Abstract
The oncostatin M receptor (OSMR) has recently emerged as an adverse prognostic factor in acute myeloid leukemia (AML) and several non-hematological malignancies. In this perspective, we discuss how oncostatin M (OSM) and its receptor OSMR regulate tumor cells as well as mesenchymal and endothelial cells, which are key components of hematopoietic stem cell and tumor stem cell niches, and how these mechanisms could explain the poor prognosis associated with high expression of OSM and OSMR in hematological and non-hematological malignancies.
1 Introduction
High expression of oncostatin M (OSM) receptor OSMR has recently emerged as a poor prognosis factor in several malignancies such as acute myeloblastic leukemia (AML), gliomas, pancreatic, gastric and kidney carcinomas. In this perspective, we perform a larger in silico survey of the prognostic value of high OSMR and OSM transcripts in 33 malignancies and discuss the mechanisms involved in the adverse effects of high OSM-mediated signaling in malignant cells and the tumor microenvironment, and potential treatments to target this signaling pathway.
OSM is an inflammatory cytokine of the interleukin-6 (IL-6) family. OSM binds to two different receptors made up of glycoprotein-130 (GP130, gene IL6ST in humans Il6st in mice) complexed with either transmembrane OSM receptor (OSMR, gene OSMR in humans or Osmr in mice) or with leukemia inhibitory factor receptor (LIFR) (1, 2). Both OSMR and LIFR have very long intracellular domains enabling the docking of many kinases and adaptors, eliciting similar but not identical signaling in response to OSM (3, 4). OSM is mostly produced by activated myeloid cells (granulocytes, monocytes and macrophages) as well as all cells of the osteoblast lineage (3, 5, 6), and astrocytes, neurons, microglia in the brain (7). In contrast, its receptor OSMR is not expressed by leukocytes or hematopoietic stem and progenitor cells (HSPC) but expressed by cells of the mesenchymal and endothelial lineages in the bone marrow (6) and other tissues. OSM is a regulator of both hematopoiesis and skeleton homeostasis mostly via the OSMR: GP130 receptor complex (6) and plays important roles in inflammatory responses in skeletal (8, 9) and cardiac (10) muscles, lung (11), liver (12, 13), intestine (14), adipose (15), skin (16, 17) and joints (18). Adding to the complexity of OSMR protein roles, OSMR protein complexed with the IL-31 receptor α chain IL31RA acts as a receptor for IL-31 (19). Recent literature now highlights important roles of both OSMR and OSM in the pathogenesis and response to treatment of several malignancies as detailed below.
2 OSM and OSMR in hematological malignancies
In a recent paper, high throughput proteomic analysis on more than 550 newly diagnosed AML patients demonstrated that high plasma concentration of soluble OSMR protein was a strong independent predictor of poor survival and early mortality (20). These clinical data suggest an oncogenic role of OSMR-mediated signaling and are consistent with the anti-proliferative effect of OSMR gene inactivation in mice and humans. Indeed, Osmr-/- mice display mild anemia and thrombocytopenia (21–23) with decreased HSPC cycling in the bone marrow, increased HSPC chemotactic response, increased HSPC mobilization into the blood in response to G-CSF or CXCR4 antagonists, as well as decreased expression of genes associated with cell cycling, lipid metabolism, and erythropoiesis in hematopoietic stem cells (HSC) (5). Another recent report has shown that biallelic loss-of-function of the OSM gene in humans causes profound anemia, thrombocytopenia and neutropenia (24). Therefore, OSM and OSMR-mediated signaling contributes to increased HSPC cycling and retention within the bone marrow, enabling sufficient erythropoiesis and thrombopoiesis output. Interestingly, the hematopoietic effects of OSMR are mediated via the hematopoietic environment as OSMR mRNA is undetectable to very low in mouse and human HSPC (5) or in AML blasts (20). Reville et al. highlighted that high OSMR protein production and transcript expression was limited to mesenchymal stromal cells (MSCs) while undetectable in AML blasts in cultures of leukemic marrow aspirates (20). However, their culture system did not enable the survival and growth of bone marrow endothelial cells, which similar to MSCs, express high levels of OSMR transcripts and protein (5, 6) and are key functional regulatory elements of HSC niches and leukemia stem cell niches (25–30). Therefore, the source of sOSMR protein in the blood of AML patients could be endothelial cells as well as MSCs and the deregulation of these niche cell types in the bone marrow may be involved in the mechanisms of poorer outcome in patients with high sOSMR protein.
