 Guanlin Dai
Guanlin Dai Ping Wang
Ping Wang Danqing Wang
Danqing Wang- 1Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children, Sichuan University, Ministry of Education, Chengdu, Sichuan, China
Background: BRCA1 and BRCA2 genes are well-established tumor suppressors, crucial for maintaining genomic stability through their roles in DNA repair. Pathogenic variants in BRCA1/2 genes are implicated in increased susceptibility to breast and ovarian cancers. However, variant interpretation remains challenging due to the large size of BRCA1/2 (>80 kb) and the broad spectrum of variant forms, particularly for rare or recently identified variants lacking adequate population, functional or segregation data.
Case presentation: This report describes a case of high-grade serous ovarian carcinoma in a patient with a strong family history of cancer. Both her mother and sister died of ovarian cancer. Genetic testing identified the germline variant BRCA1 c.5193 + 2dupT both in the patient’s tumor and peripheral blood samples, without other abnormalities detected in genomic homologous recombination deficiency assessment. Her daughter was identified as an unaffected carrier of this variant. Unfortunately, the BRCA1 status of deceased relatives could not be determined due to the unavailability of samples. Functional studies, including minigene splicing assay and transcript analysis, demonstrated that this variant induces a splicing error, specifically, an aberrant skipping of exon 18, resulting in dysfunction of the BRCA1-encoded protein. These findings provide a mechanistic explanation for the observed cancer susceptibility in this family.
Conclusion: This case highlights a rare germline variant, BRCA1 c.5193 + 2dupT, in a family with a strong cancer history. In vitro functional assays confirmed that this variant induces exon 18 skipping through aberrant splicing, leading to dysfunction of BRCA1-encoded protein. To our knowledge, this is the first functional characterization of the variant BRCA1 c.5193 + 2dupT, and our data provide novel insights for risk assessment and precision treatment strategies in carriers of this variant.
1 Introduction
Breast cancer is the most common malignancy in women worldwide. Ovarian cancer, although less frequent, remains a significant cause of cancer-related death due to late-stage diagnosis. Data from the 2021 global burden of disease, injuries, and risk factors study shows that the global incidence of breast cancer is approximately 2,000,000 cases annually and increases year by year (1). Meanwhile, ovarian cancer accounts for an estimated 200,000 new cases and 100,000 deaths globally each year, ranking the first in mortality among gynecological malignancies (1). BRCA1 and BRCA2 genes are critical tumor suppressor genes, playing an important role in homologous recombination mechanism of DNA repair. Variants in these genes can lead to genomic instability, promote tumor cell proliferation and prevent normal cell differentiation, thereby facilitating tumor development (2). Studies have shown that BRCA1/2 gene variants are relatively common in breast and ovarian cancers (3). Germline variants in BRCA1/2 gene account for 80% to 90% in cases of hereditary breast and ovarian cancer (HBOC) (3–5). Carriers of pathogenic BRCA1 variants have a cumulative risk of up to 60% for breast cancer and 59% for ovarian cancer by age 70. Pathogenic BRCA2 variant carriers, have 55% and 16% risks, respectively. In contrast, the lifetime risks of breast and ovarian cancer in general population are approximately 12% and 1.3%, respectively (5, 6). However, not all BRCA1/2 variants impair the encoded protein function and variant interpretation remains challenging due to the large size of BRCA1/2 (>80 kb) the broad spectrum of variant forms.
The American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guideline categorizes variants into five classes including pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign based on population data, computational predictions, functional studies, and familial co-segregation data (7, 8). Accurate identification and interpretation of BRCA1/2 variants are crucial for risk assessment of breast, ovarian and other cancers in women, and serve as important biomarkers for precision treatment. Current sequencing technologies, including Sanger sequencing and next-generation sequencing (NGS), can accurately detect point variants, small insertions, deletions, and rearrangements (9, 10). Advances in sequencing technologies continue to expand the molecular spectrum and drive genomics research. However, the recently identified variants often lack sufficient population and functional data, making their clinical significance unclear and limiting guidance for the clinical management which may result in missed opportunities for early intervention or targeted therapy.
In this report, we detected a highly conserved intronic variant BRCA1 c.5193 + 2dupT in a family with a cancer history. We studied the mRNA splicing pattern by constructing a minigene vector in vitro, followed by cell transfection and transcript analysis. This is the first report to conduct functional assays in vitro to validate the pathogenicity of the variant BRCA1 c.5193 + 2dupT, strictly in accordance with the ACMG/AMP variant classification guidelines.
2 Case presentation
In November 2023, a 61-year-old female was referred to our department with newly diagnosed ovarian cancer for further systematic treatment, following a recent surgical procedure. She had been diagnosed with high-grade serous ovarian carcinoma (HGSOC) at the local hospital in September 2023 and underwent tumor reduction surgery. Postoperatively, genetic testing was performed using the BRCA1 and BRCA2 Gene Mutation Detection Kit, a commercial panel targeting the BRCA1 and BRCA2 genes using combinatorial probe-anchor synthesis sequencing technology. Genomic homologous recombination deficiency (HRD) was assessed with the HRD Detection Kit, which qualitatively detects HRD through high-throughput sequencing and a genomic scar analysis algorithm. Library preparation was carried out with reagents supplied in the kits, and sequencing was performed on the DNBSEQ-T7 platform (BGI Biotechnology Co., Ltd., Wuhan, China). The variant BRCA1 c.5193 + 2dupT (GRCh37/hg19) was detected in both tumor and peripheral blood samples of this patient. No other pathogenic or likely pathogenic variants were identified.
