 Ryan M. Finnegan1,2,3
Ryan M. Finnegan1,2,3 Ahmed M. Elshazly2,3,4
Ahmed M. Elshazly2,3,4 Nipa H. Patel2,3
Nipa H. Patel2,3 Liliya Tyutyunyk-Massey2Tammy H. Tran2
Liliya Tyutyunyk-Massey2Tammy H. Tran2 Vishnu Kumarasamy5Erik S. Knudsen5
Vishnu Kumarasamy5Erik S. Knudsen5 David A. Gewirtz2,3*
David A. Gewirtz2,3*- 1Departments of Microbiology & Immunology, Virginia Commonwealth University, Richmond, VA, United States
- 2Departments of Pharmacology & Toxicology, Virginia Commonwealth University, Richmond, VA, United States
- 3Massey Cancer Center, Virginia Commonwealth University, Richmond, VA, United States
- 4Department of Pharmacology and Toxicology, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, Egypt
- 5Department of Molecular and Cellular Biology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, United States
Anti-estrogens or aromatase inhibitors in combination with cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors are the current standard of care for estrogen receptor-positive (ER+) Her-2 negative metastatic breast cancer. Although these combination therapies prolong progression-free survival compared to endocrine therapy alone, the growth-arrested state of residual tumor cells is clearly transient. Tumor cells that escape what might be considered a dormant or quiescent state and regain proliferative capacity often acquire resistance to further therapies. Our studies are based upon the observation that breast tumor cells arrested by Fulvestrant + Palbociclib enter into states of both autophagy and senescence from which a subpopulation ultimately escapes, potentially contributing to recurrent disease. Autophagy inhibition utilizing pharmacologic or genetic approaches only moderately enhanced the response to Fulvestrant + Palbociclib in ER+ MCF-7 breast tumor cells, slightly delaying proliferative recovery. In contrast, the BET inhibitor/degrader, ARV-825, prolonged the growth arrested state in both p53 wild type MCF-7 cells and p53 mutant T-47D cells and significantly delayed proliferative recovery. In addition, ARV-825 added after the Fulvestrant + Palbociclib combination promoted apoptosis and demonstrated efficacy in resistant RB deficient cell lines. These studies indicate that administration of BET inhibitors/degraders, which are currently being investigated in multiple clinical trials, may potentially improve standard of care therapy in metastatic ER+ breast cancer patients and may further prolong progression-free survival.
1 Introduction
The number of new cases of invasive breast cancer in women is projected to be over 287,000 in 2022, resulting in over 43,000 deaths; in fact, rates of breast cancer have been gradually increasing by 0.5% per year since the mid-2000s (1). Amongst these cases, approximately 73% are comprised of hormone-receptor–positive breast cancer (2). The first line hormonal therapy for this subtype of breast cancer utilizes selective estrogen receptor modulators (SERMs) such as tamoxifen, aromatase inhibitors (AIs), and selective estrogen receptor degraders (SERDs), such as Fulvestrant (3). Tamoxifen, a SERM that has been utilized in the clinic for decades, is usually prescribed to treat hormone receptor positive, early-stage breast cancer after surgery to reduce disease recurrence. Despite reducing recurrence for up to five years, resistance to hormonal therapy is a major drawback to the success of these therapies and most cases eventually result in metastatic disease progression (4, 5).
The current standard of care for metastatic ER-positive/Her2 negative breast cancer utilizes the combination of either the estrogen receptor degrader Fulvestrant or aromatase inhibitors such as letrozole with CDK4/6 inhibitors such as Palbociclib (6, 7). Fulvestrant binds and destabilizes the estrogen receptor, resulting in destruction of the receptor by the cells’ normal degradation pathways (8). CDK 4/6 inhibitors, such as Palbociclib, interfere with cell cycle progression by suppressing the CDK-cyclin complex mediated phosphorylation of Rb, thereby allowing the dephosphorylated form of Rb to bind to the transcriptional regulator, E2F, blocking G1/S cell cycle transition (7, 9). In patients whose disease has progressed on prior endocrine therapy, the combination of Fulvestrant with Palbociclib had extended progression-free survival in breast cancer patients from 4.6 to 11.2 months (10). While these treatments represent remarkable improvements, escape from the tumor suppressive effects of even the combination therapies occurs frequently, with the consequence that the patients unfortunately succumb to this disease.
Disease recurrence in breast cancer has been linked to escape from tumor dormancy. Senescence, originally thought to be permanent form of growth arrest, is now recognized as a transient arrest that could be a form of tumor dormancy (11, 12). Senescent tumor cells secrete tumor promoting factors, which has led to preclinical studies of the capacity of senolytics for their elimination. Our laboratory, along with others, have reported on the promotion and senescence by cancer therapies as well as clearance of senescent tumor cell populations by senolytic agents (12–16). Several senolytic agents such as BCl2/BCL-xL inhibitors Navitoclax (ABT-263), and Venatoclax (ABT-199), Panobinostat and Fisetin are currently in preclinical studies and/or in clinical trials with (17–20).
Almost invariably, autophagy also accompanies the induction of senescence by chemotherapy and radiation (21). Autophagy or “self-eating” is an evolutionarily conserved catabolic process through which cellular cargo is sequestered within a double membrane vesicle and ultimately undergoes lysosomal degradation. Many antitumor therapies, such as chemotherapy and radiation, have been shown to promote the cytoprotective form of autophagy, whereby autophagy inhibition sensitizes the tumors to the therapy (22, 23). It is well established in the literature that endocrine therapies display protective autophagy and consequently the autophagy inhibitors chloroquine and hydroxychloroquine show positive outcomes in combination with endocrine therapies, such as fulvestrant or tamoxifen, in preclinical studies (24). Additional studies have investigated the induction of autophagy by palbociclib and the impact of autophagy inhibition on drug sensitivity in breast tumor cells (25). These studies, however, did not involve the current standard of care combination of fulvestrant + Palbociclib.
In general, studies have focused on either autophagy or senescence as separate and distinct responses which might be manipulated for therapeutic benefit. However, autophagy and senescence often occur in parallel in response to cancer therapy (21, 26). In the current work, we demonstrate that Palbociclib and Fulvestrant promote both autophagy and senescence, suggesting that either inhibition of autophagy or the addition of senolytics might enhance and/or prolong the therapeutic response. However, neither pharmacologic inhibition of autophagy nor genetic silencing of the autophagy regulatory gene, ATG5, produced more than a modest improvement in the response to Fulvestrant + Palbociclib, particularly with respect to proliferative recovery. In contrast to the relative lack of impact of autophagy inhibition, the BET degrader, ARV-825, significantly prolonged growth arrest and delayed proliferative recovery in both p53 wildtype MCF-7 cells and p53 mutant T-47D cells independent of Rb status. Consistent with recent studies utilizing BET inhibition (27), these observations suggest that the utilization of BET inhibitors/degraders in sequence with Fulvestrant + Palbociclib may provide a therapeutic advantage for breast cancer patients undergoing standard of care therapy.
2 Methods
2.1 Cell lines
MCF7 cells were generously gifted by Dr. Charles Clevenger, at Virginia Commonwealth University. MCF7 and T47D cells were cultured in DMEM medium supplemented with 10% (v/v) fetal bovine serum (Thermo Scientific, SH30066.03), 100 U/mL penicillin G sodium (Invitrogen, 15140–122), and 100 μg/mL streptomycin sulfate (Invitrogen, 15140–122).
The ATG5-knockdown was generated as follows: Mission shRNA bacterial stocks of ATG5 were purchased from Sigma Aldrich. Lentivirus was produced in HEK 293T cells, which were co-transfected using EndoFectinTM Lenti Transfection Reagent (GeneCopoeia, 1001-01) with a packaging mixture of psPAX2 and pMD2.G constructs (Addgene). Media containing the virus was used to infect the MCF7 cells. Puromycin (1 μg/ml) was used as a selection marker to enrich for the infected cells.
Lentiviral packaging of H2B-GFP vector, pLenti0.3UbCGWH2BC1-PatGFP was carried out in 293FT cells. Exponentially growing MCF7 and T47D cells were infected with the H2B-GFP lentivirus in the presence of polybrene (4µg/ml). GFP positive cells were selected using FACS Aria cell sorter.
2.2 Drug treatment
Fulvestrant (Millipore Sigma, I4409), Palbociclib (LC Laboratories, P-7788), Bafilomycin A1 (Millipore Sigma, 196000), and ARV-825 (MedChemExpress, HY-16954) were dissolved in DMSO. Chloroquine (Millipore Sigma, C6628) was dissolved in sterile 1X PBS (Thermo Fisher Scientific, 10010).
For sensitization studies, cells were exposed to Fulvestrant (100 nM), Palbociclib (1 uM) or the combination for 6 days.
For early autophagy inhibition studies, cells were pre-treated with CQ (10 uM) or Baf (2.5 nM) for 2 h prior to exposure with Fulvestrant, Palbociclib or the combination. CQ or Baf were given in combination with the respective conditions for an addition 48 h, CQ or Baf was removed and Fulvestrant, Palbociclib or the combination were given for an additional 4 days post-CQ or Baf removal to meet the 6-day drug treatment regimen.
For late autophagy inhibition studies, cells were treated with Fulvestrant, Palbociclib or the combination for 6 days and CQ (10 uM) or Baf (2.5 nM) was given for 48 h post-anti-estrogen and CDK 4/6 inhibition therapy.
For senolytic exposure, cells were treated with Fulvestrant, Palbociclib or the combination for 6 days and the respective senolytic was given for 48 h post-anti-estrogen and CDK 4/6 inhibition therapy. ARV-825 was administered for 96 h post-anti-estrogen and CDK 4/6 inhibition therapy.
2.3 Cell viability
Trypan blue exclusion was utilized to assess cell viability. Cells were plated at 20,000 cells per well in a 6-well plate and treated with the respective conditions. On the indicated days, cells were trypsinized, stained with 0.4% trypan blue (Sigma, T01282), and counted on the indicated days using a hemocytometer. Growth curves were generated from the collected data.
2.4 Clonogenic survival assay
Cells were plated at a density of 200 cells per well in 6-well plates and treated with the respective conditions. Media was replenished every other day until colonies form. Colonies were washed with 1X phosphate-saline buffer (PBS, Life Technologies), fixed with 100% methanol and stained with 0.1% crystal violet (Sigma). The number of colonies formed were counted.
2.5 Promotion of apoptosis
The extent of apoptotic cell death was measured using Annexin V-FITC/Propidium iodide staining. On the indicated day, cells were trypsinized, washed with 1X PBS and stained according to manufacturer protocol (Annexin V-FITC Apoptosis Detection Kit; BD Biosciences, 556547). Fluorescence was quantified by flow cytometry using BD FACSCanto II and BD FACSDiva software at the Flow Cytometry Core Facility at Virginia Commonwealth University. For all flow cytometry experiments, 10,000 cells per replicate were analyzed and three replicates for each condition were analyzed per independent experiment unless otherwise stated. All experimental protocols were performed with cells protected from light.