As the OSMR ligand OSM is expressed by myeloid cells (particularly neutrophils and macrophages) and osteoblasts in the bone marrow (3, 5), we plotted the expression of OSM transcripts in the 9,736 tumor and 8,587 normal tissue samples contained in The Cancer Genome Atlas (TCGA) database using Gene Expression Profiling and Interactive Analysis (GEPIA) website (31, 32) (Figure 1A). OSM transcripts were significantly higher in AML samples than normal bone marrow samples (“LAML” in Figure 1A). Overall survival (OS) Kaplan-Meier plot from AML patients with 50% highest content and 50% lowest content in OSM transcripts is shown in Figure 1B. Although the p values of p=0.055 for differences were just above the significance threshold of p=0.05, AML patients with highest levels of OSM transcript had a trend to shorter OS with a 1.7 hazard ratio compared to AML patients with lowest OSM transcripts. Considering that high sOSMR protein is significantly associated with poorer prognosis in AML (20), confirmation of a significant association between poor prognosis and high OSM transcripts in a larger cohort of AML patients is warranted. In regard to the alternative OSMR ligand interleukin-31 (IL-31), IL31 transcripts were detected at least 2 orders of magnitude lower compared to OSM transcripts (Figure 1C) and there was no difference in OS between high and low IL31 expressing AML with the 50% cut-off (Figure 1D). Therefore, it is likely that the adverse effect of high OSM transcript and sOSMR protein in AML is mediated via its ligand OSM acting indirectly via bone marrow mesenchymal and endothelial cells which express both OSMR with its co-receptor GP130 (5, 22, 23) rather than IL-31-mediated signaling. One of the adverse effects of OSM-OSMR interaction may be mediated through its induction of E-selectin expression by endothelial cells (33) as E-selectin mediated signaling in AML stem cells increases their resistance to the cytotoxic effects of chemotherapy (30) (see model in Figure 1E).
Figure 1

Expression of transcripts for OSMR ligands OSM and IL-31 with overall survival plots of AML patients with highest and lowest expression OSM and IL31 transcripts. (A)OSM transcript quantification in transcripts per million in various tumors (red dots) versus paired normal tissues (green dots) extracted from the TCGA database stratified as adrenocortical carcinoma (ACC), bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), cholangial carcinoma (CHOL), colon adenocarcinoma (COAD), diffuse large B-cell lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiform (GBM), head and neck squamous cell carcinoma (HNSC), kidney chromophobe carcinoma (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), acute myeloid leukemia (LAML), lower grade glioma (LGG), hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), mesothelioma (MESO), ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PARD), rectum adenocarcinoma (READ), sarcoma (SARC), skin cutaneous melanoma (SKCM), stomach adenocarcinoma (STAD), testicular germ cell tumor (TGCT), thyroid carcinoma (THCA), thymoma (THYM), uterine corpus endometrial carcinoma (UCEC), uterine carcinosarcoma (UCS) and uveal melanoma (UVM). Bars show median value for each tumor (T) and normal tissue (N) with number of samples indicated. Significant differences in OSM transcripts per million between malignant and paired normal tissues are indicated with colored malignancy abbreviation above the chart in red for OSM overexpressed in tumor versus paired normal tissue and in green for OSM under-expressed in malignant versus paired normal tissue. (B) Kaplan-Meier plots of overall survival of AML patients with 50% highest (red plot) and 50% lowest OSM (blue plot) transcripts, log-rank test p value, hazard ratio (HR), significance of hazard ratio p(HR) and number of patients are indicated on the plot (n=106). (C)IL31 transcript in various tumors (red dots) versus paired normal matching tissues (green dots) extracted from the TCGA database and stratified as described in (A). (D) Kaplan-Meier plots of overall survival of AML with 50% highest and 50% lowest IL31 transcripts. (E) Model of OSM and OSMR effects on normal hematopoiesis (left side) and AML (right side). In normal bone marrow, low level of OSM is released by myeloid cells stimulating basal production of cytokines/growth factors by MSCs and endothelial cells expressing OSMR/GP130 receptor complex and forming HSPC niches. In the leukemic bone marrow, malignant AML blasts express and release high levels of OSM, stimulating MSCs and endothelial cells via the OSMR/GP130 receptor complex they express, to release excessive amounts of growth factors, cytokines, and sOSMR protein as well as boost endothelial E-selectin expression, all contributing to enhanced AML blast proliferation, migration and chemoresistance. Similar effects may occur in multiple myeloma, Philadelphia-negative MPN, and solid tumors. Panels (A–D) were generated by using the GEPIA website in April 2025 (32).
In respect to other hematological malignancies, OSM mRNA are reportedly high in Philadelphia-negative myeloproliferative neoplasm (Ph- MPN) cells with a constitutively active JAK2 tyrosine kinase mutations such as JAK2V617F (34), with increased OSM protein concentration in Ph- MPN patient bone marrow cells and blood plasma (34). Furthermore cytokine production by primary fibroblasts from MPN patients is increased in response to OSM via OSMR expressed by fibroblasts (34). Therefore, sOSMR and its ligand OSM may also have prognostic value in Ph- MPN. This hypothesis is consistent with the following findings: 1) Transduction of a JAK2V617F mutant in myeloid cell lines dramatically increases OSM expression (34). 2) OSM promotes HSPC proliferation, erythropoiesis and thrombopoiesis in vivo (5, 21) indirectly via bone marrow niche cells that express OSMR (5, 6). 3) In a mouse model of lymphoproliferative neoplasm driven by activating mutation of the FLT3 tyrosine kinase domain (FLT3-TKD), co-transduction with OSM cDNA switched the lymphoproliferative FLT3-TKD neoplasm to a myeloproliferative neoplasm suggesting that OSM is a potent inducer of a pro-myeloid niche (35). 4) Transplantation of mouse HSCs overexpressing OSM resulted in fatal myelofibrosis (36), a common adverse evolution of Ph- MPNs. Further along this latter point, it has recently emerged that activating JAK2V617F mutation upregulates the expression of the “don’t eat me” antigen CD24 at the surface of neutrophils enabling the accumulation of old senescent neutrophils to escape efferocytosis and accumulate in the bone marrow promoting evolution to myelofibrosis (37). As neutrophils are a major source of OSM protein in the bone marrow (5, 6) and OSM overexpression triggers myelofibrosis (36), accumulation of OSM secreted by senescent JAK2V617F neutrophils may be a driver of the evolution of polycythemia vera (the most frequent clinical manifestation of MPNs with JAK2V617F driving mutation) to myelofibrosis which has worse clinical outcome.
