 Ankur Chakravarthy1‡Elisa Pasini2‡Xun Zhao2
Ankur Chakravarthy1‡Elisa Pasini2‡Xun Zhao2 Jeffrey To2Shu Yi (Roxana) Shen1
Jeffrey To2Shu Yi (Roxana) Shen1 Sandra Fischer3Anand Ghanekar4‡Arndt Vogel1‡Robert C. Grant1‡Jennifer Knox1
Sandra Fischer3Anand Ghanekar4‡Arndt Vogel1‡Robert C. Grant1‡Jennifer Knox1 Gonzalo Sapisochin4‡Gregory J. Gores5‡Daniel De Carvalho1,6*†
Gonzalo Sapisochin4‡Gregory J. Gores5‡Daniel De Carvalho1,6*† Mamatha Bhat2,7,8,9*†‡
Mamatha Bhat2,7,8,9*†‡- 1Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
- 2Ajmera Transplant Centre, University Health Network, Toronto, ON, Canada
- 3Department of Pathology, University Health Network, Toronto, ON, Canada
- 4Division of Multi-Organ Transplant and Hepato-Pancreato-Biliary (HPB) Surgical Oncology, Department of General Surgery, University Health Network, Toronto, ON, Canada
- 5Division of Gastroenterology & Hepatology, Mayo Clinic, Rochester, MN, United States
- 6Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada
- 7Division of Gastroenterology and Hepatology, University of Toronto, Toronto, ON, Canada
- 8Ajmera Transplant Centre, Toronto General Hospital Research Institute, Toronto, ON, Canada
- 9Institute of Medical Science, University of Toronto, Toronto, ON, Canada
Hepatocellular carcinoma (HCC) is a highly lethal, aggressive malignancy. Little is known about the evolutionary trajectories of HCC and how clinical decision-making could be informed based on biopsies of the initial tumour. Here, we report the whole-exome sequencing of a unique series of resected HCC tumours and matched recurrences. This cohort included patients who received a liver transplant and who were immunosuppressed at time of recurrence, in comparison to patients who underwent liver resection for HCC and immunocompetent at time of recurrence, therefore facilitating analyses of immune selection in driving evolutionary divergence. We find extensive evolutionary divergence between baseline and recurrent tumours, with the majority of mutations in our cohort being private, in the process informing sampling guidelines for precision oncology in this disease. We also find no evidence that immunosuppression relaxes immune selection pressures, given the absence of a genomic footprint reflecting the presentation of neoantigens or altered dynamics of genomic evolution. We attribute this to the presence of genetic lesions that confer the capabilities of immune evasion in these tumours prior to transplantation, and then validate the link between immune selection pressures and the emergence of these lesions in publicly available HCC datasets. Our findings point to HCC as a cancer with extensive evolutionary divergence over time, partly defined by an irreversible, genetically determined trajectory towards immune escape.
Introduction
Hepatocellular carcinoma (HCC) is a high-fatality cancer represents and approximately 70% of all primary liver cancers and ranks as the sixth most frequently diagnosed cancer globally (1). In the background of chronic liver disease of various aetiologies, the ongoing cycle of injury and compensatory liver regeneration results in accumulation of cancer-causing mutations. HCC treatment is challenging as clinicians must take into consideration not only the characteristics of the primary tumour, but also the patient’s functional status and underlying liver disease that may limit therapeutic options (2, 3). Therapies such as resection or systemic therapy that would otherwise be feasible in other cancers cannot be tolerated by many patients. Carefully selected patients with HCC may be candidates for liver transplantation as a unique paradigm in solid organ transplant medicine, wherein transplantation is offered as curative treatment (4, 5). Despite careful patient selection, recurrence of the primary tumour occurs in 20% and is often aggressive with a median survival of 12 months (6, 7). Clinical predictors of recurrence include tumour burden on the explant (excised, diseased liver), presence of vascular invasion, and elevated alpha-fetoprotein level (AFP) (8). Post liver transplant, patients are subject to lifelong immunosuppression to mitigate risk of organ rejection. The aggressive recurrence of HCC has been attributed to post-transplant immunosuppression (9), though the exact mechanisms have remained unclear.
HCC is shaped by complex interactions between tumour cells and the immune system, with increasing evidence supporting the role of immune selection in driving tumour evolution. Studies on immune surveillance have shown that HCC undergoes a process of immunoediting, where immune pressure initially suppresses tumour progression but ultimately selects for resistant clones that escape immune destruction (10). Recent genomic analyses have demonstrated that HCC tumours with high immune infiltration exhibit increased selection for immune-evasive mutations, including alterations in antigen presentation pathways and immune checkpoint expression (11). Furthermore, transcriptomic studies have revealed that immune-excluded tumours—those with a non-inflamed microenvironment—tend to exhibit increased activation of oncogenic pathways that promote resistance to immune attack (12, 13) These findings highlight the dynamic nature of immune selection in HCC and its critical role in shaping tumour heterogeneity.
Post-transplant immunosuppression removes a key selective pressure exerted by the immune system, potentially enabling the outgrowth of aggressive tumour subclones that were previously constrained by immune surveillance. Prior studies have suggested that in the absence of immune control, HCC recurrence exhibits distinct molecular features, including increased genomic instability and resistance to immune checkpoint blockade (14, 15). Understanding these mechanisms is essential for refining therapeutic strategies, particularly in the selection of patients who may benefit from immune-based therapies or alternative treatment approaches.
The tumour microenvironment plays key roles in shaping tumour evolution and in determining treatment responses (14, 16). The recent clinical success of immunotherapy (13) in subpopulations of patients with previously intractable malignancies has also highlighted the importance of understanding the tumour microenvironment to identify those patients who will derive the most benefit from this class of targeted therapies (14, 16). Despite evidence of tumour genetics providing important prognostic information (13), biopsy of HCC is often omitted or inconsistently practiced in the clinical setting.
Here, we leverage genomic and epigenomic characterisation of a rare collection of baseline and recurrent tumours under conditions of immunosuppression and competence to specifically evaluate how immunosuppression alters the clonal evolution of HCC, and to document patterns of clonal evolution in this disease as a matter of general interest for the application of genomically-guided precision medicine.
Results
Study population
We retrieved matched formalin-fixed, paraffin-embedded (FFPE) tumour samples from 15 patients at both initial resection and at recurrence for a total of 32 samples retrieved (Table 1). One liver transplant recipient experienced two additional recurrences, enabling profiling of recurrent HCC lesions longitudinally. Of the 15 patients, 10 had received a liver transplant and were on immunosuppression at the time of recurrence, while the remaining 5 had been treated with liver resection alone prior to HCC recurrence. The two groups were comparable in mean age (58 years, p=0.74) and consisted entirely of male patients. The primary aetiologies of HCC included hepatitis B and C, with a higher prevalence of hepatitis B in the immune-compromised group (40%) and hepatitis C in the immune-competent group (60%). Alcohol-related HCC was observed in 10% of immune-compromised patients, whereas cryptogenic HCC accounted for 20% of cases in the immune-competent group. Preoperative tumour characteristics revealed no significant differences in alpha-fetoprotein (AFP) levels at the time of transplant or resection (p=0.84). However, the immune-competent group exhibited larger tumours compared to the immune-compromised group (5.8 ± 3.8 cm vs. 3.89 ± 3.9 cm, p=0.27). Tumour multiplicity varied, with 50% of immune-compromised patients presenting with two lesions, whereas 40% of immune-competent patients had four lesions (p=0.13). Histological grading indicated that all tumours in the immune-competent group were well-to-moderately differentiated, while only 10% of tumours in the immune-compromised group exhibited similar differentiation, with the remaining cases classified as moderately or poorly differentiated (p=0.91). Regarding vascular invasion and recurrence, microvascular invasion was present in 70% of immune-compromised and 80% of immune-competent patients (p=0.22). Major vessel invasion was observed in 10% of immune-compromised and 20% of immune-competent patients (p=0.26). Despite a longer median overall survival in the immune-compromised group (8.9 years vs. 6.4 years, p=0.27), the difference was not statistically significant. Similarly, the time to recurrence was longer in the immune-compromised group (42.03 months vs. 9.5 months, p=0.07), suggesting a trend towards delayed recurrence in this cohort. The most common sites of recurrence were the liver and lungs, with no significant intergroup differences (p=0.80). The primary treatment for recurrence varied between groups, with liver and lung resection each performed in 40% of immune-compromised patients, while immune-competent patients underwent a combination of surgical and non-surgical interventions, including radiofrequency ablation (RFA) and transarterial chemoembolization (TACE) (p=0.28). By the time of analysis, 70% of immune-compromised and 60% of immune-competent patients had died (p=0.45). These findings suggest that while tumour recurrence occurred later in the immune-compromised group, overall survival remained comparable between cohorts. We analysed the association between the clinical variables listed in Table 1 and overall survival using univariate survival analysis with the survival package in R. However, no statistically significant associations were identified (Supplementary Table S1).