This patient reported a typical family history of ovarian cancer. Her mother (I-1) was diagnosed with HGSOC at the age of 71 and unfortunately passed away due to this cancer at age 75. Her sister (II-6) was also diagnosed with HGSOC at age 49 and succumbed to ovarian cancer three years post-surgery. The pedigree chart is detailed in Figure 1A. As of November 2023, no other family members had reported a history of cancer. To further evaluate this variant in family members, Sanger sequencing was performed, and the patient’s daughter (III-2) was identified as an unaffected heterozygous carrier. While it was absent in other relatives (Figure 1B). Her mother (I-1) and sister (II-6) had died, and thus failed to perform sequencing.
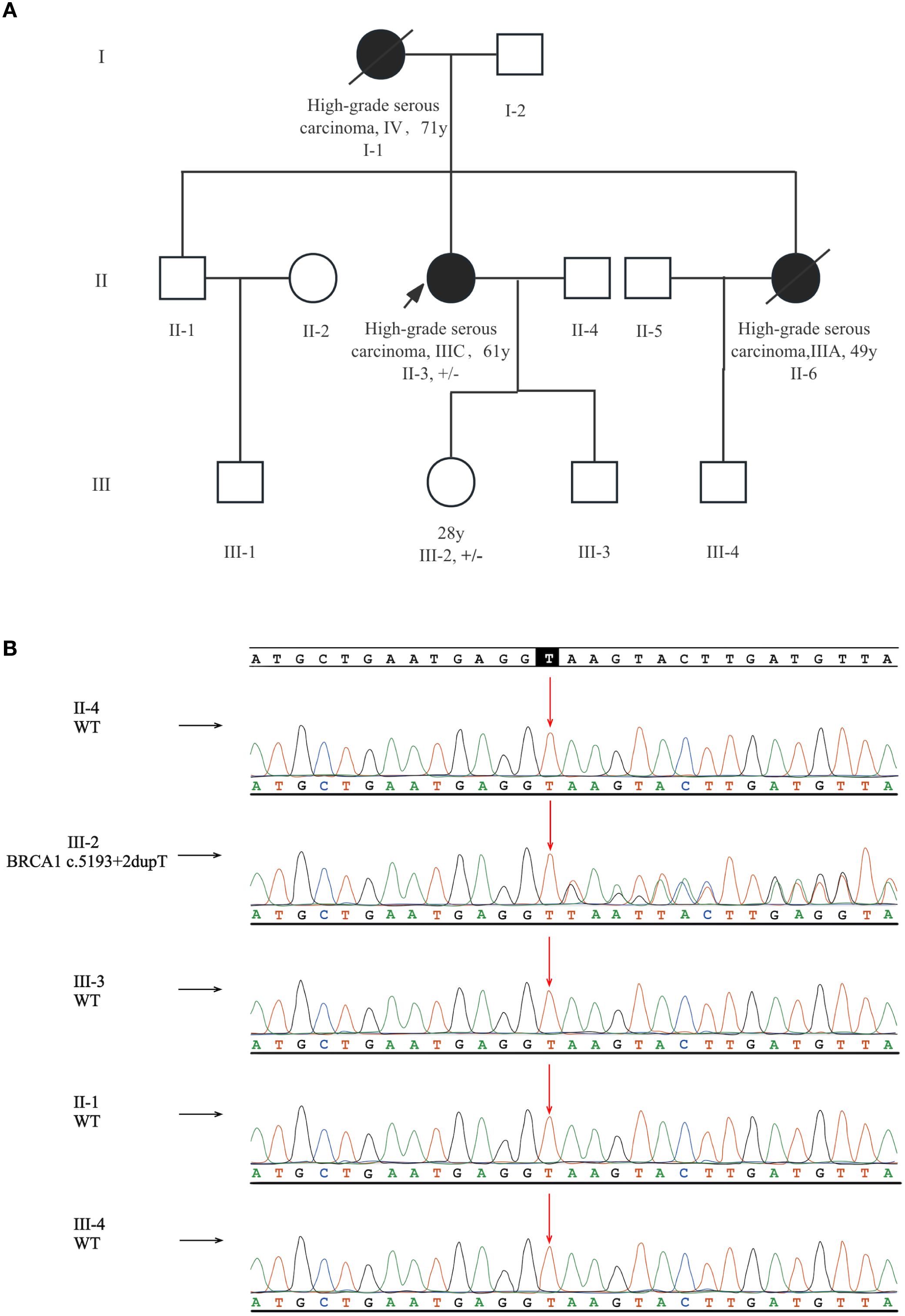
Figure 1. Pedigree and Sanger sequencing results of the family. (A) The pedigree of this family with a intronic BRCA1 variant (c.5193 + 2dupT). The proband is marked by an arrow. Patients diagnosed with ovarian cancer are denoted by solid black symbols, and deceased members are marked with a diagonal line. Carriers of the BRCA1 c.5193 + 2dupT variant are annotated in the figure (+/– heterozygous carrier). (B) The proband’s daughter was diagnosed as an unaffected carrier.
The variant BRCA1 c.5193 + 2dupT, located in intron 19, is well conserved and has not been recorded in population databases such as the Exome Aggregation Consortium, 1000 Genomes Project, and the Exome Variant Server. In the ClinVar database and previous literature, this variant was classified discordantly as pathogenic or of uncertain significance, without any functional assessment and familial co-segregation analysis. Based on existing data at that time, the pathogenicity of BRCA1 c.5193 + 2dupT was unclear and could only be classified as a VUS according to the ACMG/AMP guideline.
