 Christina-Nefeli Kontandreopoulou
Christina-Nefeli Kontandreopoulou Konstantinos Kalopisis
Konstantinos Kalopisis Nora-Athina Viniou
Nora-Athina Viniou Panagiotis Diamantopoulos
Panagiotis Diamantopoulos- First Department of Internal Medicine, Laikon General Hospital, National and Kapodistrian University of Athens, Athens, Greece
Genomic instability, microenvironmental aberrations, and somatic mutations contribute to the phenotype of myelodysplastic syndrome and the risk for transformation to AML. Genes involved in RNA splicing, DNA methylation, histone modification, the cohesin complex, transcription, DNA damage response pathway, signal transduction and other pathways constitute recurrent mutational targets in MDS. RNA-splicing and DNA methylation mutations seem to occur early and are reported as driver mutations in over 50% of MDS patients. The improved understanding of the molecular landscape of MDS has led to better disease and risk classification, leading to novel therapeutic opportunities. Based on these findings, novel agents are currently under preclinical and clinical development and expected to improve the clinical outcome of patients with MDS in the upcoming years. This review provides a comprehensive update of the normal gene function as well as the impact of mutations in the pathogenesis, deregulation, diagnosis, and prognosis of MDS, focuses on the most recent advances of the genetic basis of myelodysplastic syndromes and their clinical relevance, and the latest targeted therapeutic approaches including investigational and approved agents for MDS.
1 Introduction
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of malignant hematopoietic stem cell (HSC) disorders characterized by ineffective hematopoiesis, cytopenias, and an increased risk of transformation to acute myeloid leukemia (AML) (1, 2). Patient disease course, progression, and survival rates are highly variable, based on genomic features, risk stratification, symptom burden, and comorbidities. The incidence of MDS is higher in elderly patients, highlighting the increased risk of acquired mutations in hematopoietic stem cells (HSCs) and progenitor cells (HSPCs), which keep on accumulating with age and reach on average 5–10 mutations (3). The International Prognostic Scoring System (IPSS) (4) and the Revised IPSS (IPSS-R) (5) are the most widely used prognostic tools for predicting the risk of leukemic transformation and overall survival (OS). Patients with lower-risk MDS (LR-MDS) are managed largely with supportive care and agents targeting cytopenias (6). Patients with higher-risk MDS (HR-MDS) are treated with the hypomethylating agent (HMA), 5-azacytidine (5-AZA). Allogenic hematopoietic stem cell transplant (HSCT) remains the only curative treatment for younger, fit HR-MDS patients (7).
The genomic landscape of MDS revealed by next-generation sequencing indicates that mutations of various genes mainly involved in RNA splicing, epigenetic, signaling, transcription, DNA repair, and cohesin pathways serve as drivers for MDS. The rapid technological development for understanding the genetic mutations over the last decade has changed the knowledge of MDS pathogenesis and prognosis based on the discovery of higher-incidence genetic abnormalities in MDS. In this review, we give an outline of the molecular mutations involved in the pathogenesis of MDS, and their clinical implications in treatment outcomes and prognosis, sole or through interaction in co-occurrence. Moreover, we provide data on the currently available or under investigation targeted therapies for MDS. The paper is structured in a way that each molecular disorder is analyzed in terms of frequency, effect on normal protein function, clinical and prognostic impact, and a second section follows analyzing the already available or upcoming agents against individual molecular targets.
2 Molecular pathogenesis
The molecular pathogenesis of MDS is complex due to multiple genetic and epigenetic aberrations resulting in the dysplastic and proliferative features of MDS. These aberrant alterations are more often due to somatic mutations followed by chromosomal abnormalities and less often due to germline mutations (8, 9).
2.1 Cytogenetic (chromosomal) abnormalities
The hematopoietic cells of about 50% of MDS patients bear chromosomal abnormalities (10). Most of them affect genomic balance, such as deletion, monosomy or trisomy, and structural alterations of chromosomes, such as the translocations frequently seen in acute leukemia. In MDS patients with an abnormal karyotype, more than 680 different types of chromosomal abnormalities have been observed, with loss or deletion of chromosome 5 or 7 and mosaic trisomy 8 being the most common among them (11). Monosomy 7 is known to be associated with poor prognosis (11). MDS with isolated del (5q), also known as 5q- syndrome, is a unique subtype associated with a favorable outcome and characterized by macrocytic anemia with normal/high platelet counts (12). The frequency of 5q- syndrome is relatively high in the US and European countries, accounting for approximately 5% of MDS patients (13). Several pathogenic genes have been identified in the commonly deleted region of 5q31-5q33, including ribosomal protein S14 (RPS14), microRNA (miR)-145, and miR146a. RPS14 is believed to be responsible for macrocytic anemia (14), while miR-145 and miR-146a are involved in thrombocytosis and neutropenia through interleukin (IL)-6 regulation (15). Apart from 5q- syndrome, there are no other cytogenetically defined MDS subtypes.
2.2 Genetic mutations
As mentioned above, approximately half of MDS patients have a normal karyotype, suggesting that genetic mutations are responsible for the pathogenesis of MDS. A detailed understanding of the mutational landscape of MDS has emerged over the last years, with the advent of advanced high-throughput techniques such as multiple ligation dependent probe amplification (MLPA), high-fidelity single nucleotide polymorphism (SNP) array, next-generation sequencing (NGS), whole-genome sequencing (WGS), and whole exome sequencing (WES) (16). NGS is superior to cytogenetics in establishing clonality, especially given the high rate of a normal karyotype; thus a myeloid mutational panel can provide essential evidence of clonality, besides cytogenetics/FISH studies, useful in the differential diagnosis of cytopenia (17). Some of the recurring chromosomal abnormalities and somatic mutations observed in MDS have a distinctive gene expression profile (GEP). Nevertheless, the pathophysiology of these disorders remains not fully understood and only few in vivo models are available to help uncover the disease (18). Application of these technologies has revealed a large set of recurrently mutated genes which are organized in eight major categories corresponding to the implicated cellular processes: RNA splicing, DNA methylation, chromatin modification, the cohesin complex, transcription factors, the DNA damage response pathway, signal transduction and other pathways (19–21). Somatic mutations in MDS-related genes have been identified in healthy individuals with clonal hematopoiesis of indeterminate potential (CHIP) (22, 23). Similarly, patients with idiopathic cytopenias of undetermined significance, with low blood cell counts, normal karyotype, and absence of significant dysplasia, have somatic mutations in MDS-related genes in more than one-third of the time (24). Despite the inter-patient variation, some of the mutations have developed independent expression and prognostic value such as the splicing factor 3 subunit 1 (SF3B1) mutation with ring sideroblasts (RS) (25) in 60%-80% of RS-MDS (26) and the association of tet methylcytosine deoxygenase 2 (TET2) mutation with longer OS, good prognosis (27), phenotype of the disease (28), and an increased response to 5-AZA (29, 30). There are only a few genes in MDS that are mutated in high frequency, while many more are found mutated only in a small proportion of patients, with the overall pattern of most frequent mutations comprising SF3B1, TET2, ASXL1, SRSF2, DNMT3A, RUNX1, U2AF1, ZRSR2, STAG2, TP53, EZH2, CBL, JAK2, BCOR, IDH2, NRAS, and NF1 genes (31–34) in order of descending frequency.
3 Somatic mutations
As mentioned above mutations found in MDS have been categorized into 8 subgroups depending on their function, including RNA-splicing machinery: SF3B1, SRSF, ZRSR2, U2AF1, U2AF2, SF1, PRPF8, LUC7L2, DDX41; DNA methylation: TET2, DNMT3A, IDH1/2; chromatin modification: ASXL1, EZH2, KMT2, SUZ12, JARID2, KDM6A, PHF6, EED, EP300; signal transduction: FLT3, JAK2, MPL, GNAS, KIT, CALR, CSF3R, NOTCH1/2, KRAS, NRAS, CBL, NF1, PTPN11; transcription factors: TP53, PPM1D, RUNX1, ETV1, GATA2, ETV6, CUX1, IRF1, CEBPA, BCOR LAMB4; cohesin complex: STAG2, CTCF, SMC1A, RAD21, SMC3; DNA repair: ATM, BRCC3, DLRE1C, FANCA-L, BRCA2, RAD21; others: SETBP1, NPM1, WT1 (29,36,37,38). The molecular landscape of these mutations is shown in Table 1. Moreover, Table 2 summarizes the preclinical data on targeting selective mutations, while Supplementary Table 1 comprises the clinical trials on investigational or already approved targeted therapies in MDS.
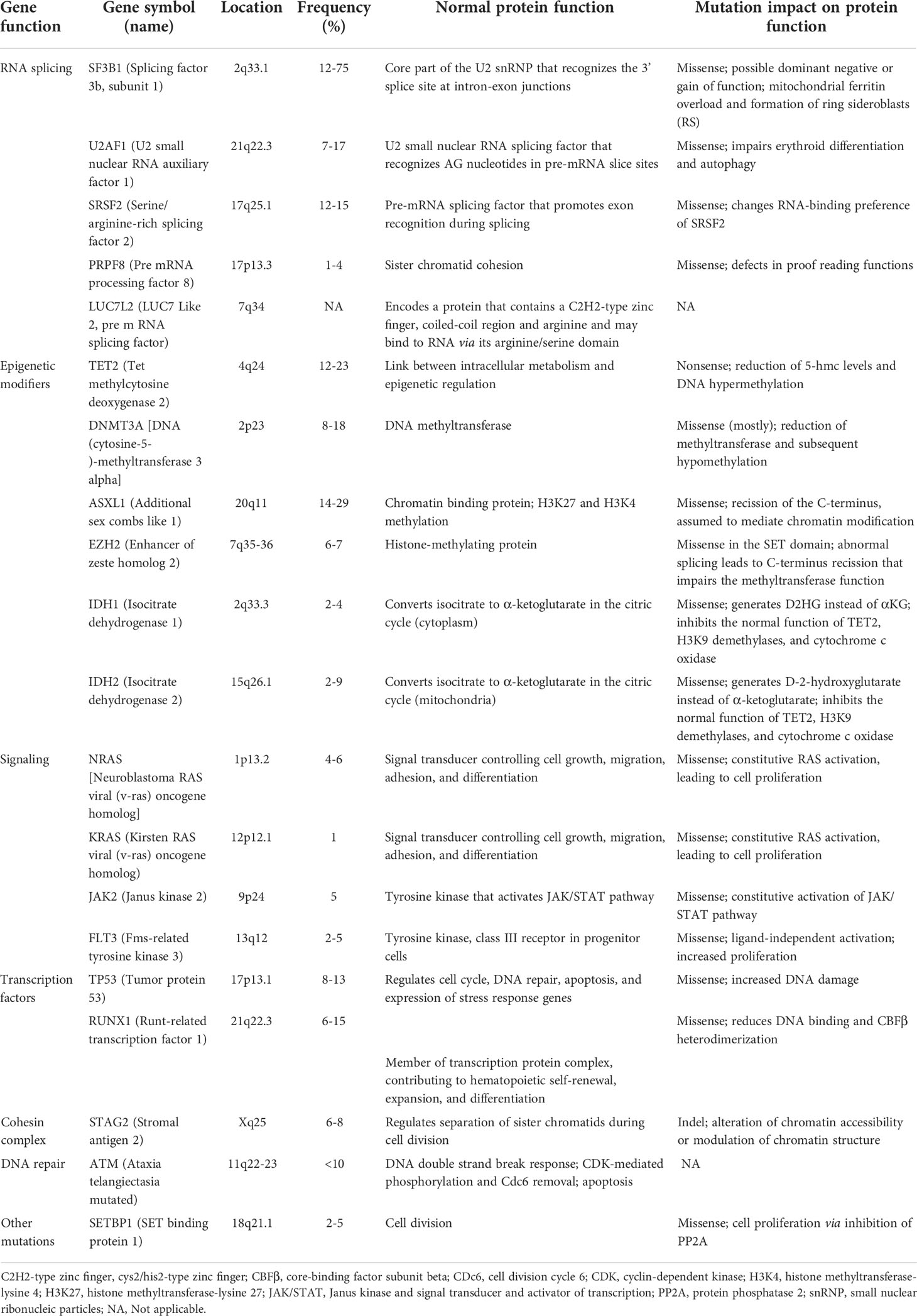
Table 1 Somatic mutations in MDS.
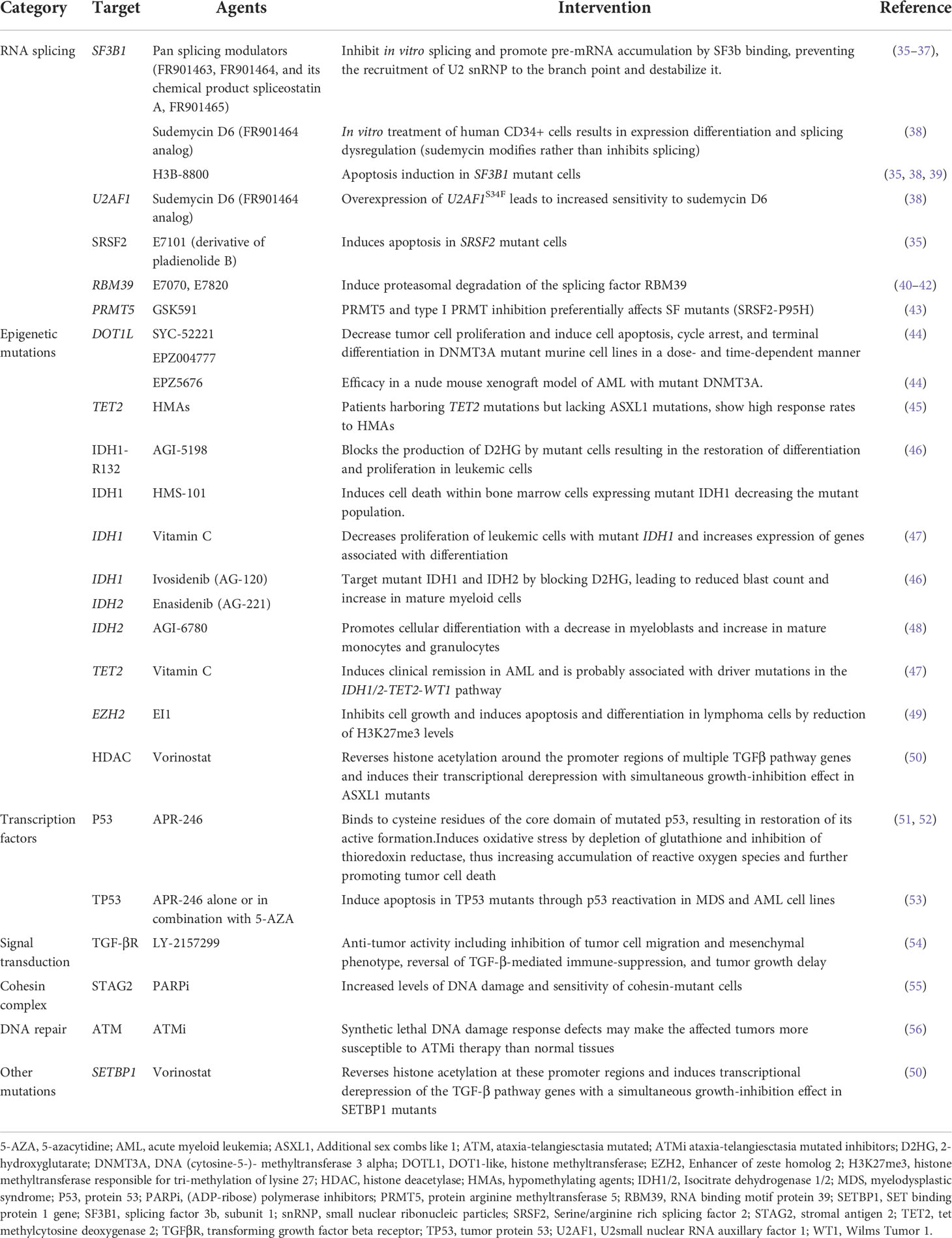
Table 2 Preclinical data on agents targeting mutations commonly found in patients with myelodysplastic syndrome.