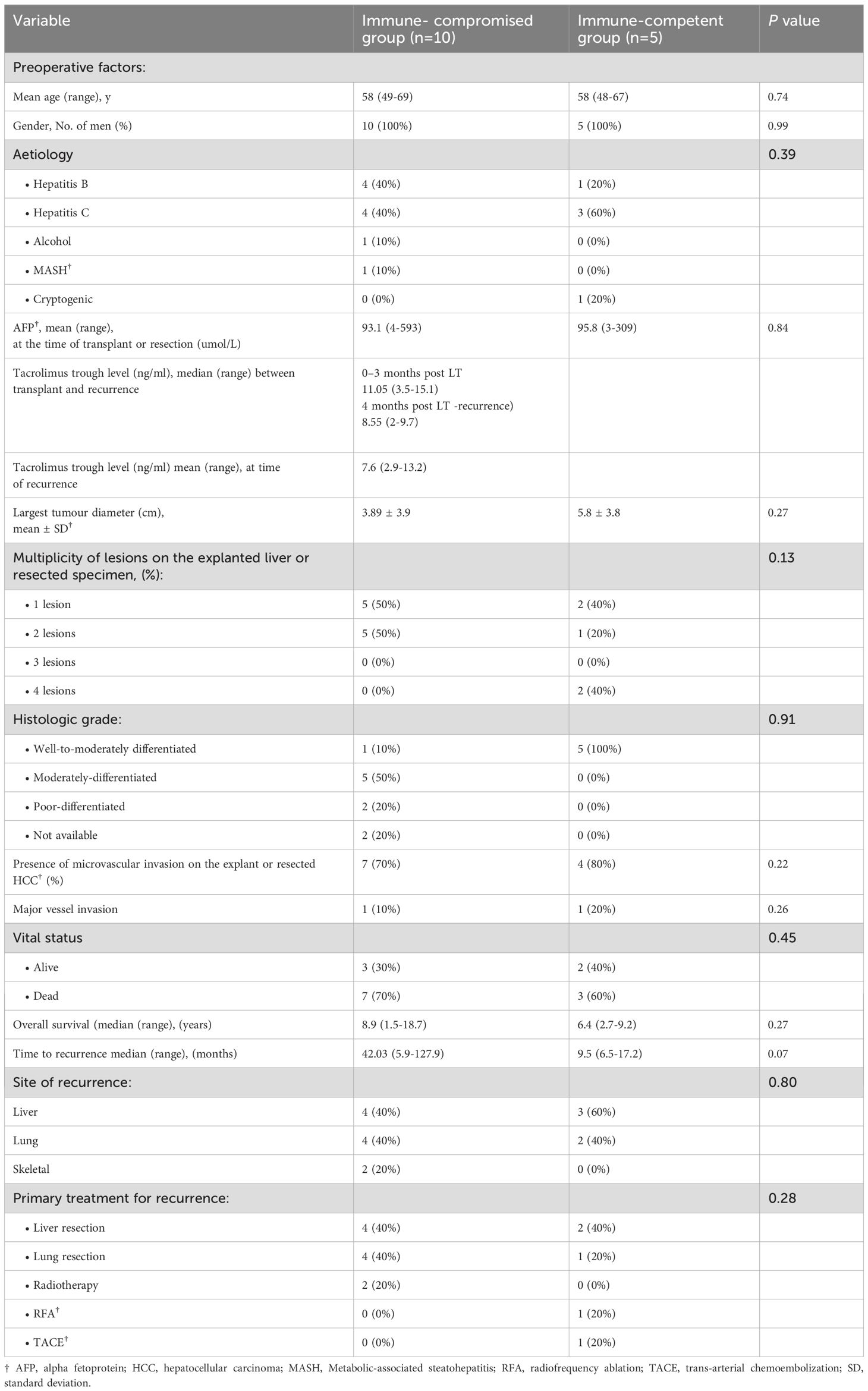
Table 1. Clinicopathological characteristics of the study participants transplanted or underwent surgical resection for HCC†.
Generation of a catalogue of somatic mutations in matched pairs of HCCs
We generated somatic mutation calls using paired, matched FFPE (Formalin-Fixed, Paraffin-Embedded) samples from 15 patients at both initial tumour resection and recurrence, resulting in a total of 32 samples retrieved (Table 1), including two additional recurrent samples from one patient with multiple post-transplant recurrences. Whole-exome sequencing (WES) was performed at high coverage (100× for tumour, 50× for matched normal). Comparison to matched normal tissue revealed a high number of somatic mutations. FFPE samples are known to introduce artefactual mutations due to chemical modifications during fixation. These false-positive variants often follow a characteristic mutational signature (17) and typically occur at low variant allele frequencies (VAF). VAF is defined as the proportion of sequencing reads supporting a given mutation. To mitigate these errors, we incorporated FFPE-specific filtering modules into our variant calling pipeline and applied an additional VAF threshold to exclude variants likely to be artefactual. Given that FFPE-induced mutations exhibit characteristic patterns of DNA damage and repair, this dual-filtering strategy effectively reduced their prevalence in the final mutation set (Supplementary Figure S1A). As expected, the filtering also decreased the overall number of detected mutations, resulting in variable retention rates of initially called variants following VAF-based exclusion (Supplementary Figures S1B, C).
Because multiple samples were derived from some patients, the dataset lacked full independence, limiting our ability to apply de novo driver discovery tools like dNdScv (18). We therefore focussed our analysis on previously characterised HCC and pan-cancer driver genes (18). Mutations in known HCC drivers were identified in 68% (22/32) of samples, though they occurred less frequently in tumours from immunocompetent patients (Figure 1A).