Traditional in-silicon prediction algorithms, including dbscSNV_ADA and dbscSNV_RF, were applied but yielded no positive results. However, SpliceAI, a deep-learning model, suggested that variant BRCA1 c.5193 + 2dupT could disrupt splicing process, causing a 2 bp loss at the splicing donor site with a score of 0.96, and a 42 bp loss at the acceptor site with a score of 0.89 (Figure 2). This could result in the aberrant transcript and the consequent loss of encoded protein function. Nevertheless, these predictions were solely based on machine learning, and are insufficient to support its pathogenicity.
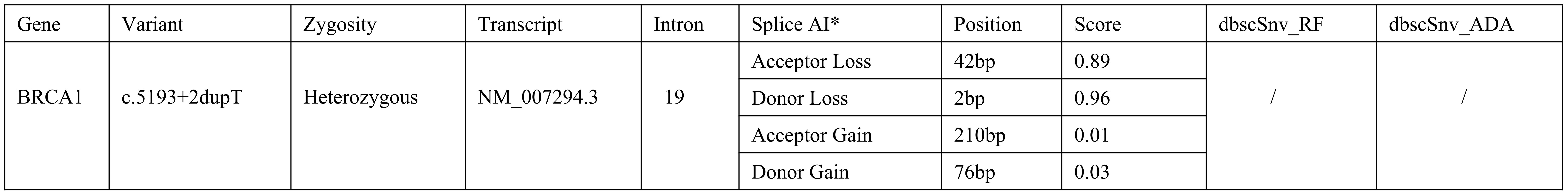
Figure 2. Bioinformatic Predictions of Splicing Disruption for BRCA1 c.5193 + 2dupT Variant. *The figure shows the SpliceAI output, including delta scores for donor and acceptor site loss. The delta scores indicate the probability of splicing disruption at the donor and acceptor sites.
To further validate these predictions, we performed a minigene splicing assay and transcript analysis. Human genomic DNA fragment including the splicing sites of exon 17 and 18 (BRCA1 RNA F: 5’-TGAATGAGGTTAAGTACTTGA; BRCA1 RNA R: 5’-TCAAGTACTTAACCTCATTCA) served as the template and was cloned into pcMINI-C vector for plasmid reconstruction (Supplementary Figure 1). Sequencing diagrams of the constructed plasmids were depicted in Figure 3A. The reconstructed plasmids were transfected into human 293T cells, and RNA samples were extracted 24 hours later for RT-PCR analysis. The aberrant transcript product in the BRCA1 c.5193 + 2dupT carrier was clearly identified with a distinct band by agarose gel electrophoresis. (Figure 3B). These products were further validated by Sanger sequencing (Figure 3C). These results indicated that the BRCA1 c.5193 + 2dupT variant can disrupt the splicing pattern, leading to the skipping of exon 18. The specific splicing patterns are shown in Figure 3D. We further organized the coding sequences of BRCA1 wild-type and variant-type of exon 18 skipping, as well as the corresponding amino acid