 Nawel Zouggari1,2†
Nawel Zouggari1,2† Camilla Trugenberger1,2†
Camilla Trugenberger1,2† Valentine Du Bois1
Valentine Du Bois1 Wenwen Wang1Giacomo G. Rossetti2Thanos D. Halazonetis2
Wenwen Wang1Giacomo G. Rossetti2Thanos D. Halazonetis2 Intidhar Labidi-Galy1,3*
Intidhar Labidi-Galy1,3*- 1Department of Medicine and Center of Translational Research in Onco-Hematology, Faculty of Medicine, University of Geneva, Geneva, Switzerland
- 2Department of Molecular and Cellular Biology, Faculty of Sciences, University of Geneva, Geneva, Switzerland
- 3Department of Oncology, Geneva University Hospitals, Geneva, Switzerland
PARP inhibitors are widely used class of drugs for the treatment of homologous recombination deficient cancers, including BRCA mutated ones. These drugs led to substantial improvement in survival, particularly for patients with BRCA mutated tumors. However, many patients eventually develop resistance to PARP inhibitors, mainly due to BRCA reversion mutations. Overcoming resistance to PARP inhibitors is an unmet medical need. Recently, it has been shown that BRCA-deficient cells are hypersensitive to the thymidine analogue 5-chloro-2’-deoxyuridine (CldU), either alone or in combination with PARP inhibitors. In this study, we show, across multiple BRCA2 mutated cell lines, that CldU sensitizes PARP inhibitor-resistant cells to PARP inhibitors. This synergy was also present in cell lines with BRCA2 reversion mutations and was associated with high levels of DNA damage and arrest in S phase. This effect, which is specific to thymidine analogue CldU, may open new avenues for the treatment of BRCA mutated cancers resistant to PARP inhibitors.
1 Introduction
Poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) induce cell death by exploiting the absence of homologous recombination in cancer cells harboring mutations in the BRCA1/BRCA2 genes (1). More precisely, cancer cells lacking the repair proteins BRCA1 and BRCA2 rely more heavily on PARP to repair their damaged DNA. Hence, inhibiting PARP leads to cell death as these cells are no longer able to repair the damage to their DNA. Studies have shown that loss of BRCA2 leads to cells being 100 to 1000 times more sensitive to PARPi, this led to their exploitation in the clinic in the context of BRCA1/2-mutated cancer (2, 3). Other mechanisms whereby PARPi induce cell death include regulation of fork reversal and non-homologous end joining (NHEJ) at collapsed forks (4). It is also thought that inhibition of PARP activity causes a delay in single-strand breaks, which will accumulate and become toxic double-strand breaks upon encounters with the replication fork (5). PARPi are the first successful example of therapy exploiting synthetic lethality in cancer. They showed survival benefit across multiple cancers with BRCA mutations (6, 7).
Despite the substantial impact that PARPi have made in the clinic, most patients with metastatic disease do eventually develop resistance, creating a major unmet medical need. For instance, the SOLO2 phase III trial exemplified how 78% of BRCA-mutated patients with relapsed ovarian cancer eventually experienced disease progression on Olaparib, indicating the development of resistance to PARPi (8). Another example is the ARIEL2 study, which showed that 60% of BRCA-mutated, high-grade ovarian carcinoma patients treated with Rucaparib ultimately experienced disease progression (9). Patients that become resistant to PARPi have poor outcome and develop cross-resistance with other DNA damage agents such as platinum (10, 11).
There are various described mechanisms to render cancer cells resistance to PARPi. The first is the restoration of the homologous recombination pathway, either through reversion mutations that restore activity to the BRCA proteins (12, 13) or via loss of 53BP1 and other resection-associated proteins (14), which will, in turn, restore the homologous recombination capacity of the cell (15). Recent analyses reported that up to 80% of prostate cancer patients with BRCA2 mutations who developed resistance to PARPi had undergone reversion mutations (16). Mutations in PARP itself can also lead to resistance to inhibitors by reducing the binding of the drug (17). Finally, loss of Poly(ADP-ribose) Glycohydrolase results in defective removal of PAR chains, potentially conferring resistance to PARPi (18).
It is expected that this resistance issue will affect approximately 40-70% of metastatic patients with BRCA mutations (19). Strategies aiming to combine PARPi with other drugs to overcome the hurdle of resistance have not yet proven to be successful. Strategies aiming to combine different PARPi with various chemotherapeutic drugs, such as PI3K inhibitors (20), ATR inhibitors (21, 22) or Polθ inhibitors (23, 24) have been explored but are yet to deliver impactful results with manageable toxicities. This illustrates the major need for strategies to overcome resistance to PARPi. Recently, it was shown that BRCA-defective cells are sensitive to treatment with the thymidine analogue CldU either alone or in combination with PARPi olaparib (5). In this study, we found that the thymidine analogue CldU conferred specific sensitivity to PARPi in BRCA2 mutated cell lines that were previously resistant, including those with reversion mutations. We show that this combination of treatments induced high levels of DNA damage in PARP inhibitor-resistant cell lines.
2 Material and methods
2.1 Cell lines
PEO1 and PEO4 serous ovarian cancer cell lines were purchased from Sigma-Aldrich. They are derived from peritoneal ascites of the same patient with a poorly differentiated serous ovarian adenocarcinoma. PEO1 cells were collected from the patient at first relapse (cisplatin-sensitive). PEO4 cells were collected after the patient demonstrated resistance to cisplatin (25). PEO1 has BRCA2 non-sense mutation (5193C>G, Y1655X) and PEO4 harbors BRCA2 reversion mutation (5193C>T, Y1655Y). C4–02 and C4–13 clones were derived in vitro from PEO1 cells through continuous exposure to cisplatin for 4 weeks (25). C4–02 exhibited BRCA2 reversion mutation (5192A>T). PEO1 and its 3 clones were cultured in RPMI1640 medium (+) l-glutamine supplemented with 2mM Sodium Pyruvate and 10% fetal bovine serum (FBS).
