 Senlin Zhang1†
Senlin Zhang1† Xinran Chu1†
Xinran Chu1† Zhiheng Li1†
Zhiheng Li1† Qi Ji1†
Qi Ji1† Na Meng2†Wei Gao1Xiao Zhang3Yixin Hu1Li Gao1Bohan Li1Ping Chen4Huihui Wan4Yu Liu5Yongping Zhang1Yuanyuan Tian6Shuiyan Wu7Yizhen Li1,6,8*
Na Meng2†Wei Gao1Xiao Zhang3Yixin Hu1Li Gao1Bohan Li1Ping Chen4Huihui Wan4Yu Liu5Yongping Zhang1Yuanyuan Tian6Shuiyan Wu7Yizhen Li1,6,8* Shaoyan Hu1,6*
Shaoyan Hu1,6* Hu Liu1*
Hu Liu1*- 1Department of Hematology and Oncology, Children’s Hospital of Soochow University, Soochow University, Suzhou, China
- 2Department of Pediatrics, Xinxiang Central Hospital, The Fourth Clinical College of Xinxiang Medical University, Xinxiang, China
- 3Department of Hematology, Children’s Hospital of Shandong University, Jinan, China
- 4Suzhou Jsuniwell Medical Laboratory, Suzhou, China
- 5Pediatric Translational Medicine Institute, Shanghai Children’s Medical Center, School of Medicine, Shanghai Jiao Tong University, National Health Committee Key Laboratory of Pediatric Hematology and Oncology, Shanghai, China
- 6Jiangsu Pediatric Hematology and Oncology Center, Suzhou, China
- 7Hematologic Intensive Care Unit, Children’s Hospital of Soochow University, Suzhou, China
- 8Pediatric Hematology and Oncology Key Laboratory of Higher Education Institutions in Jiangsu Province, Suzhou, China
Background and objective: The 5-year overall survival (OS) for paediatric T-cell acute lymphoblastic leukaemia (T-ALL) exceeds 80% under the current treatment strategies; however, some patients suffer from treatment failure. Next-generation sequencing (NGS) identified recurrent mutated genes in T-ALL that might affect diagnosis, classification, prognostic stratification, and treatment response. This study aimed to characterise the clinical features and prognosis of gene mutations in paediatric patients with T-ALL.
Methods: We enrolled 144 paediatric patients with T-ALL at our centre. Chi-square or Fisher’s exact tests were used for categorical variables, and Kaplan–Meier and log-rank tests analysed the survival rates of these patients.
Results: The most common mutations were in NOTCH1 (58.3%), FBXW7 (19.4%), and PTEN (17.4%). Of 1262 gene mutations detected, 50 had a mutation frequency of >1%. Common mutations were not correlated with 5-year OS. Patients with higher NOTCH1 mutation loads had a lower proportion of D15 minimal residual disease ≥0.01% and better survival than those with a lower load.
Conclusion: This study reported the gene mutation spectrum of Chinese paediatric T-ALL, highlighting the role of NGS in molecular classification, risk stratification, and prognosis. Additionally, we emphasised the role of the variant allele frequency of NOTCH1 mutations in the treatment response and prognosis of childhood T-ALL.
Introduction
T-cell acute lymphoblastic leukaemia (T-ALL) is an aggressive hematologic malignancy characterised by the abnormal presence of immature T-cell progenitors, representing approximately 15% of paediatric ALL cases (1, 2). This incidence is generally consistent across different regions, including China (3). The 5-years overall survival of paediatric patients with T-ALL exceeds 80% under current treatment strategies; however, some patients experience relapse and drug resistance, which leads to treatment failure (4, 5). Previously, gene mutations have been associated with risk stratification, treatment response, and prognosis in paediatric patients with T-ALL (6, 7). The NOTCH1 or FBXW7 mutation is the most common in T-ALL and has been linked to a better outcome (8–10). However, Zuurbier et al. indicated that the NOTCH1 or FBXW7 mutation could predict a better prednisone response but did not influence the prognosis of pediatric T-ALL (11). Alterations in other genes such as PHF6 and LEF1 have also been reported in paediatric T-ALL, although their prognostic significance remains controversial. While some smaller studies have suggested that PHF6 or LEF1 abnormalities may be associated with inferior outcomes, more recent evidence from larger cohorts indicates that PHF6 mutations may be linked to favourable prognosis (12, 13). Therefore, the clinical relevance of these less common genetic alterations requires further clarification. Several studies have explored the relationship between gene mutations and the clinical characteristics and prognosis of paediatric T-ALL; however, the conclusions have been inconsistent and require further investigation. Here, we report the results of gene mutation detection through next-generation sequencing (NGS) and the characteristics and conditions of survival in 144 paediatric patients with T-ALL. Additionally, we explored the relevance of gene mutations, clinical features, and prognoses to provide evidence for clinical diagnosis and potential treatment strategies.
Methods
Patients
Between April 2012 and April 2023, 144 paediatric patients (0–18 years) diagnosed at the Children’s Hospital of Soochow University and who underwent NGS were enrolled. All diagnoses were made according to the MICM criteria. Among them, 97 patients received the Chinese Children’s Leukaemia Group-ALL-2008 (CCLG-ALL-2008) protocol (14) and 47 patients received the Chinese Children’s Cancer Group ALL-2015 (CCCG-ALL-2015) protocol (15). A total of 54 and 90 patients were classified into intermediate-risk (IR) and high-risk (HR) groups, respectively. This study was approved by the Ethics Committee of Children’s Hospital of Soochow University and followed the principles of the Declaration of Helsinki. Written informed consent for participation was obtained from the parents/guardians of all the patients.
Chemotherapy protocols and risk stratification
The CCLG-ALL-2008 protocol consisted of the following components: Induction therapy: VDLD (vincristine, daunorubicin, L-asparaginase, dexamethasone); Early intensification: CAM ×2 (cyclophosphamide, cytarabine, 6-mercaptopurine); Consolidation: High-dose methotrexate (HD-MTX ×4); Delayed intensification I and II: VDLD plus CAM or D-CAM; Maintenance: 6-mercaptopurine (6-MP) and methotrexate (MTX), with or without additional agents depending on risk group. High-risk patients received intensified treatment modules (HR1–3) adapted from the BFM protocol.
The CCCG-ALL-2015 protocol included: Induction therapy: PVDL (prednisone, vincristine, daunorubicin, L-asparaginase); Early intensification: CAT ×2 (cyclophosphamide, cytarabine, 6-MP); Consolidation: High-dose methotrexate (HD-MTX×4); Continuation (Weeks 16–31): DVDL (dexamethasone, vincristine, daunorubicin, L-asparaginase); Reinduction (Weeks 32–34): High-dose cytarabine (HD-Ara-C); Continuation (Weeks 35–54): 6-MP, MTX, and DVCA (dexamethasone, vincristine, cyclophosphamide, cytarabine); Maintenance: 6-MP and MTX, with or without vincristine and dexamethasone depending on randomised group assignment. High-risk patients under the CCCG-ALL-2015 protocol were eligible for hematopoietic stem cell transplantation.
For T-ALL patients, risk stratification in both protocols was based on a combination of clinical and response-related parameters, including: response to prednisone window therapy, bone marrow morphology on Day 15 (CCLG) or Day 19 (CCCG), and again on Day 33 or 46, initial white blood cell (WBC) count, extramedullary involvement, including central nervous system (CNS) or testicular infiltration, presence of high-risk cytogenetic abnormalities, if available.