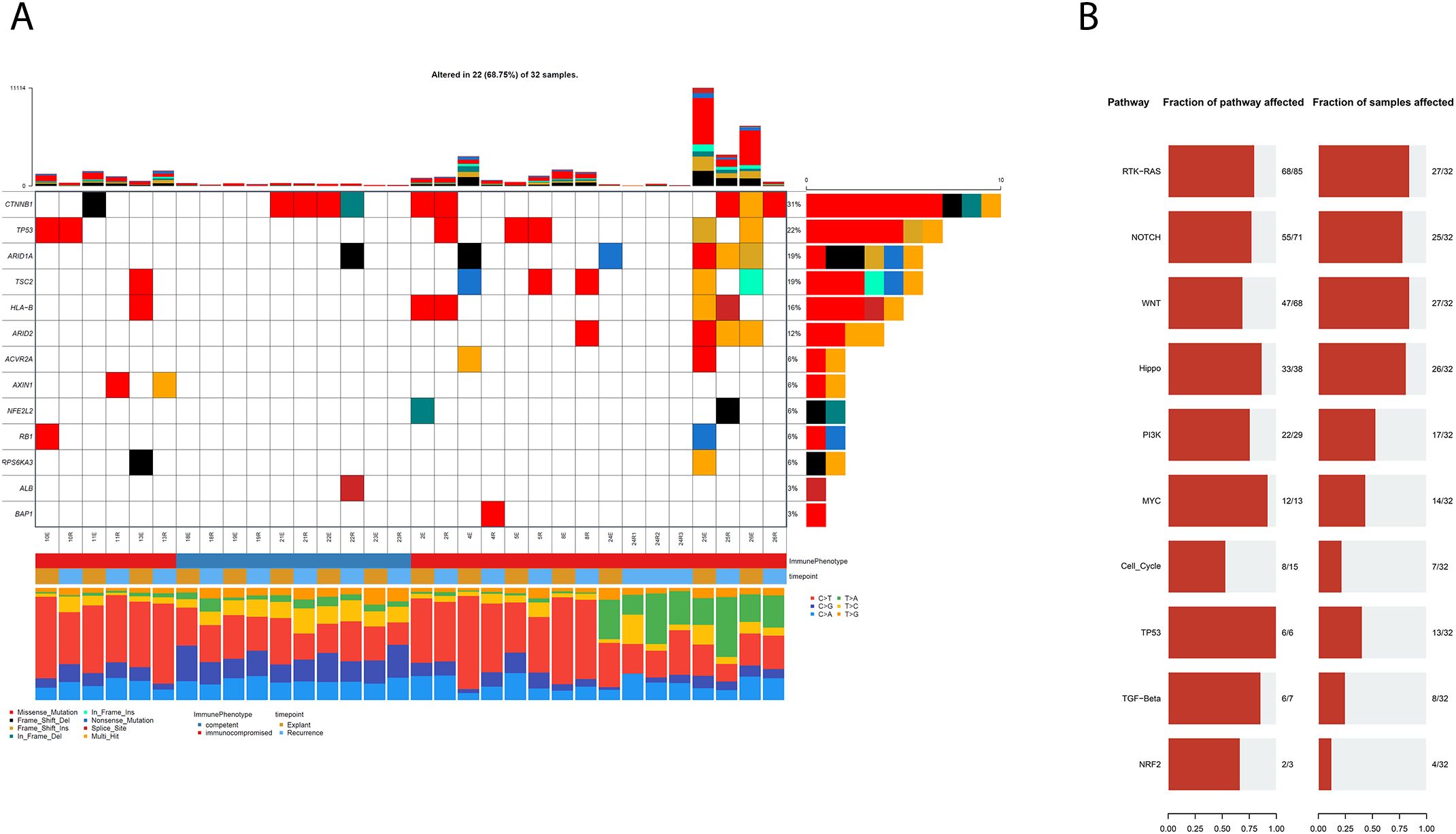
Figure 1. (A) Co-mutation Plot showing mutations in known HCC driver genes in our cohort of FFPE HCCs following FFPE specific filtering and the application of a secondary variant allele frequency filter. (B) Representations of pathways associated with observed mutations, in terms of the number of genes in a pathway that are mutated and the fraction of samples with mutations in the relevant pathways.
All samples had mutations in genes previously implicated in pan-cancer oncogenesis, suggesting that these variants may represent alternative routes to tumour development in some patients (mutation data in Supplementary Tables S2).These findings suggest that genomic assays including known pan-cancer driver genes may be clinically useful in the management of HCC using precision medicine. We identified RAS-RTK as the most frequently mutated pathway, both in terms of percentage of genes mutated within each canonical pathway as well as in the proportion of affected samples (Figure 1B). The other most mutated pathways included the NOTCH, Wnt and HIPPO signalling pathways, in concordance with established knowledge of HCC (19).
Extensive genomic divergence defines HCC evolution
In order to characterise the clonal structure and variation of mutations in our HCC cohort, we integrated the variant allele frequency and high confidence mutation calls with allele-specific absolute copy number calls generated using Sequenza. This was then used in conjunction with PyClone_VI (20), a variational bayes inferential method of the original PyClone algorithm (21), to estimate the cancer cell fraction (CCF) of each mutations and to then cluster mutations into distinct clones based on their CCF distribution.
Initial comparisons between baseline and recurrence samples revealed that the vast majority of mutations were private (i.e., found only at one timepoint, Figure 2A), indicating marked evolutionary divergence. This prompted us to assess whether the low mutational overlap observed between baseline and recurrent samples might be an artefact introduced by our stringent VAF-based filtering of formalin-fixation-related errors. We reasoned that if the observed lack of shared mutations were primarily due to filtering, then the overlap between matched baseline-recurrence pairs should be no greater than that seen between tumours from unrelated individuals. To test this, we analysed whole-exome sequencing data from 360 HCC cases in TCGA and calculated the distribution of mutational overlap across all possible pairwise combinations (n = 129,600).

Figure 2. (A) Breakdown of mutations in each patient, classified as private (found only in either the primary or recurrent tumour) or public (shared between both). The X-axis shows patient identifiers, and the Y-axis shows the percentage of all mutations found in each patient at either timepoint. (B, C) represent clone-level views of the primary and recurrent tumours for each patient, with the Y-axis indicating the percentage of all tumour mutations belonging to clones at a given Cancer Cell Fraction (X-axis). Point size and colour reflect the relative size of each clone based on mutation percentage. These results are split into panels showing: (1) immunocompetent control tumours across timepoints, (2) immunocompromised tumours across timepoints, and finally (D) longitudinal samples spanning multiple years from one immunocompromised transplant recipient.
The percentage of overlap between our matched patient samples was significantly higher in comparison to the distribution seen in the unmatched control samples, suggesting that our filtration strategy did not erroneously inflate the extent of evolutionary divergence we observed (Supplementary Figure S1D, p < 2.2e-16, Wilcoxon’s Rank Sum Test).
We subsequently characterised the clonal composition of our tumours, by clustering mutations into distinct clones based on their Cancer Cell Fractions (CCF) using a variational Bayesian inference version of the widely used PyClone algorithm. This approach is based on the principle that due to the nature of chromosomal segregation during mitosis and inheritance of pre-existing and novel variants by daughter cells, mutations that occur at similar allele frequencies are likely to co-exist in the same cellular populations. Without multi-region data, however, we decided not to perform phylogenetic reconstruction of clonal relationships within our samples, since mutation ordering is challenging in the absence of multi-region data. These findings are consistent with polyclonal seeding of recurrences in both immunocompetent and immunocompromised patients, a pattern previously described in prostate cancer metastases previously (22). The clonal structure in these tumours specifically demonstrated the presence of small clonal populations at high CCF and the large majority of mutations in a sample being present subclonally (Figure 2B). Further, in the one patient from whom we profiled multiple post-transplant recurrent samples across a timespan of years, we observed marked temporal shifts in clonal structure (Figure 2C). Taken together, the level of heterogeneity and evolution observed in these tumours suggests that repeat biopsies—ideally incorporating multi-region or spatially resolved sampling—may be necessary at recurrence to guide the selection of effective targeted therapies.
To quantitatively assess clonal architecture, we calculated a clonality score for each sample. This score was defined as the sum of each mutation’s cancer cell fraction (CCF) weighted by its frequency, generating a value between 0 (entirely subclonal) and 1 (entirely clonal). Notably, we observed no statistically significant differences in clonality between immunocompetent and immunocompromised patients, either at baseline or at recurrence, nor in the extent of change between these timepoints (Figures 3A, B). This remained true even in the longitudinal series from a single patient (Figure 3C).
