage and Proliferative Response Induced by Mercuric Chloride in Rats. Basic Clin Pharmacol Toxicol (2008) 103:109–18. doi: 10.1111/j.1742-7843.2008.00260.x
278
SakalarCYurukMKayaTAytekinMKukSCanatanH. Pronounced Transcriptional Regulation of Apoptotic and TNF–NF-Kappa-B Signaling Genes During the Course of Thymoquinone Mediated Apoptosis in HeLa Cells. Mol Cell Biochem (2013) 383:243–51. doi: 10.1007/s11010-013-1772-x
279
AliBBlundenG. Pharmacological and Toxicological Properties of Nigella Sativa. Phytother Res (2003) 17:299–305. doi: 10.1002/ptr.1309
280
AlhakamyNABadr-EldinSMA FahmyUAlruwailiNKAwanZACarusoGet al. Thymoquinone-Loaded Soy-Phospholipid-Based Phytosomes Exhibit Anticancer Potential Against Human Lung Cancer Cells. Pharmaceutics (2020) 12:761. doi: 10.3390/pharmaceutics12080761
281
SamarghandianSAzimi-NezhadMFarkhondehT. Thymoquinone-Induced Antitumor and Apoptosis in Human Lung Adenocarcinoma Cells. J Cell Physiol (2019) 234:10421–31. doi: 10.1002/jcp.27710
282
AliSAlamMKhatoonFFatimaUElasbaliAMAdnanMet al. Natural Products can be Used in Therapeutic Management of COVID-19: Probable Mechanistic Insights. Biomed Pharmacother (2022) 147:112658. doi: 10.1016/j.biopha.2022.112658
283
SinghSKMishraMKLillardJWSinghR. Thymoquinone Enhanced the Tumoricidal Activity of NK Cells Against Lung Cancer. (Am Assoc Immnol) (2018) 124–5.
284
KunduJChoiBYJeongC-HKunduJKChunK-S. Thymoquinone Induces Apoptosis in Human Colon Cancer HCT116 Cells Through Inactivation of STAT3 by Blocking JAK2-And Src−mediated Phosphorylation of EGF Receptor Tyrosine Kinase. Oncol Rep (2014) 32:821–8. doi: 10.3892/or.2014.3223
285
ParkEJChauhanAKMinK-JParkDCKwonTK. Thymoquinone Induces Apoptosis Through Downregulation of C-FLIP and Bcl-2 in Renal Carcinoma Caki Cells. Oncol Rep (2016) 36:2261–7. doi: 10.3892/or.2016.5019
286
El-GhanyARagwaMSharafNMKassemLAMahranLGHeikalOA. Thymoquinone Triggers Anti-Apoptotic Signaling Targeting Death Ligand and Apoptotic Regulators in a Model of Hepatic Ischemia Reperfusion Injury. Drug Discov Ther (2009) 3296–306.
287
El-MahdyMAZhuQWangQEWaniGWaniAA. Thymoquinone Induces Apoptosis Through Activation of Caspase-8 and Mitochondrial Events in P53-Null Myeloblastic Leukemia HL-60 Cells. Int J Cancer (2005) 117:409–17. doi: 10.1002/ijc.21205
288
AfroseSSJunaidMAkterYTaniaMZhengMKhanMA. Targeting Kinases With Thymoquinone: A Molecular Approach to Cancer Therapeutics. Drug Discov Today (2020) 25:2294–306. doi: 10.1016/j.drudis.2020.07.019
289
HirparaKVAggarwalPMukherjeeAJJoshiNBurmanAC. Quercetin and its Derivatives: Synthesis, Pharmacological Uses With Special Emphasis on Anti-Tumor Properties and Prodrug With Enhanced Bio-Availability. Anti-Cancer Agents Med Chem (Formerly Curr Med Chemistry-Anti-Cancer Agents) (2009) 9:138–61. doi: 10.2174/187152009787313855
290
LamsonDWBrignallMS. Antioxidants and Cancer, Part 3: Quercetin. Altern Med Rev: J Clin Ther (2000) 5:196–208.