sequences. Details have been provided in the Supplementary Material. Due to exon 18 skipping, transcript NM_007294.3 acquires a premature termination codon. This results in a truncated BRCA1-encoded protein (1718 amino acids) instead of the wild-type (1863 amino acids), thereby impairing protein function due to the loss of the C-terminal region. The schematic diagram is shown in Figure 3E.
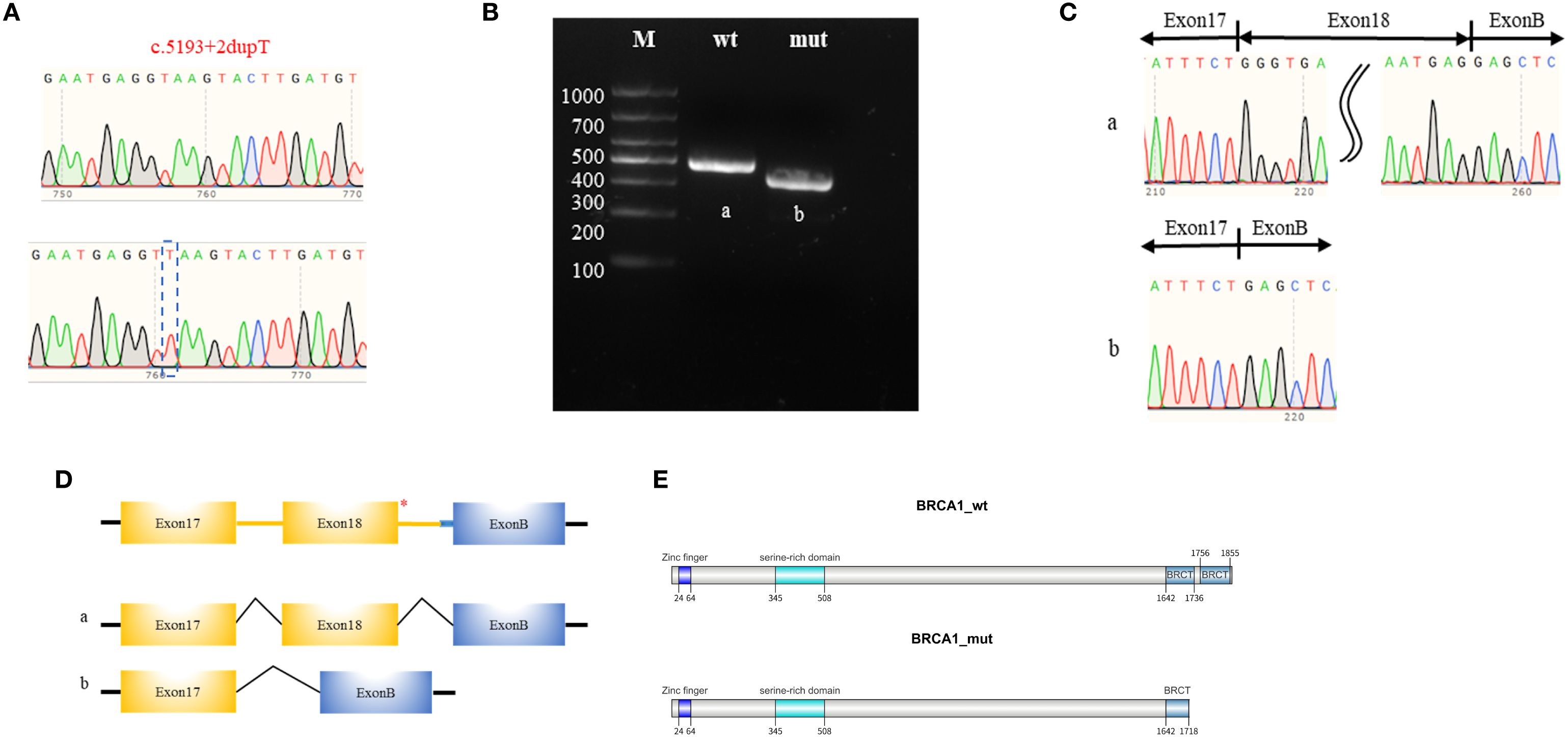
Figure 3. Minigene splicing assay and transcripts analysis. (A) Sanger sequencing chromatogram of the pcMINI-C-BRCA1-wt/mut plasmids. (B) Agarose gel electrophoresis of the transcription products, with the respective bands labeled as ‘a’ for the wild type and ‘b’ for the mutant. (C) Sanger sequencing chromatogram of the transcription bands. (D) Schematic diagram of the pcMINI-C-BRCA1 plasmids which contains a universal Exon B (57 base pairs, 57 bp). The asterisk (*) indicates the variant site. The schematic diagrams of the RNA splicing patterns for the wild-type (a) and the mutant-type (b). (E) The transcript NM_007294.3 with exon 18 skipping acquires a premature termination codon which results in the production of a truncated BRCA1 protein (1718 amino acids) instead of the wild-type protein (1863 amino acids), thereby impairing protein function due to the loss of the C-terminal region.
Based on these findings, we reclassified this variant strictly in accordance with the ACMG/AMP guidelines. This variant meets criteria PS3 (splicing error confirmed by functional assay), PM2 (absent in normal controls), PS4_P (reported in more than 2 probands), PP3 (computational support) and PP5 (previously reported in ClinVar), warranting reclassification into “likely pathogenic”. The specific basis for reclassification is summarized in Figure 4. These results provide a mechanistic explanation for the cancer susceptibility within this family.
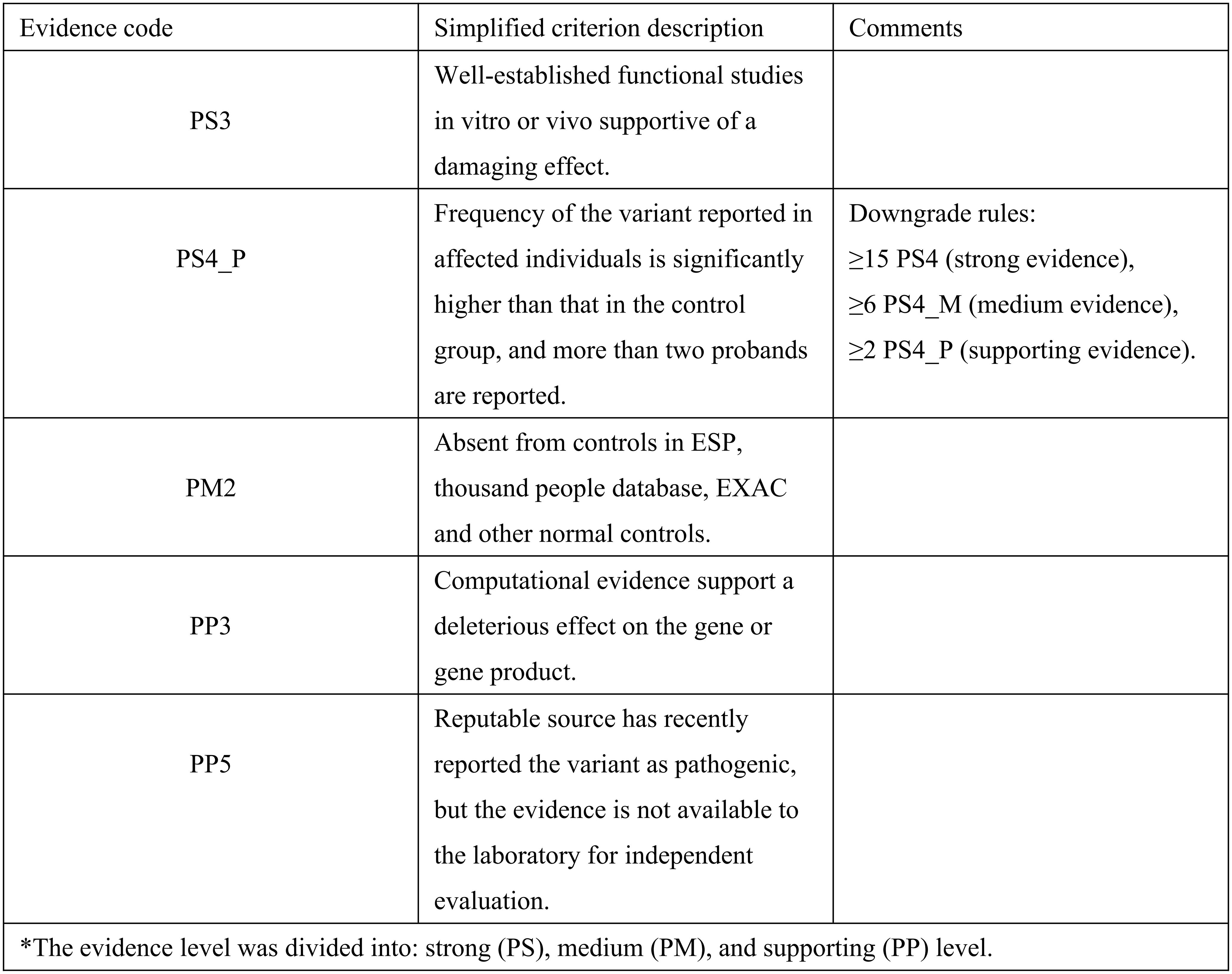
Figure 4. Evidence for the classification of BRCA1 c.5193 + 2dupT as likely pathogenic based on the ACMG/AMP variant classification guidelines.