In respect to lymphoid neoplasms, high sOSMR (19) and OSM (38) protein concentrations have also been reported in the serum of multiple myeloma patients. However, high plasma OSM had no significant prognostic value in newly diagnosed multiple myeloma patients (38) and the potential prognosis value of sOSMR concentrations in multiple myeloma has not been reported.
3 OSM and OSMR in non-hematological malignancies
Endothelial cells (39, 40) and mesenchymal cells (41) are important players in cancer stem cell niches within solid tumors. As OSMR is expressed by these cells (5, 22, 23) as well as some mucosal and glandular epithelial cells (42), OSMR and OSM may also play important roles in several non-hematological malignancies by directly acting on tumor cells or indirectly via endothelial and mesenchymal cells such as fibroblasts in the tumor environment. In support of this, high OSMR transcript content is a poor prognosis factor in glioblastoma (43, 44), pancreatic adenocarcinoma (45), and gastric cancer (46). Analysis of OSMR transcript expression in all tumor types and corresponding healthy tissues in the TCGA database (Figure 2A) showed OSMR transcripts are significantly more expressed in tumors than the paired normal tissues in esophageal carcinoma (ESCA), glioblastoma multiform (GBM), kidney renal clear cell carcinoma (KIRC), pancreatic adenocarcinoma (PAAD), stomach adenocarcinoma (STAD) and thymoma (THYM). Lower grade glioma (LGG) had higher expression of OSMR when compared to healthy tissue, but this was not significant. As samples were generally higher in solid tumor datasets compared to AML, we used a more stringent quartile cut-off and plotted OS and disease-free survival (DFS) from the 25% highest OSMR transcript expressing tumors (High OSMR) and 25% lowest OSMR expressing tumors (Low OSMR). GBM, LGG, PAAD and STAD with highest OSMR transcripts had significantly worse OS, DFS and hazard ratios than the same tumors with lowest OSMR expression (Figures 2B–E). High OSMR transcripts in KIRC had no effect on OS but significantly poorer DFS (Figure 2F). Among tumor types with similar OSMR transcripts compared to paired normal tissue, kidney renal papillary cell carcinoma (KIRP) had significantly worse OS but no difference in DFS (Figure 2G). Unexpectedly, adrenocortical carcinoma (ACC), had significantly less OSMR transcripts compared to paired normal tissue (Figure 2A) but patients with high OSMR transcripts had significantly poorer DFS and equivalent OS (Figure 2H). We could not find any significant difference in OS and DFS between high and low OSMR expression in all other tumor types contained in the TCGA database (result not shown).
Figure 2
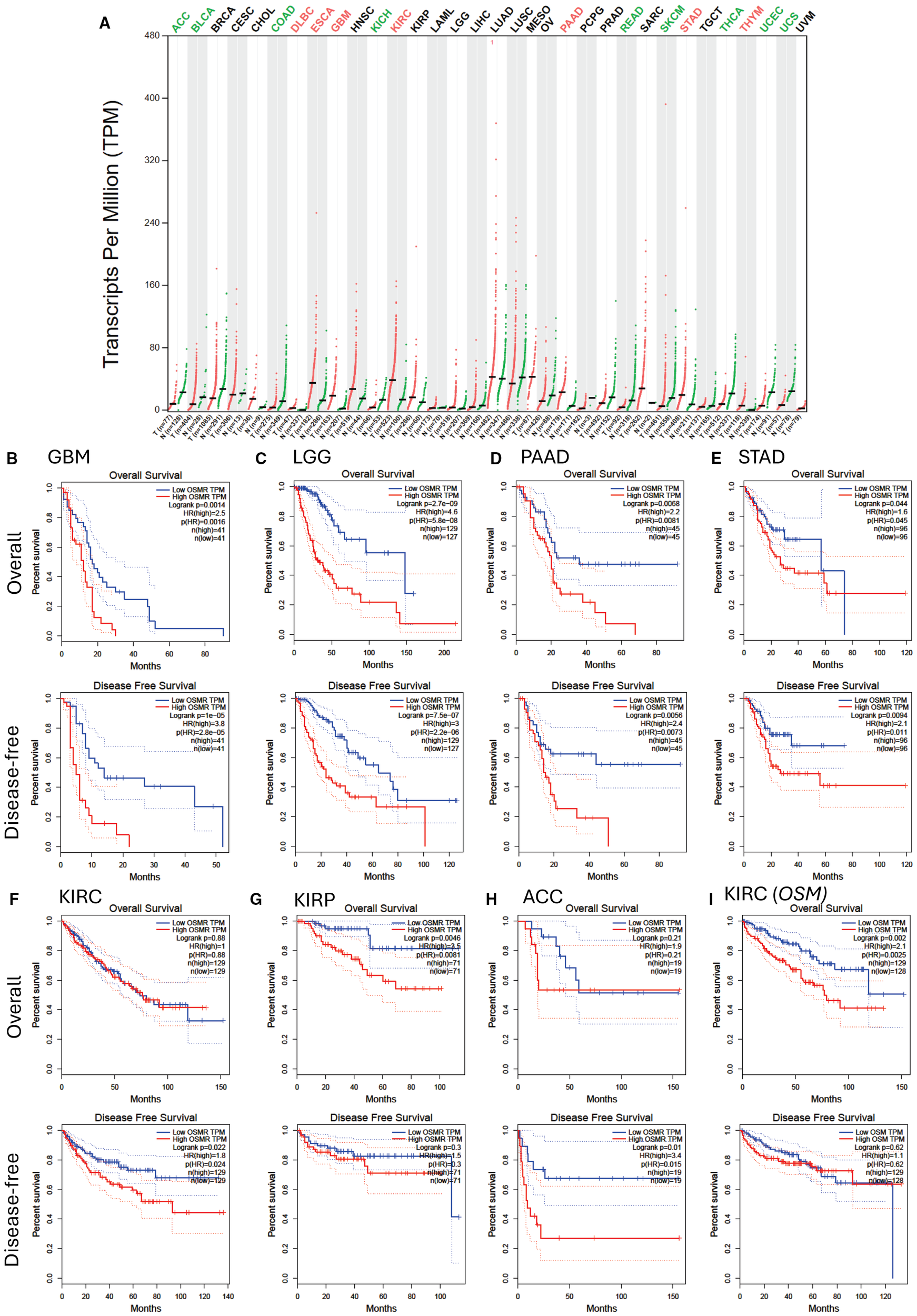
OSMR transcript expression and patient survival of highest and lowest 25% OSMR transcript expressing tumors. (A)OSMR transcript quantification in transcripts per million in various tumors (red dots) versus paired normal tissues (green dots) extracted from the TCGA database stratified as described in Figure 1A. Significant differences in OSMR transcript per million between malignant and paired normal tissues are indicated with colored malignancy abbreviations above the chart with red for OSMR transcripts significantly overexpressed in tumor versus paired normal tissue and green for OSMR transcripts significantly