3.1 RNA-splicing machinery
Spliceosome is a protein complex, composed of five small nuclear RNAs (snRNAs) and approximately 150 proteins, required for the splicing reaction (57). The complex is responsible for the removal of non-coding sequences (introns) from precursor mRNA (pre-mRNA) to form mature mRNA by ligating the coding sequences (exons). There are two types of pre-mRNA introns: U2-dependent (major spliceosome), which accounts for almost all the human introns, and U12-dependent (minor spliceosome), which accounts for less than a thousand introns (58). The U2-auxiliary factor (U2AF) protein consists of a U2AF35 (U2AF1) subunit and a U2AF65 (U2AF2) subunit. U2AF binds to SF3B1 through a splicing factor (SF1) to form a heterodimer complex. The U2AF1/U2AF2 heterodimer has a high affinity for Zinc finger RNA binding motif and serine/arginine rich 2 (ZRSR2) and serine/arginine rich splicing factor (SRSF2) (59). Any alterations to the spliceosome complex can result in a change in splicing specificity, leading to alternative splicing outcomes.
3.1.1 RNA-splicing machinery mutations
Splicing factor mutations are detected in 45%-85% of MDS, occur in a mutually exclusive manner, and are predominantly heterozygous. This indicates that homozygous splicing factor mutations are likely to be lethal to the cell (60, 61), missense, and located in restricted regions (62). SRSF2 is found mutated in a frequency of about 12%. Another 20% of MDS patients carry mutations in genes, such as U2AF1, U2AF2, SF3B1, and ZRSF2. They co-occur with epigenetic mutations, indicating cooperation of these mutations, and resulting in a specific phenotype. Mutations in SRSF2 are frequently concomitantly present with runt-related transcription factor 1 (RUNX1), isocitrate dehydrogenase 2 (IDH2), additional sex combs like 1 (ASXL1), TET2 and (STAG2) mutations, where co-mutation of TET2 and SRSF2 has been found predictive of leukemic transformation in MDS and CMML (63). Co-occurrence of mutations in ZRSR2 and TET2, as well as SF3B1 and DNA (cytosine-5-)- methyltransferase 3 alpha (DNMT3A), or U2AF1 and ASXL1 or TET2 have been noted (27). Two recent studies have used RNA sequencing to show that the common splicing factor mutations result in different alterations in splicing and affect different genes (64, 65). Enhancer of zeste homolog 2 (EZH2) in SRSF2 and U2AF1 mutant MDS cases has been found aberrantly spliced, thus representing a common target. EZH2 among other aberrantly spliced genes identified in both studies, is known to be involved in MDS pathogenesis, because of being recurrently mutated or involved in translocations. It has been also shown that SF3B1 mutations can be second hit mutations following epigenetic mutations and that they cooperate with EZH2 mutations to impair mitochondria and induce apoptosis (45). The presence of mutational hot spots and frameshift derangement could be correlated with gain or change of function of splicing factor genes leading to MDS. Concurrent spliceosome gene mutations, such as SRSF2 and SF3B1, are associated with HR-MDS, higher bone marrow blast percentage, and deregulation of RNA splicing and DNA methylation pathways (66).
The high mean variant allele frequency (VAF) of SF3B1 found in studies shows that the SF3B1 mutation is present in dominant MDS clones, suggesting that the mutation itself originates from cells actively proliferating in the MDS clones (66), while it has been also proposed that the consecutive acquirement of genetic lesions with SF3B1 in one of the mutated major clones at the early stages of MDS is implicated in AML transformation (67). Despite the mutual exclusivity of mutations in spliceosome members, mutations in SF3B1 appear to be markedly high in MDS-RS with an incidence of 60%-80% in these patients, but only 10–20% of unselected MDS. It is currently known that SF3B1-mutated MDS-RS patients, may have better outcomes compared with their wild-type partners (20, 26). SF3B1+/- mice accumulate RS in the bone marrow, which are rarely found in animal models of MDS, suggesting that SF3B1 haploinsufficiency causes biological features resembling low risk MDS-RS indicating that this mouse model could be a target for preclinical therapeutic studies (68). Clonal analysis has shown that SF3B1 mutations occur in rare lympho-myeloid HSCs (Lin− CD34+ CD38− CD90+ CD45RA−) providing a distinct clonal advantage to MDS-RS HSCs (68). However, it is widely recognized that any phenotypically defined HSC population also contains a substantial fraction of non-HSCs, and since the lymphoid lineages have not been found to be involved in the SF3B1-mutated clones, definitive evidence for recurrent SF3B1 mutations targeting true lymphomyeloid HSCs in MDS-RS is lacking (69, 70). Several studies have identified, using RNA-sequencing, deregulated gene isoforms and aberrantly spliced target genes in SF3B1 mutant MDS (71–74). The most studied gene which has been shown to be aberrantly spliced in SF3B1 mutant MDS-RS, is the mitochondrial iron exporter ATP binding cassette subfamily B member 7 (ABCB7), since a strong association between SF3B1 mutations and ABCB7 down-regulation has been found (69). Several studies have investigated genes involved in iron homeostasis and hemoglobin biosynthesis in SF3B1 mutant MDS and identified cryptic splicing events of them (72, 75, 76). As an example, SF3B1 mutations are often associated with alternative splicing of solute carrier family 25 member 37 (SLC25A37), a splice variant upregulated in MDS-RS patients with thrombocytosis, which acts as a significant importer of iron in the mitochondria (77) with a vivid role in mitochondrial iron delivery in erythroid cells (78). Somatic mutations of the pre-mRNA processing factor 8 (PRPF8) splicing gene that are also responsible for abnormal splicing involved in the iron metabolism have been also reported (79–81). Luc7-like 2 gene (LUC7L2) is located on chromosome 7q34 and has been found aberrantly spliced in MDS cases harboring SRSF2 small deletions. These mutations can be heterozygous, homozygous, and hemizygous and although they are not well characterized, it seems that they have been associated with short survival in patients with -7/del17q (82).The fact that SF3B1 mutations delineate a distinct type of MDS imply that the effects of RNA splicing factors mutations on mis-spliced target genes may open a whole new field for research.
3.1.2 Splicing as a therapeutic target
Observations of the survival dependence of splicing factor mutants on the wildtype splicing function (61, 62) have motivated the pharmacologic interest for targeting splicing. Several compounds are known to target specific steps in pre-mRNA splicing and catalysis. Pan-splicing modulators such as bacterially derived products and analogs have been shown to bind the SF3B complex and disrupt spliceosome balance (35, 83), while others such as FR901463, FR901464, and FR901465, have been shown to bind and inhibit SF3B1 from binding the pre-mRNA, thus preventing the recruitment of U2 snRNP to the branch point and destabilize it. The first that was identified was FR901464 and its chemical product, spliceostatin A (36). Meayamycins, sudemycins and thailanstatins (37, 84, 85)are analogous compounds that belong to the FR901464 family of splicing inhibitors. Specifically, in vitro treatment of human CD34+ cells with sudemycin D6 resulted in 1.030 differentially expressed genes and 18.833 dysregulated splicing events (86) leading to the conclusion that sudemycin mostly modify rather than inhibit splicing. Moreover, it has been observed that overexpression of U2AF1S34F lead to an increased sensitivity to sudemycin D6 and that this may be due to aberrant splicing caused by the drug. Other splicing modulators like FR901464 are the pladienolide inhibitors (87). E7101, a synthetic derivative of pladienolide B and more recently H3B-8800, a selective and orally bioavailable modulator of wild type and mutant SF3B seem to induce apoptosis in SRSF2 (38) and SF3B1 (38) mutants respectively. In an ongoing. phase I study (NCT02841540) of patients with myeloid cancers, different doses of H3B-8800 were compared in 84 patients with most of them harboring spliceosome mutations. The results have so far demonstrated dose-dependent target engagement, predictable pharmacokinetics profile, and safety even with prolonged dosing (35). Splicing modulators with different mechanism of action have also been identified. Idole derivatives (benzopyridoindoles) can modulate splicing (39), and small molecule kinase inhibitors (e.g., TG003 and SRPIN340) can modulate post-translational modification of serine-arginine rich proteins by blocking their interactions that are crucial for normal splicing (88).
In addition to these compounds that mostly inhibit the enzymatic activity of splicing, molecules that modify the abundance of splicing proteins are also gaining their place in the therapeutic approach of targeting RNA splicing. Recently, a subclass of sulfonamides including E7070, E7820, indisulam, tasisulam, and chloroquinoxaline sulfonamide (40, 41, 89) were shown to induce proteasomal degradation of the splicing factor RNA binding motif protein 39 (RBM39). RBM39 is a serine-arginine rich RNA binding protein, that shares great sequence with U2AF2 and is involved in the binding of the spliceosome to the 3’ exon, which is crucial for appropriate splicing (42, 90). Taking into consideration that spliceosome mutant leukemias seem to be sensitive to inhibition of protein arginine methyltransferases (PRMTs), a study identified that inhibition of arginine demethylation mediated by PRMT5 leads to preferential death of splicing factors mutant cells (91). The use of CRISR/Cas9 revealed that inhibition of the RBM39 protein leads to mis-splicing of Homeobox A Cluster (HOXA9) target genes, which constitute a network essential for AML survival (43).
Oligonucleotide-directed approaches such as RNA interference (RNAi) (92) and antisense oligonucleotides (ASOs) are also under development. A phase I trial (NCT01159028) investigated the safety and toxicity of BP1001, a liposomal growth factor receptor bound protein-2 antisense oligodeoxynucleotide (L-Grb2 AS) for patients with Philadelphia chromosome positive CML, AML, ALL, and MDS. BP1001 was well tolerated, with more than half of evaluable patients experiencing ≥50% reduction in peripheral or bone marrow blasts (93). A major challenge of using ASOs therapeutically is the improvement of their delivery systems. Recently, studies have revealed a strong correlation between splicing factor mutations and elevated levels of R-loops, DNA-RNA intermediates, and activation of the ataxia telangiectasia and Rad3-related protein (ATR) pathway (94, 95). DNA damage and cell death seem to be caused by pharmacological inhibition of ATR in CD34+ MDS cells. This can be enhanced by combination treatment with the low dose splicing modulatory compound Pladienolide B (95, 96).
3.2 Epigenetic mutations
Epigenetic modifications, such as DNA methylation and histone modification, result in inheritable changes in gene expression without changing the DNA sequence. Genes involved in DNA methylation and chromatin modification make up a second common class of mutations in MDS.
3.2.1 DNA methylation
Deregulation of CpG island methylation within gene promoters is a major epigenetic transcriptional control mechanism in human solid tissue and hematologic tumorigenesis (96, 97). Epigenetic changes in DNMT3A, IDH1/2, and TET2 are frequently reported in MDS causing hypermethylation, adhesion, and disease specific changes in HSCs.
Recurrent missense, nonsense, and frameshift mutations have been identified in DNMT3A, a methyltransferase enzyme that catalyzes the transfer of methyl groups to cytosines in CpG islands of DNA (98) DNMT3A is recurrently mutated in de novo AML and at low frequency in MDS with dominance at older age. These mutations of the methyltransferase domain are heterozygous and mostly missense or nonsense. The presence of DNMT3A mutations in nearly all bone marrow cells of MDS patients independent of blast count, has led to the hypothesis that this is an early genetic event in the disease process and may result in a clonal advantage of the mutated cells (99). It has been demonstrated that DNMT3A silencing leads to progressive damage of hematopoietic stem cell differentiation and thus, plays a crucial role in silencing of regulatory genes (100). The most commonly identified mutation is the missense R882 methyltransferase domain mutation, which occurs in almost half of DNMT3A mutated cases. Some reports have shown that the 5-year survival of AML patients with DNMT3AR882 mutation is significantly shorter than that of AML patients without such mutations (101–103). Moreover murine models harboring DNMTA3AR878H did not develop leukemia and were instead characterized by an increased percentage of circulating c-Kit-positive progenitor cells, consistent with HSPC expansion (99).
TET2, is an enzyme that hydroxylates methylated cytosines to initiate the process of DNA demethylation, by converting the methyl group at the 5-position of cytosine DNA 5-methylcytocine (5mC) to 5-hydroxy-methylcytosine (5hmC) (104). Somatic TET2 mutations in MDS are associated with advanced age, clonal hematopoiesis, and normal karyotype, suggesting that TET2 mutation is an aging-associated factor of hematopoietic cells (100). Abnormalities in TET2 with disabled TET function are observed in 15–27% of MDS, and include deletions, loss of heterozygosity, and missense, nonsense, and frameshift mutations leading to impaired DNA demethylation. TET2 mutations have most consistently been associated with favorable responses to HMAs (105, 106). This seems to be limited to patients in whom TET2 is an early, clonal mutation as opposed to a late subclonal event. On the contrary, TET2 mutations can be early events in some patients with MDS and secondary AML associated with shorter OS after HSC transplantation (107). Biochemical studies (107, 108) have demonstrated reduced levels of 5hmC in the genomic DNA of bone marrow cells from patients with TET-mutated malignancies, implying a catalytic activity of mutant TET2. Interestingly, patients with low 5hmC levels display a similar clinical phenotype independent of TET2 mutation status, suggesting that 5hmC levels rather than TET2 mutation status, may mediate the observed clinical phenotype.
TET2 activity is also affected by mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2. IDH1 and IDH2 belong to the IDH family that convert isocitrate to α-ketoglutarate (aKG) via oxidative decarboxylation. IDH2 is located in the mitochondria, whereas IDH1 in the cytosol and peroxisome. Heterozygous mutations alter the isocitrate dehydrogenase (IDH) enzymatic activity, resulting in the generation of 2-hydroxyglutarate (D2HG), an oncometabolite that inhibits the activity of several targets, including TET2. It is widely accepted that D2HG inhibits aKG-dependent dioxygenases and contributes to epigenetic alterations driving leukemogenesis (109). The connection between the two mutations became apparent when the mutant IDH enzymes were shown to catalyze the conversion of αKG to D2HG, consuming NADPH (110). D2HG is normally present at very low levels in non-mutated cells but accumulates at 100-fold higher levels in IDH-mutated AML cells (111). D2HG is structurally similar to αKG and a competitive inhibitor of αKG dependent dioxygenases including TET2 (112). The indirect interaction between IDH and TET2 mutants results in increased global expression of 5mC and impaired DNA damage repair mechanisms promoting myeloid malignancies. Mutations in IDH proteins are found in 15–20% of AML cases and less commonly in MDS patients occurring in 3–5% of cases (113). They are missense alterations affecting arginine-132 (R132) in IDH1 and either the arginine residue (R172) or the arginine-140 (R140) residue in the IDH2 protein (114). IDH1/2 mutations are generally heterozygous, suggesting a gain of function by the enzyme as a potential cancer mechanism (115). Expression of these mutations in mouse models initiate hematologic malignancy characterized by an increase in early hematopoietic progenitors, splenomegaly, anemia, hypermethylated histones, and altered DNA methylation patterns similar to those found in patients with AML with IDH1/2 mutations. It has been also shown that IDH1 mutant mice develop myeloid dysplasia via disturbance of heme biosynthesis and erythropoiesis, thus highlighting the essential role of IDH1 in normal erythropoiesis (116). Overexpression of mutant IDH enzymes can induce histone and DNA hypermethylation, as well as block cellular differentiation, which can be reversed by small molecule inhibition of the mutant enzymes (117–119). IDH mutations cooperate with other mutations to drive oncogenesis (120). They co-occur with nucleophosmin (NPM1) mutations (121), leading to better prognosis, but are almost always mutually exclusive with TET2 mutations.
3.2.2 Chromatin modifiers
Epigenetic regulation of posttranslational modifications of histone proteins at specific residues, including acetylation, methylation, and ubiquitination is catalyzed by a group of histones modifying enzymes with distinct specificity, some of which have been observed with mutations in MDS.