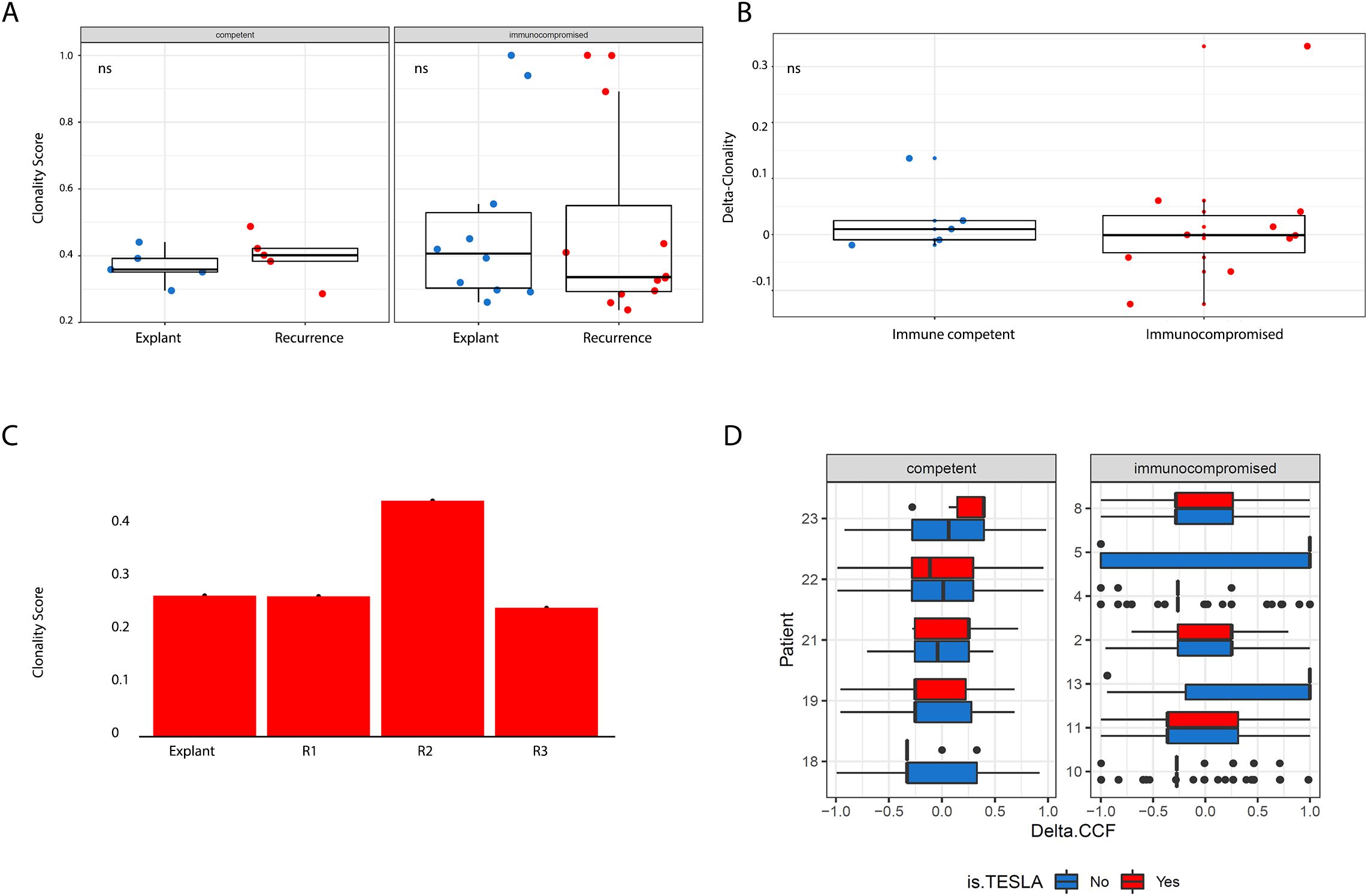
Figure 3. (A) Boxplots showing breakdowns of clonality scores (Y axis) across patient group and timepoint (X axis). More clonal tumours have higher clonality scores. We did not observe significant differences across timepoints when comparing by patient group or across patient group when comparing by timepoints. (B) Boxplots showing the magnitude of change in clonality scores (Y axis) between timepoints across patient group (X axis). We did not observe significant differences across timepoints when comparing by patient group or across patient group when comparing by timepoints. All tests pertaining to this figure were conducted using a two-sided Wilcoxon’s Rank Sum Test (C) Longitudinal series of samples profiled from a patient at multiple post-recurrence timepoints, no statistically significant variation in clonality score was observed. (D) Distribution of delta CCF for all mutations in a pair of patient samples, which revealed no significant distributional shifts. Boxplots illustrate patterns of shifts in allele frequency for all mutations present in either the primary or recurrent sample for each patient, segregated by neoantigen status (predicted neoantigens are in red, predicted non-neoantigens are in blue).
Taken together, these findings indicate that immunosuppression did not influence the evolutionary trajectory of these tumours, suggesting that immunoediting was not an active process during transplantation and subsequent recurrence. We initially hypothesised that if these tumours had not already evolved mechanisms to evade immune selection before the onset of immunosuppression, we would observe either an expansion of pre-existing neoantigens (novel proteins arising from somatic mutations that the adaptive immune system may recognise as non-self due to significant divergence from their wild-type counterparts) or a disproportionate enrichment of neoantigens in the mutational repertoire of recurrent tumours. However, the absence of these patterns suggests that immune escape had already occurred prior to transplant-associated immunosuppression.
Immunoediting entails elimination of cancer cells carrying immunogenic mutations, followed by selection and expansion of cancer cells capable of evading the immune system through a wide range of permanent, genetic, lesions or other transient mechanisms such as the upregulation of immunosuppressive checkpoint proteins (23, 24). Immunotherapy has emerged as a promising strategy through the blockade of immune checkpoints, such as CTLA-4 and PD-1, which can rebalance the Immunoediting due to immunogenic mutations in favour of tumour elimination (25).
Neoantigen calling is not an exact science, and the best state-of-the-art neoantigen calling criteria only manage to achieve about 70% sensitivity at 98% specificity based on integration of predicted affinity of binding to Class I MHC alleles and sequence similarity to pathogenic peptides and dissimilarity to the self-peptidome (26). As a result, our findings are likely to miss real changes at the expense of avoiding false positive findings.
Using the Antigen Garnish pipeline, we generated neoantigen calls and then further applied the affinity criteria developed by the TESLA consortium (the current state-of-the-art, as described earlier) to yield a stringently filtered set of candidate neoantigens for the vast majority of samples. For a subset of samples (n = 5), our HLA caller reported insufficient numbers of paired reads to call HLA type. For secondary analyses, we used more traditionally employed thresholds for binding affinity; 100nM (strong binders) and 500nm (weak binders) or less. We then quantified, for each mutation, the delta-CCF between recurrent and baseline samples and compared their distributions between candidate neoantigens and control mutations. In accordance with our general, mutanome-wide finding that there was no influence of transplant-related immunosuppression on the evolution of recurrent HCCs, we found no evidence for selective expansion of neoantigens in immunocompromised samples compared to controls (Figure 3D).
The acquisition of genetic lesions associated with immune evasion explains the lack of effect of immunosuppression on neoantigen evolution
However, these findings were consistent with multiple alternative explanations; first, any immune response that was narrowly targeted against only a few neoantigens would not affect the overall distribution, especially when the total number of candidate mutations that may produce neoantigens is high. Due to a lack of TCR sequence profiling, we were unable to estimate the breadth of the T-cell immune response and could not empirically assess whether this was the case. Secondly, another possibility is that neoantigens may be regulated epigenetically or transcriptionally. While our cohort lacked transcriptomic data, the availability of matched Illumina EPIC array profiles for most of our samples enabled us to test for differences in promoter methylation at neoantigens and non-neoantigens alike. Again, we failed to see significant differences upon lifting the immune selection pressures in the immunosuppressed patients (Supplementary Figure S1C). However, the lack of transcriptomic data prevents us from accounting for changes in the transcriptional states of neoantigens that may be regulated by mechanisms other than DNA methylation, or changes in non-genomic mechanisms that regulate anti-tumour immunity.
The remaining explanation was that the acquisition of genetic alterations associated with immune evasion had created a state of enduring immune escape in these tumours before transplantation. To test this hypothesis, we established a gold-standard catalogue of experimentally validated hits from CRISPR screens that used indel-producing sgRNAs to identify genes that, when disrupted, resulted in direct resistance to CD8 T-cell mediated killing (27), as well as genes known to be canonically involved in antigen processing and presentation.
Since the functional interpretation of missense mutations can be challenging, we reduced our analysis to mutation types typically associated with loss of function (nonsense mutations, frameshifts, other indels). We observed that 19/22 tumours in the transplant group contained at least one loss of function mutation in genes known to confer resistance to T-cell mediated destruction, as opposed to only 2/10 of the immunocompetent tumours.
Amongst the groups of genes impacted recurrently were multiple components of antigen presentation machinery (the class 2 MHC genes HLA-DRA, HLA-DQA, HLA-DQB, as well as the main transcriptional activator of MHC-II genes (CIITA), and the essential invariant MHC-I component B2Mand the primary antigen transport protein, TAP1, which is essential for the formation of functional MHC-I complexes), a significantly large repertoire of core interferon transcription factors) (IRF2/IRF8) and key signal transduction kinases amongst others (JAK2 and PRKCD, both of which are involved in post-translational modifications of STAT1, a master regulator of transcriptional activation of genes transcribed as part of the interferon response (28).