291
RobaszkiewiczABalcerczykABartoszG. Antioxidative and Prooxidative Effects of Quercetin on A549 Cells. Cell Biol Int (2007) 31:1245–50. doi: 10.1016/j.cellbi.2007.04.009
292
ChangJ-HLaiS-LChenW-SHungW-YChowJ-MHsiaoMet al. Quercetin Suppresses the Metastatic Ability of Lung Cancer Through Inhibiting Snail-Dependent Akt Activation and Snail-Independent ADAM9 Expression Pathways. Biochim Biophys Acta (BBA)-Mol Cell Res (2017) 1864:1746–58. doi: 10.1016/j.bbamcr.2017.06.017
293
HashemzaeiMDelarami FarAYariAHeraviRETabrizianKTaghdisiSMet al. Anticancer and Apoptosis−Inducing Effects of Quercetin In Vitro and In Vivo. Oncol Rep (2017) 38:819–28. doi: 10.3892/or.2017.5766
294
VafadarAShabaninejadZMovahedpourAFallahiFTaghavipourMGhasemiYet al. Quercetin and Cancer: New Insights Into its Therapeutic Effects on Ovarian Cancer Cells. Cell Biosci (2020) 10:1–17. doi: 10.1186/s13578-020-00397-0
295
YangFSongLWangHWangJXuZXingN. Quercetin in Prostate Cancer: Chemotherapeutic and Chemopreventive Effects, Mechanisms and Clinical Application Potential. Oncol Rep (2015) 33:2659–68. doi: 10.3892/or.2015.3886
296
BegumANTeraoJ. Protective Effect of Quercetin Against Cigarette Tar Extract-Induced Impairment of Erythrocyte Deformability. J Nutr Biochem (2002) 13:265–72. doi: 10.1016/S0955-2863(01)00219-4
297
DavidAVAArulmoliRParasuramanS. Overviews of Biological Importance of Quercetin: A Bioactive Flavonoid. Pharmacognosy Rev (2016) 10:84. doi: 10.4103/0973-7847.194044
298
LeeW-JHsiaoMChangJ-LYangS-FTsengT-HChengC-Wet al. Quercetin Induces Mitochondrial-Derived Apoptosis via Reactive Oxygen Species-Mediated ERK Activation in HL-60 Leukemia Cells and Xenograft. Arch Toxicol (2015) 89:1103–17. doi: 10.1007/s00204-014-1300-0
299
YounHJeongJ-CJeongYSKimE-JUmS-J. Quercetin Potentiates Apoptosis by Inhibiting Nuclear factor-kappaB Signaling in H460 Lung Cancer Cells. Biol Pharm Bull (2013) 36:944–51. doi: 10.1248/bpb.b12-01004
300
ZhangXGuoQChenJChenZ. Quercetin Enhances Cisplatin Sensitivity of Human Osteosarcoma Cells by Modulating microRNA-217-KRAS Axis. Mol Cells (2015) 38:638. doi: 10.14348/molcells.2015.0037
301
ChunOKChungS-JClaycombeKJSongWO. Serum C-Reactive Protein Concentrations are Inversely Associated With Dietary Flavonoid Intake in US Adults. J Nutr (2008) 138:753–60. doi: 10.1093/jn/138.4.753
302
García-MediavillaVCrespoIColladoPSEstellerASánchez-CamposSTuñónMJet al. The Anti-Inflammatory Flavones Quercetin and Kaempferol Cause Inhibition of Inducible Nitric Oxide Synthase, Cyclooxygenase-2 and Reactive C-Protein, and Down-Regulation of the Nuclear Factor kappaB Pathway in Chang Liver Cells. Eur J Pharmacol (2007) 557:221–9. doi: 10.1016/j.ejphar.2006.11.014
303
GuardiaTRotelliAEJuarezAOPelzerLE. Anti-Inflammatory Properties of Plant Flavonoids. Effects of Rutin, Quercetin and Hesperidin on Adjuvant Arthritis in Rat. Il Farmaco (2001) 56:683–7. doi: 10.1016/S0014-827X(01)01111-9
304
WarrenCAPaulhillKJDavidsonLALuptonJRTaddeoSSHongMYet al. Quercetin may Suppress Rat Aberrant Crypt Foci Formation by Suppressing Inflammatory Mediators That Influence Proliferation and Apoptosis. J Nutr (2009) 139:101–5. doi: 10.3945/jn.108.096271
305
XiaoXShiDLiuLWangJXieXKangTet al. Quercetin Suppresses Cyclooxygenase-2 Expression and Angiogenesis Through Inactivation of P300 Signaling. PloS One (2011) 6:e22934. doi: 10.1371/journal.pone.0022934
306
Cruz–CorreaMShoskesDASanchezPZhaoRHylindLMWexnerSDet al. Combination Treatment With Curcumin and Quercetin of Adenomas in Familial Adenomatous Polyposis. Clin Gastroenterol Hepatol (2006) 4:1035–8. doi: 10.1016/j.cgh.2006.03.020
307