under-expressed in malignant versus paired normal tissue. (B–H) Kaplan-Meier plots of overall and disease-free survival of patients with 25% highest OSMR transcript (red curves) versus lowest 25% OSMR transcript (blue curves) for (B) glioblastoma multiform (GBM, n=162), (C) low grade glioma (LGG, n=514), (D) pancreatic adenocarcinoma (PAAD, n=178), (E) stomach adenocarcinoma (STAD, n=384), (F) kidney renal clear cell carcinoma (KIRC, n=516), (G) kidney papillary cell carcinoma (KIRP, n=280), and (H) adrenocortical carcinoma (ACC, n=76). (I) Kaplan-Meier plots of overall and disease-free survival of patients with 25% highest OSM transcript (red curves) versus lowest 25% OSM transcript (blue curves) for kidney renal clear cell carcinoma (KIRC). Log-rank test p value, hazard ratio (HR), significance of hazard ratio p(HR) and number of patients are indicated on each plot. Panels were generated by using the GEPIA website in April 2025 (32).
Reciprocally, OSM mRNA expression was significantly higher in GBM, PAAD and AML compared to corresponding healthy tissue (Figure 1A). OS was significantly worse in KIRC with high OSM transcripts but there was no significant difference in DFS (Figure 2I). There was no difference in OS or DFS between high and low OSM transcript levels in GBM or PAAD patients (result not shown). We also checked that these findings were similar with a median cut-off (50%) for OSMR and OSM transcripts (Supplementary Figure 1). This was true except for KIRP and ACC which did not show significant difference for OS or DFS with a 50% cut-off.
The poorer prognosis of tumors with high OSMR transcripts is consistent with previously reported effects of OSM or OSMR gene knock-down on these tumor types. For instance, OSM protein has been reported to increase migration, invasiveness and mesenchymal phenotype of GBM cell lines (43), and OSMR protein can translocate to the mitochondrial matrix to protect GBM stem cells from irradiation (44). Likewise OSM has been found to enhance tumor growth and epithelial-to-mesenchymal transition of pancreatic ductal adenocarcinoma cell lines in vivo in immunodeficient mice via OSMR (45). Additionally, OSM produced by macrophages has been found to induce an inflammatory phenotype in PAAD-associated fibroblasts providing a pro-tumorigenic environment in PAAD (47). OSM also promotes the proliferation of gastric cancer cells in vitro and in vivo in Nude mice (46). Finally, OSM has been reported to increase migration, invasiveness and epithelial-to-mesenchymal transition of clear cell renal carcinoma cell lines in vitro (48). Importantly, deletion of the VHL gene specifically in kidney tubule cells in mice (approximately 70% of clear cell renal carcinoma have inactivating mutation of VHL) induces production of OSM which in turn pushes kidney endothelial cells into an endothelial-to-mesenchymal transition to support VHL-defective tubule cell transformation and promote macrophage polarization towards a tissue-supportive M2-like phenotype (49). This vicious loop between OSM-expressing VHL-defective tubule cells and activated endothelium expressing OSMR is functionally important as injection of neutralizing OSM antibodies into immuno-deficient mice transplanted with human clear cell renal carcinoma cells inhibited primary tumor vascularization, growth and metastasis (49).