Targeted sequencing and analysis
TRIzo (invitrogen, Cat 15596026) was used to isolate total RNA from bone marrow samples. Ribosomal RNA (rRNA) was depleted, and the stranded RNA-Seq libraries were rapidly constructed from 1 µg of purified total RNA (KAPA RNA HyperPrep Kit with RiboErase (HMR) KIT) according to the manufacturer’s instructions. After fragmentation, cDNA synthesis, adapter ligation, and library amplification, each library was sequenced on an Illumina NovaSeq 6000 platform with a 150-cycle paired-end sequencing. The raw FASTQ data were filtered using the fastp (version 0.23.4) software, which involved the removal of adapter sequences and low-quality sequencing reads. Subsequently, the filtered data were aligned to the reference genome (GRCh37) using STAR (version 2.7.10a) software. The aligned sequences were sorted using the Sambamba (version 0.6.6) software. Duplicate reads in the sorted sequences were removed using the GATK (version 4.0.5.1) software. After filtering, alignment, sorting, and duplicate removal, the resulting BAM files were used for the following analyses: (1) IKZF1 deletions were calculated using rnapeg (version 2.7.7) software; (2) the fusion genes were analysed using Arriba (version 2.4.0), Cicero (version 1.9.6), and Fusioncatcher (version 1.33) software; (3) gene expression levels were quantified using htseq-count (version 2.0.2) software. (4) INDEL analysis: INDELs (insertions and deletions) were detected using the lianti (revision 142) software. The detected mutations were annotated using annovar (updated on 2020-06-08) and snpeff (version 5.1f). All molecular testing was performed on bone marrow samples to ensure detection accuracy, as peripheral blood may contain fewer tumor cells and yield lower variant allele frequency (VAF).
Statistical analysis
All statistical analyses were performed using SPSS version 25 and R 4.3.2 software. Associations between specific gene mutations and clinical features were analysed using Fisher’s exact test or chi-square test. To account for multiple testing, false discovery rate (FDR) correction was applied. Overall survival (OS) was defined as the time from diagnosis to death. Event-free survival (EFS) was defined as the time from diagnosis (or treatment initiation) to the occurrence of any of the following events: failure to achieve complete remission (refractory disease), relapse, disease progression, or death from any cause. Cumulative incidence of treatment failure (CITF) is defined as the cumulative probability over time of experiencing either relapse or refractory disease, while accounting for death as a competing event. Median follow-up time was calculated using the reverse Kaplan–Meier method, in which censoring and event statuses are reversed. Survival functions were estimated using the Kaplan–Meier method and compared using the log-rank test. Cox regression analysis was used to identify factors associated with OS and EFS. CITF was estimated using a competing risk model and death in complete remission (CR) was treated as a competing event. Gray’s test was used for univariate comparisons between groups. Variables with P < 0.20 in univariate analysis were included in both the Cox proportional hazards model and the multivariable competing risk model. P<0.05 was considered statistically significant.
Results
Clinical characteristics of 144 childhood T-ALL
Among the 144 paediatric patients with T-ALL, 116 (80.6%) were male and 28 (19.4%) were female, with a ratio of 4.1:1. The median age at diagnosis was 8.0 years (range 1.2–16.8 years). The median white blood cell (WBC) at diagnosis was 106.33 ×109/L (range 1.05–750.20). The median haemoglobin and platelet counts at diagnosis were 98.50 g/L (range 37.00–159.00) and 63.00×109/L (range 5.00–412.00), respectively. Regarding the immunophenotype, 6 patients had early thymic precursor (ETP) (4.2%), and 138 had non-ETP (95.8%) in our cohort. At the end of follow-up, 64 patients (44.4%) underwent haematopoietic stem cell transplantation (HSCT), while 80 patients (55.6%) received chemotherapy alone. Among the 64 patients who received HSCT, 54 were transplanted in first complete remission (CR1), 9 in second complete remission (CR2) following relapse, and 1 patient underwent transplantation without achieving remission (NR). The baseline patient characteristics are summarised in Table 1.
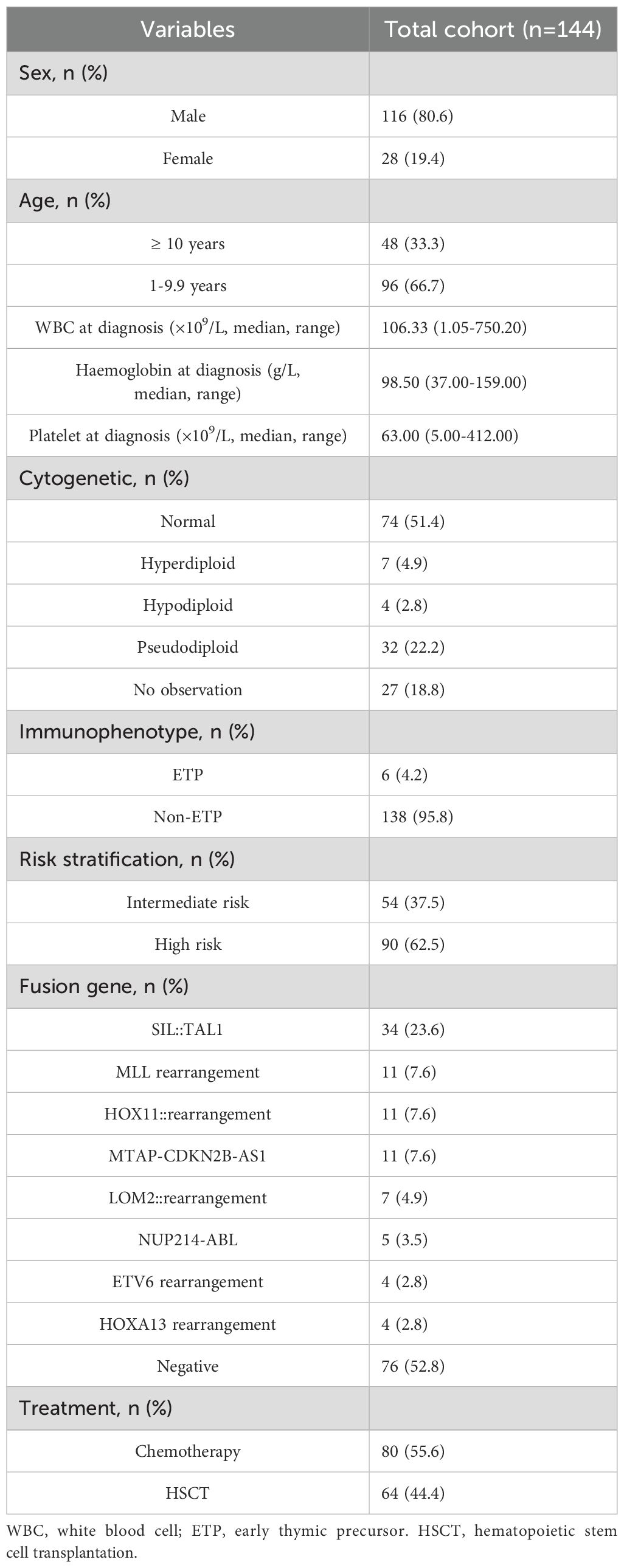
Table 1. The clinical characteristics of 144 pediatric T-ALL.