3 Discussion
In this report, we proposed and confirmed for the first time that BRCA1 c.5193 + 2dupT variant disrupts splicing pattern, causing the skipping of exon 18 in transcripts. Due to the exon 18 skipping, the transcript (NM_007294.3) acquires a premature termination codon. This results in a truncated BRCA1-encoded protein, thereby impairing protein function due to the loss of the C-terminal region. This report integrates the results from familial co-segregation analysis, computational prediction, and functional assays in vitro, providing a mechanistic explanation for the cancer susceptibility within this family.
RNA splicing is an essential biological process in eukaryotic gene expression, precisely removing introns from precursor mRNA through the recognition of cis-acting elements (11). Accurate RNA splicing relies on both canonical splice signals (CSSs) including splice donor and acceptor sites, and auxiliary splicing regulatory elements (SREs), including exonic or intronic splicing enhancers and silencers (12). Single-nucleotide variants can disrupt these signals or elements, leading to aberrant splicing events, manifesting as exon skipping, intron retention or activation of cryptic splice sites (13). These events often generate transcripts with premature termination codons, frameshift variants, or in-frame deletions/insertions, which may result in structural or functional abnormalities of the encoded proteins (13). Abnormal splicing events have been implicated in the pathogenesis of genetic disorders and cancers, accounting for up to 60% in inherited monogenic disorders (14). Although 70%-80% of pathogenic splice events are caused through CSSs disruptions, most variants outside these regions such as those affecting SREs, remain undiagnosed (15). Previously reported the BRCA1 c.5080>T variant leads to the skipping of exon 18 through disrupting a splicing enhancer (16). Our findings extend this paradigm, demonstrating that a single duplicated nucleotide within an intronic region can also interfere with the splicing process, resulting in the aberrant skipping of exon 18. These findings underscore the necessity of evaluating both canonical and non-canonical splicing regions in BRCA1 gene.
Functional analysis is crucial for interpreting the biological significance of variants, especially those in non-canonical splicing regions. In cases of HBOC, molecular profiling analysis, particularly of the BRCA1 and BRCA2 genes, assists in risk assessment and targeted therapies. However, most diagnosed variants are identified solely through DNA sequencing. Variants in non-canonical splicing regions, due to the lack of sufficient population and functional data, are often classified as VUS, which cannot provide guidance for the clinical management (17). Thus, the 9% of splicing variations previously reported in the Human Gene Mutation Database were undoubtedly underestimated (18). For patients with a potential family history of cancer, further functional analysis of variants with unknown significance is essential.
Computational prediction serves as a preliminary screening tools for functional analysis. Traditional tool, such as dbscSNV_ADA and dbscSNV_RF, are mainly based on traditional machine learning algorithms like adaptive boosting and random forest, relying on existing data of splicing sites and splicing signal. Their prediction performs well on variants at CSSs, including splicing donor (+1 and +2) and acceptor (-1 and -2) sites (19). If the variants in question fall outside the designed scope, it could yield negative results. SpliceAI, a 32-layer deep neural network trained on more diverse and extensive datasets that may include examples similar to the variant being studied, can accurately predict the impact of variants on splicing sites, especially for non-canonical or deep intronic variants (20). In contrast, traditional tools, lacking such extensive training datasets, may fail to detect splicing events in these cases. The output of SpliceAI typically includes the results with delta scores which indicate the probability of function disruption of variant sites (ranging from 0 to 1). A delta score above 0.8 typically signifies a high likelihood of splicing disruption (21). In this report, SpliceAI predicted that the BRCA1 c.5193 + 2dupT variant would cause a 2 bp loss at the donor site with a score of 0.96 and a 42 bp loss at the acceptor site with a score of 0.89. These scores suggest that the variant may severely impair splice site function, result in detrimental impact on the transcript product and encoded protein, meeting the PP3 criteria in ACMG/AMP guidelines.