2.6 Acridine orange staining
On the indicated days, cells were stained with 1 μg/ml acridine orange at 37°C for 20 min and then washed with 1X PBS. Cells were imaged using an inverted fluorescence microscope (Olympus, Tokyo, Japan) at 20X magnification. For quantification of autophagic vesicles (AVOs), on the indicated days, cells were trypsinized, harvested and washed with 1X PBS. Pellet fractions were resuspended in 1X PBS and analyzed by BD FACSCanto II and BD FACSDiva software. For all flow cytometry experiments, 10,000 cells per replicate were analyzed and three replicates for each condition were analyzed per independent experiment unless otherwise stated. All experimental protocols were performed with cells protected from light.
2.7 SA-ß-gal staining
On the indicated days, cells were stained with X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) staining as previously described by Dimri et al. (28). Cells were washed with 1X PBS and phase contrast images were taken using an inverted microscope (Olympus, Tokyo, Japan).
T47D-WT and Rb-deleted cells were treated with Palbociclib (1 µM) in combination with Fulvestrant (100 nM) for 6 days. The cells were stained for β-galactosidase to determine the senescence phenotype by using the commercially available kit (Cell Signaling; 9860) according to the manufacturer’s protocol. Cell images were taken using phase-contrast microscope at 20X magnification.
To quantify ß-gal positive senescent cells, after treatment, cells were treated with Bafilomycin A1 (100 nM) for 1 h to achieve lysosomal alkalinization, followed by staining with C12FDG (10 μM) for 2 h at 37 °C. After incubation, cells were collected and analyzed by BD FACSCanto II and BD FACSDiva software. For all flow cytometry experiments, 10,000 cells per replicate were analyzed and three replicates for each condition were analyzed per independent experiment unless otherwise stated. All experimental protocols were performed with cells protected from light.
2.8 Western blot analysis
Western blotting was performed as previously described (29). In brief, after indicated treatments, cells were trypsinized, harvested, and washed with 1X PBS. Pellets were lysed and protein concentrations were determined by the Bradford Assay (Bio-Rad Laboratories, 5000205). Protein samples were loaded and subjected to SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane, and blocked with 5% milk in 1X PBS with 0.1% Tween 20 (Fisher, BP337). The following primary antibodies were used: SQSTM1/p62 (Cell Signaling Technology, 5114T); c-Myc (Cell Signaling Technology, 5605); ATG5 (Cell Signaling Technology, 2630); LC3B (Cell Signaling Technology, 3868); BRD4 (Cell Signaling Technology, 13440S); B-actin (Cell Signaling Technology, 4970); and GAPDH (Cell Signaling Technology, 2118). The membrane was incubated overnight at 4°C with the indicated primary antibodies at a dilution of 1:1000 in 5% BSA. Secondary antibodies: Horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling, anti-mouse, 7076S; anti-rabbit, 7074S). The membrane was then washed, secondary antibody was added at a dilution of 1:2000 in 5% BSA for 2 h at room, and the membrane was washed again with 1X PBS with 0.1% Tween 20 three times. Blots were developed using Pierce enhanced chemiluminescence reagents (Thermo Scientific, 32132) on BioRad ChemiDoc System.
2.9 qRT-PCR
Fulvestrant + Palbociclib treated cells were harvested at Day 6 after drug treatment, and total RNA was extracted using RNeasy kit (QIAGEN, Germany) following the manufacturer’s instructions. cDNA was synthesized using iScript cDNA Synthesis Kit (BioRad, USA) based on the protocol that manufacture provided. cDNAs from different samples were amplified in technical triplicates using iTaq Universal SYBR® Green Supermix from BioRad in QuantStudio™ 3 Real-Time PCR System (Thermofisher, USA). QuantiTect primers were purchased from Qiagen: CXCL8: QT0000322; IL-6: QT00083720; IL-1β: QT00021385; MMP3: QT00060025; GAPDH: QT00079247. Relative mRNA expression was determined using the ΔΔCt method.
2.10 Cell proliferation assay
To determine the proliferation of T47D WT and Rb-deficient cells in real time, live cell imaging using IncuCyte S3 was performed. Cells were seeded in 96 well dish (1000 cells/well) and allowed to adhere overnight. The cells were exposed to Palbociclib (1 µM) in combination with Fulvestrant (100 nM) for 6 days and the cell division as monitored using IncuCyte that performs nuclei count. Following 6 days, the cells were released from the Palbociclib/Fulvestrant combination treatment and allowed to grow in the absence and presence of ARV825 (50 nM) for 96 H. Following 96H exposure, the cells were released from ARV825, and the cellular outgrowth was monitored. Based on the nuclei counts, the relative proliferation rate was determined. Growth cures were generated using GraphPad Prism.
2.11 Statistics
Unless otherwise indicated, all quantitative data is shown as mean ± SEM from at least three independent experiments, all of which were conducted in triplicates or duplicates. GraphPad Prism 9.0 software was used for statistical analysis. All data was analyzed using either a one- or two-way ANOVA, as appropriate, with Tukey or Sidak post hoc.3 Results
3 Results
3.1 The growth arrest response to Fulvestrant + Palbociclib in MCF7 breast tumor cells
While anti-estrogen therapy combined with CDK 4/6 inhibition is the standard of care for HR+/HER2- breast cancer patients diagnosed with locally advanced or metastatic disease progression following endocrine therapy, this combination therapy only modestly prolongs patient survival (30). In an effort to simulate the clinical treatment regimen in an in vitro environment, MCF7 cells were exposed to Fulvestrant and Palbociclib for 6 days and fresh media was replenished after drug removal on day 6. Cell viability was assessed utilizing trypan blue exclusion on the indicated days (Figure 1A). Fulvestrant initially delayed tumor cell growth and, after a delay, arrested the cells. Palbociclib, alone, and in combination with Fulvestrant, completely halted the growth of the MCF7 cells; however, the cells generally began to recover from treatment after a period of approximately 8-12 days (see Figures 3–6), which is consistent with earlier work from this and other laboratories (11–13).
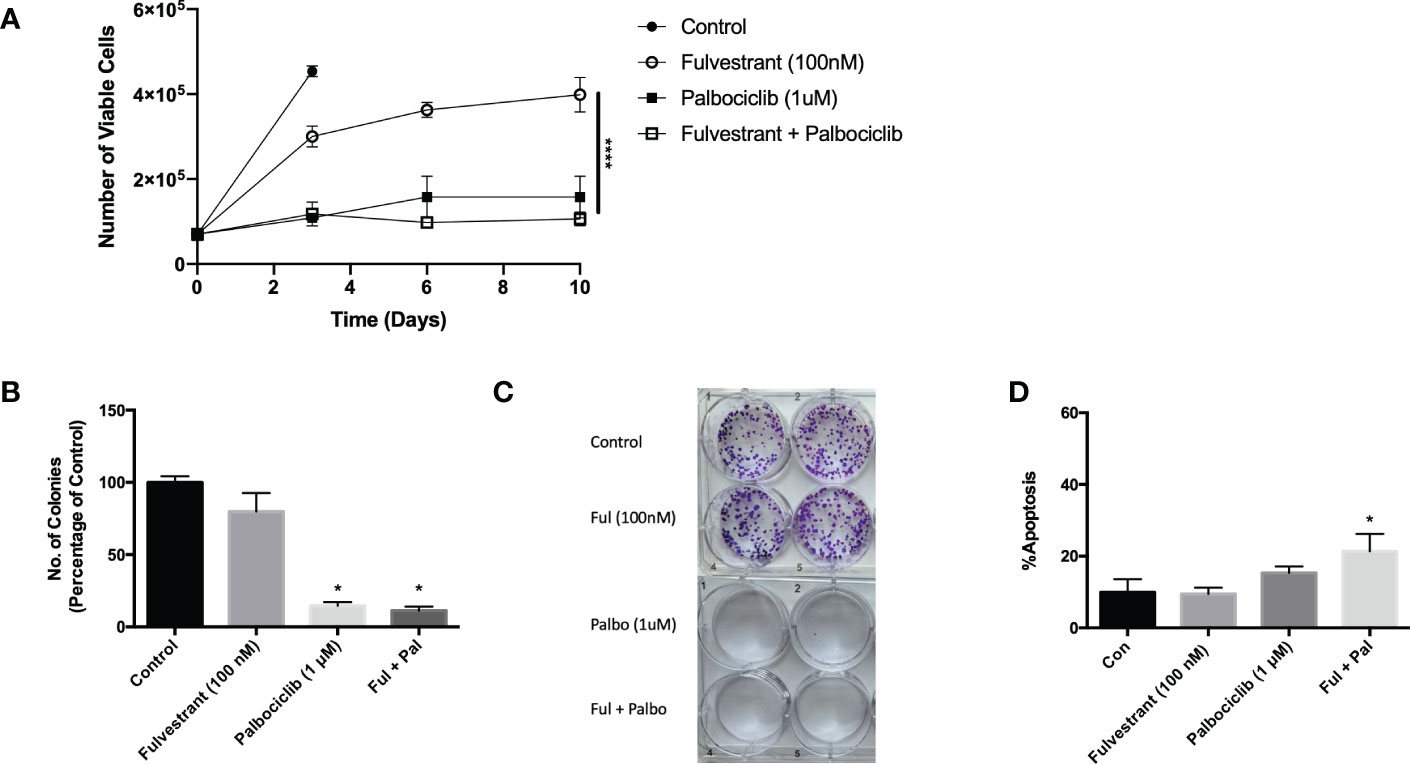
Figure 1 Fulvestrant in combination with Palbociclib sensitizes MCF7 cells. Cells were treated with Fulvestrant (100 nM), Palbociclib (1 µM) or the combination for 6 days (A-C). (A) Viability of MCF7 cells was monitored based on trypan blue exclusion at indicated days following drug exposure (n=3). ****P ≤ 0.0001 indicate statistical significance of each condition compared to Fulvestrant alone. (B) After exposure for 6 days, cells were incubated in fresh medium for 7 days. Quantification of colonies expressed as relative percentage compared to controls (n=3). (C) Representative colony formation 7 days after drug removal by crystal violet staining. (D) Apoptosis was measured using annexin V/PI staining at the end of the 6-day treatment and fluorescence was measured using flow cytometry (n=3). Unless stated otherwise, data were from three independent experiments. *P ≤ 0.05 indicate statistical significance of each condition compared to control as determined using two‐way ANOVA with Sidak's post hoc test.
The effectiveness of Palbociclib alone as well as the combination treatment in suppressing cell growth was confirmed by clonogenic survival studies (Figures 1B, C). The growth inhibitory effect of fulvestrant alone did not achieve significance, in contrast to the moderate effects in the temporal response assay; consequently, the observed effects are largely palbociclib driven. While therapy-induced tumor cell death is the desired outcome of anti-cancer treatment, there was a relatively low degree of apoptosis in MCF7 cells treated with Fulvestrant, Palbociclib or the combination (Figure 1D), which may be permissive for proliferative recovery. Consequently, one of the primary goals of the present work was to identify a strategy that might convert the growth arrest response to one of cell death, initially through efforts to block autophagy.