Gene mutations and signalling pathways in 144 T-ALL cases
The gene mutation data from 144 children with T-ALL who underwent second-generation sequencing were collected. Among them, 142 patients (98.6%) harboured at least one mutation, and 125 patients (86.8%) had two or more mutations. After applying the VAF threshold, we focused our analysis on mutations of potential clinical relevance. NGS results revealed that the top 20 genes were NOTCH1 (58.3%, 84/144), FBXW7 (19.4%, 28/144), PTEN (17.4%, 25/144), NRAS (9%, 13/144), PHF6 (9%, 13/144), USP7 (9%, 13/144), WT1 (6.9%, 10/144), IL7R (6.9%, 10/144), JAK3 (6.3%, 9/144), EZH2 (4.9%, 7/144), BCL11B (4.9%, 7/144), DNM2 (4.9%, 7/144), SPATA31E1 (4.9%, 7/144), CCND3 (4.2%, 6/144), KRAS (3.5%, 5/144), PIK3R1 (3.5%, 5/144), CDKN2A (3.5%, 5/144), STAT5B (3.5%, 5/144), SUZ12 (3.5%, 5/144), MKI67 (3.5%, 5/144) (Figure 1a). Next, we divided those gene mutations into eight groups according to the signalling pathways involved in gene mutations and their functions, including: NOTCH (62.5%, 90/144), epigenetic regulators (31.9%, 46/144), transcriptional factors (27.8%, 40/144), PI3K-AKT (25%, 36/144), RAS (18.1%, 26/144), JAK-STAT (16.7%, 24/144), cell cycle regulation (15.3%, 22/144), translation and RNA stability (3.5%, 5/144) pathways (Figure 1b). In addition, we analysed the co-occurrence of the mutated genes. As shown in Figure 1c, we found pairwise associations between NOTCH1 and PTEN (P<0.01), FBXW7 and USP7 (P<0.05).
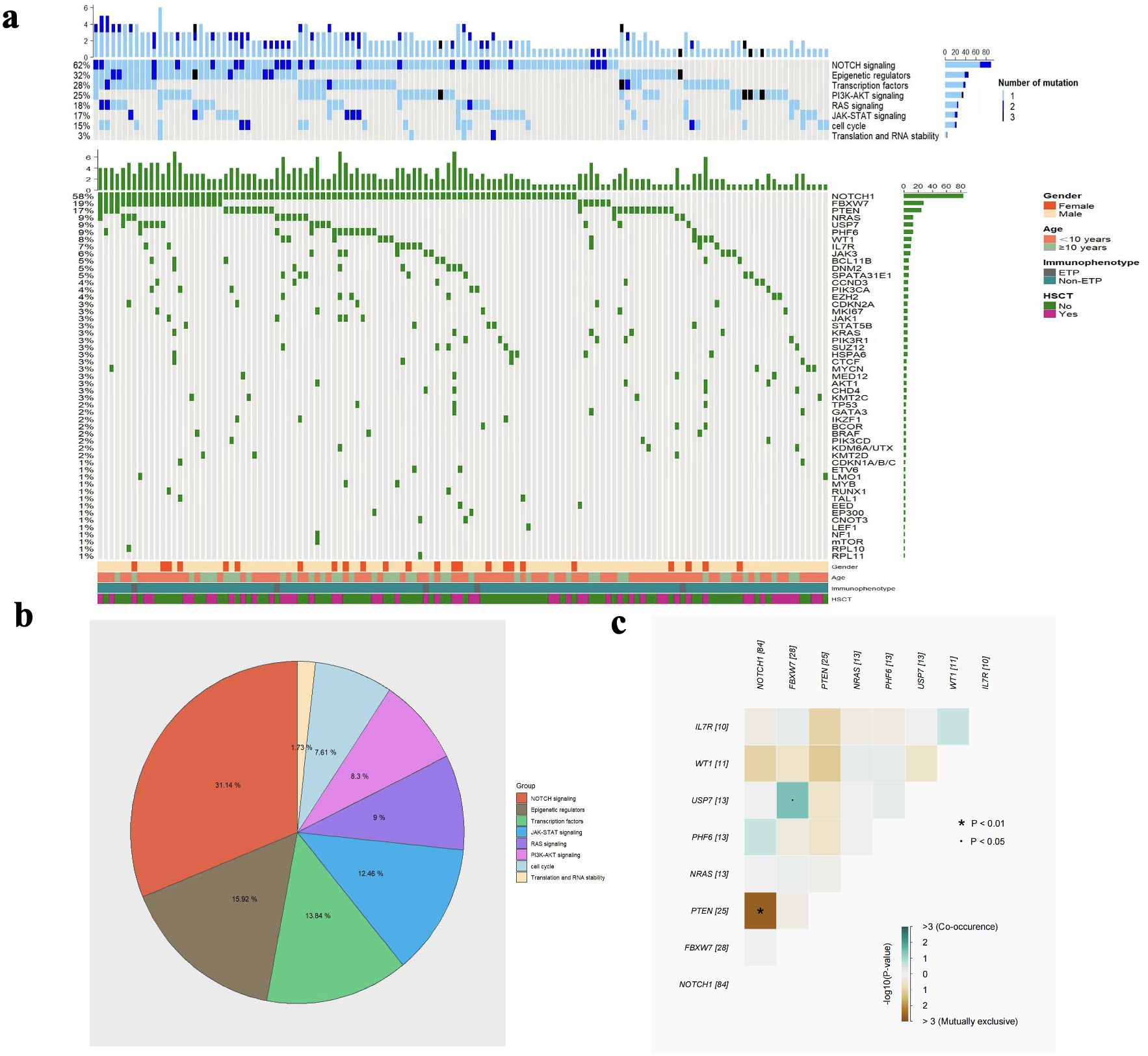
Figure 1. The heatmap of gene mutations and signalling pathways (a), the proportion of each signalling pathway (b), and the correlation analysis of co-occurring gene mutations (c).
The relationship between gene mutations and clinical features
No significant differences in gene mutation were observed between male and female groups (all P>0.050) (Figure 2a). No significant differences in gene mutation were observed between 1–9.9 years and ≥10 years groups (all P>0.050) (Figure 2b). Moreover, NOTCH1 mutations were associated with elevated WBC counts (P = 0.018) (Figure 2c). Patients with PHF6 mutations were more likely to develop lower haemoglobin levels at diagnosis (P = 0.001) (Figure 2d). Additionally, NOTCH1 mutations were associated with a higher platelet count at diagnosis (P = 0.030) (Figure 2e).