DOT1-like, histone methyltransferase (DOT1L), a histone lysine methyltransferase without a SET domain and its homologs are involved in numerous processes, including transcriptional regulation, cell cycle progression, and DNA damage repair, and are implicated in several cancers (122). EZH2 is the main catalytic member of the polycomb repressive complex 2 (PRC2) that also includes loss-of-function mutations in embryonic ectoderm development (EED), SUZ12 polycomb repressive complex 2 subunit (SUZ12) and EZH1 with a frequency less than 5%. EZH2 encodes a histone methyltransferase that is responsible for mono-, di, and tri-methylation of lysine 27 of histone 3 (H3K27me1, H3K27me2, HEK27m3). Point mutations of these methyltransferases offer high catalytic activity to the H3K27me3 in malignant cells. EZH2 also interacts with DNMT3A as mentioned above, affecting DNA methylation. Gene somatic mutations are associated with uniparental disomy of long arm of chromosome 7, del7q, and EZH2 reduced expression in CD34+ cells (123). When occurring in 7/7q-chromosome abnormalities, loss of function mutations induce overexpression of the HOX genes in MDS (124). Despite that EZH2 is commonly overexpressed in high risk MDS and AML, the observation that loss of function mutations affect patients with MDS/MPN, myelofibrosis, and various MDS entities highlights the fact that EZH2 can function as tumor suppressor and oncogene depending on the cellular context (125). Overall EZH2 mutations are associated with poor outcomes (126), but in MDS they are not associated with progression to AML (127). Moreover, EZH2 mutations co-occur with RUNX1 mutations in MDS and MDS/MPN patients (24, 27). EZH2 loss in HSCs significantly promotes RUNX1S291fs-induced MDS (128). In the MDS bone marrow, normal hematopoietic cell development is impaired via the activation of the interleukin-6 pathway amongst others, which has been shown to impair normal HSCs in vivo, hence promoting the survival and proliferation of MDS clones. Moreover, combined inactivation of EZH2 and RUNX1 in progenitor cells has been found to enhance proliferation of early thymic progenitors in mice (129). EZH2 mutations also frequently co-occur with TET2 mutations (27, 28, 130). Mice models have revealed that EZH2 or TET2 deficiency alone is sufficient to induce an MDS/MPN phenotype with lethality, while depletion of both EZH2 and TET2 synergistically induces myelodysplasia and strengthens the progression of both MDS and MDS/MPN (114).
The ASXL1 gene also plays a crucial role in recruiting polycomb repressive complex 2 (PRC2) to specific loci and normally encodes a chromatin-binding protein for the epigenetic regulation in hematopoietic cells. It belongs to a 3-member family (ASXL1/2/3) which is thought to mediate the balance between polycomb and trithorax functions and is involved in the maintenance of activation and silencing of HOX genes through chromatin remodeling (131). ASXL1 knockout mice present impaired hematopoiesis and development of MDS-like disease through decreased H3K4me3 and H3K27me3 in specific loci such as HOXA genes due to loss of interactions with the PRC2 complex. These findings indicate that wild-type ASXL1 is a major tumor suppressor in hematopoiesis and suggest loss-of-function features of ASXL1 mutations (132). ASXL1 wild-type protein can also support H3K4 methylation through O-linked N-acetylglucosamine transferase (OGT) (44), H2AK119 ubiquitination through PRC1, and H3K27 methylation in specific loci. Mutations in ASXL1 were originally identified in MDS with del20q11 (133). They involve somatic nonsense and out-of-frame insertion/deletion mutations that result in loss of function. ASXL1 is present in 15-24% of MDS patients as well as in clonal hematopoiesis of indeterminate potential (CHIP). They are three to five times more frequent in AML patients older than 60 years of age, are mutually exclusive with mutations in NPM1 in this population, and rarely found in combination with FMS related receptor tyrosine kinase 3-internal tandem duplication (FLT3-ITD (134). ASXL1 mutations are found to be associated with unfavorable prognosis and reduced survival among MDS patients transforming to AML independently of other clinical features, including age, cytogenetics, and cytopenias (135).
3.2.3 Epigenetic targeting
The HMAs which act as DNMT inhibitors, and the histone deacetylase (HDAC) inhibitors (vorinostat, romidepsin, etinostat) are the first epigenetic inhibitors that have gained FDA approval. Currently, HMAs constitute the treatment of choice for high-risk MDS patients. The incorporation of the azanucleotides, 5-azadeoxycytidine (decitabine), or 5-AZA, which is metabolized into decitabine (DEC) into the cell, essentially brings about a demethylation state which antagonizes the hypermethylation effected by the aberrant epigenome of MDS patients. HMAs has been shown to improve the overall survival of higher-risk MDS by delaying the transformation to AML (136). Hypomethylation is also effective in inducing hematopoietic recovery in ˂50% of patients with lower-risk MDS, although it does not improve survival (137, 138). The response rate of HMA in higher-risk MDS is 40-50%, and it normally takes 3-6 cycles to obtain a substantial hematological response (139). Factors predicting response to HMA are reported to be mutations of TET2 and tumor protein 53 (TP53) in high-risk groups, DNMT3A mutations, DNA hypomethylation, the absence of an ASXL1 mutation, and pharmacologic factors limiting efficacy of HMAs (high expression of cytidine deaminase (139) or low expression of deoxycytidine kinase which is a rate limiting step for DNA incorporation (140)). High (ADP-ribose) polymerase 1 (PARP1) mRNA levels have been correlated with better response (141) and higher survival rates after 5-AZA (142). In a recent study, it has been shown that the combined expression of small noncoding RNAs (sncRNAs) in MDS patients harboring SF3B1 and DNMT3A mutations can predict response to 5-AZA as well as define survival (143). Moreover, in a study of 98 MDS patients treated with 5-AZA, high methylation status of RNR subunit 1 (RRM1) was observed in responders, thus making RNR a possible prognostic factor for response to 5-AZA and for predicting success of epigenetic treatment (144).
Although inhibitors targeting TET2 mutations have not yet been developed, patients harboring TET2 mutations but lacking ASXL1 mutations showed high response rates to HMAs, indicating that they can be used as a predictor for treatment response in MDS patients (107). A recent study investigated the use of ascorbate (Vitamin C) treatment for TET2 mutant AML patients and has shown its use in enabling active demethylation of DNA (145). The use of hypomethylating agents against IDH mutations has been evaluated by treating with DEC and 5-AZA as single therapies or in combination with vorinostat or valproic acid (146). The results showed no association between IDH mutation status and treatment response. Ex vivo treatment of human AML cells with AGI-6780 has shown an outbreak of cellular differentiation with a decrease in myeloblasts and increase in mature monocytes and granulocytes (147).
Guadecitabine (SGI-110) is a next-generation HMA with a prolonged half-life. It is designed as a DEC analogue, which makes it resistant to degradation by cytidine deaminase. It is currently under clinical trials for MDS and AML (47, 48). It has been shown to be effective for higher-risk MDS, CMML, and low-blast count AML after 5-AZA failure in phase 2 trials (148). Moreover, CC-486, an oral 5-AZA compound has shown promising results regarding the toxicity and effectiveness in MDS patients and has been recently approved by FDA, as maintenance therapy in AML patients in remission after intensive chemotherapy (149, 150). Another recently approved FDA drug, ASTX727 (INQOVI®), is a combination of oral DEC and cedazuridine (151), a fluorinated tetrahydrouridine derivative specifically designed to inhibit cytidine deaminase and facilitate oral administration of hypomethylating agents (152). In a phase 1b study, it was shown that the combination of 5-AZA with pevonedistat, which is considered to suppress the proliferation of leukemic cells, is effective for AML (153). A potential survival benefit from rigosertib for a small group of high-risk MDS patients with HMAs failure within the first nine months was suggested, despite that the results from a randomized phase 3 clinical trial were not encouraging (154).
Ivosidenib (AG-120) and enasidenib (AG-221) target mutant IDH1 and IDH2 respectively, by blocking 2-hydroxyglutarate (2-HG) leading to a reduced blast count and an increase in mature myeloid cells. Overexpression of D2HG can also be targeted with AGI-5198 (selective inhibitor targeting IDH1-R132) by blocking the production of D2HG by mutant cells resulting in the restoration of differentiation and proliferation in leukemic cells (155). Another inhibitor, HMS-101, targeting IDH1 has been developed and showed to induce cell death within bone marrow cells expressing mutant IDH1 decreasing the mutant population (156). High levels of methylation of histone 3 at lysine 79 (H3K79me) are detected in AML patients bearing MLL rearrangements (MLL-r).
A recent study showed that SYC-52221 and EPZ004777 inhibitors that target DOT1L, decrease tumor cell proliferation and induce cell apoptosis, cycle arrest, and terminal differentiation in DNMT3A mutant cell lines in a dose- and time-dependent manner (46). Furthermore, the DOT1L inhibitor EPZ5676 showed promising efficacy in a nude mouse xenograft model of AML with mutant DNMT3A (44).
EZH2 has been considered a novel target for identification of small candidate compounds that can inhibit EZH2 activity. EI1, an EZH2 inhibitor in lymphoma, is currently under development and it seems to (157) inhibit cell growth and induce apoptosis and differentiation in lymphoma cells by reduction of H3K27me3 levels (158). Targeting genes which are derepressed because of EZH2 or PRC2 loss-of-function, such as HOXA9 may provide a successful therapeutic strategy. Inhibition of HOXA9 is achievable either on an epigenetic level through inhibition of expression or directly through inhibition of the HOXA9 transcription factor function, i.e., through targeting protein/protein or protein/DNA interactions. Several HOXA9 inhibitors such as HXR9 are already under clinical evaluation for patients with AML, highlighting the role of HOXA9 in leukemogenesis (159). There may also be therapeutic potential for exploring synthetic lethality, where there is mutational loss of EZH2. In mice, EZH1 has been shown to compensate for loss of EZH2 by repositioning of EZH1 to EZH2 targets (49, 158). Overall, EZH2 aberrations and PRC2 deregulation seem to be extremely prevalent in myeloid leukemia, hence, targeting EZH2 dysfunction directly or indirectly seems to be a promising approach for many patients.
Efficacy of lenalidomide on del5q in MDS is another example (160). Lenalidomide has been approved for low and intermediate risk MDS patients with 5q- (161, 162). In patients with 5q-, treatment with lenalidomide results in cytogenetic complete remissions and lower transfusion requirements (162). Inhibition of cell cycle and apoptosis, emerging from this molecule, leads to an anti-proliferative effect on leukemia blasts and a modulation effect on the bone marrow microenvironment immune cells in MDS. Recent studies have also shown that lenalidomide acts through a karyotype-dependent manner modulating the expression of genes already haploinsufficient (53, 161, 163). Moreover, patients who do not respond to lenalidomide are at a greater risk for clonal evolution to leukemia compared to responding patients (164). Surprisingly, lenalidomide was shown to have clinical activity in 20–30% of non-5q31 deleted MDS patients (165). Nevertheless, even within the subgroup of 5q- patients with a generally favorable prognosis, TP53-mutated patients tend to have decreased response rates and increased risk of evolution to leukemia (166). CSNK1A1 encodes a casein kinase 1α (Ck1α), which is a serine-threonine kinase that fine-tunes the balance between cell proliferation through wingless/integrated WNT/β-catenin signaling and apoptosis mediated by TP53. Hence, if del(5q) MDS cells acquire deletions of TP53, they become resistant to lenalidomide due to complete loss of CK1α and the inability to induce TP53-dependent apoptosis and cell death. Clinicals trials focusing on the combination of lenalidomide and 5-AZA, are in the center of the attention, showing by now favorable results (167, 168).
3.3 Signal transduction
A complex interplay of various cytokines has been implied in the regulation of differentiation of the bone marrow in MDS. Growth factors, interleukins, interferons, transforming growth factor beta (TGF-β), and tumor necrosis factor a (TNF-α) are suggested to have stimulatory and/or inhibitory actions on hematopoietic stem cells (169). The physiologic effects of a few of these cytokines are executed by the support of transcription regulators like the janus kinase and signal transducer and activator of transcription (JAK-STAT) pathway and many other pathways (170). Activating mutations of tyrosine kinase or serine/threonine kinase are commonly seen in various myeloid malignancies. Active signaling caused by these mutations, induces cell proliferation and suppresses apoptosis, contributing to the expansion of abnormal clones that have accumulated various mutations.
3.3.1 Signal transduction mutations
Normal cellular growth and differentiation are regulated by the mitogen-activated protein (MAP) and phosphatidylinositol-3 kinase (PI3K)/AKT/mTOR pathways, both of which are activated by the GTPase family proto-oncogene RAS (171, 172). The p38 MAP kinase (MAPK) pathway in HSCs can also be upregulated TGF-β which transduces gene expression signaling via phosphorylation of SMAD proteins (173). Mutations in neuroblastoma RAS viral oncogene homolog (NRAS) and kirsten rat sarcoma virus gene (KRAS) are the most frequent. Both mutations occur at known hotspots, including p.G12, p.G13, and p.Q61. NRAS is found to be mutated in 4-6% of MDS and is shown to predict a phenotype with increased marrow myeloblasts, lower OS, and increased risk of transformation to AML (174), with mutations in KRAS detected in no higher than 1% of cases (175, 176). FLT3 and v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) mutations, commonly seen in AML, occur in less than 5% in MDS (177). Neurofibromatosis type 1 (NF1), B-Raf Proto-Oncogene (BRAF), and Protein Tyrosine Phosphatase Non-Receptor Type 11 (PTPN11) are rarely mutated. Cbl proto-oncogene (CBL) mutations in MDS (5%) are involved in negative modulation and aberrant tyrosine kinase signaling (177).
The V617F JAK2 mutation encoding a cytoplasmic tyrosine kinase, has been described in 5% of MDS associated with megakaryocytic proliferation, and 50% of MDS/MPN overlapping refractory anemia with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) (37, 63) The overall prognostic significance of JAK2 is unclear in MDS, but targeting similar to that in MPN is not irrational. Mutations in signal transduction pathways occur in a mutually exclusive manner in individual MDS (24). PTPN11, is part of the RAS family pathway and rarely mutated in adult MDS. The missense mutations include N-SH2 and PTP domains and have been associated with juvenile myelomonocytic leukemia (JMML) and childhood MDS/AML (177, 178). Additionally, it has been found that PTPN11 mutations can be acquired during the progression of MDS or JMML (179).
3.3.2 Targeting signaling transduction pathways
Targeting these mutations has been achieved through direct mutational applications or other involved pathways including PI3K, MEK or MAPK. Both JAK2 and RAS mutations have been targeted with some success in other myeloid diseases, suggesting potential application in MDS as well. Selective, single-agent activity of trametinib, a MEK1/MEK2 inhibitor against RAS-mutated myeloid malignancies, has been evaluated in a phase 2 clinical trial (NCT00920140) and has shown that repeated cycles of the drug were well tolerated, with manageable or reversible toxicities (180). The clinical activity of trametinib is currently being evaluated in a phase 2 clinical trial (NCT04487106) (181), in combination with 5-AZA and venetoclax in patients with AML and HR-MDS. Trials investigating the MAPK pathway include inhibitors of MAPK signaling, namely SCIO-469, ARRY-614, and ezatiostat, with reasonable clinical responses in lower-risk MDS patients (182, 183). TGF-β signaling has been therapeutically targeted by small molecule inhibitor of the TGF-β receptor kinase, LY-2157299 (184), with encouraging preclinical results. Apart from TGF-β receptor kinase inhibition, members of TGF-β superfamily and bone morphogenetic protein ligands have also been targeted by compounds like sotatercept (ACE-011) and ACE-536 (169). The multikinase inhibitor of PI3K, Akt, and PLK1 and RAS mimetic, rigosertib results in decreased tumor cell proliferation. In a phase II dose optimization trial of oral rigosertib in patients with lower-risk MDS (54), different doses of rigosertib were compared in 82 patients, with 15 of 34 eligible patients achieving transfusion independence. In another phase II study (185), rigosertib in combination with 5-AZA led to an overall response rate of 59% in patients who had failed HMA. Another phase III trial (NCT02562443) (186) is ongoing to evaluate the clinical benefit of this subset of patients. Tipifarnib, lonafarnib and a MEK inhibitor (GSK1120212) have shown acceptable toxicity profiles in small studies, and efforts to select mutational subgroups of MDS and AML that may benefit from these inhibitors are underway (186). Although quite common in AML, FLT3 mutations occur rarely in MDS. Despite the approval of midostaurin, sorafenib, gilteritinib, and quizartinib in FLT3-mutated AML this target is less likely to play a role in MDS therapeutics.