3.2 Fulvestrant and Palbociclib-induced autophagy
In response to therapy, cancer cells upregulate multiple mechanisms in attempts to evade cell death, one of which is autophagy (31, 32). Autophagy is conventionally considered to be a cytoprotective process that allows cells to combat either intrinsic or extrinsic forms of injury; however, other functions of autophagy have been identified, specifically a cytotoxic form (33) and what has been termed as a nonprotective form (31, 34, 35). Consequently, we examined whether autophagy was induced in response to the anti-estrogen, Fulvestrant, and CDK4/6 inhibition therapy in MCF7 cells. Initially, acridine orange was utilized at day 4 to assess acidic vesicle formation. Figure 2A demonstrates basal autophagy in these cells as well as increased acidic vesicle generation in response to Fulvestrant or/and Palbociclib. Figure 2B further provides quantification of fluorescence via flow cytometry and indicates that autophagy is marginally higher for the combination treatment at day 3 and significantly higher at day 6 (although autophagy is reduced overall). To verify autophagy induction, western blot analysis in Figure 2C revealed a temporal decline in p62/SQSTM1 levels in MCF7 cells treated with Fulvestrant, Palbociclib and the drug combination.
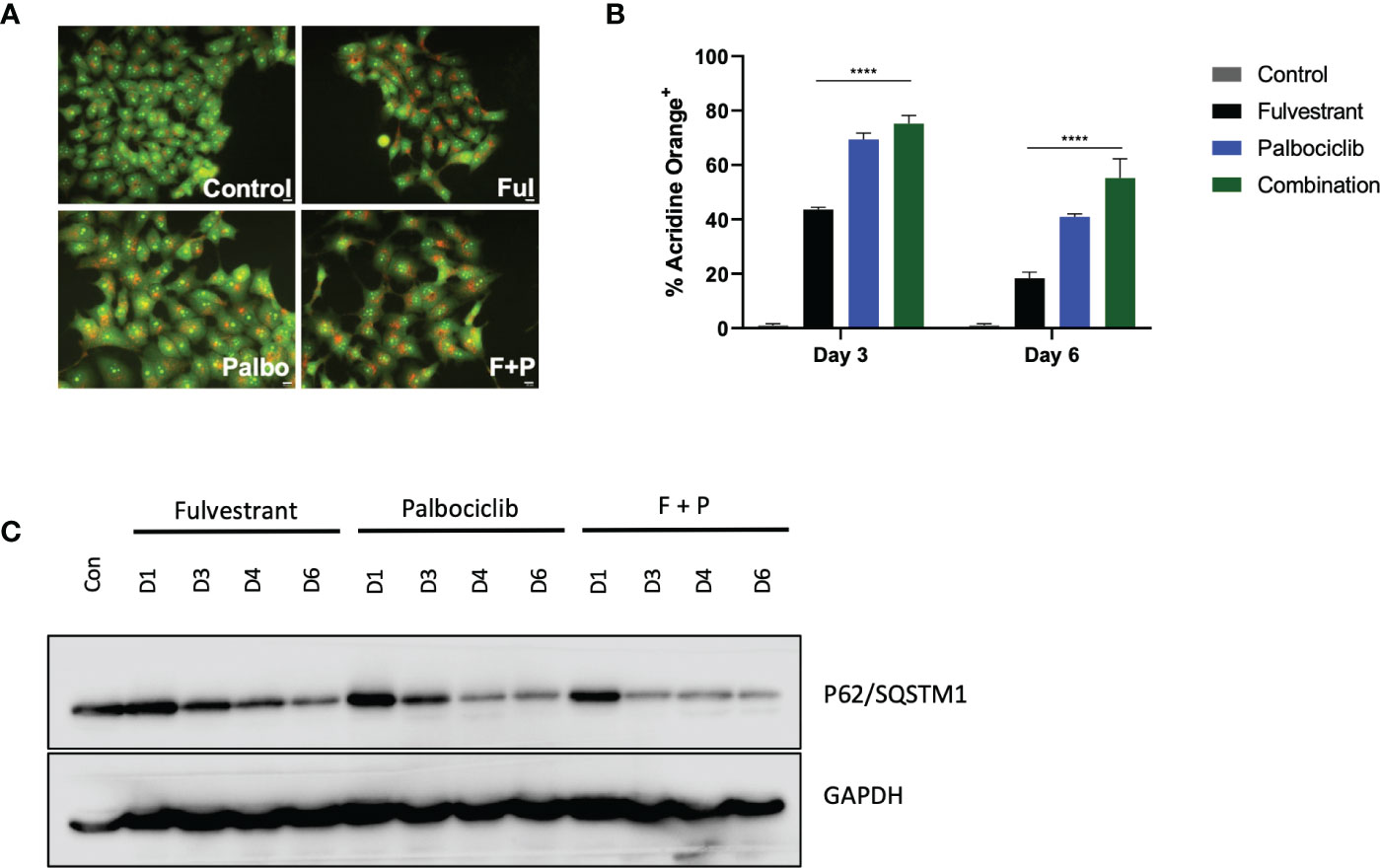
Figure 2 Fulvestrant in combination with Palbociclib induces autophagy. Cells were treated with Fulvestrant (100 nM), Palbociclib (1 µM) or the combination for 6 days (A–C). Lysates were collected at specified days (A) Cells were stained with acridine orange on day 4 and imaged using a fluorescent microscope. All images were taken at the same magnification (scale bar= 200 µm, n=3). (B) Cells were stained with acridine orange and fluorescence was quantified using flow cytometry. (C) Autophagy induction over time was assessed by degradation of p62/SQSTM1 protein levels. All images are representative fields or blots from at least three independent experiments. ****P ≤ 0.001 indicate statistical significance of each condition compared to Fulvestrant as determined using two-way ANOVA with Sidak's post hoc test.
3.3 Efforts to sensitize MCF-7 breast tumor cells via autophagy inhibition
In an effort to sensitize MCF7 cells to the Fulvestrant, Palbociclib, and the combination therapy, the lysosomotropic agents, chloroquine (CQ) and bafilomycin A1 (Baf), were utilized as pharmacological inhibitors of autophagy, based on their ability to interfere with autophagosome-lysosome fusion (36). Chloroquine and bafilomycin were added concurrently with the Fulvestrant + Palbociclib for 48 hrs; CQ and Baf were removed, and the Fulvestrant + Palbociclib treatment restored for an additional 4 days (Figure 3A). Autophagy was visualized by acridine orange staining; acridine orange fluoresces bright orange in acidic environments, such as the lysosomes, and shifts to an orange/yellow color when pH becomes less acidic (37, 38). We also observed an increased number of autophagic vacuoles upon the addition of the CQ and Baf, which is indicative of autophagic vacuole accumulation when the autophagic process is prevented from going to completion (Figure 3B) (39). The inhibition of autophagy in cells exposed to CQ and Baf was further confirmed by western blot analysis of LC3-II and p62/SQSTM1. We observed an increase in accumulation of LC3-II in the presence of CQ and Baf (due to inhibition of autolysosome formation as well as accumulation of autophagosomes) in both control and drug treated groups, indicating that CQ and Baf inhibited both basal and treatment-induced autophagy in MCF7 and T47D cells (Figures 3C, S1C). Accumulation of p62/SQSTM1 indicative of interference with p62/SQSTM1 degradation further confirmed that CQ and Baf inhibited treatment-induced autophagy. However, autophagy inhibition failed to sensitize the MCF-7 and T47D cells to Fulvestrant, Palbociclib or the combination therapy (Figures 3D–F, S1B), indicating that the autophagy was functionally nonprotective, as we have observed previously in other experimental systems (40–42). Of note is the recovery of proliferative capacity that occurs between days 8-12, which is indicative of escape from senescence (see below). Consistent with the lack of sensitization, pharmacological autophagy inhibition did not significantly promote apoptotic cell death by the combination of the anti-estrogen and CDK 4/6 inhibitor (Figure 3G).
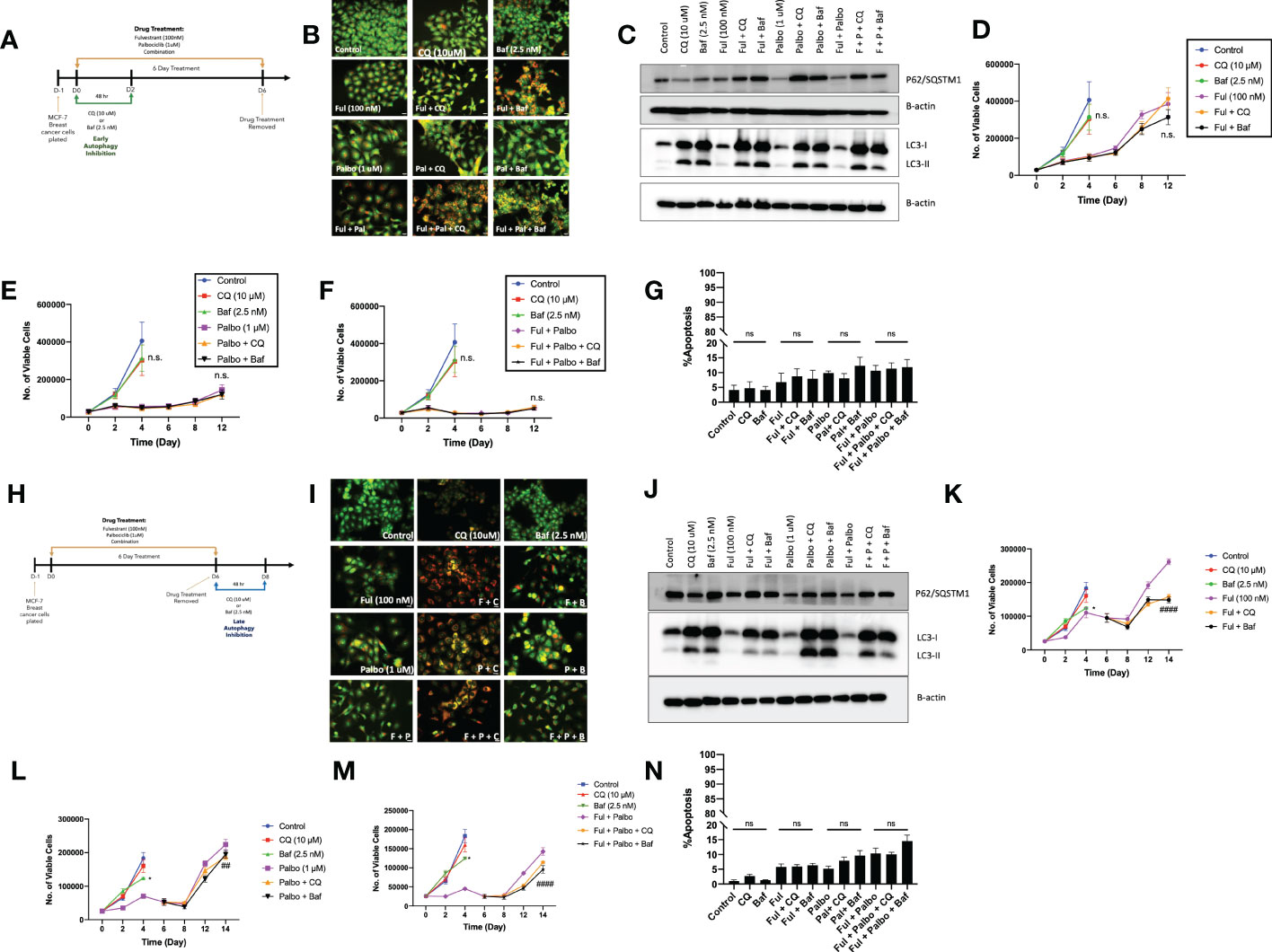
Figure 3 Autophagy inhibition does not alter sensitivity to Fulvestrant in combination with Palbociclib in MCF7 cells. Cells were pre-treated for 3 h with either CQ (10 uM) or Baf A1 (2.5 nM). CQ and Baf A1 were given for an additional 48 h, alongside the treatment with Fulvestrant (100 nM), Palbociclib (1 µM) or the combination for 6 days. (A) Schematic of in vitro treatment. (B) Cells were stained with acridine orange on day 3 and imaged using a fluorescent microscope. All images were taken at the same magnification (scale bar= 200 µm, n=3). (C) Western blot analysis at day 3 assessing accumulation of LC3 I-II and p62/SQSTM1 protein levels. (D–F) Viable cell number was counted via trypan blue exclusion on the indicated days. (G) Apoptosis was measured by annexin V/PI staining. Staining was performed on day 6 and fluorescence was measured using flow cytometry. (H) Schematic of in vitro treatment. Cells were treated with Fulvestrant (100 nM), Palbociclib (1 µM) or the combination for 6 days, drugs were removed, and cells were given an additional 48 h CQ (10 uM) or Baf A1 (2.5 nM). (I) Cells were stained with acridine orange on day 8 and imaged using a fluorescent microscope. All images were taken at the same magnification (scale bar= 200 µm, n=3). (J) Autophagy inhibition was confirmed by western blot analysis assessing accumulation of LC3 I-II and p62/SQSTM1 protein levels. (K–M) Viable cell number was counted via trypan blue exclusion on the indicated days. (N) Apoptosis was measured using annexin V/PI staining. Staining was performed on day 8 and fluorescence was measured using flow cytometry. All images are representative fields, blots, or data from at least three independent experiments. *P ≤ 0.05, ##P ≤ 0.01, and ###P ≤ 0.001, ns (not significant) indicate statistical significance of each condition compared to Fulvestrant, Palbociclib or the combination of Fulvestrant and Palbociclib as determined using two-way ANOVA with Sidak's post hoc test.