HanMSongYZhangX. Quercetin Suppresses the Migration and Invasion in Human Colon Cancer Caco-2 Cells Through Regulating Toll-Like Receptor 4/Nuclear Factor-Kappa B Pathway. Pharmacognosy Magazine (2016) 12:S237. doi: 10.4103/0973-1296.182154
308
PratheeshkumarPSonY-ODivyaSPWangLTurciosLRoyRVet al. Quercetin Inhibits Cr (VI)-Induced Malignant Cell Transformation by Targeting miR-21-PDCD4 Signaling Pathway. Oncotarget (2017) 8:52118. doi: 10.18632/oncotarget.10130
309
ChenDDanielKGChenMSKuhnDJLandis-PiwowarKRDouQP. Dietary Flavonoids as Proteasome Inhibitors and Apoptosis Inducers in Human Leukemia Cells. Biochem Pharmacol (2005) 69:1421–32. doi: 10.1016/j.bcp.2005.02.022
310
VijayababuMKanagarajPArunkumarAIlangovanRAruldhasMArunakaranJ. Quercetin-Induced Growth Inhibition and Cell Death in Prostatic Carcinoma Cells (PC-3) are Associated With Increase in P21 and Hypophosphorylated Retinoblastoma Proteins Expression. J Cancer Res Clin Oncol (2005) 131:765–71. doi: 10.1007/s00432-005-0005-4
311
DuoJYingG-GWangG-WZhangL. Quercetin Inhibits Human Breast Cancer Cell Proliferation and Induces Apoptosis via Bcl-2 and Bax Regulation. Mol Med Rep (2012) 5:1453–6. doi: 10.3892/mmr.2012.845
312
VijayababuMRArunkumarAKanagarajPArunakaranJ. Effects of Quercetin on Insulin-Like Growth Factors (IGFs) and Their Binding Protein-3 (IGFBP-3) Secretion and Induction of Apoptosis in Human Prostate Cancer Cells. J Carcinogene (2006) 5:10. doi: 10.1186/1477-3163-5-10
313
WangXYanYYangLLiMZhongX. Effect of Quercetin on the Expression of Bcl-2/Bax Apoptotic Proteins in Endometrial Cells of Lipopolysaccharide-Induced-Abortion. J Tradit Chin Med Chung i Tsa Chih Ying Wen Pan (2016) 36:737–42. doi: 10.1016/s0254-6272(17)30008-0
314
NiuGYinSXieSLiYNieDMaLet al. Quercetin Induces Apoptosis by Activating Caspase-3 and Regulating Bcl-2 and Cyclooxygenase-2 Pathways in Human HL-60 Cells. Acta Biochim Biophys Sin (2011) 43:30–7. doi: 10.1093/abbs/gmq107
315
KuharMSenSSinghN. > Role of Mitochondria in Quercetin-Enhanced Chemotherapeutic Response in Human Non-Small Cell Lung Carcinoma H-520 Cells. Anticancer Res (2006) 26:1297–303.
316
HuangK-YWangT-HChenC-CLeuY-LLiH-JJhongC-Let al. Growth Suppression in Lung Cancer Cells Harboring EGFR-C797S Mutation by Quercetin. Biomolecules (2021) 11:1271. doi: 10.3390/biom11091271
317
BabyBAntonyPVijayanR. Interactions of Quercetin With Receptor Tyrosine Kinases Associated With Human Lung Carcinoma. Natural Product Res (2018) 32:2928–31. doi: 10.1080/14786419.2017.1385015
318
LupoGCambriaMTOlivieriMRoccoCCaporarelloNLongoAet al. Anti-Angiogenic Effect of Quercetin and its 8-Methyl Pentamethyl Ether Derivative in Human Microvascular Endothelial Cells. J Cell Mol Med (2019) 23:6565–77. doi: 10.1111/jcmm.14455
319
KashyapDGargVKTuliHSYererMBSakKSharmaAKet al. Fisetin and Quercetin: Promising Flavonoids With Chemopreventive Potential. Biomolecules (2019) 9:174. doi: 10.3390/biom9050174
320
DongYYangJYangLLiP. Quercetin Inhibits the Proliferation and Metastasis of Human non-Small Cell Lung Cancer Cell Line: The Key Role of Src-Mediated Fibroblast Growth Factor-Inducible 14 (Fn14)/nuclear Factor Kappa B (NF-κb) Pathway. Med Sci Monit (2020) 26:e920537–920531. doi: 10.12659/MSM.920537
321
AlmatroodiSAAlsahliMAAlmatroudiAVermaAKAloliqiAAllemailemKSet al. Potential Therapeutic Targets of Quercetin, a Plant Flavonol, and its Role in the Therapy of Various Types of Cancer Through the Modulation of Various Cell Signaling Pathways. Molecules (2021) 26:1315. doi: 10.3390/molecules26051315
322
GulatiNLaudetBZohrabianVMMuraliRJhanwar-UniyalM. The Antiproliferative Effect of Quercetin in Cancer Cells is Mediated via Inhibition of the PI3K-Akt/PKB Pathway. Anticancer Res (2006) 26:1177–81.