4 Discussion
Kaplan-Meier plots in Figures 1, 2 and previous reports discussed above clearly indicate that the interaction of OSM with the OSMR: GP130 receptor complex plays a role in the pathobiology and response to treatment of a number of malignancies such as AML, MPNs, glioma and glioblastoma, pancreatic, gastric and renal carcinomas where high expression of OSMR or OSM transcripts has poor prognosis value. The effects of the OSM: OSMR interaction are multipronged as it can alter either the function of malignant stem cell niches as observed in malignant hematological stem cells that do not express OSMR such as AML or MPNs, or directly act on malignant cells that express OSMR with further support from tumor-associated fibroblasts and endothelial cells which also express OSMR.
As OSM protein is highly expressed by activated neutrophils, monocytes and macrophages, blood concentration of OSM protein may not be a reliable prognosis marker of cancer treatment outcome because OSM concentration is also elevated during infections, sepsis (50, 51), acute and chronic inflammation (14, 16, 52) or in response to therapeutic treatments with myelopoietic cytokines such as granulocyte colony-stimulating factor (5). On the other hand, OSMR is not expressed by leukocytes but by non-hematopoietic malignant cells, as well as endothelial and mesenchymal cells forming the tumor environment. As soluble sOSMR protein is produced by alternative splicing of the OSMR transcript in exon 8 (19), it is likely that sOSMR protein may be similarly increased in the plasma of patients with tumors expressing high levels of OSMR transcripts, although actual concentrations of plasma sOSMR were not measured in patient samples from the TCGA used for our in silico analysis.
5 Conclusion and perspectives
Blood sOSMR concentration may represent an easily measurable prognosis marker not only in AML as recently reported (20) but also in the other malignancies discussed in this perspective, which warrants further clinical studies for its prognostic potential. In regards of possible treatments of patients with high levels of sOSMR protein or OSMR or OSM gene expression, the main axis of OSMR-mediated signaling is via tyrosine phosphorylation and activation of transcription factors STAT1 and STAT3 (and in some cell types STAT5) via tyrosine kinases JAK1 and JAK2 (4, 6). Therefore, adjunct treatments with small non-selective JAK1 and JAK2 tyrosine kinase inhibitors such as ruxolitinib, tofacitinib and others (53) may improve outcome in these patients. Therapeutics that specifically target OSMR or OSM such as the humanized neutralizing anti-OSM monoclonal antibody GSK2330811 may also be of interest to treat AML patients with high sOSMR protein or OSM transcripts as GSK2330811 was found to induce thrombocytopenia, anemia and mild neutropenia in patients with diffuse cutaneous systemic sclerosis (54). Effectiveness of such neutralizing OSM antibodies in other malignancies in which high OSMR expression leads to poorer outcome remains to be established. In conclusion, elevated OSMR-mediated signaling is emerging as a poor prognosis factor in a number of malignancies such as AML, GBM, LGG, STAD, KIRC and KIRP. Consequently, OSMR gene expression and sOSMR protein blood concentration should be systematically measured in patients with these diseases and clinical trials should be undertaken to test efficacy of adjunct therapies with JAK1/2 inhibitors or neutralizing OSM antibodies to current best treatments.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
JL: Formal analysis, Methodology, Writing – review & editing, Conceptualization, Writing – original draft. KB: Writing – review & editing, Writing – original draft. KA: Writing – review & editing, Writing – original draft. IW: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. JL, KB, KA and IW are supported by the Mater Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1636570/full#supplementary-material
Glossary
- ACC
adrenocortical carcinoma
- AML
acute myeloblastic leukemia
- BLCA
bladder urothelial carcinoma
- CESC
cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL
cholangial carcinoma
- COAD
colon adenocarcinoma
- DFS
disease-free survival
- DLBC
diffuse large B-cell lymphoma
- ESCA
esophageal carcinoma
- FLT-3
Fms-like tyrosine-protein kinase-3
- GBM
glioblastoma multiform
- GEPIA
Gene expression Profiling and Interactive Analysis
- GP130
glycoprotein-130
- HNSC
head and neck squamous cell carcinoma
- HSC
hematopoietic stem cell
- HSPC
hematopoietic stem and progenitor cell
- IL
interleukin
- JAK
Janus tyrosine-kinase
- KICH
kidney chromophobe carcinoma
- KIRC
kidney renal clear cell carcinoma
- KIRP
kidney renal papillary cell carcinoma