Conventionally, most preclinical, and clinical studies designed to evaluate the effect of autophagy inhibition involve the addition of the pharmacological autophagy inhibitors concurrently or as a pre-treatment to therapy (41, 43, 44). Preliminary experiments (not shown) suggested the possibility that a more delayed approach to autophagy inhibition might prove to be a more effective sensitization strategy. Consequently, MCF7 cells were treated with Fulvestrant, Palbociclib or the combination for 6 days, and then exposed to CQ or Baf for 48 hrs post treatment (Figure 3H). Similar to the experiments in Figures 3B, I demonstrates autophagy inhibition based on interference with lysosomal acidification by acridine orange staining. Autophagy inhibition by Baf and CQ was additionally confirmed by western blot analysis of LC3-II and p62/SQSTM1. Again, similar to the data generated in Figure 3C, we observed an increase in LC3-II accumulation (Figures 3J, S1E) and accumulation of p62/SQTM1 (interference with degradation) with CQ and Baf. Figures 3K–M indicate that while addition of CQ or Baf provided a modest degree of sensitization to Fulvestrant alone, in terms of delaying proliferative recovery, sensitization of the MCF7 cells to Palbociclib or the combination treatment was barely significant. Furthermore, no sensitization was observed in T47D cells (Figure S1E). Additionally, there was minimal promotion of apoptosis with autophagy inhibition, (Figure 3N), again consistent with the autophagy being largely nonprotective in function in this experimental model system.
3.4 Efforts to sensitize MCF-7 breast tumor cells by autophagy inhibition via genetic silencing
To further confirm the absence of a pronounced sensitization to Fulvestrant, Palbociclib and the combination treatment via autophagy inhibition, the MCF-7 cells were stably transfected using short hairpin RNA for ATG5 (shATG5) or scrambled control (shControl). Knockdown of ATG5 and impairment of autophagy was confirmed by western blot analysis indicating reduced levels of ATG5 and accumulation of p62/SQSTM1 (Figure 4A). Temporal analysis of cell viability showed that autophagy deficient cells were significantly more sensitive to Fulvestrant, as was the case with pharmacologic autophagy inhibition; however, the MCF7 shATG5 cells were only slightly more sensitive to Palbociclib and the Palbociclib + Fulvestrant combination therapy when compared to shControl cells (Figures 4B–D). These data with genetic knockdown of ATG5 are consistent with the observed outcomes upon pharmacological inhibition of autophagy with CQ and Baf (Figure 3). Genetic silencing of autophagy also did not promote apoptosis in MCF-7 cells when exposed to Ful or Pablo, but exhibited statistically significant, albeit minimal apoptotic cell death with the combination treatment compared to shControl MCF7 cells (Figure 4E). Taken together, these studies indicate that the autophagy induced by the Fulvestrant, Palbociclib, and the combination treatment is largely nonprotective, and suggests the possibility that autophagy inhibition may not prove to be an effective strategy to enhance the therapeutic response (40, 42).
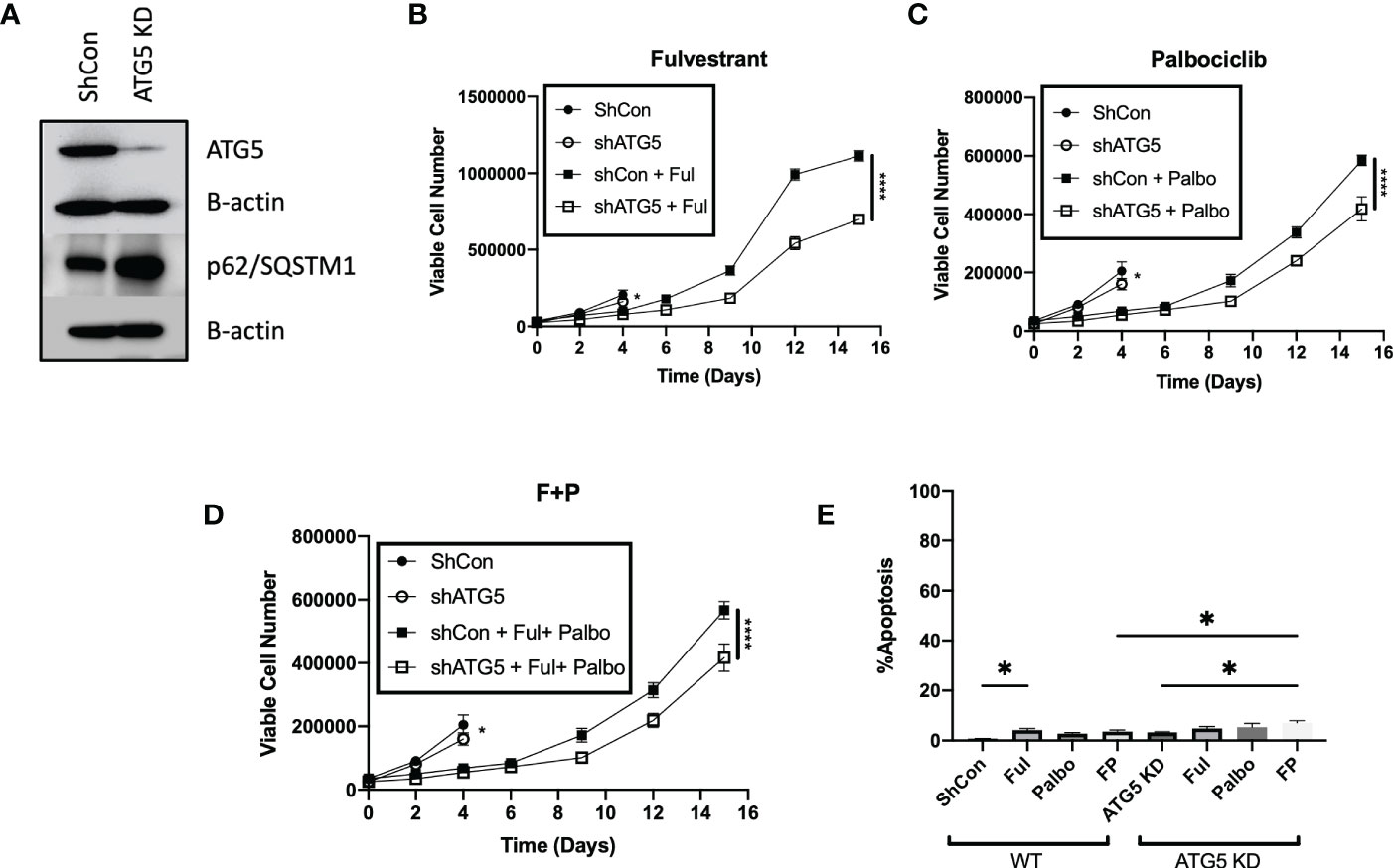
Figure 4 Genetic knockdown of autophagy only moderately increases sensitivity to Fulvestrant in combination with Palbociclib in MCF7 cells. Cells were treated with Fulvestrant (100 nM), Palbociclib (1 µM) or the combination for 6 days (A–E). Short hairpin RNA was used to knockdown ATG5. (A) Western blot analysis of ATG5 and p62/SQSTM1 protein levels. (B–D) Viable cell number was counted via trypan blue exclusion on the indicated days. (E) Apoptosis was measured using annexin V/PI staining. Staining was performed on day 8 and fluorescence was measured using flow cytometry. Unless stated, otherwise data were from three independent experiments. *P ≤ 0.05 and ****P ≤ 0.0001 compared to shControl cells treated with Fulvestrant, Palbociclib or the combination of Fulvestrant and Palbociclib.
3.5 ARV-825 extends growth delay and suppresses proliferative recovery in MCF-7 breast tumor cells treated with Fulvestrant + Palbociclib
Given that administration of Fulvestrant + Palbociclib either alone or in combination, induces a transient growth arrest, and that autophagy and senescence tend to occur in parallel, we examined senescence induction, a durable growth arrest induced by therapy (21, 29, 45–47). Previous work from our group and others has consistently shown proliferative recovery from various models of therapy induced senescence (11–16). Figure 5A demonstrates the promotion of senescence by β-gal staining with exposure to Fulvestrant alone, Palbociclib alone and the Fulvestrant and Palbociclib combination in MCF7 cells as well as morphological changes (cell enlargement and flattening) associated with senescence (45). Senescence was further confirmed using flow cytometry quantification of C12FDG fluorescence, a metabolite for SA-ß-gal. Figure 5B demonstrates increased SA-ß-gal activity in Fulvestrant, Palbociclib and combination treated MCF7 cells when compared to untreated controls. The senescence in the combination therapy appears to be largely due to the Palbociclib (46, 48). Figure 5C presents qRTPCR data indicating significant increases of the expression of IL-6, IL-8, and MMP3, components of the senescence-associated secretory phenotype (45, 49), at Day 6 post-combination treatment with Fulvestrant and Palbociclib.