This offered a potential explanation for why immunosuppression did not affect the behaviour of neoantigens; quite simply, prior genetic histories had already nullified the effect of any future modulation of immune selection pressures (Figure 4A).
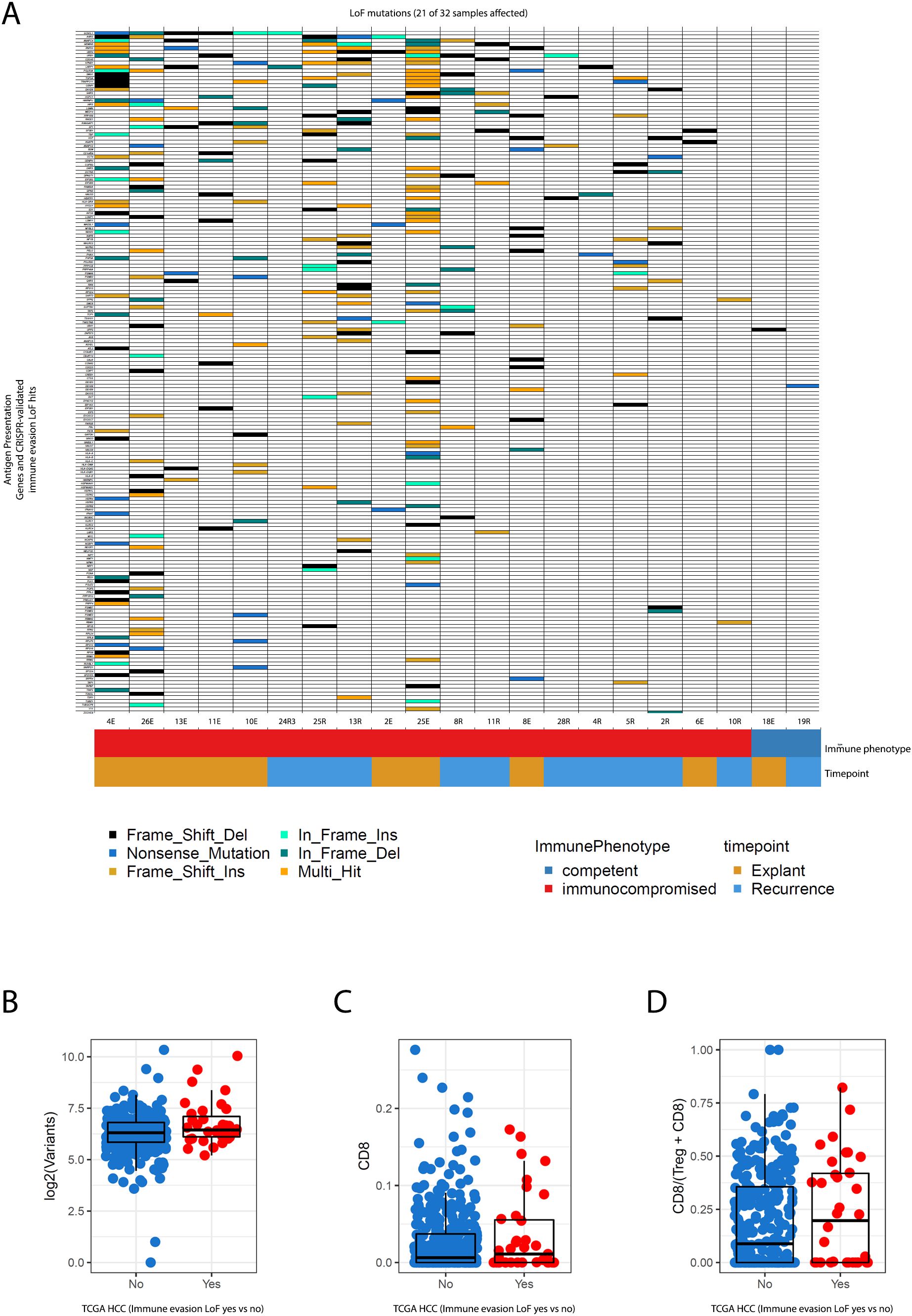
Figure 4. (A) Co-mutation plot shows patterns of loss-of-function mutations in genes experimentally validated to confer a resistance to CD8 T-cell mediated killing upon inactivation in CRISPR screens and canonically recognised antigen presentation pathway genes. These mutations are frequent and numerous in patients that went on to receive systemic immunosuppression, both at baseline, and upon recurrence. (B–D) represent relationships between parameters of immunogenicity (mutational burden, CD8 infiltration, CD8/Treg ratios) in TCGA HCC data for the same catalogue of genes. Tumours are segregated by whether they possess immune-evasion conferring LoF mutations or not. Statistical estimates are from Wilcoxon’s Rank Sum Test.
While our cohorts were limited by sample size, we reasoned that if our observation of enrichment for immune-escape genomic alterations pre-treatment was a generalisable finding, large cohorts of excised HCC would also demonstrate the same pattern. Therefore we also investigated the extent to which these mutations were present in resected primary HCC samples from the TCGA to evaluate whether genetic mechanisms of immune escape are commonly present at baseline. Using the same stringent criteria as described above, we found that nearly 10% of TCGA HCCs possess loss-of-function (LoF) mutations in at least one of the immune evasion genes surveyed, which is no doubt a conservative estimate due to our stringency. The inclusion of missense mutations in these genes further expanded this number to 55% of TCGA HCCs as a liberal upper bound.
Traditionally, selection for immune escape-related events has been associated with the presence of an inflamed antitumour immune microenvironment, often in the presence of a high burden of neoantigens (29). Indeed, we observed that most of the samples in our transplant cohort had much higher mutational burden (Figure 1A) even at baseline before exposure to immunosuppression. This higher mutational burden tracked with the presence of immune evasion LoF alterations, and was also present in the matched recurrent HCCs post-transplant.
In TCGA HCCs however, the presence of immune evasion LoF alterations was not associated with increasing mutational burden but remained strongly associated with increased CD8 infiltration as assessed using DNA methylation through MethylCIBERSORT, and higher CD8/Treg ratios (Figures 4B–D, p < 0.05 for the two latter metrics Wilcoxon’s Rank Sum test).
Conclusions
The temporal profiling of tumours under conditions of different activity in the immune microenvironment can reveal how tumour evolution is shaped by the immune system. In this study, we leveraged the availability of patients who had tumours recur under conditions of immunosuppression to observe these trends, as well as gain an understanding of evolutionary divergence in HCC tumours in general from the perspective of clinical management.
Our observation of extensive evolutionary divergence between baseline and recurrent tumours indicates the need for re-biopsy upon recurrence to ensure that the treatments selected are targeting lesions found within the tumour. Where recurrent or worsening disease is marked by multiple metastases, approaches that are based on whole-exome-sequencing of the plasma, or other approaches such as CAPP-seq with panels designed to target druggable genes, may be a useful substitute for biopsies that are hard to obtain.
The complex clonal structures of these tumours, as well as the strong propensity for subclonal mutagenesis, both point to intratumour heterogeneity being a strong factor in the clinical behaviour of these tumours. This is consistent with multi-region sequencing studies of HCC at a single time point that reveal a similar degree of heterogeneity (14, 16, 30–36).
Finally, the high burden of immune evasion mutations in neoantigen-rich tumours implicates a genomic basis for a state of immune escape that may result in the failure of immune checkpoint blockade. We therefore suggest that the management of HCC regularly involve sequencing to detect the presence of these alterations in order to carefully select patients that may respond to immune checkpoint blockade. Multiple clinical trials of ICB monotherapies in HCC have indicated overall response rates between 20-32% (37, 38) in patients. The role of checkpoint inhibitors in the treatment of hepatocellular carcinoma is evolving and expanding (39, 40). An essential step in expanding the use of CPI in unresectable HCC and recurrence is recognising the abundance of pre-existing immune evasion mutations in tumours. This in turn may facilitate more efficient use of these therapies.