- LAML
acute myeloid leukemia
- LGG
lower grade glioma
- LIHC
hepatocellular carcinoma
- LUAD
lung adenocarcinoma
- LUSC
lung squamous cell carcinoma
- MESO
mesothelioma
- MPN
myeloproliferative neoplasm
- MSC
mesenchymal stromal cell
- OS
overall survival
- OSM
oncostatin M
- OSMR
oncostatin M receptor
- sOSMR
soluble oncostatin M receptor protein
- OV
ovarian serous cystadenocarcinoma
- PAAD
pancreatic adenocarcinoma
- PCGP
pheochromocytoma and paraganglioma
- PARD
prostate adenocarcinoma
- READ
rectum adenocarcinoma
- SARC
sarcoma
- SKCM
skin cutaneous melanoma
- STAD
stomach adenocarcinoma
- STAT
signal transducer and activator of transduction
- TCGA
The Cancer Genome Atlas
- TGCT
testicular germ cell tumor
- THCA
thyroid carcinoma
- THYM
thymoma
- UCEC
uterine corpus endometrial carcinoma
- UCS
uterine carcinosarcoma
- UVM
uveal melanoma
- VHL
von Hippel Lindau protein
References
1
Mosley B De Imus C Friend D Boiani N Thoma B Park LS et al . Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J Biol Chem. (1996) 271:32635–43. doi: 10.1074/jbc.271.51.32635
2
Ichihara M Hara T Kim H Murate T Miyajima A . Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood. (1997) 90:165–73. doi: 10.1182/blood.V90.1.165
3
Walker EC McGregor NE Poulton IJ Solano M Pompolo S Fernandes TJ et al . Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. (2010) 120:582–92. doi: 10.1172/JCI40568
4
Hermanns HM . Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. (2015) 26:545–58. doi: 10.1016/j.cytogfr.2015.07.006
5
Bisht K McGirr C Lee S-Y Tseng H-W Fleming W Alexander KA et al . Oncostatin M regulates hematopoietic stem cell (HSC) niches in the bone marrow to restrict HSC mobilization. Leukemia. (2022) 36:333–47. doi: 10.1038/s41375-021-01413-z
6
Sims NA Lévesque J-P . Oncostatin M: Dual regulator of the skeletal and hematopoietic systems. Curr Osteoporos Rep. (2024) 22:80–95. doi: 10.1007/s11914-023-00837-z
7
Houben E Hellings N Broux B Oncostatin M . an underestimated player in the central nervous system. Front Immunol. (2019) 10:1165. doi: 10.3389/fimmu.2019.01165
8
Sampath SC Sampath SC Ho ATV Corbel SY Millstone JD Lamb J et al . Induction of muscle stem cell quiescence by the secreted niche factor oncostatin M. Nat Commun. (2018) 9:1531. doi: 10.1038/s41467-018-03876-8
9
Jengelley DHA Wang M Narasimhan A Rupert JE Young AR Zhong X et al . Exogenous oncostatin M induces cardiac dysfunction, musculoskeletal atrophy, and fibrosis. Cytokine. (2022) 159:155972. doi: 10.1016/j.cyto.2022.155972
10
Kubin T Pöling J Kostin S Gajawada P Hein S Rees W et al . Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. (2011) 9:420–32. doi: 10.1016/j.stem.2011.08.013
11
Headland SE Dengler HS Xu D Teng G Everett C Ratsimandresy RA et al . Oncostatin M expression induced by bacterial triggers drives airway inflammatory and mucus secretion in severe asthma. Sci Transl Med. (2022) 14:eabf8188. doi: 10.1126/scitranslmed.abf8188
12
Luo P Wang P-X Li Z-Z Zhang X-J Jiang X Gong J et al . Hepatic oncostatin M receptor β regulates obesity-induced steatosis and insulin resistance. Am J Pathol. (2016) 186:1278–92. doi: 10.1016/j.ajpath.2015.12.028
13
Foglia B Sutti S Pedicini D Cannito S Bocca C Maggiora M et al . Oncostatin M, a profibrogenic mediator overexpressed in non-alcoholic fatty liver disease, stimulates migration of hepatic myofibroblasts. Cells. (2020) 9:28. doi: 10.3390/cells9010028
14
West NR Hegazy AN Owens BMJ Bullers SJ Linggi B Buonocore S et al . Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. (2017) 23:579–89. doi: 10.1038/nm.4307
15
Elks CM Zhao P Grant RW Hang H Bailey JL Burk DH et al . Loss of oncostatin M signaling in adipocytes induces insulin resistance and adipose tissue inflammation in vivo. J Biol Chem. (2016) 291:17066–76. doi: 10.1074/jbc.M116.739110
16
Boniface K Diveu C Morel F Pedretti N Froger J Ravon E et al . Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J Immunol. (2007) 178:4615–22. doi: 10.4049/jimmunol.178.7.4615
17
Wang ECE Dai Z Ferrante AW Drake CG Christiano AM . A subset of TREM2+ dermal macrophages secretes oncostatin M to maintain hair follicle stem cell quiescence and inhibit hair growth. Cell Stem Cell. (2019) 24:654–69.e6. doi: 10.1016/j.stem.2019.01.011
18
Garcia JP Utomo L Rudnik-Jansen I Du J Zuithoff NPA Krouwels A et al . Association between oncostatin M expression and inflammatory phenotype in experimental arthritis models and osteoarthritis patients. Cells. (2021) 10:508. doi: 10.3390/cells10030508
19
Diveu C Venereau E Froger J Ravon E Grimaud L Rousseau F et al . Molecular and functional characterization of a soluble form of oncostatin M/interleukin-31 shared receptor. J Biol Chem. (2006) 281:36673–82. doi: 10.1074/jbc.M607005200