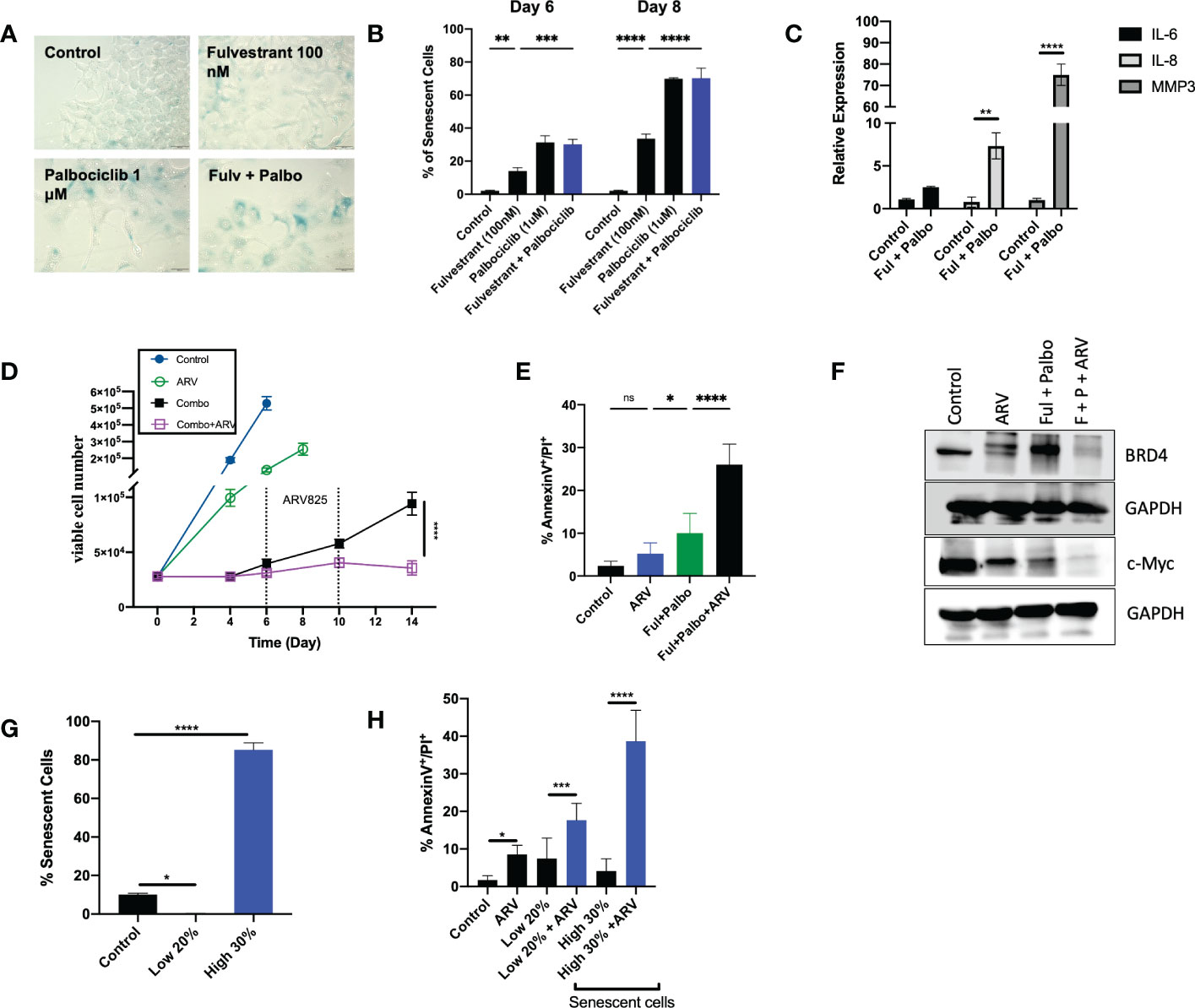
Figure 5 ARV prolongs growth arrest induced by Fulvestrant in combination with Palbociclib in MCF- cells. Cells were treated with Fulvestrant (100nM), Palbociclib (1uM) or the combination for 6 days. (A) Cells were fixed on Day 6, stained with x-gal staining solution and imaged using brightfield microscope. All images were taken with the same magnification. (B) Quantification of SA-Bgal using C12FDG at indicated timepoints. (C) qRTPCR examining SASP mRNA expression of IL-6, IL-8, and MMP3 on D6 post-combination treatment. (D) Cell viability was monitored over a period of 14 days by trypan blue exclusion. (E) Apoptosis was evaluated by flow cytometry using an APC Annexin V Apoptosis Detection Kit. (F) Western blotting for BRD4, c-Myc at day 4 of ARV treatment. (G) Sorted cells was analyzed by flow cytometry to verify purity of the SA-β-Gal positive population (high 30% and low 20% SA-β-Gal positive). Control cells were sorted for high 80% SA-β-Gal positive. (H) Sorted senescent cells were plated for 24-hours and treated with ARV-825 for 4 days followed by annexin V/PI apoptosis staining. Flow cytometry was performed post-sort for C12FDG staining to confirm senescence induction. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns (not significant) indicate statistical significance of each condition compared to control as determined using two‐way ANOVA with Sidak's post hoc test. All images are representative fields, blots, or data from three independent experiments (n = 3).
We and others have demonstrated that the senolytic ABT-263 (navitoclax), is effective at the elimination of tumor cells induced into senescence by various chemotherapeutic strategies in breast, lung, head and neck and prostate tumor cells (14–16). However, one fundamental limitation in the use of ABT-263 is thrombocytopenia, given that the target of ABT-263 is the Bcl-xL protein, upon which platelets depend for survival (50).
Inhibitors of bromodomain-containing protein 4 (BRD4), particularly ARV-825, have demonstrated antitumor activity in multiple preclinical models, and have recently been considered as potential senolytics (51). To investigate whether ARV-825 might act as a senolytic in combination with the senescence induced by the Fulvestrant + Palbociclib combination, cells were treated for 6 days with Fulvestrant (100 nM) and Palbociclib (1 µM), followed by ARV-825 (50 nM) for 96 h post-treatment. Temporal analysis of cell viability demonstrated that ARV-825, alone, moderately suppressed growth of the MCF-7 cells (Figure 5D); this is consistent with prior literature studies of the action of ARV-825 (52, 53), and with the degradation of BRD4 and the suppression of downstream c-Myc shown in Figure 5F. Our laboratory as well as others have shown that c-Myc is upregulated in breast cancer and involved in breast cancer proliferation (52, 54, 55).
The most critical observation in this work is that ARV-825 treatment sequentially after the Fulvestrant + Palbociclib combination resulted in a prolonged growth arrest with suppression of proliferative recovery (Figure 5D). This finding is consistent with the pronounced suppression of both BRD4 and c-Myc for the combination of Fulvestrant + Palbociclib and ARV-825 in Figure 5F.
Although the addition of ARV-825 to the Fulvestrant + Palbociclib combination appeared to result in prolongation of growth arrest rather than cell killing, this therapeutic strategy nevertheless demonstrated some characteristics of senolysis, specifically the promotion of apoptosis. As shown in Figure 5E, a significant increase in apoptosis was evident at Day 10 with ARV-825 treatment compared to the combination treatment alone; the percentage of apoptosis stayed relatively constant through day 14 (data not shown).
Although the combination treatment promotes substantial senescence in the MCF-7 cells (Figures 5A, B), the entire cell population is not senescent, and consequently it was necessary to address whether the ARV-825 was functioning as a senolytic and that the senescent cell population might be particularly vulnerable to the ARV-825. To address this question, cells were sorted by flow cytometry to distinguish the SA-β-Gal highly positive and low positive populations (high 30% and low 20% SA-β-Gal positive). Control cells were also sorted for SA-β-Gal positive cells (Figure 5G). Flow cytometry was performed post-sorting for C12FDG staining to confirm the senescent population (Figure 5G). Figure 5H indicates that the senescent high cell population underwent significantly more apoptosis compared to the non-senescent cells after treatment with Fulvestrant/Palbociclib + ARV-825. These data indicate that the senescence induced by the combination of an anti-estrogen and CDK4/6 inhibitor increases susceptibility to ARV-825 induced apoptotic cell death.
3.6 Sensitization by ARV-825 in p53 mutant T-47D breast tumor cells treated with the Fulvestrant + Palbociclib combination
Approximately 20% of ER positive breast cancers present with p53 mutations (56). In order to evaluate whether ARV-825 would also be effective against p53 mutant ER+ breast tumors treated with the Fulvestrant + Palbociclib combination, we assessed the temporal response by real time, live cell imaging using IncuCyte S3 in T-47D breast tumor cells. Analogous to the outcomes in MCF-7 cells (Figure 5D), we observed that the combination of Fulvestrant + Palbociclib induced senescence based on senescence associated β-galactosidase staining (Figure 6A) that was followed by proliferative recovery (Figure 5C); as in the studies with the p53 wild-type MCF-7 cells, the addition of ARV-825 resulted in prolonged growth arrest without recovery, at least over the ~ 13-day time course of this study (Figure 6C). Quantifying the extent of senescence using C12FDG staining indicated that approximately 40% of the population represented senescent cells at both day 6 and day 8 (Figure 6B). Analysis of apoptosis indicated that there was no significant difference with the combination + ARV825, compared to the combination alone (Figure 6D), despite some evidence of a decline in cell number in the temporal response study. Additionally, western blot analysis confirmed a reduction in target protein, BRD4, by ARV-825 in both control and Fulvestrant + Palbociclib (Figure 6E). Similarly, a profound suppression of downstream c-Myc by ARV-825 is evident in control cells, the combination alone and the combination + ARV825 (Figure 6E).