323
SpencerJPRice-EvansCWilliamsRJ. Modulation of Pro-Survival Akt/protein Kinase B and ERK1/2 Signaling Cascades by Quercetin and its In Vivo Metabolites Underlie Their Action on Neuronal Viability. J Biol Chem (2003) 278:34783–93. doi: 10.1074/jbc.M305063200
324
PradhanSJMishraRSharmaPKunduGC. Quercetin and Sulforaphane in Combination Suppress the Progression of Melanoma Through the Down−Regulation of Matrix Metalloproteinase-9. Exp Ther Med (2010) 1:915–20. doi: 10.3892/etm.2010.144
325
PanRHogdalLJBenitoJMBucciDHanLBorthakurGet al. Selective BCL-2 Inhibition by ABT-199 Causes on-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov (2014) 4:362–75. doi: 10.1158/2159-8290.CD-13-0609
326
RobertsAWDavidsMSPagelJMKahlBSPuvvadaSDGerecitanoJFet al. Targeting BCL2 With Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2016) 374:311–22. doi: 10.1056/NEJMoa1513257
327
MukherjeeNAlmeidaAPartykaKALuYSchwanJVLambertKet al. Combining a GSI and BCL-2 Inhibitor to Overcome Melanoma's Resistance to Current Treatments. Oncotarget (2016) 7:84594. doi: 10.18632/oncotarget.13141
328
CangSIragavarapuCSavoojiJSongYLiuD. ABT-199 (Venetoclax) and BCL-2 Inhibitors in Clinical Development. J Hematol Oncol (2015) 8:129. doi: 10.1186/s13045-015-0224-3
329
SouersAJLeversonJDBoghaertERAcklerSLCatronNDChenJet al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat Med (2013) 19:202–8. doi: 10.1038/nm.3048
330
LochmannTLFlorosKVNaseriMPowellKMCookWMarchRJet al. Venetoclax is Effective in Small-Cell Lung Cancers With High BCL-2 Expression. Clin Cancer Res (2018) 24:360–9. doi: 10.1158/1078-0432.CCR-17-1606
331
PeirsSMatthijssensFGoossensSVan de WalleIRuggeroKde BockCEet al. ABT-199 Mediated Inhibition of BCL-2 as a Novel Therapeutic Strategy in T-Cell Acute Lymphoblastic Leukemia. Blood (2014) 124:3738–47. doi: 10.1182/blood-2014-05-574566
332
DiNardoCDPratzKPullarkatVJonasBAArellanoMBeckerPSet al. Venetoclax Combined With Decitabine or Azacitidine in Treatment-Naive, Elderly Patients With Acute Myeloid Leukemia. Blood (2019) 133:7–17. doi: 10.1182/blood-2018-08-868752
333
ZhangHNimmerPMTahirSKChenJFryerRMHahnKRet al. Bcl-2 Family Proteins are Essential for Platelet Survival. Cell Death Differ (2007) 14:943–51. doi: 10.1038/sj.cdd.4402081
334
LiuZGaoW. Synergistic Effects of Bcl-2 Inhibitors With AZD9291 on Overcoming the Acquired Resistance of AZD9291 in H1975 Cells. Arch Toxicol (2020) 94:3125–36. doi: 10.1007/s00204-020-02816-0
335
GuyJ-BEspenelSLouatiSGauthierAGarciaM-AVialNet al. Combining Radiation to EGFR and Bcl-2 Blockade: A New Approach to Target Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma. J Cancer Res Clin Oncol (2021) 147:1–12. doi: 10.1007/s00432-021-03593-8
336
ThiagarajanPSWuXZhangWShiIBagaiRLeahyPet al. Transcriptomic-Metabolomic Reprogramming in EGFR-Mutant NSCLC Early Adaptive Drug Escape Linking Tgfβ2-Bioenergetics-Mitochondrial Priming. Oncotarget (2016) 7:82013. doi: 10.18632/oncotarget.13307
337
KlanovaMAnderaLSoukupJJanBSvadlenkaJBenesovaSet al. MCL1 Targeting Agent Homoharringtonine Exerts Strong Cytotoxicity Towards Diffuse Large B-Cell Lymphoma (DLBCL) Cells and Synergizes With BCL2 Targeting Agent ABT199 in Eliminating BCL2-Positive DLBCL Cells. Blood (2014) 124:3645. doi: 10.1182/blood.V124.21.3645.3645
338
LemmonMASchlessingerJFergusonKM. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harbor Perspect Biol (2014) 6:a020768. doi: 10.1101/cshperspect.a020768
339
CiardielloFCaputoRBiancoRDamianoVPomaticoGDe PlacidoSet al. Antitumor Effect and Potentiation of Cytotoxic Drugs Activity in Human Cancer Cells by ZD-1839 (Iressa), an Epidermal Growth Factor Receptor-Selective Tyrosine Kinase Inhibitor. Clin Cancer Res (2000) 6:2053–63.