CAPAN-1 is a BRCA2 mutant (6174delT) pancreatic cancer cell line. Its clones C2-5, C2–8 and C2–13 were derived in vitro through continuous exposure to cisplatin for 4 weeks. C2–5 exhibited BRCA2 reversion mutation (6006_6308del303) while C2–08 and C2–13 do not have reversion mutations (13). CAPAN-1 and its 3 clones were cultured in RPMI1640 medium (+) l-glutamine supplemented with 2mM Sodium Pyruvate and 10% fetal bovine serum (FBS). PEO1 derived clones (C4–02 and C4-13), CAPAN-1 and its clones (C2-05, C2–08 and C2-13) were generously provided by Prof. Toshiyasu Taniguchi (Tokai University school of medicine).
2.2 Drugs and chemicals
Olaparib (HY-10162), CldU (Merck, C6891) and Saruparib (HY-132167) were purchased from MedChemExpress (LUCERNA-CHEM). Thymidine (T1895) was purchased from Sigma-Aldrich, EdU (A10044) from ThermoFisher Scientific and BrdU (B23151 from Invitrogen). The stock solutions of PARPi and chemical compounds were prepared from powders dissolved in 100% dimethyl sulfoxide (DMSO) for a stock solution concentration of 10mM except for thymidine that was dissolved in water, aliquoted, and stored at −80°C for up to a maximum of 12 months. In order to minimize the cytotoxic effect of DMSO dilution solution on the cells, several intermediate dilutions were prepared to dispense 2µL of inhibitors in 2mL medium per well of a 6-well plate. The same volume of DMSO was added to control wells.
2.3 Clonogenic assay
The cytotoxic activity of drugs and their influence on cell growth, survival and their ability to form colonies were assessed using the colony formation assay. Briefly, cells were seeded in 6-well plates in 2 mL of culture medium in triplicate (1500 cells per well for CAPAN-1 and its clones, and 3000 cells for PEO1 and its clones) and incubated for 24 h (37°C, 5% CO2). Drugs were added to the medium 24h after cell seeding with pre-selected doses of tested compounds (0.001 – 10μM olaparib, 10 - 100–1000 nM saruparib, 0.05 - 5 μM CldU or their combinations) by adding 2μL of 1000 × concentrated drugs prepared in DMSO. The same volume of DMSO was added to control wells. After 48h, the medium was changed, and cells were allowed to grow and proliferate in a drug-free medium for 14–21 days until non-overlapping colonies were formed in control wells. Colonies were fixed with paraformaldehyde (PFA) 4% for 20 min, stained with 0.5% crystal violet in 20% ethanol for 20 min, thoroughly rinsed with deionized water to remove residual dye, and air-dried at room temperature. Each well was photographed using the FUSION FX6 EDGE Imaging System and number of colonies was quantified using ImageJ software® with colony counting extension. A colony of at least a size of 20 pixel2 was scored as one survivable colony and considered for the count. Results were expressed as relative survival (percentage of colonies) as the number of colonies per treatment versus colonies that appeared in the DMSO control (mean colony counts ± standard errors are reported). Graphs were generated using GraphPad Prism®, 9 software (v.9.4.1).
2.4 Flow cytometry
Following drug treatment, cells were harvested by trypsin and fixed in 70% ethanol in PBS1X overnight at −20°C. Detection of γH2AX phosphorylation was performed using the Guava Histone H2AX Phosphorylation Assay Kit (Luminex, catalogue no. FCCS100182) according to the manufacturer’s instructions. Genomic DNA was stained by incubating the cells in PBS containing RNase (Roche, catalogue no. 11119915001) and propidium iodide (Sigma-Aldrich catalogue no. 81845). DNA-γH2AX profiles were acquired by flow cytometry (CytoFLEX LX flow cytometer); more than 5,000 cells were analyzed per sample using Kaluza® software (Beckman Coulter).
2.5 Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 software (v.9.4.1). Detailed description of means or medians, error bars and the number replicates and/or cells analyzed is reported in the figure legends. For comparison of more than two groups, the two-way ANOVA with Tukey’s multiple comparisons test was used Values are presented as mean ± SEM. p<0.05 was considered significant. Detailed description of means or medians, error bars and the number replicates and/or cells analyzed is reported in the figure legends. Statistical analysis was reported on Supplementary Tables.
3 Results
3.1 BRCA2-mutant cells’ sensitivity to CldU resembles the sensitivity to PARP inhibitor
Recent findings have demonstrated that BRCA1-deficient cells exhibit marked sensitivity to chlorodeoxyuridine (CldU), both as a monotherapy and in combination with the PARP inhibitor olaparib (4). In this study, we assessed the sensitivity to CldU across eight BRCA2-mutant cancer cell lines. These included: (1) PEO1, an ovarian cancer-derived cell line, and its isogenic derivatives resistant to cisplatin, either with (PEO4; C4-02) or without BRCA2 reversion mutation (25); and (2) CAPAN-1, a pancreatic cancer-derived cell line, along with its cisplatin-resistant clones due to either BRCA2 reversion mutations (C2–05 and C2-13) or other mechanisms (C2-08) (13). Our results revealed that sensitivity to CldU partially reflected sensitivity to PARP inhibitor olaparib. Notably, PEO1 displayed pronounced sensitivity to olaparib (Figures 1A–C) and CldU (Figure 1E), whereas PEO4, C4-02, CAPAN-1, C2-05, C2-08, and C2–13 exhibited reduced sensitivity to both olaparib (Figures 1C, D) and CldU (Figures 1E, F).