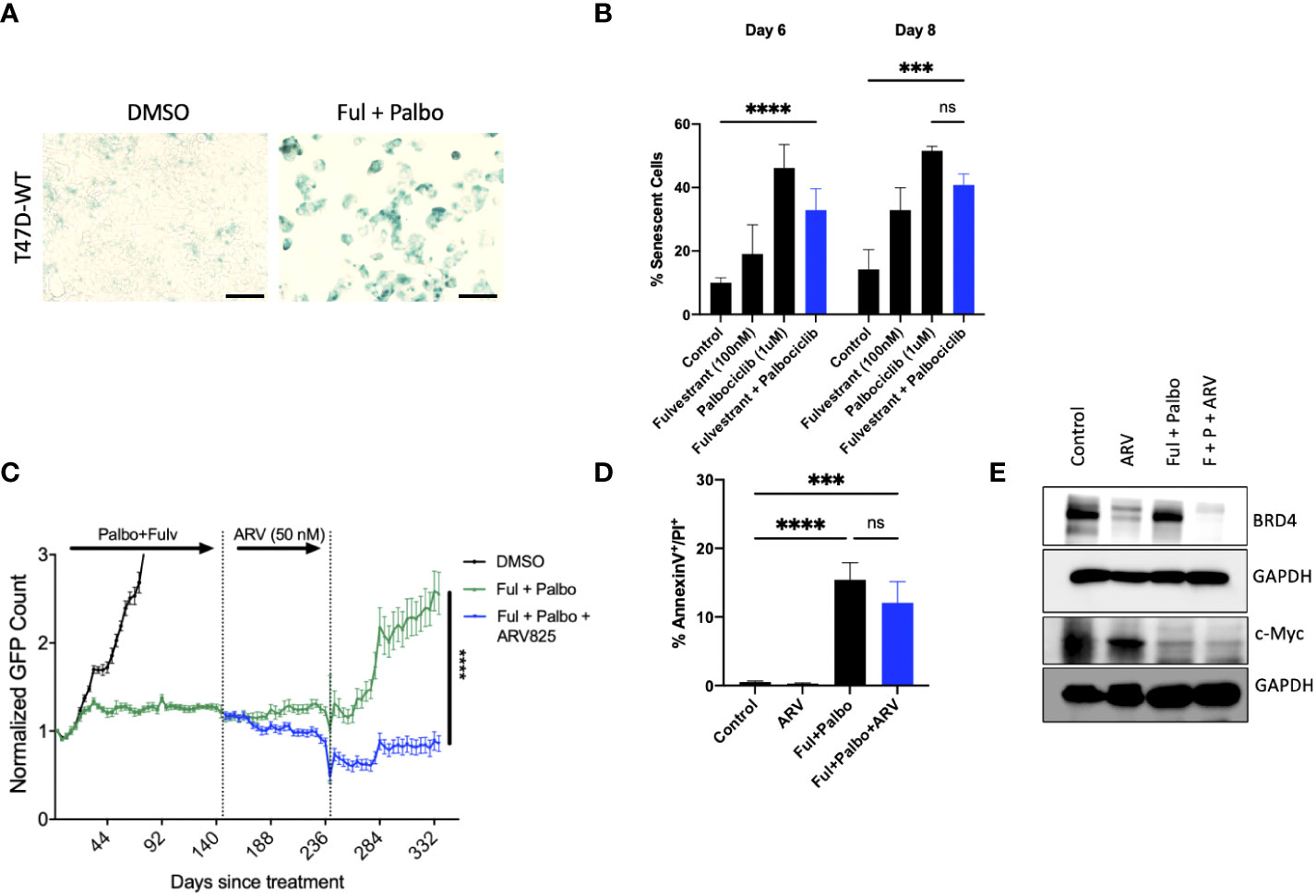
Figure 6 ARV prolongs growth arrest induced by Fulvestrant in combination with Palbociclib in p53 mutant T-47D cells. T47D-WT cells that were treated with palbociclib (1 µM) in combination with Fulvestrant (100 nM) for 6 days. (A) Cells were fixed on Day 6, stained with x-gal staining solution, and imaged using brightfield microscope. All images were taken with the same magnification. (B) Quantification of SA-Bgal using C12FDG at indicated timepoints. (C) Live cell viability was monitored via IncuCyte over a period of 14 days and normalized to GFP count. (D) Apoptosis was evaluated by flow cytometry using an APC Annexin V Apoptosis Detection Kit. (E) Western blotting for BRD4, c-Myc at day 4 of ARV treatment. Mean and SD were determined based on triplicates from 3 independent experiments. ***P ≤ 0.001, ****P ≤ 0.0001, ns (not significant) indicate statistical significance of each condition compared to control as determined using two‐way ANOVA with Sidak’s post hoc test.
3.7 Sensitization by ARV-825 in MCF-7 cells deficient in Rb and treated with the fulvestrant + palbociclib combination
One common mechanism of resistance that develops to palbociclib therapy in ER positive breast tumor cells is the loss of Rb, which prevents the cells from entering into a state of senescence (57). However, it is possible that the ARV-825 is not acting as a classical senolytic, which might make cells treated with fulvestrant + palbociclib susceptible to cell killing even where senescence is not the primary response. To address this possibility, MCF-7 and T47D cells, where Rb function was genetically deleted, were exposed to the fulvestrant + palbociclib combination, followed by ARV-825. As shown in Figure 7A, in MCF-7 Rb deleted cells, the combination treatment promotes delayed growth compared to control, followed by proliferative recovery, similar to what we observe with Rb competent MCF-7 cells. Figure 7B demonstrates the lack of senescence induction with MCF-7 Rb deleted cells, which was confirmed with quantification of senescence using C12FDG where we observe a relatively low percentage of senescent cells (Figure 7C). The addition of ARV-825 to the combination treatment of fulvestrant + palbociclib clearly promotes an increase in apoptosis over the low level of apoptosis induced by fulvestrant + palbociclib (Figure 7D). Figure 7E indicates that ARV-825 suppressed both the target protein, BRD4, as well c-Myc, which is presumably downstream of BRD4, in both control and fulvestrant + palbociclib treated cells.
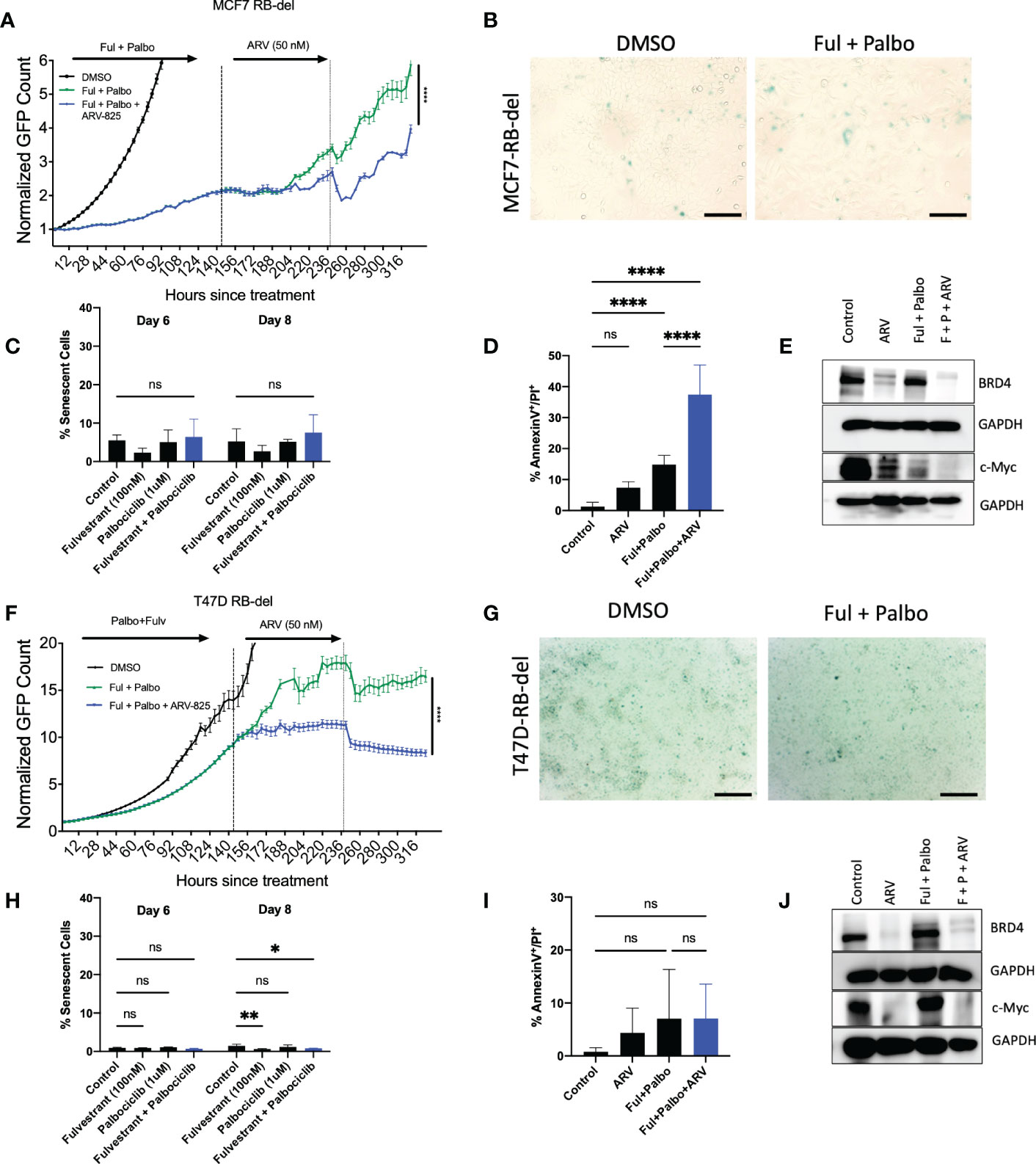
Figure 7 ARV-825 prolongs growth arrest induced by the combination of Fulvestrant and Palbociclib in resistant MCF-7 and T47D cells. Live cell imaging in MCF-7 Rb-del cells that were treated with palbociclib (1 µM) in combination with fulvestrant (100 nM) for 6 days. (A) Live cell viability of Rb-deficient MCF-7 cells was monitored via IncuCyte over a period of 14 days and normalized to GFP count. (B) MCF-7 Rb deleted cells were fixed on Day 6, stained with x-gal staining solution, and imaged using brightfield microscope. All images were taken with the same magnification. (C) Quantification of SA-Beta gal using C12FDG at indicated timepoints. (D) Apoptosis was evaluated by flow cytometry using an APC Annexin V Apoptosis Detection Kit. (E) Western blotting for BRD4, c-Myc at day 4 of ARV treatment. (F) Live cell viability of Rb-deficient T47D cells was monitored via IncuCyte over a period of 14 days and normalized to GFP count. (G) Cells were fixed on Day 6, stained with x-gal staining solution, and imaged using brightfield microscope. All images were taken with the same magnification. (H) Quantification of SA-Beta gal using C12FDG at indicated timepoints. (I) Apoptosis was evaluated by flow cytometry using an APC Annexin V Apoptosis Detection Kit. (J) Western blotting for BRD4, c-Myc at day 4 of ARV treatment. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, ns (not significant) indicate statistical significance of each condition compared to control as determined using two‐way ANOVA with Sidak's post hoc test. All images are representative fields, blots, or data from two independent experiments (n = 2).
In contrast to the MCF-7/Rb deleted cells, growth arrest by the treatment with the fulvestrant + palbociclib combination is markedly delayed in the T47D Rb deleted cells; nevertheless, the addition of ARV-825 significantly suppresses growth (Figure 7F). As was largely the case with the MCF-7/Rb deleted cells, the combination treatment fails to promote senescence (Figure 7G), as further confirmed by C12FDG quantification (Figure 7H). Furthermore, while MCF-7 Rb deficient cells demonstrated enhanced apoptosis with the combination treatment followed by ARV-825 (Figure 7D), there was no increase in apoptosis in the T47D Rb deficient cells (Figure 7I). This is likely related to the absence of functional p53, as there was also no increase in apoptosis in the Rb proficient T-47D cells (Figure 6D). Finally, the targeting of BRD4 and c-Myc by the ARV-825 was confirmed by the Western blot presented in Figure 7J.
4 Discussion
Disease recurrence, both local and distal, is an ongoing issue contributing to the majority of hormone receptor-positive breast cancer deaths and is observed in many other types of cancers, such as triple-negative breast cancer, lung, and prostate cancer. Often this recurrence can be associated with therapy-induced residual dormant tumor cell populations, that can escape and often become more aggressive in nature (58–60). While therapy-induced senescence has been studied for decades, successful utilization of senolytics in cancer treatment has not yet been implemented. Despite this, several senolytic agents have been considered and studied to modulate and eliminate senescent tumor cells.
Another potential route for modulation of drug sensitivity in cancer is autophagy inhibition (23, 61). Estrogen receptor-targeted therapies are generally the first-line treatment for hormone receptor-positive breast cancer and autophagy in response to tamoxifen has been shown quite convincingly to be cytoprotective (62). In fact, this form of autophagy has been shown to lead to the development of resistance to anti-estrogen therapies (44, 63, 64). In cases where autophagy is cytoprotective, pharmacological inhibition of autophagy may be utilized to enhance the tumor cell sensitivity to treatment. In addition to cytoprotective autophagy, we have identified a non-protective form of autophagy, which apparently plays no distinct role in promoting or suppressing the growth or sensitivity of the tumor cells in response to therapy (39, 40, 61). Clinical trials are currently underway evaluating HCQ as a pre-treatment with the combination of letrozole + Palbociclib, based on preclinical studies showing efficacy in autophagy inhibition as a pretreatment with this combination (25).