20
Reville PK Wang B Marvin-Peek J Yuan B Kuo Y-A Garza AI et al . Blood-based proteomic profiling identifies OSMR as a novel biomarker of AML outcomes. Blood. (2025) 145:3015–28. doi: 10.1182/blood.2024027244
21
Tanaka M Hirabayashi Y Sekiguchi T Inoue T Katsuki M Miyajima A . Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood. (2003) 102:3154–62. doi: 10.1182/blood-2003-02-0367
22
Minehata K Takeuchi M Hirabayashi Y Inoue T Donovan PJ Tanaka M et al . Oncostatin M maintains the hematopoietic microenvironment and retains hematopoietic progenitors in the bone marrow. Int J Hematol. (2006) 84:319–27. doi: 10.1532/ijh97.06090
23
Sato F Miyaoka Y Miyajima A Tanaka M . Oncostatin M maintains the hematopoietic microenvironment in the bone marrow by modulating adipogenesis and osteogenesis. PLoS One. (2014) 9:e116209. doi: 10.1371/journal.pone.0116209
24
Garrigue A Kermasson L Susini S Fert I Mahony CB Sadek H et al . Human oncostatin M deficiency underlies an inherited severe bone marrow failure syndrome. J Clin Invest. (2025) 135:e180981. doi: 10.1172/jci180981
25
Ding L Saunders TL Enikolopov G Morrison SJ . Endothelial and perivascular cells maintain hematopoietic stem cells. Nature. (2012) 481:457–62. doi: 10.1038/nature10783
26
Winkler IG Barbier V Nowlan B Jacobsen RN Forristal CE Patton JT et al . Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self-renewal and chemoresistance. Nat Med. (2012) 18:1651–7. doi: 10.1038/nm.2969
27
Himburg HA Termini CM Schlussel L Kan J Li M Zhao L et al . Distinct bone marrow sources of pleiotrophin control hematopoietic stem cell maintenance and regeneration. Cell Stem Cell. (2018) 23:370–81.e5. doi: 10.1016/j.stem.2018.07.003
28
Itkin T Gur-Cohen S Spencer JA Schajnovitz A Ramasamy SK Kusumbe AP et al . Distinct bone marrow blood vessels differentially regulate hematopoiesis. Nature. (2016) 532:323–8. doi: 10.1038/nature17624
29
Furuhashi K Kakiuchi M Ueda R Oda H Ummarino S Ebralidze AK et al . Bone marrow niches orchestrate stem-cell hierarchy and immune tolerance. Nature. (2025) 638:206–15. doi: 10.1038/s41586-024-08352-6
30
Barbier V Erbani J Fiveash C Davies JM Tay J Tallack MR et al . Endothelial E-selectin inhibition improves acute myeloid leukemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. (2020) 11:2042. doi: 10.1038/s41467-020-15817-5
31
Tang Z Li C Kang B Gao G Li C Zhang Z . GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. (2017) 45:W98–W102. doi: 10.1093/nar/gkx247
32
Tang Z Li C Kang B . GEPIA Gene Expression Profiling and Interactive Analysis. China: Peking University (2025). Available online at: http://gepia.cancer-pku.cn/detail.php?gene=osm (Accessed May 22, 2025).
33
Yao L Setiadi H Xia L Laszik Z Taylor FB McEver RP . Divergent inducible expression of P-selectin and E-selectin in mice and primates. Blood. (1999) 94:3820–8. doi: 10.1182/blood.V94.11.3820
34
Hoermann G Cerny-Reiterer S Herrmann H Blatt K Bilban M Gisslinger H et al . Identification of oncostatin M as a JAK2 V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J. (2012) 26:894–906. doi: 10.1096/fj.11-193078
35
Müller TA Grundler R Istvanffy R Rudelius M Hennighausen L Illert AL et al . Lineage-specific STAT5 target gene activation in hematopoietic progenitor cells predicts the FLT3+-mediated leukemic phenotype. Leukemia. (2016) 30:1725–33. doi: 10.1038/leu.2016.72
36
Schwaller J Parganas E Wang D Cain D Aster JC Williams IR et al . Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol Cell. (2000) 6:693–704. doi: 10.1016/S1097-2765(00)00067-8
37
Khatib-Massalha E Di Buduo CA Chédeville AL Ho Y-H Zhu Y Grockowiak E et al . Defective neutrophil clearance in JAK2V617F myeloproliferative neoplasms drives myelofibrosis via immune checkpoint CD24. Blood. (2025) 146:717–31. doi: 10.1182/blood.2024027455
38
Koskela K Pelliniemi TT Rajamäki A Pulkki K Remes K . Serum oncostatin M in multiple myeloma: impact on disease severity and prognosis. Eur J Hematol. (2000) 65:52–6. doi: 10.1034/j.1600-0609.2000.90167.x
39
Krishnamurthy S Dong Z Vodopyanov D Imai A Helman JI Prince ME et al . Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. (2010) 70:9969–78. doi: 10.1158/0008-5472.Can-10-1712
40
Ayob AZ Ramasamy TS . Cancer stem cells as key drivers of tumor progression. J BioMed Sci. (2018) 25:20. doi: 10.1186/s12929-018-0426-4
41
Melzer C von der Ohe J Lehnert H Ungefroren H Hass R . Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol Cancer. (2017) 16:28. doi: 10.1186/s12943-017-0595-x
42
Uhlén M Fagerberg L Hallström BM Lindskog C Oksvold P Mardinoglu A et al . The Human Protein Atlas. Sweden: OSMR (2025). Available online at: https://www.proteinatlas.org/ENSG00000145623-OSMR/single+cell (Accessed May 22, 2025).