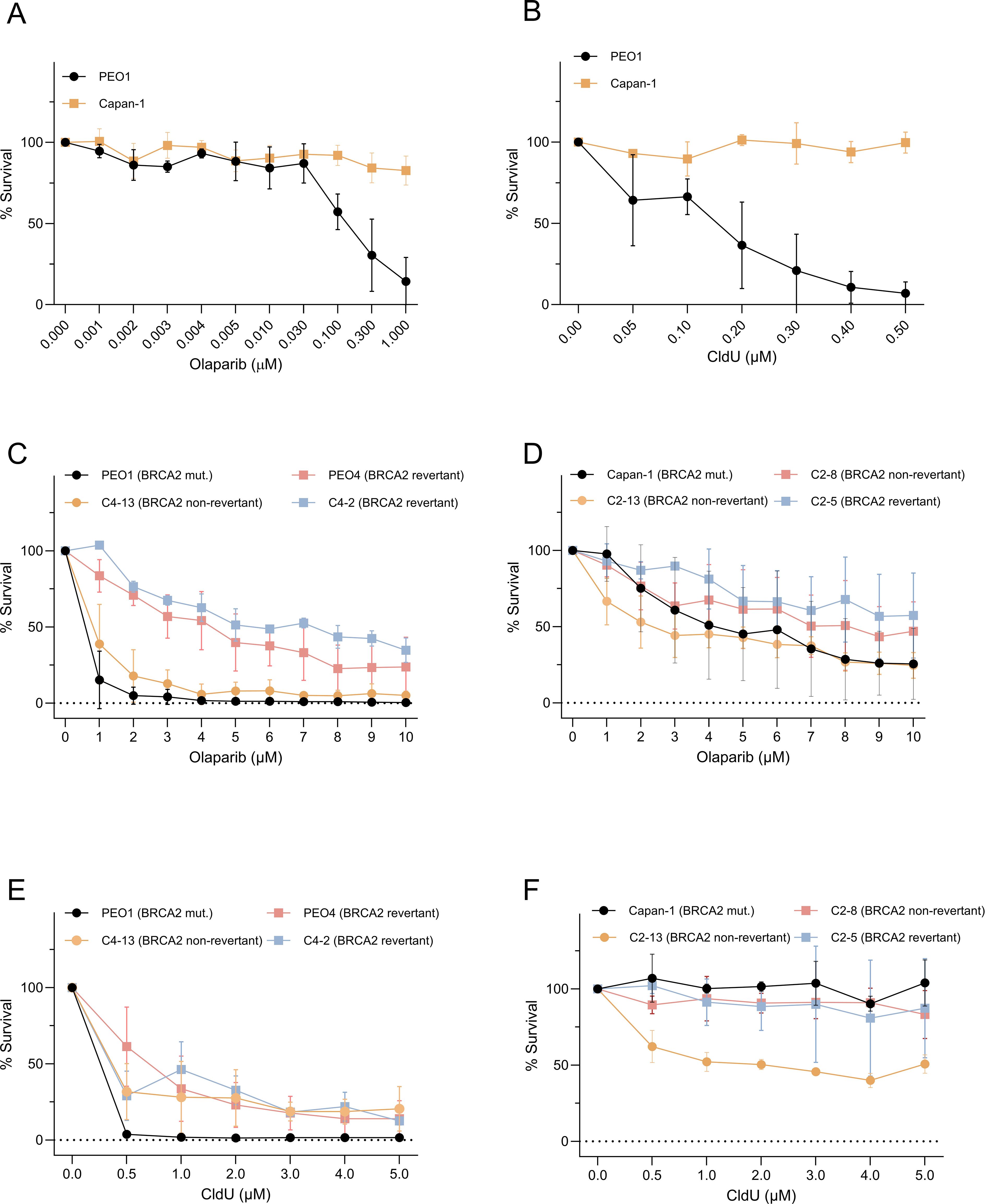
Figure 1. Clonogenic sensitivity to olaparib and CldU in BRCA2-mutant cancer cells and their PARPi-resistant derivatives. (A, B) Dose–response of BRCA2-deficient PEO1 and Capan-1 cells treated for 48 h with increasing concentrations of (A) the PARP inhibitor olaparib (0.0001–1 µM) or (B) chlorodeoxyuridine (CldU; 0.05–0.5 µM). Survival is expressed as percent of untreated control. (C, D) Olaparib sensitivity in BRCA2-mutant parental lines versus isogenic PARPi-resistant clones. (C) PEO1 (BRCA2-mutant) compared to C4-13 (non-revertant resistant) and two BRCA2-reversion derivatives (PEO4, C4-2). (D) Capan-1 (BRCA2-mutant) compared to C2-13 (non-revertant) and two BRCA2-reversion clones (C2-8, C2-5). (E, F) Corresponding clonogenic survival following 48 h CldU treatment in the same sets of PEO1-derived [(E) PEO1, PEO4, C4-2, C4-13] and Capan-1-derived [(F) Capan-1, C2-8, C2-13, C2-5] cell lines. In all panels, data are mean ± SD of three independent biological replicates; curves are normalized to untreated controls.
3.2 CldU sensitizes PARP inhibitor-resistant cells to PARP inhibitors
We next investigated whether the combination of CldU and PARPi exerts a synergistic effect in BRCA2-mutant cancer cells. Remarkably, the co-treatment with low doses of olaparib (1 μM) and CldU (0.5 μM) proved to be lethal in BRCA2-mutant PEO1 cells, as well as in its olaparib-resistant isogenic derivatives, including revertant clones PEO4 and C4-02 (Figure 2A, Supplementary Table 1 and Supplementary Figure 1A). This synergistic effect was further validated using saruparib (AZ5305), a second-generation, highly potent and PARP1-selective inhibitor with approximately 500-fold selectivity for PARP1 over PARP2 (18). Low-dose saruparib (10 nM) combined with CldU resulted in >80% cell death across the three PEO1-derived clones, all of which were resistant to saruparib monotherapy (Figure 2C, Supplementary Table 3, Supplementary Figure 1C and Supplementary Figures 2A–D). Consistently, the combination of CldU with olaparib (Figure 2B, Supplementary Table 2 and Supplementary Figure 1B) elicited a synergistic response in the BRCA2-mutant CAPAN-1 cell line and its PARP inhibitor-resistant isogenic derivatives, including the reversion-bearing C2–05 clone. Interestingly, the synergistic response between saruparib and CldU in CAPAN-1 cells was less significant (Figure 2D, Supplementary Table 4, Supplementary Figure 1D and Supplementary Figures 2E–H). This could reflect the high intrinsic resistance of these cells to both agents, in addition to saruparib being a PARP1 specific inhibitor with lower trapping potential than olaparib (26). Collectively, these findings demonstrate that CldU and PARPi act synergistically in BRCA2-mutant cancer cells, even in the context of acquired PARP inhibitor resistance, including resistance mediated by BRCA2 reversion mutations.