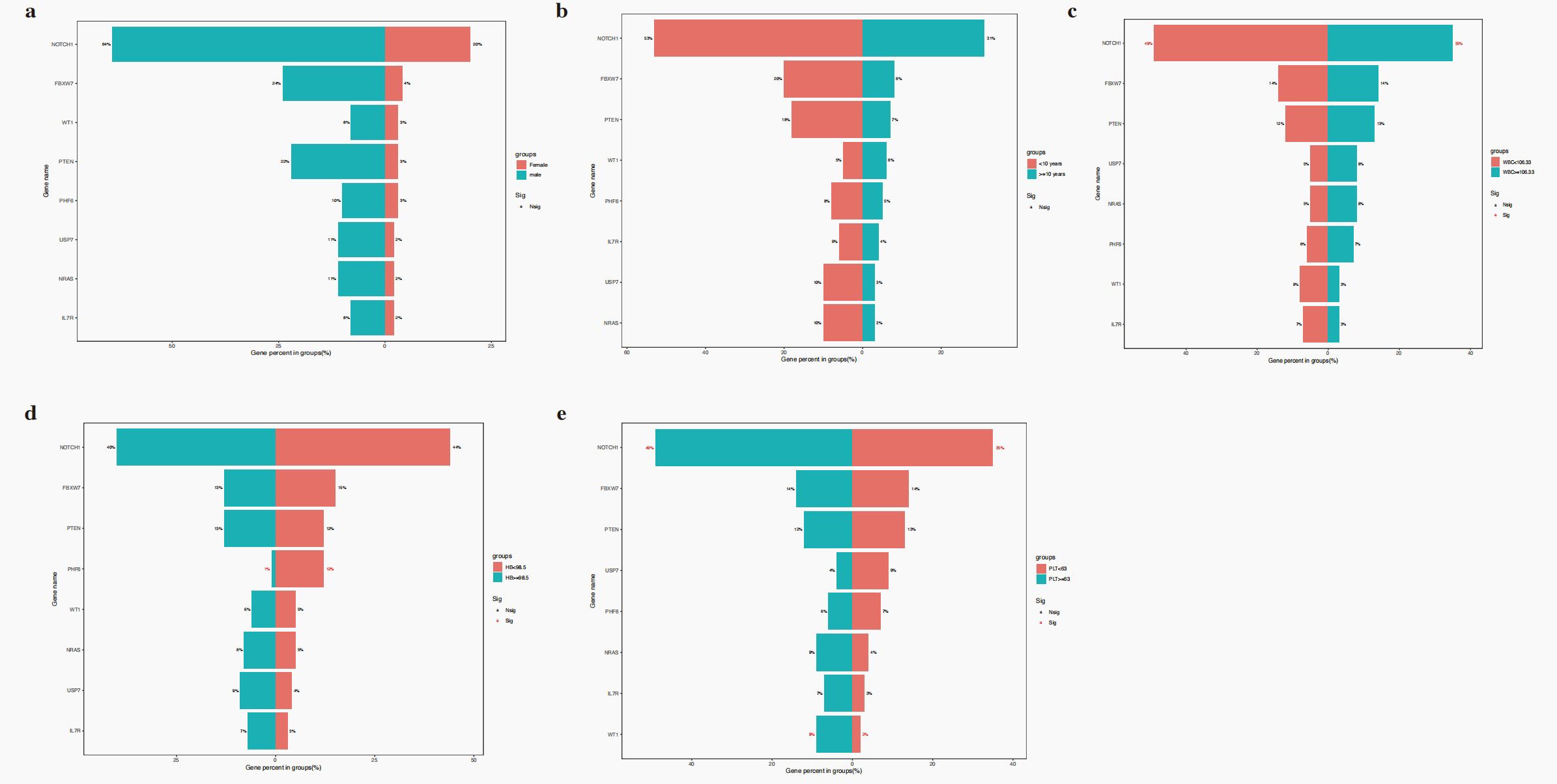
Figure 2. The relationship of gene mutations and sex (a), age (b), WBC (c), haemoglobin (d) and platelet (e).
No significant differences in gene mutation were observed between insensitive and sensitive to steroid treatment in this cohort (all P>0.050) (Figure 3a). The NOTCH1 mutation was associated with a better D15/19 BM response with a worse response (P = 0.023, Figure 3b). No gene mutations were related to other early treatment outcomes, such as D15/19 minimal residual disease (MRD), D33/46 BM response, and MRD (Figure 3c–e).
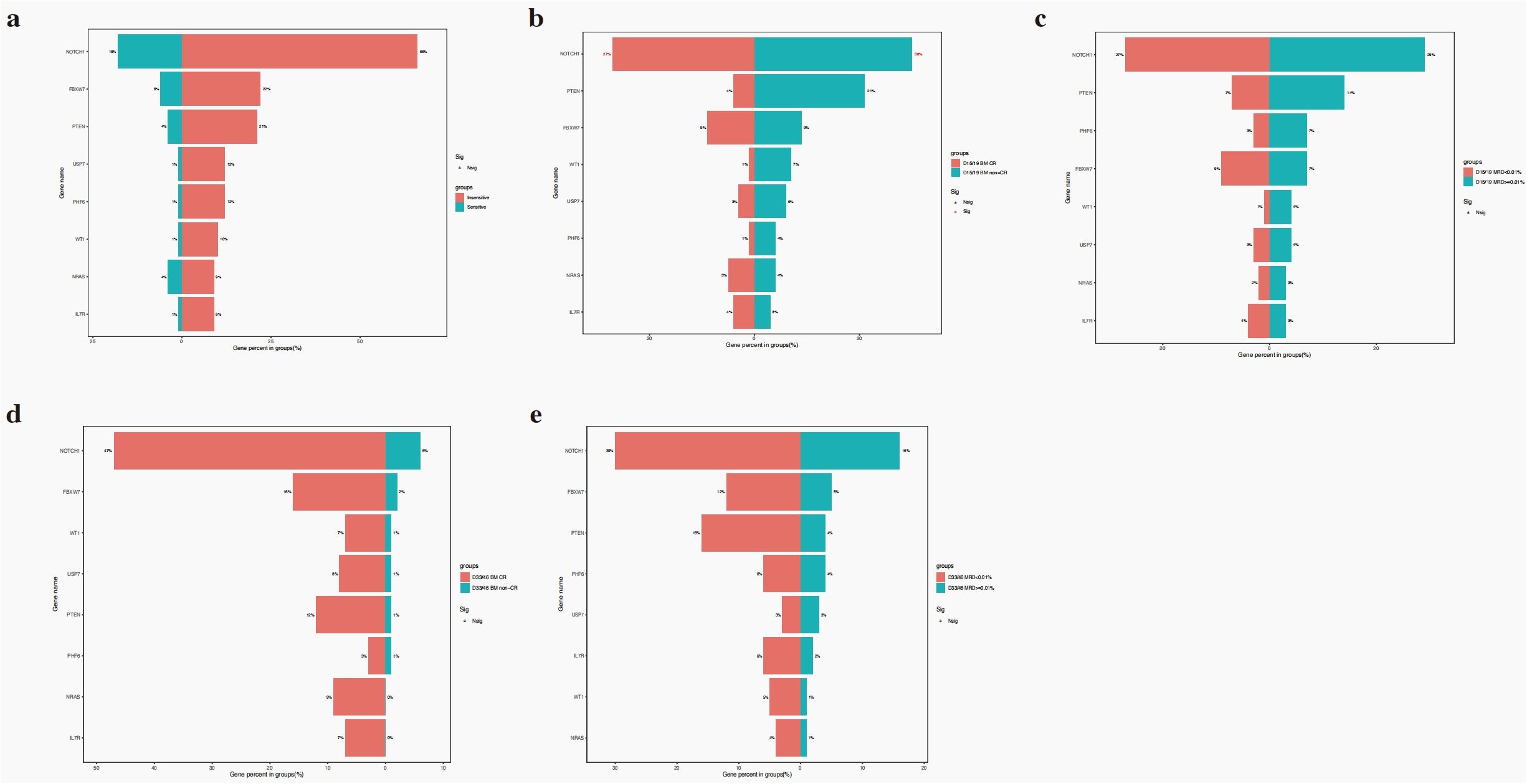
Figure 3. The relationship of gene mutations and steroid response (a), D15/19 BM response (b), D33/46 BM response (c), D15/19 MRD (d), D33/46 MRD (e).
The role of gene mutations on the prognosis of paediatric T-ALL
The latest follow-up occurred on 2023 December 31, and the median follow-up was 42 months (range, 1.0–129.0 months). The 5-year OS for 139 paediatric T-ALL was 69.90 ± 4.62% (Figure 4a). The ten most common gene mutations were NOTCH1, FBXW7, PTEN, RAS (NRAS/KRAS), PHF6, USP7, WT1, and IL7R. We explored their prognostic roles in the 5-year OS of 139 paediatric patients with T-ALL (5 of 144 patients were lost to follow-up). However, no prognostic biomarker was found among the gene mutations that influenced the survival rate of these patients (Figure 4b–i).