Although spliceAI has outperformed in this report, positive results from computational prediction alone remain insufficient for the pathogenic classification of a variant. Functional assays, such as minigene splicing assays, can directly observe the impact of variants on RNA splicing and provide authentic biological evidence. The minigene splicing assay, analyzing the splicing outcome of a single allele, is a powerful tool for evaluating allele-specific expression. It can demonstrate that the variant allele produces abnormal transcripts that are predicted to disrupt the encoded protein’s structure and function. This is a crucial step in classifying splicing variants as pathogenic and can be exemplified by the variant BRCA1 c.5193 + 2dupT in this report, where computational tools and clinical databases exhibited discordant interpretations. Our minigene splicing assay and RT-PCR analysis directly demonstrated the aberrant splicing events, resolving this ambiguity through functional evidence. Similarly, the variant BRCA1 c.5152 + 5G>C was initially classified as VUS until a minigene assay confirmed the aberrant skipping of exon 17 (22). The variant BRCA1 c.442–7 T>A, with conflicting results in multiple computational predictions, was further confirmed by minigene splicing assay to cause a 5-nt insertion before exon 8: TTTAG in the transcript (23). The variant BRCA1 c.231 G>T was predicted to have no effect in computational prediction, while minigene assay revealed the exon 6 skipping in transcripts (23). The variant c.5193 + 2T>C, located at CSS and multiple computational algorithms have predicted that this variant might disrupt the donor site. While minigene splicing assay revealed no differences in the transcription products between this variant and the wild-type allele (23). In these cases, employing additional methods to ascertain the functional impact of the variants appears to be essential. RNA sequencing can directly analyze transcripts through high-throughput sequencing and detect aberrant splicing events (24). Due to the availability and stability of RNA in tumor or blood samples, RNA sequencing is rarely included in the routine molecular diagnostics. Moreover, when multiple suspected variants are present in a single allele, minigene splicing assays are still needed to elucidate the causal relationship between variants and aberrant transcripts. In this report, the unavailability of RNA data from the proband limited our direct assessment of the variant’s impact on transcripts. Nevertheless, we conducted a minigene splicing assay to simulate the transcription process in vitro and analyzed the transcript products, thereby indirectly confirming the aberrant splicing event caused by this variant. Current ACMG/AMP guidelines prioritize functional evidence for splicing variant classification, and minigene splicing assays serve as a critical part in fulfilling these evidence requirements (8).
In this report, the unavailability of biological samples from the deceased relatives precluded us from providing rigorous segregation data, and fulfilling the stringent PS4 criteria in the ACMG/AMP guideline. The latest ACMG/AMP guideline suggests that for extremely rare variants, case-control studies may not be statistically significant and if a variant originally observed in multiple patients with the same phenotype (and absent in controls) would have qualified a downgraded PS4 criteria from the strong evidence of pathogenicity to a moderate or supporting level of evidence (36). The downgrade rules are as follows: ≥15 probands for PS4 (strong evidence), ≥6 probands for PS4_M (moderate evidence), and ≥2 probands for PS4_P (supporting evidence). Based on sporadic cases recorded in the database and confirmed cases reported in this report, the frequency of the variant BRCA1 c.5193 + 2dupT in the affected population is significantly higher than that in the control population, meeting the ACMG/AMP criteria for evidence downgrading. Therefore, PS4 can be downgraded to the supporting level of evidence (PS4_P). The pathogenicity classification evidence for this variant ultimately includes one strong (PS3), one moderate (PM2), and three supporting (PS4_P, PP3, and PP5) pieces of evidence, still qualifying it as a likely pathogenic variant according to the ACMG/AMP guideline. While the lack of genetic data weakens the segregation evidence in this report, PS4_P still provides meaningful support for its pathogenicity classification.
The BRCA1 protein, crucial for in maintaining genomic stability, primarily exerts its tumor-suppressive function by mediating the homologous recombination repair mechanism for DNA double-strand breaks (2). Splicing variants in BRCA1 can lead to homologous recombination deficiency, significantly increasing genomic instability and driving tumorigenesis (2). Germline variants in BRCA1 gene, classified as pathogenic or likely pathogenic, confer markedly elevated lifetime cancer risks, with breast cancer risk reaching 60% and ovarian cancer risk reaching 59% (5, 6). Therefore, BRCA1 gene variants can be used for risk assessment of breast, ovarian and other cancers. Given that BRCA1/2 germline variants are inherited in an autosomal dominant manner, genetic counselling and testing are recommended for the first-degree relatives of the proband with risk management based on the screening results (9, 25). Unaffected carriers of pathogenic or likely pathogenic variants in BRCA1 should undergo a standardized surveillance procedure in accordance with their age and reproductive options. Females should initiate the consistent breast self-examination starting at age 18 and undergo clinical breast examination every 6–12 months beginning at age 25 (26). From age 25 onward, regular breast imaging surveillance should be implemented, with breast MRI preferred due to the established radiation-associated risk in carriers (27). Furthermore, risk-reducing mastectomy and chemopreventive agents require individual risk-benefit assessment to thoroughly weigh the interventions’ advantages against potential risk (26). The risk management of ovarian cancer requires a comprehensive consideration integrating cancer risk reduction, fertility preservation, and management of hormone-related symptoms. Current primary screening tools include CA125 and pelvic ultrasound, and definitive surgical risk reduction via bilateral salpingo-oophorectomy is recommended between ages 35 and 40 years following completion of childbearing (26, 28).
BRCA1 gene variants are also critical biomarkers for precision treatment. Studies have shown that ovarian cancer patients with pathogenic BRCA variants are more sensitive to platinum-based chemotherapy and can benefit from treatment with poly (ADP-ribose) polymerase (PARP) inhibitors (29). The efficacy and safety of PARP inhibitors including Olaparib, niraparib, rucaparib, and talazoparib, have been demonstrated in patients with breast or ovarian cancer carrying pathogenic BRCA1/2 variants (29–32). Therefore, conducting BRCA variant testing and functional interpretation for patients with breast or ovarian cancer is beneficial for devising precision treatment plans. In addition to BRCA1/2 genes, other homologous recombination repair genes are also implicated in breast and ovarian cancers, such as PALB2, ATM, and CHEK2 (33–35). Variants in these genes may also increase the risk of breast and ovarian cancers. Therefore, genetic testing and counselling is extremely important in clinical practice. Early detection and management of breast and ovarian cancer risks can help improve patients’ prognosis and quality of life.