3.4 Transcription factors
Transcription factors involved in hematopoietic differentiation and HSC regulation are recurrently mutated in MDS. Mutations of the transcriptional factors have been reported to cause impairment of differentiation and maintenance of HSCs of MDS.
3.4.1 Transcription factors mutations
Hematopoietic differentiation involves the activation of lineage-specific gene expression programs by core transcription factors, such as GATA binding protein 2 (GATA2), RUNX1, and CCAAT Enhancer Binding Protein Alpha (CEBPA)f. Recurrent loss-of-function mutations in these molecules occur somatically in MDS and can also be inherited in the germline, thus causing familial bone marrow failure syndromes with a propensity to evolve into myeloid malignancies (28, 29). Familial presentation of MDS due to germline mutations is still unclear but, with the development of NGS techniques, the understanding of pathophysiology of these mutations is in the spotlight. About 45% of MDS/AML families has a mutation in genes including RUNX1, TERT, ANKRD26, CEBPA, DDX41, ETV6, GATA2, TERC, and TP53 (187), with these gene variants having a significant association with disease progression.
The tumor suppressor TP53 plays a key role in response to cellular stress such as DNA damage by activating several protective pathways inducing apoptosis, cell arrest, and DNA repair. It is reported as a unique mutation, being mutually exclusive with other mutations, suggesting its potential role as a driver of mutation in MDS.
TP53 mutations are seen in 8-13% of MDS, usually (30-50% of cases) in the context of a complex karyotype. They are also associated with chromosomal abnormalities such as deletions of the long arm of chromosome 5 [del(5q)] or chromosome 7 [del(7q)] and high blast percentage, higher-risk disease by IPSS, and adverse prognosis (188). In a retrospective study of patients undergoing pre-transplant genetic profiling, mutations in TP53 were associated with significantly decreased OS (24). TP53 mutated patients present an unfavorable clinical outcome and a high risk of disease progression (189). In fact, TP53 mutations can divide MDS with complex karyotype into distinct prognostic groups with specific clinical features (60, 190). Another recent study has elucidated the negative prognostic impact of TP53 mutations in the MDS transplant population, complementary to identifying RUNX1 and ASXL1 as potential markers of poor outcome. TP53 mutations in association with del(5q) at the early stage of the disease worsens treatment outcomes due to loss of p53 protein function (14). TP53 mutations seem to have no effect on response to 5-AZA (191). However, a 100% response rate in MDS patients with a TP53 mutation treated with DEC on a 10-day schedule has been demonstrated (192), though this result has not been verified in other cohorts. Moreover, differences in the TP53 allelic status have been found between low and high-risk patients, since monoallelic TP53 mutations are commonly seen in patients with lower risk MDS (163). Hence, it is important to investigate the TP53 allelic status for diagnostic and therapeutic purposes.
3.4.2 Targeting transcription factor mutations
Mutated p53 protein appears to be an important therapeutic target in MDS and AML, therefore restoration of its biological function could be highly beneficial. P53 re-activation and induction of massive apoptosis, {PRIMA-1Met (APR-246 or eprenetapopt)} is a methylated derivative of PRIMA-1, a molecule that reactivates the normal transcription of p53, leading human tumor cells to apoptosis. Mechanistically, APR-246 is a prodrug that binds to cysteine residues of the core domain of mutated p53, resulting in a change of its structure that restores its active formation. Moreover, it induces oxidative stress by depletion of glutathione and inhibition of thioredoxin reductase, thus increasing accumulation of reactive oxygen species and further promoting tumor cell death (193, 194). It has been recently reported that low doses of APR-246 alone or in combination with 5-AZA induce apoptosis in TP53 mutants through p53 reactivation in MDS and AML cell lines (53). In a phase I study including patients with AML, APR-246 monotherapy demonstrated clinical activity with corresponding activation of p53 (51). A recent phase Ib/II study determined the safety and efficacy of APR-246 in combination with 5-AZA in patients with TP53-mutant MDS or low blast count AML (NCT03072043) (52). 5-AZA and APR-246 resulted in a 73% overall response rate with 50% achieving complete remission in the MDS population with a median OS of 10.4 months. These data also support the ongoing phase III multicenter, randomized study of APR-246 in combination with 5-AZA versus 5-AZA alone in patients with TP53-mutant MDS (NCT03745716) (195).
Luspatercept (ACE-536) is a novel agent responsible for stimulating the late stage of erythropoiesis through a different mechanism from erythropoietin (196, 197), and is expected to be effective in MDS patients who are refractory to erythropoiesis stimulating agents (ESA). It is a fusion protein of the human immunoglobin G1 (IgG1) which acts through the TGF-β/SMAD pathway, binding to GDF8, GDF11, and activin β, and leading to an increase in erythroid cell maturation by promoting differentiation of erythroblasts. Since TGF-β1 accelerates the terminal maturation of erythroblasts, and luspatercept is supposed to reverse the effects of TGF-β1, it is probably working by preventing the skipping of cell divisions induced by TGF-β1 rather than by supporting maturation. It is approved for the treatment of adult patients with transfusion-dependent anemia due to MDS-RS and an unsatisfactory response to EPO-based therapy, as well as for adult patients suffering from transfusion-dependent anemia associated with beta-thalassemia (198). In lower-risk MDS patients, TGF-β signaling inhibition with luspatercept and sotatercept (ACE-011) has shown activity in clinical trials (194, 199). Interestingly, patients with an SF3B1 mutation seem to respond better to luspatercept than patients without it. A phase 3 trial with 229 randomized transfusion dependent LR-MDS-RS patients, evaluating the erythroid response and transfusion independence of luspatercept (179), showed the effectiveness of the agent for treating ESA-refractory anemia in MDS-RS patients.
3.5 The cohesin complex
Cohesin is a multi-subunit protein complex involved in the 3D shaping of the human genome and plays a critical role in chromosome maintenance, transcriptional regulation, and several DNA repair mechanisms (62, 200). The cohesin complex is known to align and stabilize replicated chromosomes prior to cell division.
3.5.1 Cohesin complex mutations
Mutations of the cohesin complex have been reported in various myeloid malignancies, including MDS. Mutations in four components of the cohesin complex are mutually exclusive, indicating that functional disruption of the whole complex is important for the pathogenesis of MDS (201). Recurrent somatic mutations in the cohesin complex are frequent genetic drivers which lead to disturbances in post-replication DNA repair, stabilization of chromosomes, and transcriptional regulation in MDS and other myeloid malignancies (1, 62). Mutations in spliceosome subunits, particularly in PRPF8 and SRF3B1 have also been thought to disrupt sister chromatid cohesion in human cells (202). In overall, it is about loss-of-function nonsense and frameshift mutations, often heterozygous, in a frequency of 12-20% in high risk MDS and secondary AML, mostly indicating poor OS (1, 30, 62). Mutations of the STAG gene have been found in 4-5% of MDS patients, making it the most mutated subunit. STAG2 mutations have been found to be very early events occurring in naïve stem cells and affecting dominant clones (154). Cohesin mutations have been shown to induce abnormal myeloid differentiation and alter the hematopoietic stem cell homeostasis in adult mouse models (203, 204). Therefore, it is assumed that cohesin mutations contribute to the development of MDS through the structural alteration of chromatin and a resulting transcriptional gene deregulation, which is critical for HSCs and their differentiation. A co-occurrence of STAG2 mutation and RAS pathways (NRAS, FLT3) mutations in 15-20% of MDS progressing to AML has also been demonstrated, indicating that the great genomic instability which characterizes the progression from MDS to AML, is associated with these mutations (205).
3.5.2 Targeting the cohesin complex
There are three categories of emerging therapeutic strategies in cohesin mutant cells: direct targeting of cohesin components or its regulators; targeting underlying dysregulated transcription or signaling events; targeting DNA damage repair (DDR) (206, 207). Synthetic lethal interaction between STAG1 and STAG2 homologous genes has been identified in several cancer cells (173). Inactivation of STAG1 leads STAG2 mutant cells to death. Depletion of the other core subunits RAD21 and structural maintenance of chromosome 3 (SMC3) reduces growth in both wild-type and STAG2 mutant cells as well. In cohesin mutant cancers, targeting DNA methylation or histone modification has an emerging therapeutic potential. It has been shown that myeloid dysplasia patients with STAG2 or RAD21 mutations had a significantly better response to DEC and 5-AZA than patients without cohesin mutations (208). Cohesin-deficient cancers cells have shown increased sensitivity to ionizing radiation, because of the cohesin’s role in DNA double-strand break repair. Therefore, using damaging agents to block the underlying DNA repair pathway of cohesin mutant cells has a potential for selective inhibition of these cells. PARP inhibitors (PARPi) benzamide and olaparib have been shown to induce synthetic lethality in cohesin-depleted colon neoplastic cell lines with intrinsic high or low PARP activity (209). Interestingly, a recent study demonstrated increased levels of DNA damage and sensitivity of cohesin-mutant cells to poly(ADP-ribose) polymerase (PARP) inhibition. In the same study, in a mouse model of MDS, STAG2 mutations arose and were driven by TET2 mutations and displayed selective depletion of cohesin mutant cells with PARP inhibition in vivo. Finally, a shift from STAG2- to STAG1- containing cohesin complexes in cohesin-mutant cells, which was associated with a longer DNA loop and increased interaction with PARP and replication protein A complex was observed (153). Understanding the mechanisms underlying increased sensitivity of cohesin-deficient cells to agents like HMA alone or with combination with other treatment strategies might lead to tailored therapeutics for cohesin mutant cancers.
3.6 DNA repair genes
Double-strand breaks (DSB) constitute the most important DNA damage in HSCs. The non-homologous ends-joining (NHEJ) and homologous recombination (HR) repair mechanisms are essentials for ensuring the genomic stability of stem cells. DNA repair is a crucial mechanism for maintaining the integrity of multi-cellular organisms and for ensuring genomic stability (210). Mutations of DNA repair genes may be associated with differences in the efficiency of DNA repair and may influence the risk of developing tumors (55).
3.6.1 DNA repair genes mutations
Ataxia-Telangiesctasia Mutated (ATM) belongs to the phosphatidylinositol 3-kinase-related kinase family. It participates in DNA DSB repair, metabolic regulation, cell cycle checkpoints, and chromatin remodeling (211). It has five major autophosphorylation sites: Ser367, Ser1893, Ser1981, Ser2996, and Thr1885. Once ATM is stimulated by DNA damage, its dimerized form dissociates and converts to monomers, leading to phosphorylation of the above-mentioned residues (212). ATM acts as a cell cycle checkpoint following exposure to stress (213). Somatic mutations have been found in 4% of all samples in a pan-cancer analysis, including missense, loss-of-function and truncating alterations (214).
3.6.2 Targeting DNA damage repair
To study the role of ATM in different tumor cell types, recombinase technology techniques have been applied to elucidate how therapies that target ATM may affect cancer compared to normal tissues. Although, at first glance, targeting a tumor suppressor gene could not enhance cancer cell killing, recently a CRISPR screen revealed key FANCONI anemia/BRCA pathway genes as strong synthetic lethality with ATM in the presence of ATM inhibitors (ATMi). Synthetic lethal DNA damage response defects may make the affected tumors more susceptible to ATMi therapy than normal tissues. Growing evidence suggests that ATM inactivation may also potentiate the effects of PARPi (215). It has been shown that deficiency of ATM in addition to BRCA also confers PARPi sensitivity (216). Moreover, experimental investigations have linked the functional status of p53 and the ability for ATM inactivation to confer radiosensitization (56). It has been previously shown that in HR-MDS and AML, ATM activates NF-κB and that its pharmacological inhibition induces apoptosis in malignant myeloblasts that are freshly isolated from the BM of these patients (217).
3.7 Other mutations
SET binding protein 1 gene (SETBP1) encodes a protein with 1542 amino-acid residues. SETBP1 is a 170-kDa nuclear protein that binds to SET protein, a protein previously noted to be associated with acute undifferentiated leukemia (218).
3.7.1 SETBP1
The interaction of SETBP1 with the SET nuclear oncoprotein involved in cell division is hypothesized to inhibit various tumor suppressor effects and promote cell proliferation (219). Somatic mutations in SETBP1 have recently been demonstrated in MDS and mainly affect 858–874 residues. SETBP1 binds directly to the promoters of HOXA9 and HOXA10 resulting in increased expression. Overexpression of SETBP1 causes inhibition of protein phosphatase 2 (PP2A), leading to cell proliferation. Co-occurrence of SETBP1 has been noticed with -7/del(7q) and, although it is associated with intermediate prognosis, cooccurrence of SETBP1 with this chromosomal rearrangement might indicate a poor prognostic impact of SETBP1 in MDS (220, 221). In particular, SETBP1 mutations have been found overrepresented in patients with isolated i (16) (q10) (41-54%) as compared to cases with other cytogenetic rearrangements and were mutually exclusive with TP53 mutations (222). SETBP1 mutations occur in a frequency of 2-5%, usually in LR-MDS, and are correlated with significantly shorter survival (219). They are somatic gain-of-function mutations and are enriched in patients with ASXL1 mutations, giving a higher risk of transformation to AML (223). It has been previously shown that expression of a constitutively active TGFβ type I receptor (ALK5-TD) inhibits leukemic proliferation of MDS/AML cells with concurrent ASXL1 and SETBP1 mutations.
3.7.2 Targeting SETBP1
The exact mechanisms of SETBP1 mediated leukemogenesis are largely unknown and there are no drugs available to specifically target SETBP1. However, aberrantly reduced acetylation of histone H3 and H4 lysine residues has been found around the promoter regions of multiple TGF-β pathway genes. The HDAC inhibitor vorinostat reverses histone acetylation at these promoter regions and induces transcriptional derepression of the TGF-β pathway genes with a simultaneous growth-inhibition effect in ASXL1 mutants, indicating that HDAC inhibitors will be promising therapeutic drugs for MDS and AML with ASXL1 and SETBP1 mutations (224).
3.8 B-cell lymphoma 2 (BCL-2)
BCL-2 and its relatives are functionally classified as either antiapoptotic or proapoptotic. Most cells express a variety of antiapoptotic and proapoptotic BCL-2 proteins, and the regulation of their interactions dictates survival or commitment to apoptosis (50).
3.8.1 The role of BCL2 in HR-MDS
The anti-apoptotic protein BCL-2 provides protection against various proapoptotic stresses by blocking cell death induction (225). In HR-MDS harboring ASXL1, RUNX1, TP53 or EZH2, clinical remission depends on the quantity of the drug required to overcome the apoptotic resistance. This is due to the finding that the balance between pro-survival and pro-apoptotic proteins of the BCL-2 family is substantially disturbed during disease progression (50). Data from murine models have shown an interaction between ASXL1, RUNX1, EZH2, TP53 and BCL-2 expression, suggesting that BCL-2 inhibition might fail to kill progenitors in MDS that harbor any of these mutations (226, 227).
3.8.2 Targeting BCL-2
Since selective BCL-2 inhibition is a promising treatment strategy in hematologic malignancies, the therapeutic impact of venetoclax (formerly known as ABT-199) has been tested in HR-MDS harboring the above mutations. Unlikely to what had been observed in murine models, it has been shown that HR-MDS patient samples specifically undergo cell death in response to venetoclax even when harboring mutations in ASXL1, RUNX1, TP53 or EZH2 (228). The rationale of combining venetoclax with 5-AZA was linked to the existing role of hypomethylating agents in MDS and the promising results observed for the venetoclax/5-AZA combination in older patients with AML (229). The combination of venetoclax and 5-AZA produces a rapid and high rate of bone marrow blast clearance. Data from ongoing phase 3 clinical trials (NCT04401748, NCT04628026) (230, 231) will determine whether combining venetoclax with 5-AZA decreases leukemia stem cell populations, and whether sensitivity in subpopulations correlates with intracellular levels of BCL-2 family proteins. The results of these studies may have a major impact on the way HR-MDS will be managed in the future.