The current work evaluated the potential of utilizing autophagy inhibition, before and after treatment, to sensitize ER positive MCF-7 breast tumor cells to the combination of Fulvestrant + Palbociclib. This treatment promotes significant growth arrest and both autophagy and senescence. induction after 6 days of treatment with Fulvestrant and Palbociclib. Early administration of pharmacological autophagy inhibitors did not improve tumor cell responsiveness to the combination treatment, leading to the conclusion that the autophagy was non-protective. Slight sensitization was evident with late addition of pharmacological autophagy inhibitors as well as with genetic knockdown. The latter was most pronounced with Fulvestrant, which may relate to previous studies demonstrating that ER targeted therapies are generally cytoprotective (65, 66). However, the autophagy for the combination treatment appears to be largely nonprotective, indicating that autophagy inhibition is unlikely to become a clinically useful therapeutic strategy (61, 67). This does not rule out the possibility that the autophagy induced by aromatase inhibitors in combination with cdk4/6 inhibitors could be cytoprotective and amenable to autophagy inhibition in the clinic.
Our studies further examined the incorporation of a BET degrader, ARV-825, into the combination treatment. We initially screened a variety of agents from different drug classes that had been reported to have senolytic activity and found that this BET inhibitor was the most promising agent. Although we had published previous studies using ABT-263 (navitoclax), we chose not to continue with this class of drugs due to the thrombocytopenia associated with the administration of Bcl-xL targeted agents. BET inhibition has demonstrated efficacy in many clinical trials consisting of both hematological malignancies as well as solid tumors (68). BET degrader ARV-825 has been used in pre-clinical studies with different cancer types, and we hypothesized that ARV-825 could potentially improve ER+ breast cancer tumor response following Fulvestrant + Palbociclib treatment (59–62). ARV-825 suppressed tumor growth for both the ER positive p53 wt MCF-7 cells and the ER positive p53 mutant T-47D cells, significantly delaying proliferative recovery. We also observed significant induction of apoptosis in the MCF-7 cells treated with Fulvestrant + Palbociclib followed by ARV-825, but not in T47D cells, which may be due to T-47D p53 mutational status. The senescent MCF-7 cell population appears to be more susceptible to ARV-825 induced apoptosis, although a low degree of apoptosis is also observed in non-senescent cells exposed to the ARV-825. While the combination of Fulvestrant + Palbociclib rapidly induced growth arrest in MCF-7 Rb deficient cells, the growth was delayed in the T47D Rb deficient cells. The observed growth arrest was confirmed to be independent of senescence, as Rb loss is a well-known factor of resistance to CDK 4/6 inhibitors (69, 70).
The observed growth arrest is consistent with the degradation of BRD4 and the suppression of downstream c-Myc as well as with previous studies by our laboratory and others demonstrating c-Myc to be upregulated in ER+ breast cancer and involved in breast cancer proliferation (52, 54, 55). These findings are supported by an analysis of three patient database sets, demonstrating that high expression of BRD4 that can be observed across multiple subtypes of breast cancer is correlated with overall lower recurrence-free survival when compared to patients with low BRD4 expression levels (71).
Taken together, the current studies indicate that administration of BET inhibitors/degraders may potentially improve the standard of care therapy in metastatic ER+ breast cancer patients and may further prolong progression-free survival. Further validation of these findings in cell culture will require studies in tumor-bearing animals. The BET inhibitors, ABBV-075 and ABBV-744, have shown promise in preclinical studies and will be tested in the future both in cell culture and in tumor-bearing animal models (72–74).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
RF, LM, DG contributed to conception and design of the study. RF, AE, NP, TT, VK contributed to methodology. RF contributed to data curation. RF wrote the first draft of the manuscript. RF, NP, VK wrote sections of the manuscript. All authors contributed to the article and approved the submitted version. Research in Dr. Gewirtz’s laboratory is supported by grants # CA268819 and CA239706 from the National Cancer Institute/National Institutes of Health and Grant # W81XWH 19-1-0490 from the Department of Defense Congressionally Directed Breast Cancer Research Program.
Funding
Research in Dr. Gewirtz’s laboratory is supported by grants # CA268819 and CA239706 from the National Cancer Institute/National Institutes of Health and Grant # W81XWH 19-1-0490 from the Department of Defense Congressionally Directed Breast Cancer Research Program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.966441/full#supplementary-material
Supplementary Figure 1 | Autophagy inhibition does not alter sensitivity to Fulvestrant in combination with Palbociclib in T47D cells. Cells were pre-treated for 3 h with either CQ (10 uM) or Baf A1 (2.5 nM). CQ and Baf A1 were given for an additional 48 h, alongside the treatment with Fulvestrant (100 nM), Palbociclib (1 µM) or the combination for 6 days. (A) Schematic of in vitro treatment of early addition of CQ and Baf. (B) Percent cell viability was measured using MTS viability assay. (C) Western blot analysis at day 6 assessing accumulation of LC3 I-II. (D) Schematic of in vitro treatment of late addition of CQ and Baf. (E) Percent cell viability was measured using MTS viability assay. (F) Western blot analysis at day 6 assessing accumulation of LC3 I-II.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA. Cancer J Clin (2022) 72:7–33. doi: 10.3322/caac.21708
2. Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LAG, et al. US Incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst (2014) 106. doi: 10.1093/jnci/dju055
3. Coates AS, Winer EP, Goldhirsch A, Gelber RD, Gnant M, Piccart-Gebhart M, et al. Panel members tailoring therapies–improving the management of early breast cancer: St gallen international expert consensus on the primary therapy of early breast cancer 2015. Ann Oncol Off J Eur Soc Med Oncol (2015) 26:1533–46. doi: 10.1093/annonc/mdv221
4. Wilson S, Chia SK. Treatment algorithms for hormone receptor-positive advanced breast cancer: Applying the results from recent clinical trials into daily practice–insights, limitations, and moving forward. Am Soc Clin Oncol Educ Book Am Soc Clin Oncol Annu Meet. (2013) e20–7. doi: 10.1200/EdBook_AM.2013.33.e20
5. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet Lond Engl (2011) 378:771–84. doi: 10.1016/S0140-6736(11)60993-8
6. D’Amico P, Cristofanilli M. Standard of care in hormone receptor-positive metastatic breast cancer: Can we improve the current regimens or develop better selection tools? JCO Oncol Pract (2022) 18:331–4. doi: 10.1200/OP.21.00707
7. Murphy CG. The role of CDK4/6 inhibitors in breast cancer. (2019) 20:52. doi: 10.1007/s11864-019-0651-4
8. Lai AC, Crews CM. Induced protein degradation: An emerging drug discovery paradigm. Nat Rev Drug Discovery (2017) 16:101–14. doi: 10.1038/nrd.2016.211
9. Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: Beyond cell cycle arrest. Trends Cell Biol (2018) 28:911–25. doi: 10.1016/j.tcb.2018.07.002
10. Ettl J, Im S-A, Ro J, Masuda N, Colleoni M, Schnell P, et al. Hematologic adverse events following palbociclib dose reduction in patients with hormone receptor-Positive/Human epidermal growth factor receptor 2-negative advanced breast cancer: Pooled analysis from randomized phase 2 and 3 studies. Breast Cancer Res BCR (2020) 22:27. doi: 10.1186/s13058-020-01263-0
11. Saleh T, Tyutyunyk-Massey L, Gewirtz DA. Tumor cell escape from therapy-induced senescence as a model of disease recurrence after dormancy. Cancer Res (2019) 79:1044–6. doi: 10.1158/0008-5472.CAN-18-3437
12. Roberson RS, Kussick SJ, Vallieres E, Chen S-YJ, Wu DY. Escape from therapy-induced accelerated cellular senescence in P53-null lung cancer cells and in human lung cancers. Cancer Res (2005) 65:2795–803. doi: 10.1158/0008-5472.CAN-04-1270
13. Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol (2019) 162:202–12. doi: 10.1016/j.bcp.2018.12.013
14. Ahmadinejad F, Bos T, Hu B, Britt E, Koblinski J, Souers AJ, et al. Senolytic-mediated elimination of head and neck tumor cells induced into senescence by cisplatin. Mol Pharmacol (2022) 101:168–80. doi: 10.1124/molpharm.121.000354
15. Saleh T, Carpenter VJ, Tyutyunyk-Massey L, Murray G, Leverson JD, Souers AJ, et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-XL -BAX interaction. Mol Oncol (2020) 14:2504–19. doi: 10.1002/1878-0261.12761
16. Carpenter V, Saleh T, Min Lee S, Murray G, Reed J, Souers A, et al. Androgen-deprivation induced senescence in prostate cancer cells is permissive for the development of castration-resistance but susceptible to senolytic therapy. Biochem Pharmacol (2021) 193:114765. doi: 10.1016/j.bcp.2021.114765
17. Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of navitoclax (ABT-263), a novel bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol Off J Am Soc Clin Oncol (2011) 29:909–16. doi: 10.1200/JCO.2010.31.6208
18. Juárez-Salcedo LM, Desai V, Dalia S. Venetoclax: Evidence to date and clinical potential. Drugs Context (2019) 8:212574. doi: 10.7573/dic.212574
19. Samaraweera L, Adomako A, Rodriguez-Gabin A, McDaid HM. A novel indication for panobinostat as a senolytic drug in NSCLC and HNSCC. Sci Rep (2017) 7:1900. doi: 10.1038/s41598-017-01964-1
20. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
21. Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, et al. The autophagy-senescence connection in chemotherapy: Must tumor cells (Self) eat before they sleep? J Pharmacol Exp Ther (2012) 343:763–78. doi: 10.1124/jpet.112.197590
22. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther (2011) 10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047
23. Xiao M, Benoit A, Hasmim M, Duhem C, Vogin G, Berchem G, et al. Targeting cytoprotective autophagy to enhance anticancer therapies. Front Oncol (2021) 11:626309. doi: 10.3389/fonc.2021.626309
24. Cook KL, Wärri A, Soto-Pantoja DR, Clarke PA, Cruz MI, Zwart A, et al. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2014) 20:3222–32. doi: 10.1158/1078-0432.CCR-13-3227
25. Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin e negative cancers. Nat Commun (2017) 8:15916. doi: 10.1038/ncomms15916