Accurate interpretation and reclassification of the variant BRCA1 c.5193 + 2dupT has significant clinical implications for this family. It provides a mechanistic explanation for their observed cancer susceptibility and enables more informed decisions regarding surveillance, risk-reducing interventions, and targeted therapies. This case underscores the importance of integrating functional studies with genetic counselling and highlights the need for further research to assess the clinical significance of VUS in BRCA1/2 genes. This study also provides a novel research approach under the condition that clinical samples are still lacking for the discovery of rare BRCA1/2 variants in the clinic. Future research should focus on developing comprehensive databases and functional assays to address the challenges posed by VUS, ultimately improving clinical outcomes for patients and families affected by hereditary cancer syndromes.
4 Conclusion
In conclusion, this study provides functional evidence for the likely pathogenicity of the variant BRCA1 c.5193 + 2dupT, emphasizing the importance of integrating computational predictions with functional validation in variant interpretation. These findings have significant clinical implications for the carriers, enabling more informed risk assessment and management strategies. Future research should focus on developing comprehensive databases and functional assays to address the challenges posed by VUSs, ultimately improving clinical outcomes for patients and families affected by hereditary cancer syndromes.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by medical ethics committee of West China Second Hospital of Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
GD: Conceptualization, Visualization, Formal Analysis, Writing – original draft, Data curation. PW: Supervision, Writing – review & editing, Conceptualization, Formal Analysis. DW: Formal Analysis, Project administration, Writing – review & editing, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Project of Science and Technology Department of Sichuan Province (2020YFS0131).
Acknowledgments
We would like to extend our sincere gratitude to this patient and her relatives who participated in this report for their invaluable contributions and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1623700/full#supplementary-material
Abbreviations
HBOC, hereditary breast and ovarian cancer; ACMG/AMP, The American College of Medical Genetics and Genomics/Association for Molecular Pathology; VUS, variant of uncertain significance; SRE, splicing regulatory elements; HGSOC, high-grade serous ovarian carcinoma.
References
1. Sun P, Yu C, Yin L, Chen Y, Sun Z, Zhang T, et al. Global, regional, and national burden of. female cancers in women of child-bearing age, 1990–2021: analysis of data from the global burden of disease study 2021. eClinicalMedicine. (2024) 74:102713. doi: 10.1016/j.eclinm.2024.102713
2. Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. (2017) 49:1476–86. doi: 10.1038/ng.3934
3. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. (2017) 317:2402. doi: 10.1001/jama.2017.7112
4. Gabai-Kapara E, Lahad A, Kaufman B, Friedman E, Segev S, Renbaum P, et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci U.S.A. (2014) 111:14205–10. doi: 10.1073/pnas.1415979111
5. Golmard L, Delnatte C, Laugé A, Moncoutier V, Lefol C, Abidallah K, et al. Breast and ovarian cancer predisposition due to de novo BRCA1 and BRCA2 mutations. Oncogene. (2016) 35:1324–7. doi: 10.1038/onc.2015.181
6. Engel C, Fischer C, Zachariae S, Bucksch K, Rhiem K, Giesecke J, et al. Breast cancer risk in BRCA1/2 mutation carriers and noncarriers under prospective intensified surveillance. Int J Cancer. (2020) 146:999–1009. doi: 10.1002/ijc.32396
7. Walker LC, de la HM, Wiggins GAR, Lindy A, LM V, MT P, et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Gene. (2023) 110:1046–67. doi: 10.1016/j.ajhg.2023.06.002
8. Harrison SM and Rehm HL. Is “likely pathogenic” really 90% likely? Reclassification data in ClinVar. Genome Med. (2019) 11:72. doi: 10.1186/s13073-019-0688-9
9. Yoshida R. Hereditary breast and ovarian cancer (HBOC): review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer. (2021) 28:1167–80. doi: 10.1007/s12282-020-01148-2
10. Yildiz Tacar S, Bozgeyik E, Seber ES, Yetisyigit T, Tozkir H, Avci O, et al. Next generation sequencing analysis of BRCA1 and BRCA2 identifies novel variations in breast cancer. Life Sci. (2020) 261:118334. doi: 10.1016/j.lfs.2020.118334
11. Carazo F, Romero JP, and Rubio A. Upstream analysis of alternative splicing: a review of computational approaches to predict context-dependent splicing factors. Brief Bioinform. (2019) 20:1358–75. doi: 10.1093/bib/bby005
12. Fraile-Bethencourt E, Díez-Gómez B, Velásquez-Zapata V, Acedo A, Sanz DJ, and Velasco EA. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PloS Genet. (2017) 13:e1006691. doi: 10.1371/journal.pgen.1006691
13. Shao Y and Zhang R. Identifying six single nucleotide variants in the COL17A1 gene that alter RNA splicing: database analysis and minigene assays. Sci Rep. (2025) 15:11387. doi: 10.1038/s41598-025-95851-9
14. Bradley RK and Anczuków O. RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer. (2023) 23:135–55. doi: 10.1038/s41568-022-00541-7