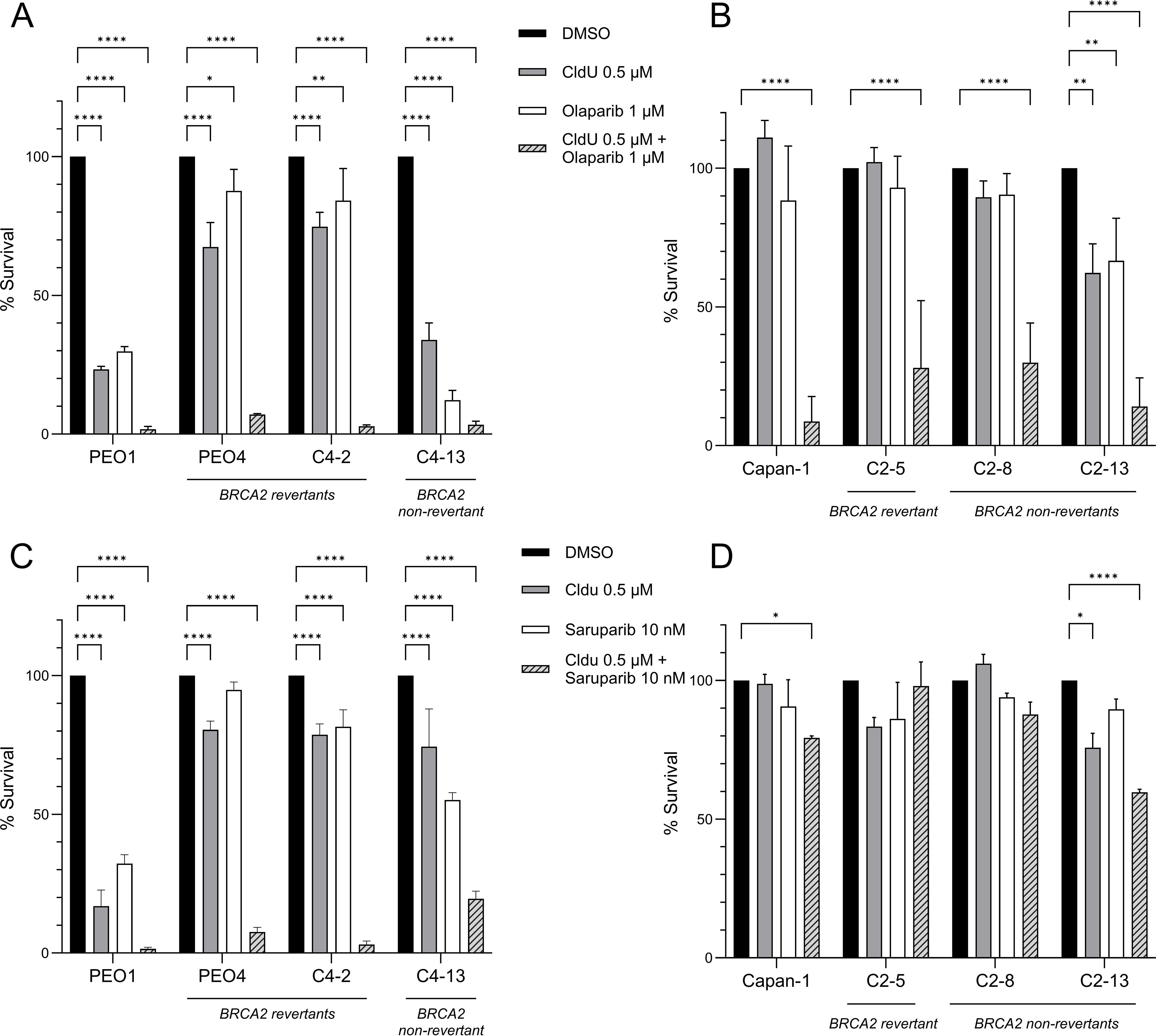
Figure 2. Synergistic cytotoxicity of CldU and PARP inhibitors in BRCA2-deficient and PARPi-resistant cell lines. (A, B) Clonogenic survival of parental BRCA2-mutant cells and their isogenic PARPi-resistant derivatives after 48 h treatment with vehicle (DMSO), CldU (0.5 µM), olaparib (1 µM), or the combination. (A) PEO1 (BRCA2-mutant), PEO4 and C4-2 (BRCA2-revertant resistant), and C4-13 (non-revertant resistant). Data are mean ± SD of three technical replicates from one representative experiment (n = 3 independent repeats). (B) Capan-1 (BRCA2-mutant), C2–5 and C2-8 (BRCA2-revertant resistant), and C2-13 (non-revertant resistant). Data are mean ± SD of three independent biological replicates. (C, D) Clonogenic survival of the same cell panels treated for 48 h with CldU (0.5 µM), the PARP-1 selective inhibitor saruparib (10 nM), or their combination. (C) PEO1 lineage (PEO1, PEO4, C4-2, C4-13); mean ± SD of three technical replicates from one representative experiment (n = 3). (D) Capan-1 lineage (Capan-1, C2-5, C2-8, C2-13); mean ± SD of three technical replicates from one representative experiment (n=3). Statistical significance was assessed using GraphPad Prism 10.5.0 software by two-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. In all panels, the striped bars (combination) reveal pronounced loss of clonogenic survival in both parental and PARPi-resistant clones, indicating strong synergy between CldU and either olaparib or saruparib.
3.3 The synergistic effect of CldU and PARP inhibitor is specific
CldU is a thymidine analogue with a chemical structure closely resembling that of native thymidine. It is commonly used in molecular biology to label newly synthesized DNA, as it is incorporated into DNA but not RNA. Other thymidine analogues, such as 5-ethynyl-2′-deoxyuridine (EdU) and 5-bromo-2′-deoxyuridine (BrdU), serve similar roles in tracking DNA synthesis (Figure 3A). To determine whether the observed synergy between CldU and PARPi is unique to CldU or shared among thymidine analogues, we evaluated the cytotoxic effects of olaparib (1 μM) in combination with thymidine or its analogues (CldU, BrdU, and EdU) at an equivalent concentration (0.5 μM) in PEO1 and PEO4 cell lines. Our results demonstrated that the synergistic interaction with olaparib was specific to CldU (Figures 3B-E and Supplementary Figure 3). In contrast, EdU exhibited intrinsic cytotoxicity across all conditions, independent of olaparib co-treatment (Figures 3B–E and Supplementary Figure 3). This result is consistent with a recent report showing that EdU induces DNA damage in mammalian cells, that is repaired by nucleotide excision repair (27). Neither thymidine nor BrdU alone, nor in combination with olaparib, exhibited significant cytotoxic effects. These findings suggest that the synergy between CldU and PARPi is not a general property of thymidine analogues, but rather a specific feature of CldU.