340
SirotnakFMZakowskiMFMillerVAScherHIKrisMG. Efficacy of Cytotoxic Agents Against Human Tumor Xenografts is Markedly Enhanced by Coadministration of ZD1839 (Iressa), an Inhibitor of EGFR Tyrosine Kinase. Clin Cancer Res (2000) 6:4885–92.
341
WilliamsKJTelferBStratfordIWedgeS. ZD1839 (‘Iressa’), a Specific Oral Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor, Potentiates Radiotherapy in a Human Colorectal Cancer Xenograft Model. Br J Cancer (2002) 86:1157–61. doi: 10.1038/sj.bjc.6600182
342
ZhouCWuY-LChenGFengJLiuX-QWangCet al. Erlotinib Versus Chemotherapy as First-Line Treatment for Patients With Advanced EGFR Mutation-Positive non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol (2011) 12:735–42. doi: 10.1016/S1470-2045(11)70184-X
343
DouillardJ-YShepherdFAHirshVMokTSocinskiMAGervaisRet al. Molecular Predictors of Outcome With Gefitinib and Docetaxel in Previously Treated non-Small-Cell Lung Cancer: Data From the Randomized Phase III INTEREST Trial. J Clin Oncol (2010) 28:744–52. doi: 10.1200/JCO.2009.24.3030
344
PaezJGJännePALeeJCTracySGreulichHGabrielSet al. EGFR Mutations in Lung Cancer: Correlation With Clinical Response to Gefitinib Therapy. Science (2004) 304:1497–500. doi: 10.1126/science.1099314
345
FukuokaMWuY-LThongprasertSSunpaweravongPLeongS-SSriuranpongVet al. Biomarker Analyses and Final Overall Survival Results From a Phase III, Randomized, Open-Label, First-Line Study of Gefitinib Versus Carboplatin/Paclitaxel in Clinically Selected Patients With Advanced non–Small-Cell Lung Cancer in Asia (IPASS). J Clin Oncol (2011) 29:2866–74. doi: 10.1200/JCO.2010.33.4235
346
WheelerDLDunnEFHarariPM. Understanding Resistance to EGFR Inhibitors—Impact on Future Treatment Strategies. Nat Rev Clin Oncol (2010) 7:493–507. doi: 10.1038/nrclinonc.2010.97
347
AntonicelliACafarottiSIndiniAGalliARussoACesarioAet al. EGFR-Targeted Therapy for non-Small Cell Lung Cancer: Focus on EGFR Oncogenic Mutation. Int J Med Sci (2013) 10:320. doi: 10.7150/ijms.4609
348
CardarellaSOginoANishinoMButaneyMShenJLydonCet al. Clinical, Pathologic, and Biologic Features Associated With BRAF Mutations in non–Small Cell Lung Cancer. Clin Cancer Res (2013) 19:4532–40. doi: 10.1158/1078-0432.CCR-13-0657
349
HymanDMPuzanovISubbiahVFarisJEChauIBlayJ-Yet al. Vemurafenib in Multiple Nonmelanoma Cancers With BRAF V600 Mutations. New Engl J Med (2015) 373:726–36. doi: 10.1056/NEJMoa1502309
350
PlanchardDBesseBGroenHJSouquetP-JQuoixEBaikCSet al. An Open-Label Phase 2 Trial of Dabrafenib Plus Trametinib in Patients With Previously Treated BRAF V600E–mutant Metastatic non-Small Cell Lung Cancer. Lancet Oncol (2016) 17:984. doi: 10.1016/S1470-2045(16)30146-2
351
PlanchardDKimTMMazieresJQuoixERielyGBarlesiFet al. Dabrafenib in Patients With BRAFV600E-Positive Advanced Non-Small-Cell Lung Cancer: A Single-Arm, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol (2016) 17:642–50. doi: 10.1016/S1470-2045(16)00077-2
352