26. Patel NH, Sohal SS, Manjili MH, Harrell JC, Gewirtz DA. The roles of autophagy and senescence in the tumor cell response to radiation. Radiat. Res (2020) 194:103–15. doi: 10.1667/RADE-20-00009
27. Kharenko OA, Patel RG, Calosing C, van der Horst EH. Combination of ZEN-3694 with CDK4/6 inhibitors reverses acquired resistance to CDK4/6 inhibitors in ER-positive breast cancer. Cancer Gene Ther (2022) 29:859–69. doi: 10.1038/s41417-021-00375-9
28. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U. S. A. (1995) 92:9363–7. doi: 10.1073/pnas.92.20.9363
29. Sharma K, Goehe RW, Di X, Hicks MA, Torti SV, Torti FM, et al. Novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin d and the vitamin d analog, EB 1089. Autophagy (2014) 10:2346–61. doi: 10.4161/15548627.2014.993283
30. Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-Receptor-Positive advanced breast cancer. N Engl J Med (2015) 373:209–19. doi: 10.1056/NEJMoa1505270
31. Gewirtz DA. The four faces of autophagy: Implications for cancer therapy. Cancer Res (2014) 74:647–51. doi: 10.1158/0008-5472.CAN-13-2966
32. Li X, Zhou Y, Li Y, Yang L, Ma Y, Peng X, et al. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed Pharmacother. Biomedecine Pharmacother. (2019) 119:109415. doi: 10.1016/j.biopha.2019.109415
33. Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic autophagy in cancer therapy. Int J Mol Sci (2014) 15:10034–51. doi: 10.3390/ijms150610034
34. Zeng X, Kinsella TJ. Impact of autophagy on chemotherapy and radiotherapy mediated tumor cytotoxicity: “To live or not to live ”. Front Oncol (2011) 1:30. doi: 10.3389/fonc.2011.00030
35. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334:1573–7. doi: 10.1126/science.1208347
36. Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy (2012) 8:200–12. doi: 10.4161/auto.8.2.18554
37. Fu W, Li X, Lu X, Zhang L, Li R, Zhang N, et al. A novel acridine derivative, LS-1-10 inhibits autophagic degradation and triggers apoptosis in colon cancer cells. Cell Death Dis (2017) 8:e3086. doi: 10.1038/cddis.2017.498
38. Zhao H, Cai Y, Santi S, Lafrenie R, Lee H. Chloroquine-mediated radiosensitization is due to the destabilization of the lysosomal membrane and subsequent induction of cell death by necrosis. Radiat. Res (2005) 164:250–7. doi: 10.1667/rr3436.1
39. Chakradeo S, Sharma K, Alhaddad A, Bakhshwin D, Le N, Harada H, et al. Yet another function of P53–the switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: Implications for autophagy inhibition as a therapeutic strategy. Mol Pharmacol (2015) 87:803–14. doi: 10.1124/mol.114.095273
40. Patel NH, Xu J, Saleh T, Wu Y, Lima S, Gewirtz DA. Influence of nonprotective autophagy and the autophagic switch on sensitivity to cisplatin in non-small cell lung cancer cells. Biochem Pharmacol (2020) 175:113896. doi: 10.1016/j.bcp.2020.113896
41. Saleh T, Tyutyunyk-Massey L, Patel NH, Cudjoe EK, Alotaibi M, Gewirtz DA. Studies of non-protective autophagy provide evidence that recovery from therapy-induced senescence is independent of early autophagy. Int J Mol Sci (2020) 21:E1427. doi: 10.3390/ijms21041427
42. Bristol ML, Emery SM, Maycotte P, Thorburn A, Chakradeo S, Gewirtz DA. Autophagy inhibition for chemosensitization and radiosensitization in cancer: Do the preclinical data support this therapeutic strategy? J Pharmacol Exp Ther (2013) 344:544–52. doi: 10.1124/jpet.112.199802
43. Gąsiorkiewicz BM, Koczurkiewicz-Adamczyk P, Piska K, Pękala E. Autophagy modulating agents as chemosensitizers for cisplatin therapy in cancer. Invest. New Drugs (2021) 39:538–63. doi: 10.1007/s10637-020-01032-y
44. Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat (2008) 112:389–403. doi: 10.1007/s10549-007-9873-4
45. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer (2015) 15:397–408. doi: 10.1038/nrc3960
46. Whittle JR, Vaillant F, Surgenor E, Policheni AN, Giner G, Capaldo BD, et al. Dual targeting of CDK4/6 and BCL2 pathways augments tumor response in estrogen receptor-positive breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2020) 26:4120–34. doi: 10.1158/1078-0432.CCR-19-1872
47. Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: An emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol (2008) 76:947–57. doi: 10.1016/j.bcp.2008.06.024
48. Jost T, Heinzerling L, Fietkau R, Hecht M, Distel LV. Palbociclib induces senescence in melanoma and breast cancer cells and leads to additive growth arrest in combination with irradiation. Front Oncol (2021) 11:740002. doi: 10.3389/fonc.2021.740002
49. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
50. Zhang P, Zhang X, Liu X, Khan S, Zhou D, Zheng G. PROTACs are effective in addressing the platelet toxicity associated with BCL-XL inhibitors. Explor Targeting Anti-Tumor Ther (2020) 1:259–72. doi: 10.37349/etat.2020.00017
51. Wakita M, Takahashi A, Sano O, Loo TM, Imai Y, Narukawa M, et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun (2020) 11:1935. doi: 10.1038/s41467-020-15719-6
52. Liao X, Qian X, Zhang Z, Tao Y, Li Z, Zhang Q, et al. ARV-825 demonstrates antitumor activity in gastric cancer via MYC-targets and G2M-checkpoint signaling pathways. Front Oncol (2021) 11:753119. doi: 10.3389/fonc.2021.753119
53. Lu Q, Ding X, Huang T, Zhang S, Li Y, Xu L, et al. BRD4 degrader ARV-825 produces long-lasting loss of BRD4 protein and exhibits potent efficacy against cholangiocarcinoma cells. Am J Transl Res (2019) 11:5728–39.
54. Fallah Y, Brundage J, Allegakoen P, Shajahan-Haq AN. MYC-driven pathways in breast cancer subtypes. Biomolecules (2017) 7:E53. doi: 10.3390/biom7030053
55. Fornari FA, Jarvis DW, Grant S, Orr MS, Randolph JK, White FK, et al. Growth arrest and non-apoptotic cell death associated with the suppression of c-myc expression in MCF-7 breast tumor cells following acute exposure to doxorubicin. Biochem Pharmacol (1996) 51:931–40. doi: 10.1016/0006-2952(96)00050-0
56. Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, et al. Mechanisms of estrogen receptor antagonism toward P53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci U. S. A. (2010) 107:15081–6. doi: 10.1073/pnas.1009575107
57. Kumarasamy V, Nambiar R, Wang J, Rosenheck H, Witkiewicz AK, Knudsen ES. RB loss determines selective resistance and novel vulnerabilities in ER-positive breast cancer models. Oncogene (2022) 41:3524–38. doi: 10.1038/s41388-022-02362-2
58. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife (2019) 8:e43653. doi: 10.7554/eLife.43653
59. Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res (2009) 15:677–83. doi: 10.1158/1078-0432.CCR-08-1754
60. Wu C-Y, Lee C-L, Wu C-F, Fu J-Y, Yang C-T, Wen C-T, et al. Circulating tumor cells as a tool of minimal residual disease can predict lung cancer recurrence: A longitudinal, prospective trial. Diagnostics (2020) 10:144. doi: 10.3390/diagnostics10030144
61. Gewirtz DA. The switch between protective and nonprotective autophagy, implications for autophagy inhibition as a therapeutic strategy in cancer. Biology (2020) 9:E12. doi: 10.3390/biology9010012
62. Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, et al. A role for macroautophagy in protection against 4-Hydroxytamoxifen-Induced cell death and the development of antiestrogen resistance. Mol Cancer Ther (2008) 7:2977–87. doi: 10.1158/1535-7163.MCT-08-0447
63. Cook KL, Shajahan AN, Clarke R. Autophagy and endocrine resistance in breast cancer. Expert Rev Anticancer Ther (2011) 11:1283–94. doi: 10.1586/era.11.111
64. Amaral C, Augusto TV, Tavares-da-Silva E, Roleira FMF, Correia-da-Silva G, Teixeira N. Hormone-dependent breast cancer: Targeting autophagy and PI3K overcomes exemestane-acquired resistance. J Steroid Biochem Mol Biol (2018) 183:51–61. doi: 10.1016/j.jsbmb.2018.05.006
65. Cook KL, Clarke PAG, Parmar J, Hu R, Schwartz-Roberts JL, Abu-Asab M, et al. Knockdown of estrogen receptor-α induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J Off Publ. Fed. Am Soc Exp Biol (2014) 28:3891–905. doi: 10.1096/fj.13-247353
66. Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy (2009) 5:400–3. doi: 10.4161/auto.5.3.7784
67. Mele L, Del Vecchio V, Liccardo D, Prisco C, Schwerdtfeger M, Robinson N, et al. The role of autophagy in resistance to targeted therapies. Cancer Treat Rev (2020) 88:102043. doi: 10.1016/j.ctrv.2020.102043
68. Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: A systematic study of clinical trials. Front Pharmacol (2020) 11:621093. doi: 10.3389/fphar.2020.621093
69. McCartney A, Migliaccio I, Bonechi M, Biagioni C, Romagnoli D, De Luca F, et al. Mechanisms of resistance to CDK4/6 inhibitors: Potential implications and biomarkers for clinical practice. Front Oncol (2019) 9:666. doi: 10.3389/fonc.2019.00666
70. Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: Prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res BCR (2014) 16:207. doi: 10.1186/bcr3652
71. Ali A, Shafarin J, Unnikannan H, Al-Jabi N, Jabal RA, Bajbouj K, et al. Co-Targeting BET bromodomain BRD4 and RAC1 suppresses growth, stemness and tumorigenesis by disrupting the c-MYC-G9a-FTH1axis and downregulating HDAC1 in molecular subtypes of breast cancer. Int J Biol Sci (2021) 17:4474–92. doi: 10.7150/ijbs.62236
72. Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, et al. First-in-Human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with Relapsed/Refractory solid tumors. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:6309–19. doi: 10.1158/1078-0432.CCR-19-0578
73. Sheppard GS, Wang L, Fidanze SD, Hasvold LA, Liu D, Pratt JK, et al. Discovery of n-Ethyl-4-[2-(4-Fluoro-2,6-Dimethyl-Phenoxy)-5-(1-Hydroxy-1-Methyl-Ethyl)Phenyl]-6-Methyl-7-Oxo-1H-Pyrrolo[2,3-c]Pyridine-2-Carboxamide (ABBV-744), a BET bromodomain inhibitor with selectivity for the second bromodomain. J Med Chem (2020) 63:5585–623. doi: 10.1021/acs.jmedchem.0c00628
Keywords: cancer, autophagy, senescence, fulvestrant, Palbociclib, ARV-825
Citation: Finnegan RM, Elshazly AM, Patel NH, Tyutyunyk-Massey L, Tran TH, Kumarasamy V, Knudsen ES and Gewirtz DA (2023) The BET inhibitor/degrader ARV-825 prolongs the growth arrest response to Fulvestrant + Palbociclib and suppresses proliferative recovery in ER-positive breast cancer. Front. Oncol. 12:966441. doi: 10.3389/fonc.2022.966441
Received: 10 June 2022; Accepted: 15 December 2022;
Published: 18 January 2023.
Edited by:
Massimo Broggini, Mario Negri Institute for Pharmacological Research (IRCCS), ItalyReviewed by:
Olesya Kharenko, Zenith Epigenetics Ltd, CanadaSara Ravaioli, Maria Cecilia Hospital, Italy
Daniele Vergara, University of Salento, Italy
Copyright © 2023 Finnegan, Elshazly, Patel, Tyutyunyk-Massey, Tran, Kumarasamy, Knudsen and Gewirtz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Gewirtz, ZGF2aWQuZ2V3aXJ0ekB2Y3VoZWFsdGgub3Jn