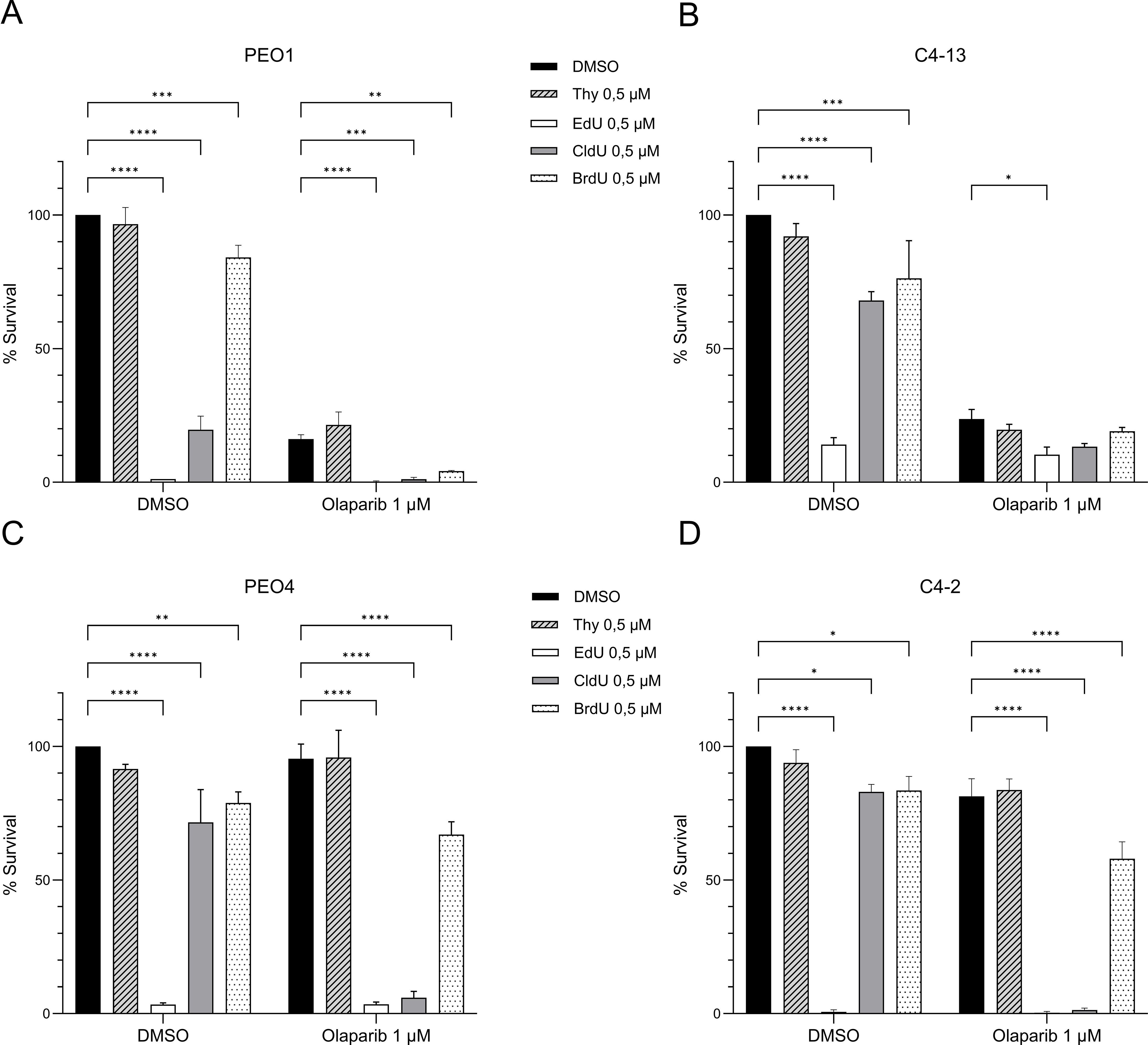
Figure 3. CldU is the most potent and selective thymidine analogue for synergizing with PARP inhibition. (A–D) Clonogenic survival of BRCA2-mutant PEO1 cells and isogenic derivatives following 48-hour treatment with thymidine analogues (Thymidine, EdU, CldU, or BrdU; 0.5 µM) alone or combined with olaparib (1 µM). (A) PEO1 parental cells. (B) C4-13 (PARPi-resistant, BRCA2 non-revertant). (C) PEO4 (PARPi-resistant, BRCA2-revertant). (D) C4-2 (PARPi-resistant, BRCA2-revertant). Data represent the mean ± SD of three technical replicates from one representative experiment (n=3). Statistical significance was assessed using GraphPad Prism 10.5.0 software by two-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Experiments were repeated independently three times for panels (A, C), and twice for panels (B, D).
3.4 CldU combination with PARP inhibitor induce DNA damage
Finally, we sought to determine whether the combination of CldU and PARP inhibition induces DNA damage in BRCA2-mutant cancer cells. As expected, treatment with olaparib alone triggered DNA damage in PARP-sensitive PEO1 cells (Figure 4A). In contrast, olaparib monotherapy did not elicit substantial DNA damage in PARP-resistant PEO4 and C4–02 cells (Figure 4B and Supplementary Figure 4). Notably, co-treatment with CldU and olaparib resulted in marked DNA damage in these resistant cell lines (Supplementary Figure 4). Furthermore, the combination of CldU and olaparib induced early S-phase cell cycle arrest in both PEO1 and PEO4 cells (Supplementary Figure 4), consistent with replication stress-associated DNA damage. In parallel, EdU treatment led to DNA damage across all conditions (Figures 4A, B), independent of BRCA2 status, underscoring its inherent cytotoxicity. Collectively, these findings demonstrate that CldU and olaparib cooperate to induce DNA damage in PARPi-resistant cells, supporting a synergistic mechanism of action.