Our findings align with studies in other cancers demonstrating that immune selection pressures drive tumour evolution and resistance to immunotherapy (25). Similar patterns of immune escape have been observed in metastatic melanoma and non-small cell lung cancer, where tumours acquire genetic alterations that impair antigen presentation and T-cell recognition (24, 29, 36). Comparing HCC with these malignancies highlights the broader implications of immune-driven tumour evolution and reinforces the need for multi-modal treatment approaches (41, 42).
Our results suggest that tumour-intrinsic factors, rather than host immunity, may be the dominant force driving HCC recurrence, explaining why immunosuppression failed to alter tumour evolution. One potential mechanism is inherent resistance to immune editing, where tumours had already undergone immunoediting prior to transplantation, selecting for clones that could evade immune-mediated destruction (13, 43). Previous studies have shown that tumours frequently develop genetic and epigenetic mechanisms that disable antigen presentation and modulate immune checkpoint expression, rendering them resistant to immune attack irrespective of immunosuppressive status (15). Given that immune checkpoint inhibitors (ICIs) have shown efficacy in a subset of HCC patients with intact antigen presentation pathways (44), our findings suggest that only select patients may benefit from these therapies. Future research should explore combination approaches, integrating ICIs with epigenetic modulators or kinase inhibitors to enhance anti-tumour immunity, particularly in immune-excluded tumours (45). Furthermore, the role of HCV-driven oncogenesis in shaping immune evasion landscapes remains an important avenue for investigation, as regional disparities in HCV prevalence and antiviral access could impact both HCC evolution and treatment strategies (46, 47). A deeper understanding of these molecular interactions could inform tailored therapeutic interventions and precision oncology approaches for HCC patients across different disease contexts.
Our contextualisation of how immune escape works in HCC is a useful contribution to this end, especially when these patterns are reflected in much larger cohorts of resected HCC. Our pilot study points to the importance of extensive spatial and temporal genomic characterisation of HCC to guide personalised therapy.
Limitations
While our study provides valuable insights into HCC evolution under immune selection pressures, it is limited by sample size and the absence of transcriptomic data. Future studies should integrate RNA sequencing and spatial transcriptomics to assess how immune-modulatory changes at the transcriptional level influence tumour progression (31).
Methods
Patient population
Patients who underwent liver transplant or surgical resection for hepatitis B/C or alcohol-induced HCC between 2002 and 2016 were included in our study. Ten patients who underwent liver transplant (n=10) or surgical resection (n=5) were included in our study. Patients with a maximum of 4 viable lesions of the primary tumour were included. For both groups, archived formalin-fixed, paraffin-embedded (FFPE) samples available were retrieved. In case of more than one HCC lesion, only the largest dominant lesion was retrieved to obtain DNA. DNA was obtained from the FFPE samples using the QIAamp DNA FFPE Tissue Kit (QIAGEN) following manufacturer’s instructions. Characteristics of the tumours on the explant or resected lesions, including grade, size, number of tumours, the presence of microvascular invasion and associated AFP at the time of transplant or resection were documented. For the recurrent lesions, the date of recurrence, the number of lesions identified, the AFP peak and the type of therapy used to treat the recurrent lesions were also retrieved. For the transplant cohort, the trough level of the immunosuppressant medication from the time of transplant to the time of recurrence was also documented. Patient clinicopathological characteristics are summarised in Table 1.
Statistical analysis
Comparative statistical analyses of clinical characteristics between the immune-compromised and immune-competent groups were performed using non-parametric tests. Continuous variables were summarised as means with standard deviations or medians with interquartile ranges, and group differences were assessed using the Wilcoxon rank-sum test. Categorical variables were compared using Fisher’s exact test due to the small sample size. The association between clinical variables and overall survival was explored using univariate Cox proportional hazards models with the coxph() function in the survival package (https://cran.r-project.org/package=survival (48)in R (v3.8-3).
Details of the statistical methods are outlined in the Methods section, described in-line in the text, and summarised in the figure legends. Proportions were analysed using Fisher’s exact test, and comparisons of distributions were performed using the Wilcoxon rank-sum test.
Briefly, differences in clonality scores were evaluated using two-sided Wilcoxon rank-sum tests. The extent of mutational overlap between paired baseline-recurrent samples versus unrelated TCGA controls was assessed using Wilcoxon tests. Associations between immune escape mutations and markers of immune infiltration (e.g., CD8+ T-cell levels, CD8/Treg ratios) in the TCGA dataset were also evaluated using Wilcoxon rank-sum test.
Ethics statement
This study was conducted in accordance with the Declaration of Helsinki and approved by the University Health Network (UHN) Research Ethics Board (REB#22-5006). Given the retrospective nature of this study, a waiver of consent was granted for the collection of retrospective data and tissue samples. While retrospective studies offer valuable insights, they are inherently subject to potential selection biases, including the availability of archived specimens and variations in clinical follow-up. To mitigate these biases, we implemented standardised inclusion criteria and validated our findings against publicly available datasets where applicable.
Whole exome sequencing and variant calling
Methylation profiling: DNA was extracted from FFPE samples using QIAamp DNA FFPE Tissue Kit (Qiagen) following manufacture’s protocol and quantified using Qubit 4 fluorometer (ThermoFisher). DNA (500ng) was bisulfite-converted and processed on Infinium Human Methylation EPIC BeadChips (Illumina Inc.) (41).
Whole exome sequencing: Exome capture was performed using Agilent exome capture kit on extracted DNA (10 ng). Libraries were prepared using Agilent SureSelect XT-HS kit and hybridised to Agilent human all exon v7 panel. Sequencing included 125 cycle paired-end reads from Illumina HiSeq 2500 at 100X for tumour samples and 50X for normal tissue, followed by exome alignment and mutation calling (49).
Fastq files were aligned to hg19 using BWA-mem (50) and were processed using Samtools (51) to remove duplicate reads for all samples. For each sample, we sequenced the explant/baseline sample, the recurrent sample, and matched normal tissue, sourced from adjacent pathologically normal liver tissue at the time of explant resection. Somatic mutations (both SNVs and short Indels) were then called using MuTect2 (52).
To mitigate artefactual mutations in FFPE samples, the FFPE module in MuTect2 was applied, followed by additional filtering to remove mutations with a variant allele frequency below 10% based on MuTect2’s empirical VAF output.
The neoantigen pipeline used, Antigen Garnish (53), required hg38 input to accurately call neoantigens. We therefore used Picard (http://broadinstitute.github.io/picard/) to lift over variant calls to hg38 using the UCSC chain file. Our hg19 callset was annotated using Annovar (53) to annotate these mutations for downstream analyses and functional annotation using the maftools R package (54), and we used snpEff 4.5 (55) to annotate VCFs for neoantigen calling from our lifted over callsets. Mutational signature analysis was performed using DeconstructSigs (53) using the updated COSMIC mutational signatures database for reference (54).
Copy number calling and clonality analyses
We used Sequenza-utils to create input files for Sequenza (56) from tumour and normal BAM files, followed by segmentation and probabilistic modelling to estimate purity, ploidy, and absolute copy number combinations that best fit the observed data. The absolute copy numbers from the top-scoring solution, along with variant allele frequencies estimated during SNV calling, were used to cluster variants into clones using PyClone-VI, specifying an upper limit of 15 subclones and 15,000 iterations.
Neoantigen calling and estimation
Class I HLA types were called from the normal tissue samples for each patient using OptiType (57) at four-digit resolution.
The Antigen Garnish (53) package was used to estimate the Class I MHC binding potential of nonameric peptide (composed of nine amino acids) sequences containing somatic mutations, based on hg38-lifted and annotated VCF files generated in previous steps. Peptides meeting the stringent affinity criteria established by the TESLA consortium, a gold standard for immunogenic neoantigens, were selected. The distribution and clonal dynamics of predicted neoantigens were then compared to non-neoantigens to assess the impact of immunosuppression on neoantigen evolution.