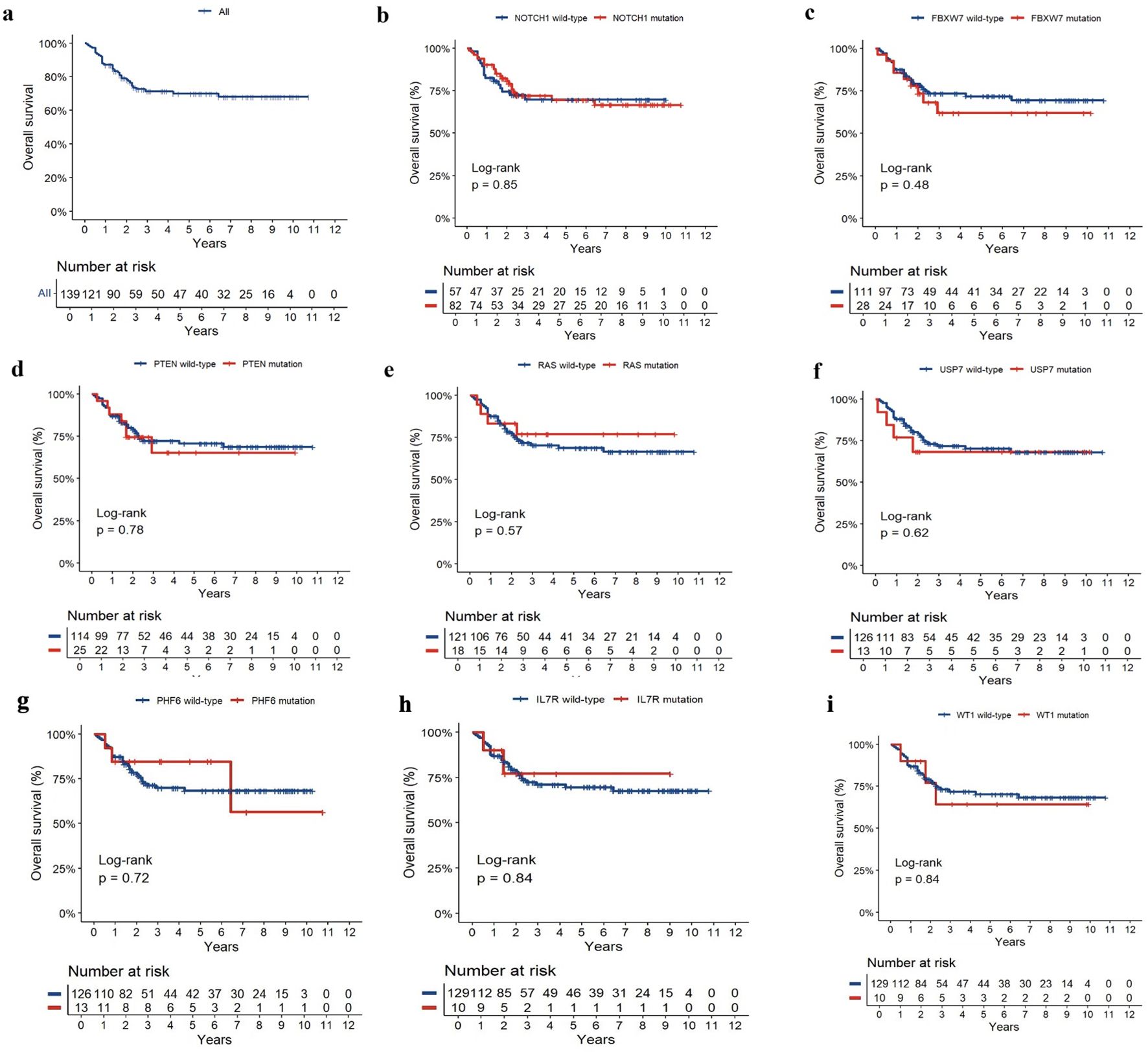
Figure 4. The curves of OS (a) and the prognosis of NOTCH1 (b), FBXW7 (c), PTEN (d), RAS (NRAS/KRAS) (e), USP7 (f), PHF6 (g), WT1 (h), and IL7R (i) mutation in pediatric T-ALL.
The role of NOTCH1 mutation load in the treatment response, prognosis of childhood T-ALL
Of 84 patients with NOTCH1 mutations, 71 had variant allele frequencies (VAF) data. Based on the maximally selected log-rank statistic, a VAF of 12.850 was the cutoff point for differentiating patient prognosis. Patients were categorised into high (52 patients) and low (19 patients) mutation load groups. Regarding clinical characteristics, the high mutation load group had higher bone marrow blasts at diagnosis, and a lower proportion underwent allogeneic HSCT. Other clinical features were not significantly different between the two groups (all P>0.05). The clinical features of the 71 patients are summarised in Table 2, and the detailed results with false discovery rate (FDR) correction are provided in Supplementary Table 1. Regarding the association between NOTCH1 mutation load and early treatment response, we found that a high NOTCH1 mutation load have a lower proportion of MRD ≥1% on D15 (Table 3). Additionally, we examined the impact of the NOTCH1 mutation load on the 5-year OS, EFS and CITF in the cohort of 71 paediatric patients with T-ALL. Our results indicated that patients with a high NOTCH1 mutation load have higher 5-year OS (Figure 5a) and EFS (Figure 5b) than those with a low NOTCH1 mutation load (P = 0.02 and 0.043, respectively). No statistically significant difference was observed for CITF between the two groups (Figure 5c), although a trend toward lower incidence was noted in the high mutation load group (Gray’s test, P = 0.067). Table 4 presents the univariate and multivariate analyses of the factors affecting the 5-year OS, EFS, and CITF. In the univariate analyses, we found that platelet count less than and equal to 50×109 was a risk factor for 5-year OS, BM not CR after induction was a risk factor for 5-year EFS, while a high NOTCH1 mutation load was a protective factor for 5-year OS and EFS. In the multivariate analyses, a high NOTCH1 mutation load was an independent protective factor for the 5-year OS (P = 0.042) and 5-year EFS (P = 0.0104). However, high VAF was not significantly associated with CITF in our univariate and multivariate analysis. We further validated the role of VAF in NOTCH1 mutations on paediatric T-ALL OS. The training set consisted of 50 patients treated with the CCLG-ALL-2008 protocol, and 21 patients who received the CCCG-ALL-2015 protocol were used for the validation set. A VAF of 0.1594 was identified as the cutoff point for differentiating prognosis among patients treated with CCLG-ALL-2008 protocol (83.0 ± 8.39 vs 65.0 ± 11.82, P = 0.23, Figure 5c). When this cutoff was applied to patients who received CCCG-ALL-2015, we still found superior survival in the high mutation load group (100% vs 71.4 ± 17.1%, P = 0.041, Figure 5d).