43
Natesh K Bhosale D Desai A Chandrika G Pujari R Jagtap J et al . Oncostatin-M differentially regulates mesenchymal and proneural signature genes in gliomas via STAT3 signaling. Neoplasia. (2015) 17:225–37. doi: 10.1016/j.neo.2015.01.001
44
Sharanek A Burban A Laaper M Heckel E Joyal J-S Soleimani VD et al . OSMR controls glioma stem cell respiration and confers resistance of glioblastoma to ionizing radiation. Nat Commun. (2020) 11:4116. doi: 10.1038/s41467-020-17885-z
45
Polak KL Tamagno I Parameswaran N Smigiel J Chan ER Yuan X et al . Oncostatin-M and OSM-receptor feed-forward activation of MAPK induces separable stem-like and mesenchymal programs. Mol Cancer Res. (2023) 21:975–90. doi: 10.1158/1541-7786.MCR-22-0715
46
Yu Z Li Z Wang C Pan T Chang X Wang X et al . Oncostatin M receptor, positively regulated by SP1, promotes gastric cancer growth and metastasis upon treatment with Oncostatin M. Gastric Cancer. (2019) 22:955–66. doi: 10.1007/s10120-019-00934-y
47
Lee BY Hogg EKJ Below CR Kononov A Blanco-Gomez A Heider F et al . Heterocellular OSM-OSMR signaling reprograms fibroblasts to promote pancreatic cancer growth and metastasis. Nat Commun. (2021) 12:7336. doi: 10.1038/s41467-021-27607-8
48
Wei S Chen Y Shi X Zuo L Zhang L . OSM may serve as a biomarker of poor prognosis in clear cell renal cell carcinoma and promote tumor cell invasion and migration. Int J Genomics. (2023) 2023:6665452. doi: 10.1155/2023/6665452
49
Nguyen-Tran HH Nguyen TN Chen CY Hsu T . Endothelial reprogramming stimulated by oncostatin M promotes inflammation and tumorigenesis in VHL-deficient kidney tissue. Cancer Res. (2021) 81:5060–73. doi: 10.1158/0008-5472.Can-21-0345
50
Gong Y Yan X Sun X Chen T Liu Y Cao J . Oncostatin M is a prognostic biomarker and inflammatory mediator for sepsis. J Infect Dis. (2020) 221:1989–98. doi: 10.1093/infdis/jiaa009
51
Salim SY AlMalki N Macala KF Wiedemeyer A Mueller TF Churchill TA et al . Oncostatin M receptor type II knockout mitigates inflammation and improves survival from sepsis in mice. Biomedicines. (2023) 11:483. doi: 10.3390/biomedicines11020483
52
Setiadi H El-Banayosy AM Long JW Maybauer MO Mihu MR El Banayosy A . Oncostatin M for characterizing the inflammatory burden and outcome of V-V ECMO in ARDS patients. Artif Organs. (2023) 47:1885–92. doi: 10.1111/aor.14619
53
Shawky AM Almalki FA Abdalla AN Abdelazeem AH Gouda AM . A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics. (2022) 14:1001. doi: 10.3390/pharmaceutics14051001
54
Denton CP Del Galdo F Khanna D Vonk MC Chung L Johnson SR et al . Biological and clinical insights from a randomized phase 2 study of an anti-oncostatin M monoclonal antibody in systemic sclerosis. Rheumatol (Oxford). (2022) 62:234–42. doi: 10.1093/rheumatology/keac300
Summary
Keywords
oncostatin M, prognosis factor, tumor microenvironment, acute myeloid leukemia, myeloproliferative neoplasm, glioblastoma, glioma, pancreatic adenocarcinoma
Citation
Lévesque J-P, Bisht K, Alexander KA and Winkler IG (2025) Role and prognostic value of oncostatin M and its receptor OSMR in acute myeloid leukemia, myeloproliferative neoplasms and non-hematological malignancies. Front. Oncol. 15:1636570. doi: 10.3389/fonc.2025.1636570
Received
28 May 2025
Accepted
02 September 2025
Published
17 September 2025
Volume
15 - 2025
Edited by
Fabrizio Carta, University of Florence, Italy
Reviewed by
Zongsi Zhu, Wenzhou Medical University, China
Kalyan Sethi, National Institute of Pharmaceutical Education and Research, India
Tina Bhardwaj, Lifecell International Pvt Ltd, India
Updates
Copyright
© 2025 Lévesque, Bisht, Alexander and Winkler.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Pierre Lévesque, jp.levesque@mater.uq.edu.au
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.