Analysis of DNA methylation profiles at neoantigens
We generated Illumina EPIC array data for our samples that were processed uniformly using the minfi R package via the ssNoob normalisation pipeline (58). We then summarised the methylation at upstream regulatory region probes (formally designated as TSS200, TSS1500, and 1st Exon according to Illumina convention) using the average and the median, given the general association of these probes with repression of gene expression. We then examined methylation shifts at predicted neoantigens and non-neoantigens as a function of timepoint and immunosuppression status.
Analysis of immune evasion genes
We defined a catalogue of immune evasion genes by combining gene sets of antigen presentation genes from the Kyoto Encyclopaedia of Genes and Genomes KEGG) (59), and genes with loss-of-function hits implicated in resistance to CD8 T-cell mediated destruction in CRISPR screens Patel et al (27). To define loss of function mutations in our whole exome dataset, we only included variants classified as nonsense, frameshifts, other indels and nonstop mutations. Proportions were compared using a Fisher’s Exact Test. TCGA mutation calls were obtained from the MC3 callset via SAGE Synapse and were restricted to the same categories of variants. Associations of LoF mutations in immune evasion genes with immune infiltration (CD8 levels and CD8/Treg ratios) and mutational burdens were tested using Wilcoxon’s Rank Sum Tests.
Data availability statement
Methylation and Whole Exome Sequencing data supporting this study’s findings are available upon reasonable request from the corresponding author, in compliance with institutional ethics regulations. External researchers interested in accessing the data should submit a formal request via email to the corresponding author, including a brief description of the intended research use, institutional ethics approval (if applicable), and a signed data-sharing agreement. Upon approval, the data will be made available through a secure repository under controlled access to ensure compliance with patient confidentiality and ethical guidelines.
Ethics statement
The studies involving humans were approved by The University Health Network (UHN) Research Ethics Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
AC: Investigation, Writing – original draft, Formal Analysis, Software. EP: Investigation, Writing – original draft, Methodology, Project administration. XZ: Writing – original draft, Data curation. JT: Writing – original draft, Methodology. SS: Methodology, Writing – original draft. SF: Methodology, Writing – review & editing. AG: Methodology, Writing – review & editing. AV: Writing – review & editing. RG: Writing – review & editing. JK: Writing – review & editing. GS: Writing – review & editing. GG: Writing – review & editing. DD: Writing – review & editing, Funding acquisition, Investigation, Methodology, Resources, Writing – original draft. MB: Funding acquisition, Investigation, Writing – original draft, Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a grant from the Canadian Liver Foundation to MB and DD, as well as an Early Researcher Award from the Government of Ontario and Terry Fox Research Institute (TFRI) New Investigator Award to MB and Roche Canada. AC was funded by a CIHR Banting Postdoctoral Fellowship.
Acknowledgments
The authors acknowledge the help of Erin Winter, Peregrina Peralta, Amirhossein Azhie and Catherine Chen in the retrieval of patient samples and clinical details.
Conflict of interest
MB reports receiving grants or investigator-initiated trials from Novo Nordisk, Natera, Paladin, Roche, Oncoustics, Medo-AI, Ironshore Therapeutics; and speakers fees from Paladin.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1537087/full#supplementary-material
Supplementary Figure 1 | (A) Analysis of artefactual mutational signatures and the percent of mutations mapping to them with caller-specific FFPE filtering, and caller specific FFPE filtering combined with an additional VAF filter (removal of mutations at less than 10% VAF), respectively. (B, C) are bar graphs that show the total numbers of mutations and percentages before and after the additional VAF filter was applied to our call set. (D) is a boxplot showing the percentage of overlap in mutanomes (Y axis) in our matched patient cohort when compared to a distribution of overlaps in mutanomes between unmatched patient samples derived from different individuals in the TCGA HCC cohort (X axis). P < 2.2e-16, Wilcoxon’s Rank Sum Test. (E) Boxplots show distributions of mean CpG methylation beta values at upstream regulatory regions (Y axis) of genes predicted to encode neoantigens (red) or not (blue) by timepoint (primary tumour/recurrence) (X axis), each panel represents one patient, with the immunocompetent/compromised status of the patients labelled.
References
1. Calderon-Martinez E, Landazuri-Navas S, Vilchez E, Cantu-Hernandez R, Mosquera-Moscoso J, Encalada S, et al. Prognostic scores and survival rates by etiology of hepatocellular carcinoma: A review. J Clin Med Res. (2023) 15:200–7. doi: 10.14740/jocmr4902, PMID: 37187717
2. Singal AG, Llovet JM, Yarchoan M, Mehta N, Heimbach JK, Dawson LA, et al. AASLD Practice Guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology. (2023) 78:1922–65. doi: 10.1097/hep.0000000000000466, PMID: 37199193
3. Reig M, Forner A, Rimola J, Ferrer-Fàbrega J, Burrel M, Garcia-Criado Á, et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J Hepatol. (2022) 76:681–93. doi: 10.1016/j.jhep.2021.11.018, PMID: 34801630
4. Bhat M, Ghali P, Dupont B, Hilzenrat R, Tazari M, Roy A, et al. Proposal of a novel MELD exception point system for hepatocellular carcinoma based on tumor characteristics and dynamics. J Hepatol. (2017) 66:374–81. doi: 10.1016/j.jhep.2016.10.008, PMID: 27751840
5. Lingiah VA, Niazi M, Olivo R, Paterno F, Guarrera JV, and Pyrsopoulos NT. Liver transplantation beyond Milan criteria. J Clin Transl Hepatol. (2020) 8:69–75. doi: 10.14218/jcth.2019.00050, PMID: 32274347
6. Mehta N, Dodge JL, Roberts JP, and Yao FY. Validation of the prognostic power of the RETREAT score for hepatocellular carcinoma recurrence using the UNOS database. Am J Transplant. (2018) 18:1206–13. doi: 10.1111/ajt.14549, PMID: 29068145
7. Sposito C, Citterio D, Virdis M, Battiston C, Busset Dit Droz M, Flores M, et al. Therapeutic strategies for post-transplant recurrence of hepatocellular carcinoma. World J Gastroenterol. (2022) 28:4929–42. doi: 10.3748/wjg.v28.i34.4929, PMID: 36160651
8. Mehta N, Heimbach J, Harnois DM, Sapisochin G, Dodge JL, Lee D, et al. Validation of a risk estimation of tumor recurrence after transplant (RETREAT) score for hepatocellular carcinoma recurrence after liver transplant. JAMA Oncol. (2017) 3:493–500. doi: 10.1001/jamaoncol.2016.5116, PMID: 27838698
9. Di Marco L, Romanzi A, Pivetti A, Maria De N, Ravaioli F, Salati M, et al. Suppressing, stimulating and/or inhibiting: The evolving management of HCC patient after liver transplantation. Crit Rev Oncol Hematol. (2025) 207:104607. doi: 10.1016/j.critrevonc.2024.104607, PMID: 39725094
10. Dunn GP, Old LJ, and Schreiber RD. The three es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803, PMID: 15032581
11. McGranahan N and Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. (2017) 168:613–28. doi: 10.1016/j.cell.2017.01.018, PMID: 28187284
12. Sangro B, Sarobe P, Hervás-Stubbs S, and Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2021) 18:525–43. doi: 10.1038/s41575-021-00438-0, PMID: 33850328
13. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. (2020) 382:1894–905. doi: 10.1056/NEJMoa1915745, PMID: 32402160
14. Zhai W, Lim TK, Zhang T, Phang ST, Tiang Z, Guan P, et al. The spatial organization of intra-tumour heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. Nat Commun. (2017) 8:4565. doi: 10.1038/ncomms14565, PMID: 28240289
15. Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. (2022) 19:151–72. doi: 10.1038/s41571-021-00573-2, PMID: 34764464