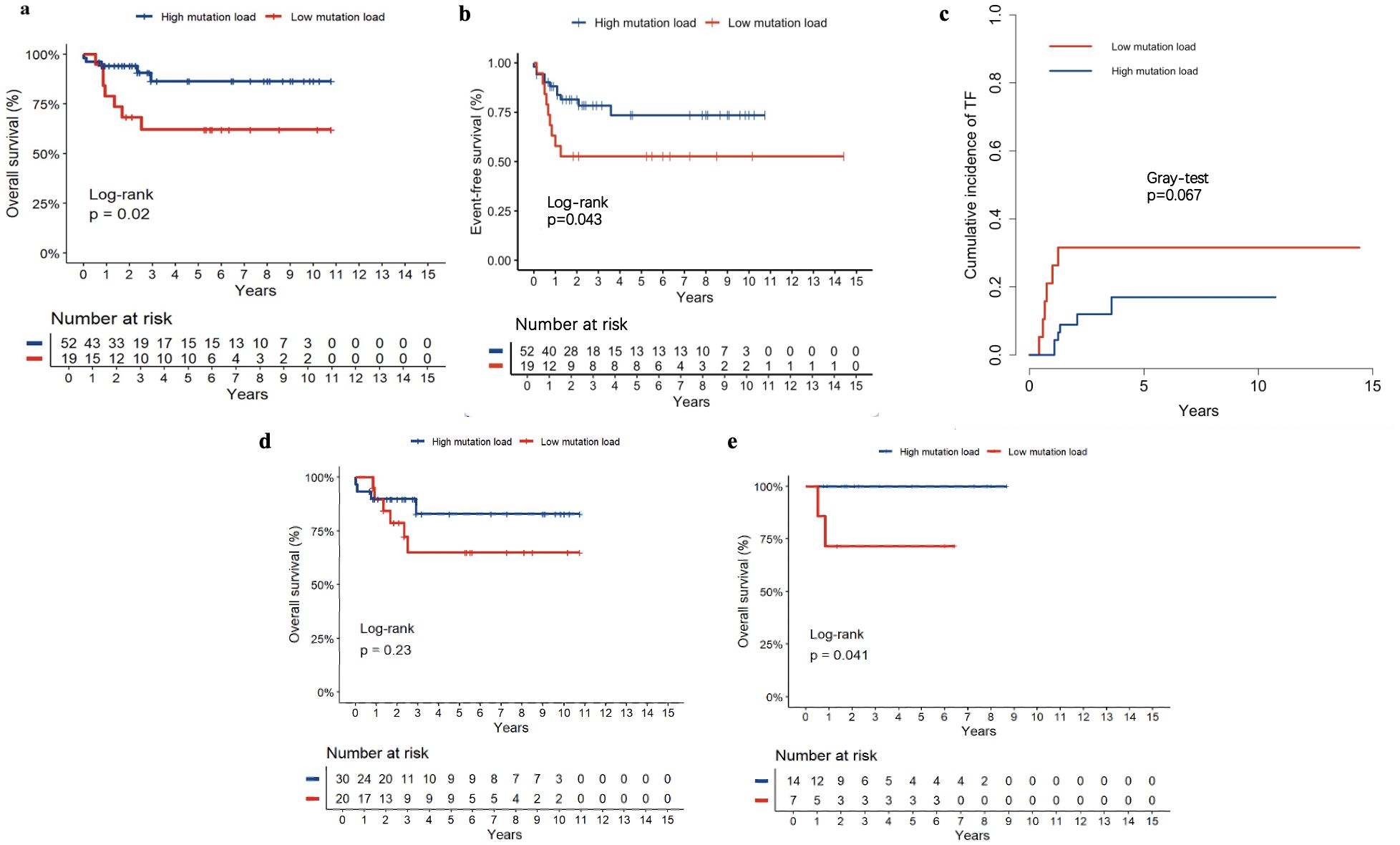
Figure 5. The 5-year OS (a), EFS (b) and CITF (c) of high and low NOTCH1 mutation load, The role of VAF on the OS of the training set (CCLG-ALL-2008, (d) and validation set (CCCG-ALL-2015, (e).
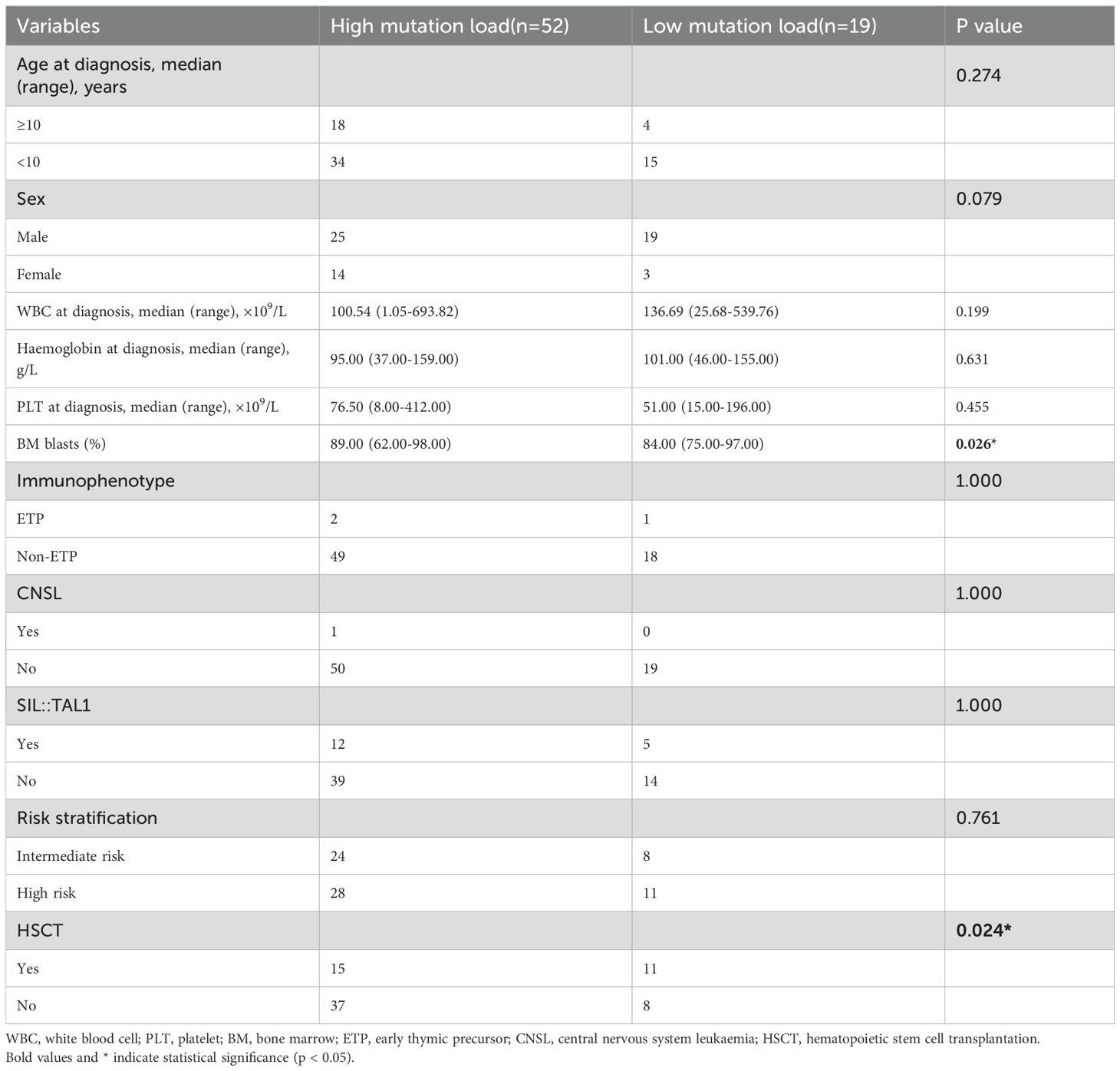
Table 2. The clinical characteristics of 52 high mutation load and 19 low mutation load patients.