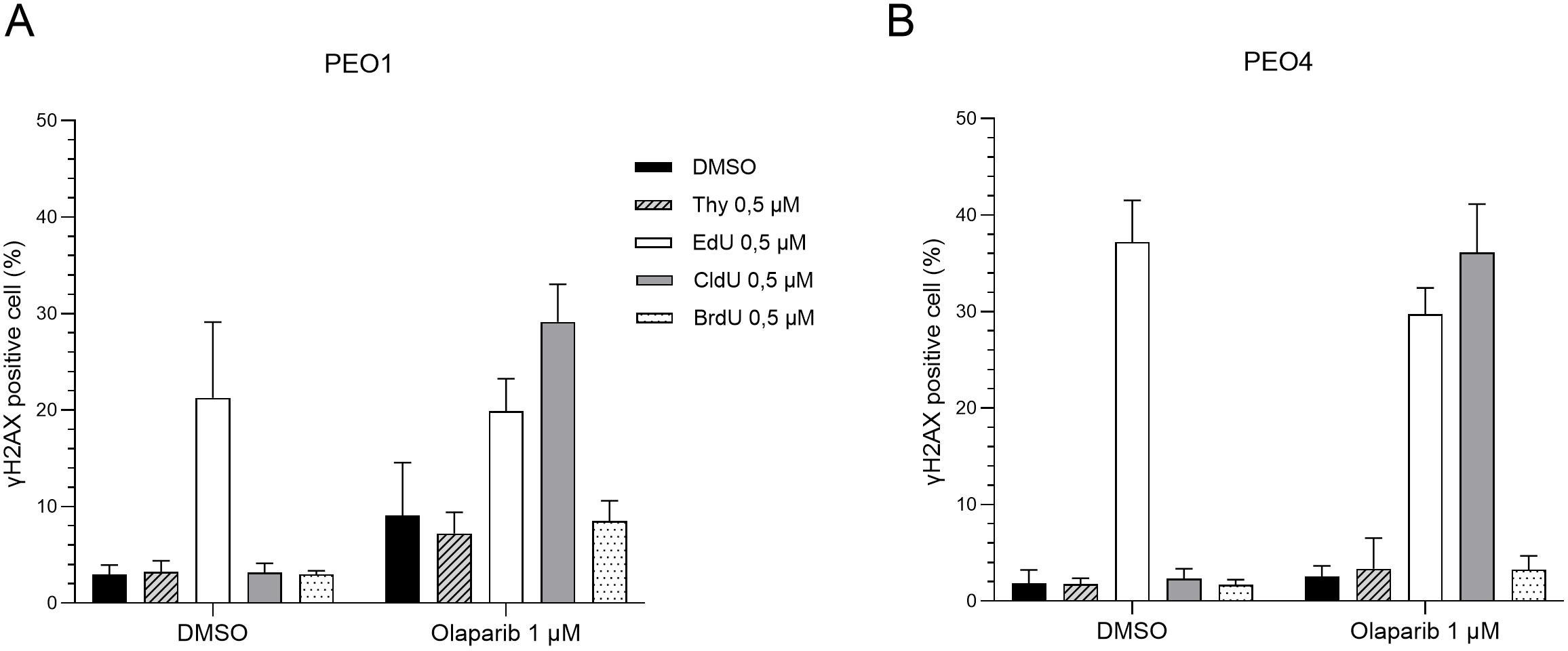
Figure 4. The chlorine group in CldU drives enhanced DNA damage in BRCA2-mutant cells under PARP inhibition. Quantification of γH2AX-positive cells (marker of DNA damage) by flow cytometry after 48-hour treatment with thymidine analogues (Thymidine, EdU, CldU, or BrdU; 0.5 µM), alone or combined with olaparib (1 µM). (A) PEO1 (BRCA2-mutant parental line). (B) PEO4 (PARPi-resistant, BRCA2-revertant). Data show the percentage of γH2AX-positive cells from three independent experiments. Increased DNA damage in CldU-treated groups highlights the role of the chlorine modification under PARP inhibition.
4 Discussion
PARP inhibitors (PARPi) have significantly advanced the treatment of cancers harboring BRCA1 or BRCA2 mutations by exploiting deficiencies in homologous recombination-mediated DNA repair. However, resistance to PARPi remains a major clinical challenge. Reversion mutations in BRCA1/BRCA2—observed in up to 80% of patients who develop resistance to PARPi—can restore protein function, thereby reinstating DNA repair capability and leading to therapeutic resistance and poor outcomes (16, 17). Strategies to overcome PARPi resistance are actively being explored. In this study, we demonstrate that the thymidine analogue chlorodeoxyuridine (CldU) sensitizes PARPi-resistant cancer cells to PARP inhibition. This cytotoxic effect is thought to result from the accumulation of single-stranded DNA gaps initiated by uracil DNA glycosylase-mediated base excision repair. When combined with PARPi-induced replication stress and compromised fork protection in BRCA-deficient cells, this leads to lethal levels of DNA damage. Notably, even cells harboring BRCA reversion mutations, which partially restore homologous recombination, remain sensitive to the combination of CldU and PARPi. This suggests that the mechanism of cytotoxicity may bypass conventional BRCA-mediated repair pathways. Although the exact mechanism of cell death remains to be fully elucidated, our findings point to a potentially novel vulnerability in PARPi-resistant cancers. Of note, we observed that the combination of CldU and olaparib was synergetic across all cell lines derived from both PEO1 and CAPAN-1, while the synergistic effect between saruparib and CldU in CAPAN-1 cells was less significant. Elucidating whether this is due to intrinsic differences in DNA repair between cell lines, replication stress response, or PARP trapping efficiency (26) need to be addressed in the future.