16. Ding X, He M, Chan AWH, Song QX, Sze SC, Chen H, et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology. (2019) 157:1630–45.e6. doi: 10.1053/j.gastro.2019.09.005, PMID: 31560893
17. Guo Q, Lakatos E, Al Bakir I, Curtius K, Graham TA, and Mustonen V. The mutational signatures of formalin fixation on the human genome. Nat Commun. (2022) 13:4487. doi: 10.1101/2021.03.11.434918, PMID: 36068219
18. Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Loo Van P, et al. Universal patterns of selection in cancer and somatic tissues. Cell. (2017) 171:1029–41.e21. doi: 10.1016/j.cell.2017.09.042, PMID: 29056346
19. Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. (2017) 169:1327–41.e23. doi: 10.1016/j.cell.2017.05.046, PMID: 28622513
20. Gillis S and Roth A. PyClone-VI: scalable inference of clonal population structures using whole genome data. BMC Bioinf. (2020) 21:571. doi: 10.1186/s12859-020-03919-2, PMID: 33302872
21. Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. (2014) 11:396–8. doi: 10.1038/nmeth.2883, PMID: 24633410
22. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. (2015) 520:353–7. doi: 10.1038/nature14347, PMID: 25830880
23. Schreiber RD, Old LJ, and Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486, PMID: 21436444
24. Chen DS and Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349, PMID: 28102259
25. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. (2017) 171:934–49.e16. doi: 10.1016/j.cell.2017.09.028, PMID: 29033130
26. Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. (2020) 183:818–34.e13. doi: 10.1016/j.cell.2020.09.015, PMID: 33038342
27. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. (2017) 548:537–42. doi: 10.1038/nature23477, PMID: 28783722
28. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604, PMID: 15864272
29. Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. (2018) 9:3220. doi: 10.1038/s41467-018-05570-1, PMID: 30104673
30. Losic B, Craig AJ, Villacorta-Martin C, Martins-Filho SN, Akers N, Chen X, et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat Commun. (2020) 11:291. doi: 10.1038/s41467-019-14050-z, PMID: 31941899
31. Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Méndez-González J, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. (2015) 61:1945–56. doi: 10.1002/hep.27732, PMID: 25645722
32. Hernandez-Meza G, von Felden J, Gonzalez-Kozlova EE, Gonzalez-Kozlova EE, Garcia-Lezana T, Peix J, et al. DNA methylation profiling of human hepatocarcinogenesis. Hepatology. (2021) 74:183–99. doi: 10.1002/hep.31659, PMID: 33237575
33. Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Moura Castro M, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology. (2017) 153(3):812–26. doi: 10.1053/j.gastro.2017.06.007, PMID: 28624577
34. Ruiz de Galarreta M, Bresnahan E--, Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. (2019) 9:1124–41. doi: 10.1158/2159-8290.CD-19-0074, PMID: 31186238
35. Ho WJ and Wood LD. Opposing roles of the immune system in tumors. Science. (2021) 373:1306–7. doi: 10.1126/science.abl5376, PMID: 34529483
36. Eghbali S and Heumann TR. Next-generation immunotherapy for hepatocellular carcinoma: mechanisms of resistance and novel treatment approaches. Cancers (Basel). (2025) 17(2):236. doi: 10.3390/cancers17020236, PMID: 39858016
37. D’Alessio A, Rimassa L, Cortellini A, and Pinato DJ. PD-1 blockade for hepatocellular carcinoma: current research and future prospects. J Hepatocell Carcinoma. (2021) 8:887–97. doi: 10.2147/jhc.S284440, PMID: 34386437
38. Bloom M, Podder S, Dang H, and Lin D. Advances in immunotherapy in hepatocellular carcinoma. Int J Mol Sci. (2025) 26:1936. doi: 10.3390/ijms26051936, PMID: 40076561
39. Cheng AL, Hsu C, Chan SL, Choo SP, and Kudo M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J Hepatol. (2020) 72:307–19. doi: 10.1016/j.jhep.2019.09.025, PMID: 31954494
40. Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2–3 study. Lancet Oncol. (2021) 22:977–90. doi: 10.1016/s1470-2045(21)00252-7, PMID: 34143971
41. Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, and Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. (2020) 17:139–52. doi: 10.1038/s41575-019-0229-4, PMID: 31792430
42. Zheng J, Wang S, Xia L, Sun Z, Chan KM, Bernards R, et al. Hepatocellular carcinoma: signaling pathways and therapeutic advances. Signal Transduction Targeted Ther. (2025) 10:35. doi: 10.1038/s41392-024-02075-w, PMID: 39915447
43. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2, PMID: 28434648
44. Wang Z, Wang Y, Gao P, and Ding J. Immune checkpoint inhibitor resistance in hepatocellular carcinoma. Cancer Lett. (2023) 555:216038. doi: 10.1016/j.canlet.2022.216038, PMID: 36529238
45. Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017, PMID: 28187290
46. Polaris Observatory HCV Collaborators . Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol. (2017) 2:161–76. doi: 10.1016/s2468-1253(16)30181-9, PMID: 28404132
47. Guntipalli P, Pakala R, Kumari Gara S, Ahmed F, Bhatnagar A, Coronel Endaya MK, et al. Worldwide prevalence, genotype distribution and management of hepatitis C. Acta Gastroenterol Belg. (2021) 84:637–56. doi: 10.51821/84.4.015, PMID: 34965046
48. A Package for Survival Analysis in R. R package version 3.8-3 (2024). Available online at: https://CRAN.R-project.org/package=survival. (Accessed March 2025).
49. Feber A, Guilhamon P, Lechner M, Fenton T, Wilson GA, Thirlwell C, et al. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. (2014) 15:R30. doi: 10.1186/gb-2014-15-2-r30, PMID: 24490765
50. Li H and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324, PMID: 19451168
51. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352, PMID: 19505943
52. Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L. Calling somatic SNVs and Indels with Mutect2. bioRxiv. (2019) 861054. doi: 10.1101/861054
53. Richman LP, Vonderheide RH, and Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. (2019) 9:375–82.e4. doi: 10.1016/j.cels.2019.08.009, PMID: 31606370
54. Mayakonda A, Lin DC, Assenov Y, Plass C, and Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. (2018) 28:1747–56. doi: 10.1101/gr.239244.118, PMID: 30341162
55. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). (2012) 6:80–92. doi: 10.4161/fly.19695, PMID: 22728672
56. Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. (2015) 26:64–70. doi: 10.1093/annonc/mdu479, PMID: 25319062
57. Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, and Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. (2014) 30:3310–6. doi: 10.1093/bioinformatics/btu548, PMID: 25143287
58. Fortin JP, Triche TJ Jr., and Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. (2017) 33:558–60. doi: 10.1093/bioinformatics/btw691, PMID: 28035024
Keywords: hepatocellular carcinoma, tumour evolution, immune selection, immune evasion, whole-exome sequencing
Citation: Chakravarthy A, Pasini E, Zhao X, To J, Shen SYR, Fischer S, Ghanekar A, Vogel A, Grant RC, Knox J, Sapisochin G, Gores GJ, De Carvalho D and Bhat M (2025) Evolutionary dynamics of recurrent hepatocellular carcinoma under divergent immune selection pressures. Front. Oncol. 15:1537087. doi: 10.3389/fonc.2025.1537087
Received: 29 November 2024; Accepted: 07 July 2025;
Published: 04 August 2025.
Edited by:
Jiang Chen, Zhejiang University, ChinaReviewed by:
Mohammad Imran K. Khan, Columbia University, United StatesMansoor-Ali Vaali-Mohammed, King Saud University, Saudi Arabia
Yin Tang, Institute for Systems Biology (ISB), United States
Copyright © 2025 Chakravarthy, Pasini, Zhao, To, Shen, Fischer, Ghanekar, Vogel, Grant, Knox, Sapisochin, Gores, De Carvalho and Bhat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel De Carvalho, ZGFuaWVsLmRlY2FydmFsaG9AdWhuLmNh; Mamatha Bhat, bWFtYXRoYS5iaGF0QHVobi5jYQ==
†These authors share senior authorship
‡ORCID: Ankur Chakravarthy, orcid.org/0000-0002-2434-6781
Elisa Pasini, orcid.org/0000-0002-2434-6781
Anand Ghanekar, orcid.org/0000-0003-2966-4145
Arndt Vogel, orcid.org/0000-0003-0560-5538
Robert C. Grant, orcid.org/0000-0002-9821-0377
Gonzalo Sapisochin, orcid.org/0000-0001-9527-8723
Gregory J. Gores, orcid.org/0000-0003-3151-009X
Mamatha Bhat, orcid.org/0000-0003-1960-8449