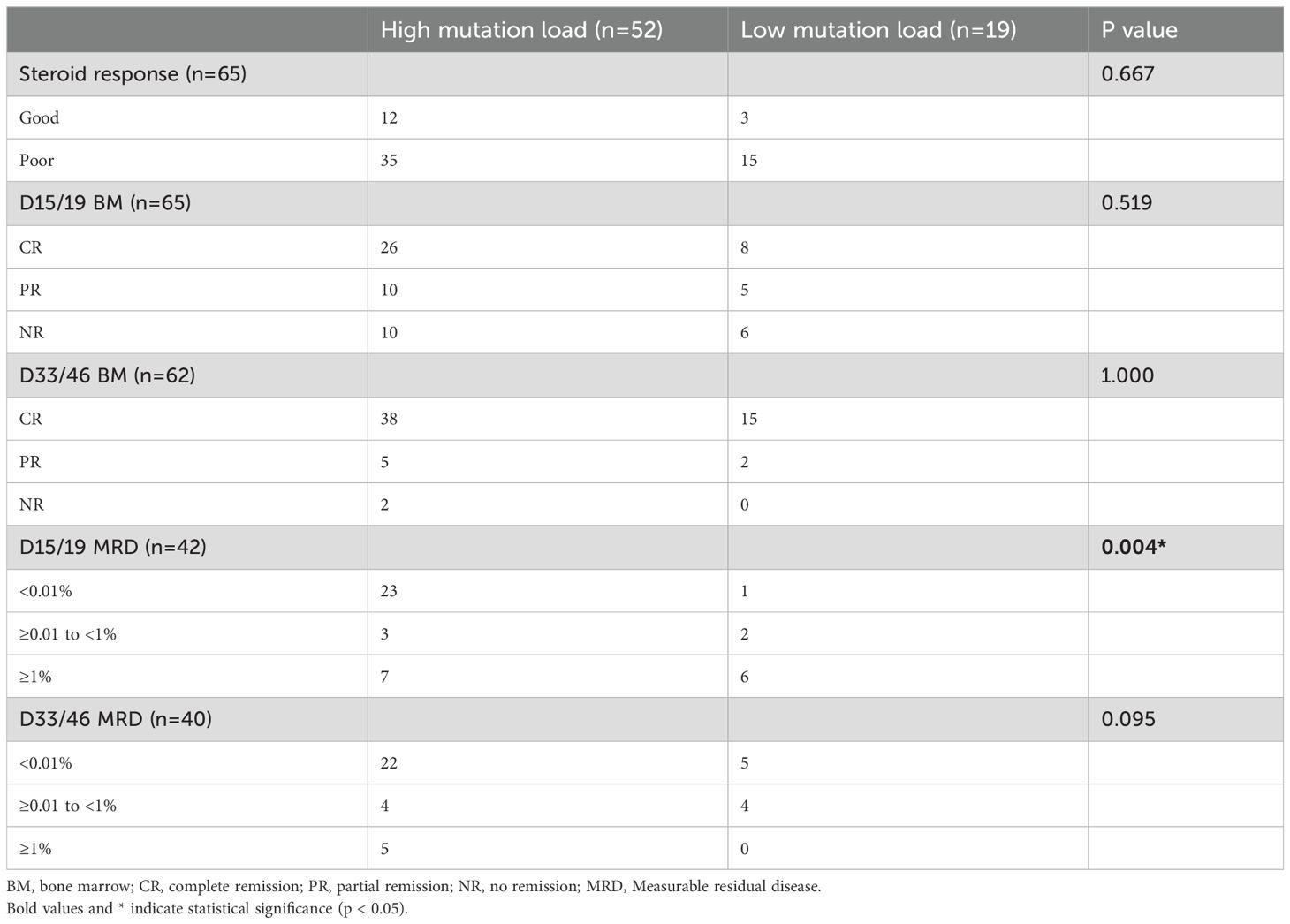
Table 3. The relationship between NOTCH1 mutation load and early treatment response.
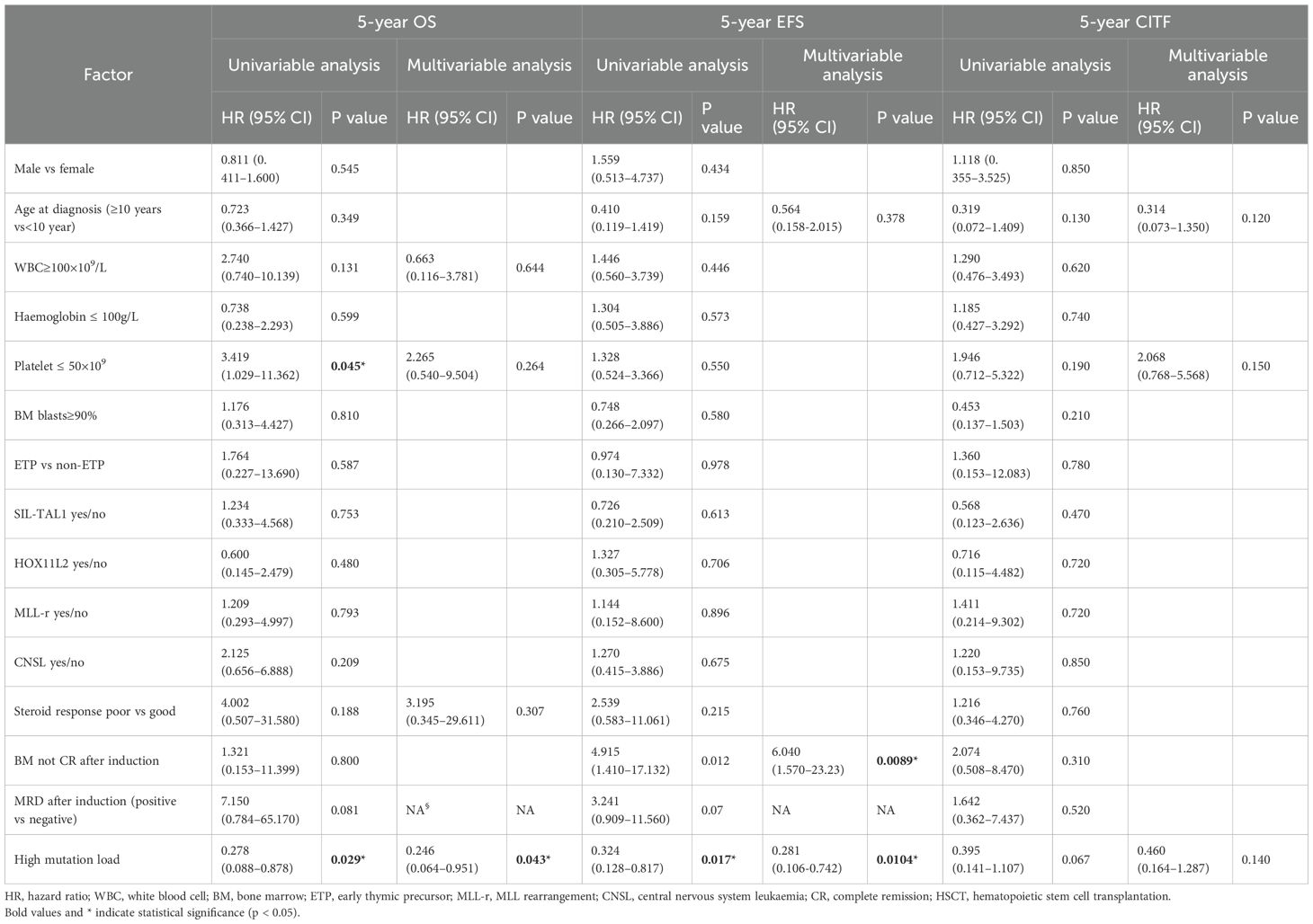
Table 4. The results of univariable and multivariable cox regression model for 5-years OS and EFS, competing risk regression model for 5-years CIR in childhood T-ALL.