3.9 The role of immune checkpoints
Immune checkpoints play an important role in the regulation of immune homeostasis by balancing the stimulatory and inhibitory signals that mediate T-cell immune responses (232). Under normal conditions, immune checkpoints regulate self-tolerance and protect tissues from damage by conducting the immune system response to pathogenic infection.
3.9.1 Immune checkpoints in MDS
Leukemic cells express several checkpoint inhibitor receptors as well as ligands making them potential direct targets for inhibition. Deregulated immune microenvironment has been recognized as a key pathogenic driver of MDS and AML, causing high rate of apoptosis in LR-MDS and immunosuppression in HR-MDS and AML (233). Several cytokines that are abnormally expressed in MDS can induce their expression, which together represent an immune checkpoint involved in regulating T cells in the peripheral tissues (234). Immune checkpoint molecules, including programmed cell death-1 (PD-1) and programmed cell death ligand-1 (PD-L1), play important roles in oncogenesis by maintaining an immunosuppressive tumor microenvironment. Recently, both molecules have been examined in MDS and AML. Abnormal inflammatory signaling, genetic and epigenetic alterations, interactions between cells, and treatment of patients, all have been involved in deregulating PD-1/PD-L1 signaling in these two diseases. Patients with autoimmune diseases seem to develop MDS in a ratio of 1.5-3.5, indicating that immune deregulation could be implicated in the pathogenesis of MDS.
3.9.2 Immune checkpoints inhibitors (ICIs)
Following the successes with ICI1s in solid tumors (235–237) these agents are being evaluated in hematologic malignancies, including AML and MDS (238). Epigenetic modification through PD-1 inhibition is a new approach in HR-MDS. Abnormal upregulation of PD-L1, PD-L2, PD-1, and cytotoxic T-lymphocyte antigen 4 (CTLA4) in CD34+ cells in MDS patients compared to healthy controls has been reported, suggesting that as a potential mechanism triggering HR-MDS (239). Higher percentage of PD-L1 expression is presented on the HR-MDS blasts compared to LR-MDS (240).
Pidilizumab (MDV9300, formerly CT-011) is a humanized monoclonal IgG1 antibody to PD-1. In a phase I study, pidilizumab was relatively safe, with no dose-limiting or treatment-related adverse events (241). Other relevant PD-1 inhibitors include nivolumab and pembrolizumab that are still being evaluated in phase 1b and 2 trials (NCT01953692, NCT02397720) respectively in MDS/AML patients after HMA failure (242, 243). Several clinical trials are trying to evaluate the combination of PD-1/PDL-1 inhibitors with HMAs, showing mixed responses (NCT03094637, NCT02508870, NCT02775903) (244–249)
Ipilimumab is a human IgG1 isotype monoclonal antibody that binds the CTLA-4, a T-cell receptor that downregulates T cells (250). This agent blocks the inhibitory signal, allowing cytotoxic T cell mediated antitumor immune response. Two studies are actively investigating the role of single agent ipilimumab in relapsed/refractory MDS (NCT02530463, NCT01757639). Preliminary results from the phase II study (NCT02530463) that evaluated multiple cohorts (nivolumab alone, ipilimumab alone, nivolumab with ipilimumab) in MDS patients who had failed first-line therapy with an HMA and combination cohorts (5-AZA with nivolumab, 5-AZA with ipilimumab, and 5-AZA with nivolumab and ipilimumab) in previously untreated intermediate 2/high-risk MDS are available (251). The same study included 15 intermediate/high risk MDS patients failing HMA who received single agent nivolumab in a parallel cohort with no responses noted.
3.9.3 CD47 and the “don’t eat me” signal
Cluster of differentiation 47 (CD47) is a cell surface glycoprotein belonging to the immunoglobulin superfamily, binding to various proteins including integrin, thrombospondin-1, and signal regulatory protein α (SIRPα). The interaction of CD47 with SIRPα triggers a “don’t eat me” signal to the macrophages, inhibiting phagocytosis (252). Thus, overexpression of CD47 enables tumor cells to evade immune surveillance via the blockade of phagocytic mechanisms. CD47 blockade alone is not sufficient to trigger macrophage anti-tumor activity. Macrophages also need a pro-phagocytic signal. Accumulating data suggest that CD47-SIRPα is a key immune checkpoint in different cancers including hematological malignancies. CD47 monoclonal antibodies (mAbs) not only block CD47 from engaging SIRPα, but also engage the activating Fc gamma receptor (FcγR) on macrophages. Together they deliver a potent phagocytic signal to macrophages (253). Magrolimab is a monoclonal antibody that blocks CD47 to escape immune surveillance and macrophage-mediated clearance (254). Clinical trials have shown that magrolimab, is safe when administered as monotherapy (255). However, the mechanism underlying this observed protection has not been fully defined. Moreover, a synergy between magrolimab and 5-AZA has also been observed. At present, clinical trials designed to examine whether such a synergy can be seen in patients, and to assess the tolerability and clinical activity of the combination (NCT03248479, NCT04313881) (256, 257) show promising results.
4 Germline mutations
The molecular recognition of germline predisposition to hematopoietic neoplasms was formalized in the 2016 revision of the WHO classification of myeloid neoplasms and AML. Germline variants in genes such as CEBPA, DDX41, RUNX1, ETV6 and GATA2 should be considered in patients when mutations in these genes are seen in tumor sequencing tests, especially when the mutations are biallelic or consistent with their presence in the germline (264). Estimates suggest that 4-10% of children and young adults with MDS or AML and 4% of adults with AML carry inherited mutations affecting cancer susceptibility genes 259.
4.1 CEBPA
The CEBPA gene is located on chromosome 19q13.1 and is essential for cellular growth arrest. The inheritance of a germline CEBPA mutation predisposes the development of AML with autosomal dominant inheritance. Familial CEBPA AML shares characteristics with somatic CEBPA AML (260). It has become clear that familial AML with germline CEBPA mutations involves the inheritance of a single copy of mutated CEBPA encoding a granulocyte differentiation factor (261) and that more than 10% of AML with biallelic CEBPA mutations carry a N-terminal frameshift CEBPA germline mutation with acquisition of a C-terminal somatic mutation as a second event in the development of AML (262).
4.2 DDX41
DDX41 is a receptor belonging to the DEAD/H-box helicase family, encoded by a gene comprising 17 exons on chromosome 5 (263). It takes part in pre-mRNA splicing, mRNA export, transcriptional and translational regulation, ribosome biogenesis and RNA decay (264). DDX41 mutations have been identified as germline mutations in families with multiple cases of late-onset MDS and/or AML. The majority of germline mutations are frameshift mutations suggesting loss of function with DDX41 acting as a tumor suppressor, and there is a common somatic missense mutation found in a majority of germline mutated tumors. Clinically, DDX41 mutations lead to the development of high-risk MDS (265). Patients with DDX41 mutations who develop MDS/AML usually present with leukopenia with or without other cytopenias and macrocytosis, in addition to hypocellular bone marrow with prominent erythroid dysplasia, and a normal karyotype, often leading to erythroleukemia (266). The prognosis of these patients is generally poor. In a limited number of patients, cases with DDX41 mutation may respond to lenalidomide (267), while in general DDX41-related myeloid malignancies have been reported to present good outcome (268).
4.3 RUNX1
RUNX1 is located on chromosome band 21q22 and is associated with the development of normal hematopoiesis. It is a key regulator of hematopoietic and bone marrow differentiation. In a study of familial platelet disease with propensity to myeloid malignancies (FPD/AML), an autosomal dominant syndrome characterized by platelet abnormalities and susceptibility to MDS/AML that is caused by the genetic mutation of RUNX1, the authors identified germline RUNX1 mutations in five families with a history of MDS/AML (269). Germline RUNX1 mutations include partial and whole gene deletions and frameshift, stop-gain, and missense mutations and are likely to be an initial factor in the development of AML, in addition to other genetic abnormalities. Different families with germline RUNX1 mutations exhibit varying risks of developing MDS and AML (11-100%). The median age of patients with such mutations at the onset of MDS/AML is 33 years, which is younger than that of sporadic MDS/AML (270, 271).
4.4 ETV6
The ETV6 gene is located on chromosome 12. It encodes an ETS family transcription factor with three functional domains necessary for hematopoiesis and the vascular development. Several studies have described germline ETV6 variations as a susceptibility factor for hematologic malignancies (272–274). Since the first description of germline ETV6 mutations, 18 families have been reported. The common phenotype is mild to moderate thrombocytopenia with a variable predisposition to acute lymphoblastic leukemia (ALL), AML, and MDS (275).
4.5 GATA2
The GATA2 gene, located on chromosome 3, encodes a zinc finger transcription factor that contains two zinc fingers and a nuclear localization signal. This protein is central in the production and maintenance of HSCs in embryonic and adult hematopoietic processes (276). It has been found that GATA2 germline mutations account for 15% of advanced primary MDS cases and 7% of all cases. Germline mutations in GATA2 can lead to GATA2 deficiency characterized by a complex multi-system disorder that can present with many manifestations including variable cytopenias, bone marrow failure, MDS/AML, and severe immunodeficiency. Penetrance and expressivity within families is often variable. GATA2 mutational status does not negatively affect the outcome of MDS or HSCT. Moreover, findings have suggested that abnormal clonal hematopoiesis is a common event in symptomatic germline mutated GATA2 patients with MDS and those with hypocellular marrows without overt morphologic evidence of dysplasia, possibly indicating a pre-MDS stage warranting close monitoring for disease progression (277).
5 Conclusions
The genetic and biologic heterogeneity of MDS poses significant challenges in developing new clinical therapeutics. A plethora of molecular pathway disruptions have been used to explain the disease phenotype, but no exclusive genetic drivers can explain all of its aspects. Novel agents targeting the molecular pathways involved in MDS pathogenesis are currently under development while others are already clinically available. In particular, the emerging discovery of small molecules as inhibitors of mutations in genes implicated in cellular processes such as RNA splicing, DNA methylation, chromatin remodeling, signal, and transcription pathways has aroused significant hope in the management of heterogeneous hematological malignancies. Elucidation of expression of mutated genes at the pre-clinical stage and further identification of drug targets will further lead to a better understanding and control of the stages of disease progression. In this context, the genomic profile should be incorporated into a more individualized approach either in first line treatment or after failure/progression. Individualized gene expression and genome wide association studies would further promise to develop personalized therapeutic approaches with pharmaceutical tailoring of targeted drugs.
Author contributions
C-NK collected and analyzed data and drafted the manuscript. KK collected and analyzed data. N-AV critically reviewed the manuscript. PD designed and directed the project and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
PD reports honoraria from Pfizer, Sandoz, and Roche. N-AV reports investigational grants and personal fees for presentations and advisory roles from Celgene/Genesis Pharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.989483/full#supplementary-material
References
1. Shastri A, Will B, Steidl U, Verma A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood. (2017) 129(12):1586–94. doi: 10.1182/blood-2016-10-696062
2. Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med (2012) 366(12):1090–8. doi: 10.1056/NEJMoa1106968
3. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell (2012) 150:264–78. doi: 10.1016/j.cell.2012.06.023
4. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G. Et. al. Int Scoring System Evaluating Prognosis Myelodysplastic Syndromes. Blood; 89;6: 2079 - 2088. Erratum Blood (1998) 91:1100.
5. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. (2012) 120(12):2454–65. doi: 10.1182/blood-2012-03-420489
6. Patnaik MM, Tefferi A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T)–”2021 update on diagnosis, risk-stratification, and management. Am J Hematol (2021) 96:379–94. doi: 10.1002/ajh.26090
7. Alessandrino EP, Della Porta MG, Bacigalupo A, Van Lint MT, Falda M, Onida F, et al. WHO classification and WPSS predict posttransplantation outcome in patients with myelodysplastic syndrome: A study from the gruppo italiano trapianto di midollo osseo (GITMO). Blood (2008) 112:895–902. doi: 10.1182/blood-2008-03-143735
8. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol (2011) 29:504–15. doi: 10.1200/JCO.2010.31.1175
10. Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol (2008) 87:515–26. doi: 10.1007/s00277-008-0483-y
11. Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. (2007) 110(13):4385–95. doi: 10.1182/blood-2007-03-082404
12. Boultwood J, Pellagatti A, McKenzie AN, Wainscoat JS. Advances in the 5q- syndrome. Blood (2010) 116:5803–11. doi: 10.1182/blood-2010-04-273771
13. Bernasconi P, Klersy C, Boni M, Cavigliano PM, Calatroni S, Giardini I, et al. Incidence and prognostic significance of karyotype abnormalities in de novo primary myelodysplastic syndromes: a study on 331 patients from a single institution. Leukemia (2000) 19:1424–31. doi: 10.1038/sj.leu.2403806
14. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, et al. Identification of RPS14 as a 5qsyndrome gene by RNA interference screen. Nature (2008) 451:335–9. doi: 10.1038/nature06494
15. Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med (2010) 1):59–66. doi: 10.1038/nm.2063
16. Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet (2017) 49:204–12. doi: 10.1038/ng.3742
17. Ibrar W, Zhang W, Cox JL, Cushman-Vokoun A, Fu K, Greiner TC, et al. The utility of a myeloid mutation panel for the diagnosis of myelodysplastic syndrome and myelodysplastic/myeloproliferative neoplasm. Int J Lab Hematol (2021) 43(6):1501–9. doi: 10.1111/ijlh.13659
18. Tothova Z, Krill-Burger JM, Popova KD, Landers CC, Sievers QL, Sievers Q.L, Yudovich D, et al. Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell (2017) 21:547–555.e548. doi: 10.1016/j.stem.2017.07.015
19. Nagata Y, Maciejewski JP. The functional mechanisms of mutations in myelodysplastic syndrome. Leukemia. (2019) 33(12):2779–94. doi: 10.1038/s41375-019-0617-3
20. Lukackova R, Gerykova Bujalkova M, Majerova L, Mladosievicova B. Molecular genetic methods in the diagnosis of myelodysplastic syndromes. A review. BioMed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2014) 158(3):339–45. doi: 10.5507/bp.2013.084
21. Kulasekararaj AG, Mohamedali AM, Mufti GJ. Recent advances in understanding the molecular pathogenesis of myelodysplastic syndromes. Br J Haematol (2013) 162(5):587–605. doi: 10.1111/bjh.12435
22. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Agerelated clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
23. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
24. Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood (2015) 126:2355–61. doi: 10.1182/blood-2015-08-667063
25. Malcovati L, Stevenson K, Papaemmanuil E, Neuberg D, Bejar R, Boultwood J, et al. SF3B1-mutant MDS as a distinct disease subtype: a proposal from the international working group for the prognosis of MDS. Blood. (2020) 136(2):157–70. doi: 10.1182/blood.2020004850
26. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Chronic myeloid disorders working group of the international cancer genome consortium. somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med (2011) 365(15):1384–95. doi: 10.1056/NEJMoa1103283
27. Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al. Groupe francophone des myélodysplasies. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood. (2009) 114(15):3285–91. doi: 10.1182/blood-2009-04-215814
28. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet (2009) 41(7):838–42. doi: 10.1038/ng.391
29. Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem cell transplantation. J Clin Oncol (2014) 32(25):2691–8. doi: 10.1200/JCO.2013.52.3381
30. Saunthararajah Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematol Am Soc Hematol Educ Program. (2013) 2013:511–21. doi: 10.1182/asheducation-2013.1.511
31. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med (2011) 364(26):2496–506. doi: 10.1056/NEJMoa1013343
32. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. (2013) 122(22):3616–27. doi: 10.1182/blood-2013-08-518886
33. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. (2014) 28(2):241–7. doi: 10.1038/leu.2013.336
34. Bejar R. Implications of molecular genetic diversity in myelodysplastic syndromes. Curr Opin Hematol (2017) 24(2):73–8. doi: 10.1097/MOH.0000000000000313
35. Steensma D, Wermke M, Klimek V, Greenberg P, Font P, Komrokji R, et al. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML). Blood (2019) 134:673. doi: 10.1182/blood-2019-123854
36. Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, et al. Spliceostatin a targets SF3b and inhibits both splicing and nuclear retention of premRNA. Nat Chem Biol (2007) 3:576–58. doi: 10.1038/nchembio.2007.18
37. Albert BJ, McPherson PA, O'Brien K, Czaicki NL, Destefino V, Osman S, et al. Meayamycin inhibits pre-messenger RNA splicing and exhibits picomolar activity against multidrug-resistant cells. Mol Cancer Ther (2009) 8:2308–18. doi: 10.1158/1535-7163.MCT-09-0051
38. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell (2016) 201630:404–17. doi: 10.1016/j.ccell.2016.08.006
39. Soret J, Bakkour N, Maire S, Durand S, Zekri L, Gabut M, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc Natl Acad Sci U S A. (2005) 102(24):8764–9. doi: 10.1073/pnas.0409829102
40. Funahashi Y, Sugi NH, Semba T, Yamamoto Y, Hamaoka S, Tsukahara-Tamai N, et al. Sulfonamide derivative, E7820, is a unique angiogenesis inhibitor suppressing an expression of integrin alpha2 subunit on endothelium. Cancer Res (2002) 62:6116–23.