Importantly, while CldU is not approved for clinical use and is currently limited to research applications as a DNA synthesis marker, clinically approved nucleoside analogues such as gemcitabine, cytarabine, and trifluridine share structural similarities. Some of these, particularly gemcitabine, have shown synergistic activity with PARPi in preclinical models of non-small-cell lung cancer (28) and in a clinical trial that enrolled pancreatic cancer patients (29). Next-generation antibody drug conjugates combining dual payloads that target DNA damage, for instance topoisomerase 1 inhibitor and PARPi, are currently investigated (30) and could be a therapeutic approach to reduce the toxicities of such combinations. Our work also confirmed that another thymidine analogue, EdU, is cytotoxic and induces DNA damage in mammalian cancer cells (27, 31), independent of BRCA2 status. Overall, our findings prompt further investigation into nucleotide analogues for the treatment of PARPi-resistant cancers.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
NZ: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. CT: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. VD: Data curation, Project administration, Visualization, Writing – review & editing. WW: Data curation, Writing – review & editing. GR: Writing – original draft, Writing – review & editing, Data curation, Supervision, Conceptualization, Investigation, Visualization. TH: Conceptualization, Funding acquisition, Resources, Supervision, Writing – review & editing. IL: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The study was supported by la ligue genevoise contre le cancer (#LGC-2205) and the Office of the Assistant Secretary of Defense for Health Affairs through the Ovarian Cancer Research Program under Award No. W81XWH-22-1-0558 to I.L.-G. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Acknowledgments
The authors thank Prof. Toshiyasu Taniguchi, Tokai University School of Medicine, for giving access to cell lines established in his lab derived from CAPAN-1 (clones C2-5, C2–8 and C2-13) and PEO1 (clones C4–02 and C4-13).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1626301/full#supplementary-material
Supplementary Figure 1 | Representative colony formation assays showing the effects of combined CldU and PARP inhibition on BRCA2-mutant and PARPi-resistant cell lines. (A, B) Representative images from clonogenic survival assays following 48-hour treatment with olaparib (1 µM) plus CldU (0.5 µM) in: (A) PEO1 (BRCA2-mutant) and its isogenic derivatives (PEO4, C4-2, C4-13) (B) Capan-1 (BRCA2-mutant) and its isogenic derivatives (C2-8, C2-13, C2-5) (C, D) Representative images from clonogenic survival assays following 48-hour treatment with saruparib (10 nM) plus CldU (0.5 µM) in: (C) PEO1 and its isogenic derivatives (PEO4, C4-2, C4-13) (D) Capan-1 and its isogenic derivatives (C2-8, C2-13, C2-5). For each condition, one well from triplicate experiments is shown.
Supplementary Figure 2 | Effect of saruparib and CldU combination treatment on clonogenic survival in BRCA2-mutant and PARPi-resistant cell lines. Clonogenic survival of BRCA2-mutant PEO1 and Capan-1 cells, and their isogenic derivatives (PEO4, C4-2, C4-13, C2-5, C2-8, C2-13), after 48-hour treatment with saruparib alone (10 nM, 100 nM, or 1 µM) or in combination with CldU (0.5 µM). Data represent one experiment with a single well per treatment condition.
Supplementary Figure 3 | Representative colony formation assays showing the effects of thymidine analogues combined with PARP inhibition. (A) Chemical structures of the thymidine analogues used in this study (sourced from PubChem, public domain). (B–E) Representative images from clonogenic survival assays following 48-hour treatment with thymidine analogues (Thymidine, EdU, CldU, or BrdU; 0.5 µM), alone or in combination with olaparib (1 µM), in: (A) PEO1 (BRCA2-mutant parental line) (B) C4-13 (PARPi-resistant, BRCA2 non-revertant) (C) PEO4 (PARPi-resistant, BRCA2-revertant) (D) C4-2 (PARPi-resistant, BRCA2-revertant). For each condition, one well from triplicate experiments is shown.
Supplementary Figure 4 | Representative flow cytometry analysis of cell cycle distribution and DNA damage following treatment with thymidine analogues and PARP inhibition. (A–D) Representative flow cytometry plots showing cell cycle distribution and γH2AX staining after 48-hour treatment with thymidine analogues (Thymidine, EdU, CldU, or BrdU; 0.5 µM), alone or combined with olaparib (1 µM), in: (A) PEO1 (BRCA2-mutant parental line) (B) PEO4 (PARPi-resistant, BRCA2-revertant) (C) C4.13 (PARPi resistant, BRCA2 non-revertant) (D) C4.2 (PARPi resistant, BRCA2-revertant) Data represent the percentage of γH2AX-positive cells from one representative independent experiment.
Supplementary Table 1 | statistical tests of Figure 2A. Test used: Two-way ANOVA with Tukey’s multiple comparisons test.
Supplementary Table 2 | statistical tests of Figure 2B. Test used: Two-way ANOVA with Tukey’s multiple comparisons test.
Supplementary Table 3 | statistical tests of Figure 2C. Test used: Two-way ANOVA with Tukey’s multiple comparisons test.
Supplementary Table 4 | statistical tests of Figure 2D. Test used: Two-way ANOVA with Tukey’s multiple comparisons test.