FramptonGMAliSMRosenzweigMChmieleckiJLuXBauerTMet al. Activation of MET via Diverse Exon 14 Splicing Alterations Occurs in Multiple Tumor Types and Confers Clinical Sensitivity to MET Inhibitors. Cancer Discovery (2015) 5:850–9. doi: 10.1158/2159-8290.CD-15-0285
353
AwadMMOxnardGRJackmanDMSavukoskiDOHallDShivdasaniPet al. MET Exon 14 Mutations in non–Small-Cell Lung Cancer are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and C-Met Overexpression. J Clin Oncol (2016) 34:721–30. doi: 10.1200/JCO.2015.63.4600
354
PaikPKDrilonAFanP-DYuHRekhtmanNGinsbergMSet al. Response to MET Inhibitors in Patients With Stage IV Lung Adenocarcinomas Harboring MET Mutations Causing Exon 14 Skipping. Cancer Discovery (2015) 5:842–9. doi: 10.1158/2159-8290.CD-14-1467
355
CiardielloFBiancoRDamianoVDe LorenzoSPepeSDe PlacidoSet al. Antitumor Activity of Sequential Treatment With Topotecan and Anti-Epidermal Growth Factor Receptor Monoclonal Antibody C225. Clin Cancer Res (1999) 5:909–16.
356
LiuBFangMSchmidtMLuYMendelsohnJFanZ. Induction of Apoptosis and Activation of the Caspase Cascade by Anti-EGF Receptor Monoclonal Antibodies in DiFi Human Colon Cancer Cells do Not Involve the C-Jun N-Terminal Kinase Activity. Br J Cancer (2000) 82:1991–9. doi: 10.1054/bjoc.2000.1201
357
XuLHausmannMDietmaierWKellermeierSPeschTStieber-GunckelMet al. Expression of Growth Factor Receptors and Targeting of EGFR in Cholangiocarcinoma Cell Lines. BMC Cancer (2010) 10:1–11. doi: 10.1186/1471-2407-10-302
358
HuYHongYXuYLiuPGuoD-HChenY. Inhibition of the JAK/STAT Pathway With Ruxolitinib Overcomes Cisplatin Resistance in non-Small-Cell Lung Cancer NSCLC. Apoptosis (2014) 19:1627–36. doi: 10.1007/s10495-014-1030-z
359
EngelmanJAZejnullahuKMitsudomiTSongYHylandCParkJOet al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science (2007) 316:1039–43. doi: 10.1126/science.1141478
Summary
Keywords
NSCLC, B cell lymphoma 2, Bax, apoptosis, signaling, targeted therapy
Citation
Alam M, Alam S, Shamsi A, Adnan M, Elasbali AM, Al-Soud WA, Alreshidi M, Hawsawi YM, Tippana A, Pasupuleti VR and Hassan MI (2022) Bax/Bcl-2 Cascade Is Regulated by the EGFR Pathway: Therapeutic Targeting of Non-Small Cell Lung Cancer. Front. Oncol. 12:869672. doi: 10.3389/fonc.2022.869672
Received
04 February 2022
Accepted
21 February 2022
Published
25 March 2022
Volume
12 - 2022
Edited by
Mohd Wasim Nasser, University of Nebraska Medical Center, United States
Reviewed by
Rajesh Sinha, University of Alabama at Birmingham, United States; Aditya Padhi, RIKEN Yokohama, Japan; Somasish Dastidar, Manipal Academy of Higher Education, India
Updates
Copyright
© 2022 Alam, Alam, Shamsi, Adnan, Elasbali, Al-Soud, Alreshidi, Hawsawi, Tippana, Pasupuleti and Hassan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Md. Imtaiyaz Hassan, mihassan@jmi.ac.in; Visweswara Rao Pasupuleti, pvrao@ums.edu.my
This article was submitted to Molecular and Cellular Oncology, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.