41. Haritunians T, Gueller S, O'Kelly J, Ilaria R Jr, Koeffler HP. Novel acyl sulfonamide LY573636-sodium: effect on hematopoietic malignant cells. Oncol Rep (2008) 20(5):1237–42.
42. Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science (2017) 356(6336):eaal3755. doi: 10.1126/science.aal3755
43. Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell (2019) 35:369–384.e7. doi: 10.1016/j.ccell.2019.01.010
44. Asada S, Kitamura T. Aberrant histone modifications induced by mutant ASXL1 in myeloid neoplasms. Int J Hematol (2019) 110:179–86. doi: 10.1007/s12185-018-2563-7
45. Hsu J, Reilly A, Hayes BJ, Clough CA, Konnick EQ, Torok-Storb B, et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood (2019) 134:186–98. doi: 10.1182/blood.2018884338
46. Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood. (2016) 128:971–81. doi: 10.1182/blood-2015-11-684225
47. Kantarjian HM, Roboz GJ, Kropf PL, Yee KWL, O’Connell CL, Tibes R, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: Phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol (2017) 18:1317–26. doi: 10.1016/S1470-2045(17)30576-4
48. Garcia-Manero G, Roboz G, Walsh K, Kantarjian H, Ritchie E, Kropf P, et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: Phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol (2019) 6:e317–27. doi: 10.1016/S2352-3026(19)30029-8
49. Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood (2015) 126:1172–83. doi: 10.1182/blood-2015-03-634428
50. Invernizzi R, Pecci A, Bellotti L, Ascari E. Expression of p53, bcl-2 and ras oncoproteins and apoptosis levels in acute leukaemias and myelodysplastic syndromes. Leuk Lymphoma. (2001) 42:481–9. doi: 10.3109/10428190109064605
51. Deneberg S, Cherif H, Lazarevic V, Andersson PO, von Euler M, Juliusson G, et al. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J (2016) 6(7):e447. doi: 10.1038/bcj.2016.60
52. Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, et al. Phase1b/2 safety and efficacy of APR-246 w/Azacytidine for tx of TP53 mutant myeloid neoplasms J Clin Oncol (2021) 39(14):1584–94. doi: 10.1200/JCO.20.02341.
53. Giagounidis A, Mufti GJ, Fenaux P, Germing U, List A, MacBeth KJ. Lenalidomide as a disease-modifying agent in patients with del(5q) myelodysplastic syndromes: Linking mechanism of action to clinical outcomes. Ann Hematol (2014) 93:1–11. doi: 10.1007/s00277-013-1863-5
54. Raza A, AI-Kali A, Tibes R, Spitzer G, Gaddh M, Tycko B, et al. Rigosertib oral in transfusion dependent lower risk myelodysplastic syndromes (LR-MDS): optimization of dose and rate of transfusion independence (TI) or transfusion reduction (TR) in a single-arm phase 2 study. Blood. (2017) 130(suppl 1):1689.
55. Iarmarcovai G, Bonassi S, Botta A, Baan RA, Orsière T. Genetic polymorphismsand micronucleus formation: A review of the literature. Mutat Res (2008) 658:215–33. doi: 10.1016/j.mrrev.2007.10.001
56. Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Beckta JM, Valerie NC, Tokarz M, et al. ATM Kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin Cancer Res (2013) 19:3189–200. doi: 10.1158/1078-0432.Ccr-12-3408
57. Visconte V, Nakashima M O, J Rogers H. Mutations in splicing factor genes in myeloid malignancies: Significance and impact on clinical features. Cancers (Basel). (2019) 11(12):1844. doi: 10.3390/cancers11121844
58. Maciejewski JP, Padgett RA. Defects in spliceosomal machinery: A new pathway of leukaemogenesis. Br J Haematol (2012) 158:165–73. doi: 10.1111/j.1365-2141.2012.09158.x
59. Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Rep (2008) 9:1087–93. doi: 10.1038/embor.2008.189
60. Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med (2016) 22(6):672–8. doi: 10.1038/nm.4097
61. Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med (2016) 22(9):976–86. doi: 10.1038/nm.4165
62. Ogawa S. Splicing factor mutations in AML. Blood (2014) 123:3216–7. doi: 10.1182/blood-2014-04-566752
63. Pagliuca S, Gurnari C, Visconte V. Molecular targeted therapy in myelodysplastic syndromes: New options for tailored treatments. Cancers (Basel). (2021) 13(4):784. doi: 10.3390/cancers13040784
64. Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood. (2018) 132(12):1225–40. doi: 10.1182/blood-2018-04-843771
65. Shiozawa Y, Malcovati L, Gallì A, Sato-Otsubo A, Kataoka K, Sato Y, et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun (2018) 9(1):3649. doi: 10.1038/s41467-018-06063-x
66. Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell (2014) 25:794–808. doi: 10.1016/j.ccr.2014.03.036
67. Mian SA, Rouault-Pierre K, Smith AE, Seidl T, Pizzitola I, Kizilors A, et al. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun (2015) 6:10004. doi: 10.1038/ncomms10004
68. Visconte V, Tabarroki A, Zhang L, Parker Y, Hasrouni E, Mahfouz R, et al. Splicing factor 3b subunit 1 (Sf3b1) haploinsufficient mice display features of low risk myelodysplastic syndromes with ring sideroblasts. J Hematol Oncol (2014) 7:89. doi: 10.1186/s13045-014-0089-x
69. Gerstung M, Pellagatti A, Malcovati L, Giagounidis A, Porta MG, Jädersten M, et al. Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat Commun (2015) 6:5901. doi: 10.1038/ncomms6901
70. Mian SA, Smith AE, Kulasekararaj AG, Kizilors A, Mohamedali AM, Lea NC, et al. Spliceosome mutations exhibit specific associations with epigenetic modifiers and protooncogenes mutated in myelodysplastic syndrome. Haematologica. (2013) 98(7):1058–10664. doi: 10.3324/haematol.2012.075325
71. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer associated SF3B1 hotspot mutations induce cryptic 3' splice site selection through use of a different branch point. Cell Rep (2015) 13:1033–45. doi: 10.1016/j.celrep.2015.09.053
72. Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia. (2015) 29:1798. doi: 10.1038/leu.2015.178
73. Dolatshad H, Pellagatti A, Liberante FG, Llorian M, Repapi E, Steeples V, et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia (2006) 30:2322–31. doi: 10.1038/leu.2016.149
74. Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. (2012) 119:3203–10. doi: 10.1182/blood-2011-12-399774
75. Conte S, Katayama S, Vesterlund L, Karimi M, Dimitriou M, Jansson M, et al. Aberrant splicing of genes involved in haemoglobin synthesis and impaired terminal erythroid maturation in SF3B1 mutated refractory anaemia with ring sideroblasts. Br J Haematol (2015) 171:478–90. doi: 10.1111/bjh.13610
76. Jin S, Su H, Tran NT, Song J, Lu SS, Li Y, et al. Splicing factor SF3B1K700E mutant dysregulates erythroid differentiation via aberrant alternative splicing of transcription factor TAL1. PloS One (2017) 12:e0175523. doi: 10.1371/journal.pone.0175523
77. Visconte V, Avishai N, Mahfouz R, Tabarroki A, Cowen J, Sharghi-Moshtaghin R, et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia (2015) 29:188–95. doi: 10.1038/leu.2014.170
78. Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol (2009) 29:1007–16. doi: 10.1128/MCB.01685-08
79. Harada H, Harada Y. Recent advances in myelodysplastic syndromes: Molecular pathogenesis and its implications for targeted therapies. Cancer Sci (2015) 106(4):329–36. doi: 10.1111/cas.12614
80. Pellagatti A, Boultwood J. The molecular pathogenesis of the myelodysplastic syndromes. Eur J Haematol (2015) 95(1):3–15. doi: 10.1111/ejh.12515
81. MacRae AJ, Mayerle M, Hrabeta-Robinson E, Chalkley RJ, Guthrie C, Burlingame AL, et al. Prp8 positioning of U5 snRNA is linked to 50 splice site recognition. Rna (2018) 24:769–77. doi: 10.1261/rna.065458.117
82. Hershberger CE, Hosono N, Singh J, Dietrich RC, Gu X, Makishima H. The role of LUC7L2 in splicing and MDS. Blood (2016) 128:5504. doi: 10.1182/blood.V128.22.5504.5504
83. Awada H, Thapa B, Visconte V. The genomics of myelodysplastic syndromes: Origins of disease evolution, Biological Pathways, and Prognostic Implications Cells. (2020) 9(11):2512. doi: 10.3390/cells9112512.
84. Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol (2011) 6:582–9. doi: 10.1021/cb100356k
85. Liu X, Biswas S, Berg MG, Antapli CM, Xie F, Wang Q, et al. Genomics guided discovery of thailanstatins a, b, and c as pre-mRNA splicing inhibitors and antiproliferative agents from burkholderia thailandensis MSMB43. J Nat Prod (2013) 76:685–693. doi: 10.1021/np300913h
86. Shirai CL, White BS, Tripathi M, Tapia R, Ley JN, Ndonwi M, et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat Commun (2017) 8:14060. doi: 10.1038/ncomms14060
87. Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, et al. Pladienolides, new substances from culture of streptomyces platensis mer-11107. III. In Vitro Vivo antitumor activities. J Antibiot (Tokyo). (2004) 57:188–96. doi: 10.7164/antibiotics.57.188
88. Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem (2008) 283(3):1223–7. doi: 10.1074/jbc.R700034200
89. Ozawa Y, Sugi NH, Nagasu T, Owa T, Watanabe T, Koyanagi N, et al. E7070, a novel sulphonamide agent with potent antitumour activity in vitro and in vivo. Eur J Cancer (2001) 37:2275–82. doi: 10.1016/S0959-8049(01)00275-1
90. Faust TB, Yoon H, Nowak RP, Donovan KA, Li Z, Cai Q, et al. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat Chem Biol (2020) 16:7–14. doi: 10.1038/s41589-019-0378-3
91. Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, et al. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell (2019) 36(2):194–209.e9. doi: 10.1016/j.ccell.2019.07.003
92. Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, et al. RNA Splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell (2016) 19:599–612. doi: 10.1016/j.stem.2016.08.003
93. Ohanian M, Ravandi F, Borthakur G, Garcia-Manero G, Andreeff M, Jabbour E, et al. Phase I study of BP1001 (Liposomal Grb2 antisense) in patients with hematologic malignancies. J Clin Oncol (2016) 34:7010. doi: 10.1200/JCO.2016.34.15_suppl.7010
94. Flach J, Jann JC, Knaflic A, Riabov V, Streuer A, Altrock E, et al. Replication stress signaling is a therapeutic target in myelodysplastic syndromes with splicing factor mutations. Haematologica. (2021) 106(11):2906–17. doi: 10.3324/haematol.2020.254193
95. Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, et al. Spliceosome mutations induce r loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res (2018) 78(18):5363–74. doi: 10.1158/0008-5472.CAN-17-3970
96. Issa JP. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am (2010) 24(2):317–30. doi: 10.1016/j.hoc.2010.02.007
97. Toyota M, Issa JP. Epigenetic changes in solid and hematopoietic tumors. Semin Oncol (2005) 32(5):521–30. doi: 10.1053/j.seminoncol.2005.07.003
98. Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, et al. DNA Methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol Off J Am Soc Clin Oncol (2010) 28(4):605–13. doi: 10.1200/JCO.2009.23.4781
99. Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. (2013) 27(6):1275–82. doi: 10.1038/leu.2013.58
100. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet (2011) 44(1):23–31. doi: 10.1038/ng.1009
101. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. New Engl J Med (2010) 363:2424–33. doi: 10.1056/nejmoa1005143
102. Ferreira HJ, Heyn H, Vizoso M, Moutinho C, Vidal E, Gomez A, et al. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene. (2015) 35:3079–82. doi: 10.1038/onc.2015.359
103. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med (2016) 22:1488–95. doi: 10.1038/nm.4210
104. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A. Kosmider O et al: Mutation in TET2 in myeloid cancers. N Engl J Med (2009) 360:2289–301. doi: 10.1056/NEJMoa0810069
105. Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia (2011) 25:1147–52. doi: 10.1038/leu.2011.71
106. Bejar R, Lord A, Stevenson K, Bar-Natan M, Pérez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood (2014) 124:2705–12. doi: 10.1182/blood-2014-06-582809
107. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. (2010) 468(7325):839–843. doi:10.1038/nature09586
108. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA (2011) 108(35):14566–71. doi: 10.1073/pnas.1112317108
109. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discovery (2013) 3(7):730–41. doi: 10.1158/2159-8290.CD-13-0083
110. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. (2009) 462:739–44. doi: 10.1038/nature08617
111. Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med (2010) 207:339–44. doi: 10.1084/jem.20092506
112. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
113. Yonal-Hindilerden I, Daglar-Aday A, Hindilerden F, Akadam-Teker B, Yilmaz C, Nalcaci M, et al. The clinical significance of IDH mutations in essential thrombocythemia and primary myelofibrosis. J Clin Med Res (2016) 8:29–39. doi: 10.14740/jocmr2405w
114. Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood (2010) 12:2122–6. doi: 10.1182/blood-2009-11-250878
115. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. New Engl J Med (2009) 361(11):1058–66. doi: 10.1056/NEJMoa0903840
116. Gu Y, Yang R, Yang Y, Zhao Y, Wakeham A, Li WY, et al. IDH1 mutation contributes to myeloid dysplasia in mice by disturbing heme biosynthesis and erythropoiesis. Blood. (2021) 137(7):945–58. doi: 10.1182/blood.2020007075
117. Richarson AD, Scott DA, Zagnitko O, Aza-Blanc P, Chang CC, Russler-Germain DA. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Elife. (2016) 5:e10860. doi: 10.7554/eLife.10860
118. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
119. Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood (2015) 125:296–303. doi: 10.1182/blood-2013-10-533604
120. Ok CY, Loghavi S, Sui D, Wei P, Kanagal-Shamanna R, Yin CC, et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica (2019) 104:305–11. doi: 10.3324/haematol.2018.191148
121. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell (2011) 20:11–24. doi: 10.1016/j.ccr.2011.06.001
122. Sarno F, Nebbioso A, Altucci L. DOT1L: a key target in normal chromatin remodelling and in mixed-lineage leukaemia treatment. Epigenetics. (2020) 15(5):439–53. doi: 10.1080/15592294.2019.1699991
123. Jerez A, Sugimoto Y, Makishima H, Verma A, Jankowska AM, Przychodzen B, et al. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood. (2012) 119(25):6109–17. doi: 10.1182/blood-2011-12-397620
124. Xu F, Liu L, Chang C-K, He Q, Wu L-Y, Zhang Z, et al. Genomic loss of EZH2 leads to epigenetic modifications and overexpression of the HOX gene clusters in myelodysplastic syndrome. Oncotarget (2016) 7:8119–30. doi: 10.18632/oncotarget.6992
125. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet (2010) 42:665–7. doi: 10.1038/ng.620
126. Abuhadra N, Mukherjee S, Al-Issa K, Adema V, Hirsch CM, Advani A, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes (MDS): Clonal architecture and impact on outcomes. Leuk. Lymphoma (2019) 60:1587–90. doi: 10.1080/10428194.2018.1543885
127. Rinke J, Chase A, Cross NCP, Hochhaus A, Ernst T. EZH2 in myeloid malignancies. Cells. (2020) 9(7):1639. doi: 10.3390/cells9071639
128. Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun (2014) 5:4177. doi: 10.1038/ncomms5177
129. Booth CAG, Barkas N, Neo WH, Boukarabila H, Soilleux EJ, Giotopoulos G, et al. Ezh2 and Runx1 mutations collaborate to initiate lympho-myeloid leukemia in early thymic progenitors. Cancer Cell (2018) 33:274–91. doi: 10.1016/j.ccell.2018.01.006
130. Rinke J, Muller JP, Blaess MF, Chase A, Meggendorfer M, Schaefer V, et al. Molecular characterization of EZH2 mutant patients with myelodysplastic/myeloproliferative neoplasms. Leukemia (2017) 31:1936–43. doi: 10.1038/leu.2017.190
131. Park UH, Yoon SK, Park T, Kim EJ, Um SJ. Additional sex comb-like (ASXL) proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor {gamma}. J Biol Chem (2011) 286(2):1354–63. doi: 10.1074/jbc.M110.177816
132. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell (2012) 22:180–93. doi: 10.1016/j.ccr.2012.06.032
133. Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol (2009) 145(6):788–800. doi: 10.1111/j.1365-2141.2009.07697.x
134. Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN favorable genetic category. Blood. (2011) 118(26):6920–6929. doi: 10.1182/blood-2011-08-368225
135. Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol (2011) 29:2499–506. doi: 10.1200/JCO.2010.33.4938
136. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. International vidaza high-risk MDS survival study group. efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol (2009) 10(3):223–32. doi: 10.1016/S1470-2045(09)70003-8
137. Garcia-Manero G, Jabbour E, Borthakur G, Faderl S, Estrov Z, Yang H, et al. Randomized open-label phase II study of decitabine in patients with low- or intermediate-risk myelodysplastic syndromes. J Clin Oncol (2013) 31:2548–53. doi: 10.1200/JCO.2012.44.6823
138. Musto P, Maurillo L, Spagnoli A, Gozzini A, Rivellini F, Lunghi M, et al. Azacitidine for the treatment of lower risk myelodysplastic syndromes: a retrospective study of 74 patients enrolled in an Italian named patient program. Cancer (2010) 116:1485–94. doi: 10.1002/cncr.24894
139. Mahfouz RZ, Jankowska A, Ebrahem Q, Gu X, Visconte V, Tabarroki A, et al. Increased cda expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin Cancer Res (2013) 19:938–48. doi: 10.1158/1078-0432.CCR-12-1722
140. Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PloS One (2011) 6:e23372. doi: 10.1371/journal.pone.0023372
141. Diamantopoulos PT, Kontandreopoulou CN, Symeonidis A, Kotsianidis I, Pappa V, Galanopoulos A, et al. Hellenic MDS study group. bone marrow PARP1 mRNA levels predict response to treatment with 5-azacytidine in patients with myelodysplastic syndrome. Ann Hematol (2019) 98(6):1383–92. doi: 10.1007/s00277-019-03650-w
142. Diamantopoulos P, Zervakis K, Zervakis P, Sofotasiou M, Vassilakopoulos T, Kotsianidis I, et al. Poly (ADP-ribose) polymerase 1 mRNA levels strongly correlate with the prognosis of myelodysplastic syndromes. Blood Cancer J (2017) 7(2):e533. doi: 10.1038/bcj.2016.127
143. Hrustincova A, Krejcik Z, Kundrat D, Szikszai K, Belickova M, Pecherkova P, et al. Circulating small noncoding RNAs have specific expression patterns in plasma and extracellular vesicles in myelodysplastic syndromes and are predictive of patient outcome. Cells. (2020) 9(4):794. doi: 10.3390/cells9040794
144. Kontandreopoulou CN, Diamantopoulos PT, Giannopoulos A, Symeonidis A, Kotsianidis I, Pappa V, et al. Hellenic MDS study group. bone marrow ribonucleotide reductase mRNA levels and methylation status as prognostic factors in patients with myelodysplastic syndrome treated with 5-azacytidine. Leuk Lymphoma. (2022) 63(3):729–37. doi: 10.1080/10428194.2021.1998484
145. Das AB, Kakadia PM, Wojcik D, Pemberton L, Browett PJ, Bohlander SK, et al. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J (2019) 9:82. doi: 10.1038/s41408-019-0242-4
146. DiNardo CD, Patel KP, Garcia-Manero G, Luthra R, Pierce S, Borthakur G, et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk. Lymphoma (2014) 55:1925–9. doi: 10.3109/10428194.2013.855309
147. Molenaar R, Patel B, Khurshed M, Przychodzen B, Sanikommu SR, Hetty E ET AL. Ex vivo experiments show that IDH1/2-mutant inhibitors can be safely used as adjuvants to regular chemotherapy in IDH1/2-mutated acute myeloid leukemia. Blood (2015) 126(23):3788. doi: 10.1182/blood.V126.23.3788.3788
148. Sébert M, Renneville A, Bally C, Peterlin P, Beyne-Rauzy O, Legros L, et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure. Haematologica (2019) 104:1565–71. doi: 10.3324/haematol.2018.207118
149. Savona MR, Kolibaba K, Conkling P, Kingsley EC, Becerra C, Morris JC, et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am J Hematol (2018) 93:1199–206. doi: 10.1002/ajh.25216
151. Savona MR, McCloskey JK, Griffiths EA, Yee KWL, Al-Kali A, Zeidan AM, et al. Clinical efficacy and safety of oral Decitabine/Cedazuridine in 133 patients with myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML). Blood (2020) 136:37–8. doi: 10.1182/blood-2020-133855
152. Ferraris D, Duvall B, Delahanty G, Mistry B, Alt J, Rojas C ET AL. Design, synthesis, and pharmacological evaluation of fluorinated tetrahydrouridine derivatives as inhibitors of cytidine deaminase. J Med Chem (2014) 57(6):2582–8. doi: 10.1021/jm401856k
153. Swords RT, Coutre S, Maris MB, Zeidner JF, Foran JM, Cruz J, et al. Pevonedistat, a first-inclass NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood (2018) 131:1415–24. doi: 10.1182/blood-2017-09-805895
154. Garcia-Manero G, Fenaux P, Al-Kali A, Baer MR, Sekeres MA, Roboz GJ, et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol (2016) 17:496–508. doi: 10.1016/S1470-2045(16)00009-7
155. Losman J-A, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-Hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science (2013) 339:1621–5. doi: 10.1126/science.1231677
156. Chaturvedi A, Cruz MMA, Jyotsana N, Sharma A, Yun H, Görlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood (2013) 122:2877–87. doi: 10.1182/blood-2013-03-491571
157. Bravo GM, Lee E, Merchan B, Kantarjian HM, García-Manero G. Integrating genetics and epigenetics in myelodysplastic syndromes: advances in pathogenesis and disease evolution. Br J Haematol (2014) 166(5):646–59. doi: 10.1111/bjh.12957
158. Gu Z, Liu Y, Cai F, Patrick M, Zmajkovic J, Cao H, et al. Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discovery (2019) 9:1228–47. doi: 10.1158/2159-8290.CD-19-0152
159. Lambert M, Alioui M, Jambon S, Depauw S, Van Seuningen I, David-Cordonnier MH. Direct and indirect targeting of HOXA9 transcription factor in acute myeloid leukemia. Cancers (2019) 11:837. doi: 10.3390/cancers11060837
160. Mallo M, Del Rey M, Ibáñez M, Calasanz MJ, Arenillas L, Larráyoz MJ, et al. Response to lenalidomide in myelodysplastic syndromes with del(5q): influence of cytogenetics and mutations. Br J Haematol (2013) 162(1):74–86. doi: 10.1111/bjh.12354
161. Abou Zahr A, Saad Aldin E, Komrokji RS, Zeidan AM. Clinical utility of lenalidomide in the treatment of myelodysplastic syndromes. J Blood Med (2015) 6:1–16. doi: 10.2147/JBM.S50482
162. Fink EC, Ebert BL. The novel mechanism of lenalidomide activity. Blood (2015) 126:2366–9. doi: 10.1182/blood-2015-07-567958
163. Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature (2015) 523:183–8. doi: 10.1038/nature14610
164. Göhring G, Giagounidis A, Büsche G, Kreipe HH, Zimmermann M, Hellström-Lindberg E, et al. Patients with del(5q) mds who fail to achieve sustained erythroid or cytogenetic remission after treatment with lenalidomide have an increased risk for clonal evolution and aml progression. Ann Hematol (2010) 89:365–374. doi: 10.1007/s00277-009-0846-z
165. Raza A, Reeves JA, Feldman EJ, Dewald GW, Bennett JM, Deeg HJ, et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood. (2008) 111:86–93. doi: 10.1182/blood-2007-01-068833
166. Jädersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Göhring G, et al. Tp53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol (2011) 29:1971–9. doi: 10.1200/JCO.2010.31.8576
167. Sekeres MA, List AF, Cuthbertson D, Paquette R, Ganetzky R, Latham D, et al. Phase I combination trial of lenalidomide and azacitidine in patients with higher-risk myelodysplastic syndromes. J Clin Oncol (2010) 28:2253–8. doi: 10.1200/JCO.2009.26.0745
168. Sekeres MA, Tiu RV, Komrokji R, Lancet J, Advani AS, Afable M, et al. Phase 2 study of the lenalidomide and azacitidine combination in patients with higher-risk myelodysplastic syndromes. Blood. (2012) 120:4945–51. doi: 10.1182/blood-2012-06-434639
169. Bachegowda L, Gligich O, Mantzaris I, Schinke C, Wyville D, Carrillo T, et al. Signal transduction inhibitors in treatment of myelodysplastic syndromes. J Hematol Oncol (2013) 6:50. doi: 10.1186/1756-8722-6-50
170. Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. biomark Res (2013) 1(1):5. doi: 10.1186/2050-7771-1-5
171. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. (2003) 3:11–22. doi: 10.1038/nrc969
172. Akutagawa J, Huang TQ, Epstein I, Chang T, Quirindongo-Crespo M, Cottonham CL, et al. Targeting the PI3K/ akt pathway in murine MDS/MPN driven by hyperactive ras. Leukemia. (2016) 30:1335–43. doi: 10.1038/leu.2016.14
173. Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. (2008) 112:3434–43. doi: 10.1182/blood-2008-02-139824
174. Al-Kali A, Quintás-Cardama A, Luthra R, Bueso-Ramos C, Pierce S, Kadia T, et al. Prognostic impact of RAS mutations in patients with myelodysplastic syndrome. Am J Hematol (2013) 88:365–9. doi: 10.1002/ajh.23410
175. Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica. (2007) 92(6):744–52. doi: 10.3324/haematol.10869
176. Christiansen DH, Andersen MK, Desta F, Pedersen-Bjergaard J. Mutations of genes in the receptor tyrosine kinase (RTK)/RAS-BRAF signal transduction pathway in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. (2005) 19(12):2232–40. doi: 10.1038/sj.leu.2404009
177. Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet (2003) 34:148–50. doi: 10.1038/ng1156
178. Loh ML, Martinelli S, Cordeddu V, Reynolds MG, Vattikuti S, Lee CM, et al. Acquired PTPN11 mutations occur rarely in adult patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk. Res (2005) 29:459–62. doi: 10.1016/j.leukres.2004.10.001
179. Chen C-Y, Lin L-I, Tang J-L, Tsay W, Chang H-H, Yeh Y-C, et al. Acquisition of JAK2, PTPN11, and RAS mutations during disease progression in primary myelodysplastic syndrome. Leukemia (2006) 20:1155–8. doi: 10.1038/sj.leu.2404190
180. Borthakur G, Popplewell L, Boyiadzis M, Foran J, Platzbecker U, Vey N, et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer. (2016) 122(12):1871–9. doi: 10.1002/cncr.29986
181. Azacitidine, venetoclax, and trametinib for the treatment of relapsed or refractory acute myeloid leukemia or higher-risk myelodysplastic syndrome. Available from: https://clinicaltrials.gov/ct2/show/NCT04487106.
182. Sokol L, Cripe L, Kantarjian H, Sekeres MA, Parmar S, Greenberg P, et al. Randomized, dose-escalation study of the p38alpha MAPK inhibitor SCIO-469 in patients with myelodysplastic syndrome. Leukemia. (2013) 27:977–80. doi: 10.1038/leu.2012.264
183. Garcia-Manero G, Khoury HJ, Jabbour E, Lancet J, Winski SL, Cable L, et al. A phase I study of oral ARRY-614, a p38 MAPK/Tie2 dual inhibitor, in patients with low or intermediate-1 risk myelodysplastic syndromes. Clin Cancer Res (2015) 21:985–94. doi: 10.1158/1078-0432.CCR-14-1765
184. Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget. (2017) 9(6):6659–77. doi: 10.18632/oncotarget.23795
185. Navada SC, Garcia-Manero G, Atallah EL, Nabeel Rajeh M, Shammo J, Griffiths E, et al. Phase 2 expansion study of oral rigosertib combined with azacitidine (AZA) in patients (Pts) with higher-risk (HR) myelodysplastic syndromes (MDS): efficacy and safety results in HMA treatment naïve & relapsed (rel)/refractory (ref) patients. Blood. (2018) 132:230. doi: 10.1182/blood-2018-99-119259
186. Navada SC, Fruchtman SM, Odchimar-Reissig R, Demakos EP, Petrone ME, Zbyszewski PS, et al. A phase ½ study of rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia. Leuk Res. (2018) 64:10–6. doi: 10.1016/j.leukres.2017.11.006.
187. Rio-Machin A, Vulliamy T, Hug N, Walne A, Tawana K, Cardoso S, et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun (2020) 11:1–12. doi: 10.1038/s41467-020-14829-5
188. Shallis RM, Ahmad R, Zeidan AM. The genetic and molecular pathogenesis of myelodysplastic syndromes. Eur J Haematol (2018) 101(3):260–71. doi: 10.1111/ejh.13092
189. Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med (2020) 26:1549–56. doi: 10.1038/s41591-020-1008-z
190. Haase D, Stevenson KE, Neuberg D, Maciejewski JP, Nazha A, Sekeres MA, et al. International working group for MDS molecular prognostic committee. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia. (2019) 33(7):1747–58. doi: 10.1038/s41375-018-0351-2
191. Bally C, Adès L, Renneville A, Sebert M, Eclache V, Preudhomme C, et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res (2014) 38:751–5. doi: 10.1016/j.leukres.2014.03.012
192. Welch JS, Petti AA, Ley TJ. Decitabine in TP53-mutated AML. N Engl J Med (2017) 376:797–8. doi: 10.1056/NEJMc1616062
193. Maslah N, Salomao N, Drevon L, Verger E, Partouche N, Ly P, et al. Synergistic effects of PRIMA-1Met (APR-246) and azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica (2019) 105(6):1539–51. doi: 10.3324/haematol.2019.218453
194. Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol (2021) 39(14):1584–94. doi: 10.1200/JCO.20.02341
195. APR-246 and azacytidine for the treatment of TP53 mutant mutant myelodysplastic syndromes (MDS). Available from: https://clinicaltrials.gov/NCT03745716/APR-246.