References
1. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. (2011) 5:387–93. doi: 10.1016/j.molonc.2011.07.001
2. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. (2005) 434:917–21. doi: 10.1038/nature03445
3. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature. (2005) 434:913–7. doi: 10.1038/nature03443
4. Liptay M, Barbosa JS, and Rottenberg S. Replication fork remodeling and therapy escape in DNA damage response-deficient cancers. Front Oncol. (2020) 10:670. doi: 10.3389/fonc.2020.00670
5. Serrano-Benitez A, Wells SE, Drummond-Clarke L, Russo LC, Thomas JC, Leal GA, et al. Unrepaired base excision repair intermediates in template DNA strands trigger replication fork collapse and PARP inhibitor sensitivity. EMBO J. (2023) 42:e113190. doi: 10.15252/embj.2022113190
6. Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. (2020) 383:2345–57. doi: 10.1056/NEJMoa2022485
7. Geyer CE Jr., Garber JE, Gelber RD, Yothers G, Taboada M, Ross L, et al. Overall survival in the OlympiA phase III trial of adjuvant olaparib in patients with germline pathogenic variants in BRCA1/2 and high-risk, early breast cancer. Ann Oncol. (2022) 33:1250–68. doi: 10.1016/j.annonc.2022.09.159
8. Poveda A, Floquet A, Ledermann JA, Asher R, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomzsed, placebo-controlled, phase 3 trial. Lancet Oncol. (2021) 22:620–31. doi: 10.1016/S1470-2045(21)00073-5
9. Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicenter, open-label, phase 2 trial. Lancet Oncol. (2017) 18:75–87. doi: 10.1016/S1470-2045(16)30559-9
10. Cecere SC, Giannone G, Salutari V, Arenare L, Lorusso D, Ronzino G, et al. Olaparib as maintenance therapy in patients with BRCA 1–2 mutated recurrent platinum sensitive ovarian cancer: Real world data and post progression outcome. Gynecol Oncol. (2020) 156:38–44. doi: 10.1016/j.ygyno.2019.10.023
11. Harter P, Marth C, Mouret-Reynier MA, Cropet C, Lorusso D, Guerra-Alia EM, et al. Efficacy of subsequent therapies in patients with advanced ovarian cancer who relapse after first-line olaparib maintenance: results of the PAOLA-1/ENGOT-ov25 trial. Ann Oncol. (2025) 36:185–96. doi: 10.1016/j.annonc.2024.10.828
12. Li H, Liu ZY, Wu N, Chen YC, Cheng Q, and Wang J. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer. (2020) 19:107. doi: 10.1186/s12943-020-01227-0
13. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. (2008) 451:1116–20. doi: 10.1038/nature06633
14. Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. (2013) 3:68–81. doi: 10.1158/2159-8290.CD-12-0049
15. Kim DS, Camacho CV, and Kraus WL. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp Mol Med. (2021) 53:42–51. doi: 10.1038/s12276-021-00557-3
16. Seed G, Beije N, Yuan W, Bertan C, Goodall J, Lundberg A, et al. Elucidating acquired PARP inhibitor resistance in advanced prostate cancer. Cancer Cell. (2024) 42:2113–2123 e4. doi: 10.1016/j.ccell.2024.10.015
17. Mweempwa A and Wilson MK. Mechanisms of resistance to PARP inhibitors - an evolving challenge in oncology. Cancer Drug Resist. (2019) 2:608–17. doi: 10.20517/cdr.2019.50
18. Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. (2018) 33:1078–1093 e12. doi: 10.1016/j.ccell.2018.05.008
19. Kim D and Nam HJ. PARP inhibitors: clinical limitations and recent attempts to overcome them. Int J Mol Sci. (2022) 23:1–18. doi: 10.3390/ijms23158412
20. Konstantinopoulos PA, Barry WT, Birrer M, Westin SN, Cadoo KA, Shapiro GI, et al. Olaparib and alpha-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. (2019) 20:570–80. doi: 10.1016/S1470-2045(18)30905-7
21. Mahdi H, Hafez N, Doroshow D, Sohal D, Keedy V, Do KT, et al. Ceralasertib-mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (Olaparib combinations). JCO Precis Oncol. (2021) 5:1432–42. doi: 10.1200/PO.20.00439
22. Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. (2020) 11:3726. doi: 10.1038/s41467-020-17127-2
23. Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun. (2021) 12:3636. doi: 10.1038/s41467-021-23463-8
24. Zhou J, Gelot C, Pantelidou C, Li A, Yucel H, Davis RE, et al. A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat Cancer. (2021) 2:598–610. doi: 10.1038/s43018-021-00203-x
25. Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. (2009) 69:6381–6. doi: 10.1158/0008-5472.CAN-09-1178
26. Petropoulos M, Karamichali A, Rossetti GG, Freudenmann A, Iacovino LG, Dionellis VS, et al. Transcription-replication conflicts underlie sensitivity to PARP inhibitors. Nature. (2024) 628:433–41. doi: 10.1038/s41586-024-07217-2
27. Wang L, Cao X, Yang Y, Kose C, Kawara H, Lindsey-Boltz LA, et al. Nucleotide excision repair removes thymidine analog 5-ethynyl-2’-deoxyuridine from the mammalian genome. Proc Natl Acad Sci U.S.A. (2022) 119:e2210176119. doi: 10.1073/pnas.2210176119
28. Jiang Y, Dai H, Li Y, Yin J, Guo S, Lin SY, et al. PARP inhibitors synergize with gemcitabine by potentiating DNA damage in non-small-cell lung cancer. Int J Cancer. (2019) 144:1092–103. doi: 10.1002/ijc.31770
29. Bendell J, O'Reilly EM, Middleton MR, Chau I, Hochster H, Fielding A, et al. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumors and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol. (2015) 26:804–11. doi: 10.1093/annonc/mdu581
30. Yin G, Calarese D, Yam A, Rubas W, Bajjuri K, Li X, et al., in: Enhancing Topo1i ADC efficacy: development of homogeneous dual-payload ADCs combining Topo1i with microtubule inhibitors or PARP inhibitors in AACR annual meeting, Cancer Res. Chicago (2025) 85(8_Supplement_1):2870. doi: 10.1158/1538-7445.AM2025-2870
Keywords: BRCA mutation, PARP inhibitor, resistance, reversion mutation, thymidine analogue, CldU, cancer
Citation: Zouggari N, Trugenberger C, Du Bois V, Wang W, Rossetti GG, Halazonetis TD and Labidi-Galy I (2025) CldU sensitizes BRCA2 reverse-mutated cells to PARP inhibitors. Front. Oncol. 15:1626301. doi: 10.3389/fonc.2025.1626301
Received: 10 May 2025; Accepted: 28 October 2025;
Published: 19 November 2025.
Edited by:
Giovanna Damia, Mario Negri Institute for Pharmacological Research (IRCCS), ItalyReviewed by:
Alessandra Brambati, University of Colorado Anschutz Medical Campus, United StatesOriol Mirallas, The University of Texas MD Anderson Cancer Center, United States
Copyright © 2025 Zouggari, Trugenberger, Du Bois, Wang, Rossetti, Halazonetis and Labidi-Galy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Intidhar Labidi-Galy, aW50aWRoYXIubGFiaWRpLWdhbHlAaHVnLmNo
†These authors have contributed equally to this work