15. Lord J, Gallone G, Short PJ, McRae JF, Ironfield H, Wynn EH, et al. Pathogenicity and selective constraint on variation near splice sites. Genome Res. (2019) 29:159–70. doi: 10.1101/gr.238444.118
16. Wu X, Wu L, Kong B, Liu J, Yin R, Wen H, et al. The first nationwide multicenter prevalence study of germline BRCA1 and BRCA2 mutations in chinese ovarian cancer patients. Int J Gynecological Cancer. (2017) 27:1650–7. doi: 10.1097/IGC.0000000000001065
17. Sinha S and Wang SM. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput Struct Biotechnol J. (2020) 18:723–36. doi: 10.1016/j.csbj.2020.03.013
18. Whiley PJ, Guidugli L, Walker LC, Healey S, Thompson BA, Lakhani SR, et al. Splicing and multifactorial analysis of intronic BRCA1 and BRCA2 sequence variants identifies clinically significant splicing aberrations up to 12 nucleotides from the intron/exon boundary. Hum Mutat. (2011) 32:678–87. doi: 10.1002/humu.21495
19. Ghosh R, Oak N, and Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. (2017) 18:225. doi: 10.1186/s13059-017-1353-5
20. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176:535–548.e24. doi: 10.1016/j.cell.2018.12.015
21. De Sainte Agathe JM, Filser M, Isidor B, Besnard T, Gueguen P, Perrin A, et al. SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Hum Genomics. (2023) 17:7. doi: 10.1186/s40246-023-00451-1
22. De Juan Jiménez I, García Casado Z, Palanca Suela S, Esteban Cardeñosa E, López Guerrero JA, Segura Huerta Á, et al. Novel and recurrent BRCA1/BRCA2 mutations in early onset and familial breast and ovarian cancer detected in the Program of Genetic Counseling in Cancer of Valencian Community (eastern Spain). Relationship of family phenotypes with mutation prevalence. Familial Cancer. (2013) 12:767–77. doi: 10.1007/s10689-013-9622-2
23. Dong Z, Wang Y, Zhang J, Zhu F, Liu Z, Kang Y, et al. Analyzing the effects of BRCA1/2 variants on mRNA splicing by minigene assay. J Hum Genet. (2023) 68:65–71. doi: 10.1038/s10038-022-01077-2
24. Mertes C, Scheller IF, Yépez VA, Çelik MH, Liang Y, Kremer LS, et al. Author Correction: Detection of aberrant splicing events in RNA-seq data using FRASER. Nat Commun. (2022) 13:3474. doi: 10.1038/s41467-020-20573-7
25. Yadav S, Boddicker NJ, Na J, Polley EC, Hu C, Hart SN, et al. Contralateral breast cancer risk among carriers of germline pathogenic variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2. JCO. (2023) 41:1703–13. doi: 10.1200/JCO.22.01239
26. Daly MB, Pal T, Maxwell KN, Churpek J, Kohlmann W, AlHilli Z, et al. NCCN guidelines® Insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 2.2024: featured updates to the NCCN guidelines. J Natl Compr Cancer Network. (2023) 21:1000–10. doi: 10.6004/jnccn.2023.0051
27. Wehbe A, Katlin F, Sharma E, Hans M, Graichen MK, Bychkovsky BL, et al. Breast imaging recommendations for young females (age < 40 years) with ≥ 20% lifetime breast cancer risk: practice patterns at a specialized clinic. Breast Cancer Res Treat. (2025) 212(3):467–74. doi: 10.1007/s10549-025-07738-y
28. Liu J, Berchuck A, Backes FJ, Cohen J, Grisham R, Leath CA, et al. NCCN guidelines® Insights: ovarian cancer/fallopian tube cancer/primary peritoneal cancer, version 3.2024: featured updates to the NCCN guidelines. J Natl Compr Cancer Network. (2024) 22:512–9. doi: 10.6004/jnccn.2024.0052
29. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. (2017) 377:523–33. doi: 10.1056/NEJMx170012
30. Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 390:1949–61. doi: 10.1016/S0140-6736(17)32702-2
31. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
32. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tab lets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. (2017) 18:1274–84. doi: 10.1016/S1470-2045(17)30639-3
33. Weitzel JN, Neuhausen SL, Adamson A, Tao S, Ricker C, Maoz A, et al. Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA -negative Hispanics with breast cancer. Cancer. (2019) 125:2829–36. doi: 10.1002/cncr.32083
34. Parsons MT, Tudini E, Li H, Hahnen E, Wappenschmidt B, Feliubadaló L, et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum Mutation. (2019) 40:1557–78. doi: 10.1002/humu.23818
35. Caminsky NG, Mucaki EJ, Perri AM, Lu R, Knoll JHM, and Rogan PK. Prioritizing variants in complete hereditary breast and ovarian cancer genes in patients lacking known BRCA mutations. Hum Mutation. (2016) 37:640–52. doi: 10.1002/humu.22972
36. On behalf of the Clinical Genome Resource Sequence Variant Interpretation Working Group, SE B, AN AT, FJ C, GR C, MS G, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. (2020) 12:3. doi: 10.1186/s13073-019-0690-2
Keywords: BRCA1, germline variant, splicing error, hereditary breast and ovarian cancer, variant of uncertain significance
Citation: Dai G, Wang P and Wang D (2025) Case Report: Functional validation of a rare variant BRCA1 c.5193 + 2dupT in a family with cancer history. Front. Oncol. 15:1623700. doi: 10.3389/fonc.2025.1623700
Received: 06 May 2025; Accepted: 12 September 2025;
Published: 30 September 2025.
Edited by:
Gianluca Tedaldi, AUSL Romagna, Cesena, ItalyReviewed by:
Prem Prakash, Meharry Medical College, United StatesSungchan Gwark, Ewha Womans University Seoul Hospital, Republic of Korea
Yinlong Yang, Fudan University, China
Copyright © 2025 Dai, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danqing Wang, ZGFucWluZzEyMDNAMTYzLmNvbQ==; Ping Wang, d2FuZ3BpbmdfODg2QDEyNi5jb20=
†These authors have contributed equally to this work