196. Fenaux P, Platzbecker U, Mufti GJ, Garcia-Manero G, Buckstein R, Santini V, et al. The MEDALIST trial: results of a phase 3, randomized, double-blind, placebo-controlled study of luspatercept to treat anemia in patients with very low-, low-, or intermediate-risk myelodysplastic syndromes (MDS) with ring sideroblasts (RS) who require red blood cell (RBC) transfusions [abstract]. Blood (2018) 132(Suppl):1. doi: 10.1182/blood-2018-99-110805
197. Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dosefinding study with long-term extension study. Lancet Oncol (2017) 18:1338–47. doi: 10.1016/S1470-2045(17)30615-0
198. Attie KM, Allison MJ, McClure T, Boyd IE, Wilson DM, Pearsall AE, et al. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am J Hematol (2014) 89(7):766–70. doi: 10.1002/ajh.23732
199. Komrokji R, Garcia-Manero G, Ades L, Prebet T, Steensma DP, Jurcic JG, et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol (2018) 5:e63–72. doi: 10.1016/S2352-3026(18)30002-4
200. Tothova Z, Valton AL, Gorelov RA, Vallurupalli M, Krill-Burger JM, Holmes A, et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight (2021) 6(3):e142149. doi: 10.1172/jci.insight.142149
201. Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet (2013) 45:1232–7. doi: 10.1038/ng.2731
202. Sundaramoorthy S, Vázquez-Novelle MD, Lekomtsev S, Howell M, Petronczki M. Functional genomics identifies a requirement of pre-mRNA splicing factors for sister chromatid cohesion. EMBO J (2014) 33(22):2623–42. doi: 10.15252/embj.201488244
203. Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med (2015) 212:1833–50. doi: 10.1084/jem.20151323
204. Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R. Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell (2015) 17:675–688. doi: 10.1016/j.stem.2015.09.017
205. Martín-Izquierdo M, Abáigar M, Hernández-Sánchez JM, Tamborero D, López-Cadenas F, Ramos F, et al. Co-Occurrence of cohesin complex and ras signaling mutations during progression from myelodysplastic syndromes to secondary acute myeloid leukemia. Haematologica. (2021) 106(8):2215–23. doi: 10.3324/haematol.2020.248807
206. Chang CR, Wu CS, Hom Y, Gartenberg MR. Targeting of cohesin by transcriptionally silent chromatin. Genes Dev (2005) 19(24):3031–42. doi: 10.1101/gad.1356305
207. Antony J, Chin CV, Horsfield JA. Cohesin mutations in cancer: Emerging therapeutic targets. Int J Mol Sci (2021) 22(13):6788. doi: 10.3390/ijms22136788
208. Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood (2014) 124:1790–8. doi: 10.1182/blood-2014-04-567057
209. McLellan JL, O’Neil NJ, Barrett I, Ferree E, van Pel DM, Ushey K, et al. Synthetic lethality of cohesins with PARPs and replication fork mediators. PloS Genet (2012) 8:e1002574. doi: 10.1371/journal.pgen.1002574
210. Hopfner KP. DNA Double-strand breaks come into focus. Cell (2009) 139:25–7. doi: 10.1016/j.cell.2009.09.017
211. Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM signaling network in development and disease. Front Genet (2013) 4:37. doi: 10.3389/fgene.2013.00037
212. Kozlov SV, Graham ME, Jakob B, Tobias F, Kijas AW, Tanuji M, et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J Biol Chem (2011) 286:9107–19. doi: 10.1074/jbc.M110.204065
213. Guleria A, Chandna S. ATM Kinase: Much more than a DNA damage responsive protein. DNA Repair (Amst) (2016) 39:1–20. doi: 10.1016/j.dnarep.2015.12.009
214. García MEG, Kirsch DG, Reitman ZJ. Targeting the ATM kinase to enhance the efficacy of radiotherapy and outcomes for cancer patients. Semin Radiat Oncol (2022) 32(1):3–14. doi: 10.1016/j.semradonc.2021.09.008
215. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science (2017) 355:1152–8. doi: 10.1126/science.aam7344
216. Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, Barros AC, et al. ATM Orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat Commun (2019) 10:87. doi: 10.1038/s41467-018-07729-2
217. Grosjean-Raillard J, Tailler M, Adès L, Perfettini JL, Fabre C, Braun T, et al. ATM Mediates constitutive NF-kappaB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. (2009) 28(8):1099–109. doi: 10.1038/onc.2008.457
218. Adachi Y, Pavlakis GN, Copeland TD. Identification and characterization of SET, a nuclear phosphoprotein encoded by the translocation break point in acute undifferentiated leukemia. J Biol Chem (1994) 269(3):2258–62. doi: 10.1016/S0021-9258(17)42162-4
219. Shou LH, Cao D, Dong XH, Fang Q, Wu Y, Zhang Y, et al. Prognostic significance of SETBP1 mutations in myelodysplastic syndromes, chronic myelomonocytic leukemia, and chronic neutrophilic leukemia: a meta-analysis. PloS One (2017) 12:e0171608. doi: 10.1371/journal.pone.0171608
220. Fernandez-Mercado M, Pellagatti A, Di Genua C, Larrayoz MJ, Winkelmann N, Aranaz P, et al. Mutations in SETBP1 are recurrent in myelodysplastic syndromes and often coexist with cytogenetic markers associated with disease progression. Br J Haematol (2013) 163(2):235–9. doi: 10.1111/bjh.12491
221. Hou HA, Kuo YY, Tang JL, Chou WC, Yao M, Lai YJ, et al. Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. Am J Hematol (2014) 89(2):181–6. doi: 10.1002/ajh.23611
222. Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. (2013) 27:1852–60. doi: 10.1038/leu.2013.133
223. Adema V, Larrayoz MJ, Calasanz MJ, Palomo L, Patino-Garcia A, Agirre X, et al. Correlation of myelodysplastic syndromes with i(17)(q10) and TP53 and SETBP1 mutations. Br J Haematol (2015) 171:137–41. doi: 10.1111/bjh.13355
224. Saika M, Inoue D, Nagase R, Sato N, Tsuchiya A, Yabushita T, et al. ASXL1 and SETBP1 mutations promote leukaemogenesis by repressing TGFβ pathway genes through histone deacetylation. Sci Rep (2018) 8(1):15873. doi: 10.1038/s41598-018-33881-2
225. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell (2010) 37:299–310. doi: 10.1016/j.molcel.2010.01.025
226. Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, et al. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood. (2014) 123:541–53. doi: 10.1182/blood-2013-05-500272
227. Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. (2013) 123:3876–88. doi: 10.1172/JCI68557
228. Reidel V, Kauschinger J, Hauch RT, Müller-Thomas C, Nadarajah N, Burgkart R, et al. Selective inhibition of BCL-2 is a promising target in patients with high-risk myelodysplastic syndromes and adverse mutational profile. Oncotarget (2018) 9(25):17270–81. doi: 10.18632/oncotarget.24775
229. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. New Eng J Med (2020) 383(7):617–29. doi: 10.1056/NEJMoa2012971
230. Study of venetoclax tablet with intravenous or subcutaneous azacytidine to assess change in disease activity in adult participants with newly diagnosed higher risk myelodysolastic syndrome (Verona). Available from: https://www.clinicaltrials.gov/ct2/show/NCT04401748.
231. Phase III study induction consolidation chemotherapy venetoclax patients newly diagnosed AML MDS-EB2. Available from: https://www.clinicaltrials.gov/ct2/show/NCT04628026.
232. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8(6):467–77. doi: 10.1038/nri2326
233. Mekinian A, Grignano E, Braun T, Decaux O, Liozon E, Costedoat-Chalumeau N, et al. Systemic infammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: A french multicentre retrospective study. Rheumatology (2015) 55:291–300:2. doi: 10.1093/rheumatology/kev294
234. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol (2015) 23:32–8. doi: 10.1016/j.coph.2015.05.011
235. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12(4):252–64. doi: 10.1038/nrc3239
236. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
237. Sui X, Ma J, Han W, Wang X, Fang Y, Li D, et al. The anticancer immune response of anti-PD-1/PD-L1 and the genetic determinants of response to anti-PD-1/PD-L1 antibodies in cancer patients. Oncotarget. (2015) 6(23):19393–404. doi: 10.18632/oncotarget.5107
238. Alatrash G, Daver N, Mittendorf EA. Targeting immune checkpoints in hematologic malignancies. Pharmacol Rev (2016) 68(4):1014–25. doi: 10.1124/pr.116.012682
239. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia (2014) 28(6):1280–8. doi: 10.1038/leu.2013.355
240. Dail M, Yang L, Green C, Ma C, Robert A, Kadel EE, et al. Distinct patterns of PD-L1 and PD-L2 expression by tumor and non-tumor cells in patients with MM. MDS (2016) 128(22):1340–0. doi: 10.1182/blood.V128.22.1340.1340
241. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res (2008) 14(10):3044–51. doi: 10.1158/1078-0432.CCR-07-4079
242. A trial of pembrolizumab (MK-3475) in participants with blood cancers (MK-3475-013/KEYNOTE-013).
243. Nivolumab and azacytidine with or without ipilimumab in treating patients with refractory/relapsed or newly diagnosed acute myeloid leukemia.
244. An efficacy and safety study of azacytidine subcutaneous in combination with durvalumab (MEDI4736) in previously untreated adults with higher-risk myelodysplastic syndromes (MDS) or in elderly patients with acute myeloid leukemia (AML).
245. A study of atezolizumab administered alone or in combination with azacytidine in participants with myelodysplastic syndromes. Available from: https://clinicaltrials.gov/ct2/show/NCT02508870.
246. Azacitidine and pembrolizumab in treating patients with myelodysplastic syndrome. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03094637.
247. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efcacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label. phase II study. Cancer Discovery (2019) 9(3):370–83. doi: 10.1158/2159-8290.CD-18-0774
248. Zeidner JF, Vincent BG, Esparza S, Ivanova A, Moore DT, Foster MC, et al. Final clinical results of a phase II study of high dose cytarabine followed by pembrolizumab in relapsed/refractory AML. Blood (2019) 134(Supplement 1):831. doi: 10.1182/blood-2019-126065
249. Ravandi F, Assi R, Daver N, Benton CB, Kadia T, Thompson PA, et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a single-arm, phase 2 study. Lancet Haematol (2019) 6(9):e480–8. doi: 10.1016/S2352-3026(19)30114-0
250. Boddu P, Kantarjian H, Garcia-Manero G, Allison J, Sharma P, Daver N. Te Emerging role of immune checkpoint-based approaches in AML and MDS. Leukemia Lymphoma (2018) 59(4):790–802. doi: 10.1080/10428194.2017.1344905
251. Garcia-Manero G, Montalban-Bravo G, Berdeja JG, Abaza Y, Jabbour E, Essell Jc, et al. A phase II study evaluating the combination of nivolumab or ipilimumab with azacitidine in patients with previously treated or untreated myelodysplastic syndromes. American society of hematology. Blood (2016) 128(22):344. doi: 10.1182/blood.V128.22.344.344
252. Jiang Z, Sun H, Yu J, Tian W, Song Y. Targeting CD47 for cancer immunotherapy. J Hematol Oncol (2021) 14(1):180. doi: 10.1186/s13045-021-01197-w
253. . Abstract Book Citations: Authors, Title, HemaSphere, 2022; 6:(S3):pages. The individual abstract DOIs can be found.
254. Lee P, Yim R, Yung Y, Chu HT, Yip PK, Gill H. Molecular targeted therapy and immunotherapy for myelodysplastic syndrome. Int J Mol Sci (2021) 22(19):10232. doi: 10.3390/ijms221910232
255. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): An update. Mutat Res Rev Mutat Res (2016) 769:47–62. doi: 10.1016/j.mrrev.2016.04.009
256. Magrolmab monotherapy or magrolimab pwith azacytidine in participants with hematological malignancies. Available from: https://clinicaltrials.gov/ct2/show/NCT03248479.
258. Bejar R, Greenberg PL. The impact of somatic and germline mutations in myelodysplastic syndromes and related disorders. J Natl Compr Canc Netw (2017) 15(1):131–5. doi: 10.6004/jnccn.2017.0010
259. Churpek JE. Familial myelodysplastic syndrome/acute myeloid leukemia. Best Pract Res Clin Haematol (2017) 30:287– 9. doi: 10.1016/j.beha.2017.10.002
260. Boada M, Catalán AI, Ottati C, Bentancour F, Lens D, Guillermo C, et al. Germline CEBPA mutation in familial acute myeloid leukemia. Hematol Rep (2021) 13(3):9114. doi: 10.4081/hr.2021.9114
261. Wen XM, Hu JB, Yang J, Qian W, Yao DM, Deng ZQ, et al. CEBPA methylation and mutation in myelodysplastic syndrome. Med Oncol (2015) 32:192. doi: 10.1007/s12032-015-0605-z
262. Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller BU. Somatic CEBPA mutations are a frequent second event in. doi: 10.1200/JCO.2008.16.5563
263. Jiang Y, Zhu Y, Qiu W, Liu YJ, Cheng G, Liu ZJ, et al. Structural and functional analyses of human DDX41 DEAD domain. Protein Cell (2017) 8(1):72–6. doi: 10.1007/s13238-016-0351-9
264. Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell (2015) 27(5):658–70. doi: 10.1016/j.ccell.2015.03.017
265. Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, et al. Novel germline DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood. (2016) 127(8):1017–23. doi: 10.1182/blood-2015-10-676098
266. Cheah JJC, Hahn CN, Hiwase DK, Scott HS, Brown AL. Myeloid neoplasms with germline DDX41 mutation. Int J Hematol (2017) 106(2):163–74. doi: 10.1007/s12185-017-2260-y
267. Sébert M, Passet M, Raimbault A, Rahmé R, Raffoux E, Sicre de Fontbrune F, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood. (2019) 134(17):1441–4. doi: 10.1182/blood.2019000909
268. Owen CJ, Toze CL, Koochin A, Forrest DL, Smith CA, Stevens JM, et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood. (2008) 112(12):4639–45. doi: 10.1182/blood-2008-05-156745
269. Cavalcante de Andrade Silva M, Krepischi ACV, Kulikowski LD, Zanardo EA, Nardinelli L, Leal AM, et al. Deletion of RUNX1 exons 1 and 2 associated with familial platelet disorder with propensity to acute myeloid leukemia. Cancer Genet (2018) 222-223:32–7. doi: 10.1016/j.cancergen.2018.01.002
270. Harada Y, Harada H. Molecular mechanisms that produce secondary MDS/AML by RUNX1/AML1 point mutations. J Cell Biochem (2011) 112(2):425–32. doi: 10.1002/jcb.22974
271. Poggi M, Canault M, Favier M, Turro E, Saultier P, Ghalloussi D, et al. Germline variants in ETV6 underlie reduced platelet formation, platelet dysfunction and increased levels of circulating CD34+ progenitors. Haematologica. (2017) 102(2):282–94. doi: 10.3324/haematol.2016.147694
272. Moriyama T, Metzger ML, Wu G, Nishii R, Qian M, Devidas M, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol (2015) 16(16):1659–66. doi: 10.1016/S1470-2045(15)00369-1
273. Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet (2015) 47(2):180–5. doi: 10.1038/ng.3177
274. Feurstein S, Godley LA. Germline ETV6 mutations and predisposition to hematological malignancies. Int J Hematol (2017) 106(2):189–95. doi: 10.1007/s12185-017-2259-4
275. Ding LW, Ikezoe T, Tan KT, Mori M, Mayakonda A, Chien W, et al. Mutational profiling of a MonoMAC syndrome family with GATA2 deficiency. Leukemia. (2017) 31(1):244–5. doi: 10.1038/leu.2016.256
276. Wang X, Muramatsu H, Okuno Y, Sakaguchi H, Yoshida K, Kawashima N, et al. GATA2 and secondary mutations in familial myelodysplastic syndromes and pediatric myeloid malignancies. Haematologica. (2015) 100(10):e398–401. doi: 10.3324/haematol.2015.127092
Keywords: MDS, genetic mutations, spliceosome, epigenetics, transcription factors, cohesin complex, DNA repair, targeted therapy
Citation: Kontandreopoulou C-N, Kalopisis K, Viniou N-A and Diamantopoulos P (2022) The genetics of myelodysplastic syndromes and the opportunities for tailored treatments. Front. Oncol. 12:989483. doi: 10.3389/fonc.2022.989483
Received: 08 July 2022; Accepted: 14 September 2022;
Published: 20 October 2022.
Edited by:
Jignesh D. Dalal, Case Western Reserve University, United StatesReviewed by:
Zhongbo Hu, St. Jude Children’s Research Hospital, United StatesYamilet Huerta, Case Western Reserve University, United States
Copyright © 2022 Kontandreopoulou, Kalopisis, Viniou and Diamantopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina-Nefeli Kontandreopoulou, ZWxpbmFfa250QGhvdG1haWwuY29t