Discussion
In this study, we report a spectrum of gene mutations and their clinical features and prognostic roles in paediatric patients with T-ALL. NOTCH1 mutation (58.3%) and NOTCH1 signalling pathway (62.5%) were the most common gene mutations and pathways in this study, respectively. Additionally, we found that certain gene mutations were associated with different clinical features in children with T-ALL. However, we did not identify any genetic mutations that could influence the prognosis of paediatric patients with T-ALL.
Several studies have investigated the clinical characteristics of various genetic mutations. In adult patients with T-ALL, Asnafi et al. found that the NOTCH1 mutation group had lower WBC counts than the wild-type group (16). Our results are consistent with these findings. We observed that NOTCH1 mutations were associated with higher platelet counts. Steroid response is recognised as a strong predictor of treatment response and outcomes in children with ALL (17, 18). The most commonly reported genetic mechanism associated with the steroid response in paediatric T-ALL is the activation of IL-7R signalling. A study published in Leukemia found that IL-7R mutations contribute to steroid resistance in children with T-ALL (19). However, our study did not observe such an association between IL-7R mutation and steroid response, and this discrepancy warrants further investigation.
Mutations in NOTCH1 and FBXW7 result in the abnormal activation of the NOTCH1 signalling pathway, which is crucial in the pathogenesis of T-ALL (20). Previously, the reported incidence rate of NOTCH1 mutation in pediatric T-ALL was more than 50% (21, 22). In this cohort, 58.3% of paediatric patients with T-ALL had NOTCH1 mutations, and 19.4% had FBXW7 mutations. Several studies suggested that NOTCH1 or FBXW7 mutations may predict a better outcome in pediatric T-ALL (8, 9, 23). Recently, a large cohort of 1300 childhood T-ALL cases indicated that most NOTCH1 variants, except intronic single nucleotide variant and intragenic losses, were associated with superior OS and EFS (24). While NOTCH1 or FBXW7 mutations were related to superior survival in most studies, they were not correlated with survival in this study. We speculate that this difference may be attributable to the diversity of treatment strategies across different centres. Most patients with NOTCH1 mutations at our centre underwent HSCT (37 of 84, 44%), which may account for the favourable role of this mutation. In another centre with the same treatment protocol, the survival rate of patients with NOTCH1 mutation did not significantly increase (25). Therefore, the diversity of protocols from different regions in paediatric T-ALL may have contributed to this difference. More high-quality studies are warranted to investigate the prognostic role of NOTCH1 or FBXW7 mutations in paediatric patients with T-ALL, particularly in China.
PTEN is one of the most frequently expressed oncosuppressors and is commonly inactivated in human cancers, including T-ALL (26). It is a negative regulator of the phosphatidylinositol-3 kinase (PI3K)/Akt or mechanistic target of rapamycin (mTOR) signalling pathway (27). In previous studies, PTEN mutations were detected in 11–27% of pediatric T-ALL cases (28). However, the prognostic value of PTEN in paediatric T-ALL remains unclear. In a cohort from the MRC UKALL2003 trial, the prognoses of patients with PTEN abnormalities and the wild-type group were not significantly different (29). Our results align with these findings. However, among children with T-ALL treated with ALL IC-BFM protocols, PTEN mutation was associated with poor treatment response and unfavourable clinical outcomes (30). Pölönen et al. reported that deletions or losses were associated with worse outcomes (24). The role of PTEN in T-ALL requires further investigation and subdivision.
RAS genes are members of the small GTPase family and include three separate genes: NRAS, KRAS, and HRAS (31). The reported incidence rates of NRAS and KRAS were 9.38% and 3.13% in a cohort of 64 Korean children with T-ALL (6). Similar results were observed in our 144 paediatric T-ALL cohort, with NRAS and KRAS present at 9.0% and 3.5%, respectively. Perentesis et al. indicated that RAS mutations were present in approximately 15% of paediatric cases and were not associated with inferior outcomes in 870 children with ALL (32). In this study, the survival rates of patients with RAS mutations were consistent with their results. However, the prognostic role of RAS mutations in paediatric T-ALL has rarely been investigated, is inconsistent, and needs further exploration. Van Vlierberghe et al. indicated that paediatric and adult patients with T-ALL, with or without PHF6 mutations, did not influence the overall outcome (33), which is consistent with our results.
Changes in VAF resulting from different gene mutations play an important role in predicting disease status and assessing prognosis in adult myelodysplastic syndrome and acute myeloid leukaemia (34). Zhang et al. reported that a high burden of IKZF1 mutations (VAF>0.20) is associated with inferior OS in AML (35). Another study indicated that TP53 VAF>40% is an independent risk factor for lower OS in patients with myelodysplastic syndromes (MDS) (36). These studies emphasised the role of VAF in the prognosis of haematologic malignancies, whereas few studies have explored its significance in ALL, especially T-ALL. Our results indicated that a higher NOTCH1 mutation load was associated with better D15 response and superior survival compared to a lower mutation load, emphasising the role of VAF in patients with NOTCH1 mutation in T-ALL. However, the limited number of patients necessitates large-scale, high-quality prospective studies to further explore the role of VAF in paediatric T-ALL.
This study has limitations. First, the long accrual period and use of different treatment protocols may have introduced heterogeneity that affects the precision of our estimates. Second, as a retrospective analysis with a relatively small sample size, it is susceptible to selection and information bias. Third, MRD status was unavailable for many patients, precluding robust evaluation of its association with prognosis. Fourth, VAF of this study was derived from RNA sequencing data, which can be influenced by both the proportion of mutant transcripts and gene expression levels, making it challenging to disentangle these effects. Fifth, as only RNA NGS was performed, some diagnostic mutations may have been missed. Finally, the assessed links between gene mutations and clinical features were exploratory and underpowered; thus, they should be regarded as hypothesis-generating rather than definitive. Confirmation in larger, independent cohorts is required, and well-designed, multicentre prospective studies are warranted to further clarify how clinical features and diverse genetic alterations relate to outcomes.
In conclusion, several correlations were found between clinical features and gene mutations. Higher VAF in NOTCH1 mutations with superior survival rates were detected in our study. Using NGS technology analysis can improve the accurate prediction of risk stratification and prognosis in paediatric T-ALL, helping to identify potential targets for immunotherapy and guiding treatment to improve the long-term outcomes of children with T-ALL.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
This study was approved by the Ethics Committee of Children’s Hospital of Soochow University and followed the principles of the Declaration of Helsinki. Written informed consent for participation was obtained from the parents/guardians of all the patients. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SZ: Conceptualization, Data curation, Methodology, Writing – original draft, Formal analysis, Investigation, Software. XC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. ZL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. QJ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. NM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. WG: Data curation, Formal analysis, Methodology, Writing – original draft. XZ: Data curation, Formal analysis, Investigation, Software, Writing – original draft. YH: Conceptualization, Data curation, Investigation, Software, Writing – original draft. LG: Conceptualization, Investigation, Writing – original draft. BL: Conceptualization, Data curation, Investigation, Writing – original draft. PC: Conceptualization, Investigation, Software, Writing – original draft. HW: Data curation, Investigation, Writing – original draft. YL: Data curation, Investigation, Writing – review & editing. YZ: Data curation, Investigation, Project administration, Writing – review & editing. YT: Data curation, Investigation, Writing – original draft. SW: Data curation, Investigation, Project administration, Writing – original draft. YZL: Supervision, Funding acquisition, Project administration, Writing – review & editing. SH: Funding acquisition, Resources, Supervision, Validation, Visualization, Writing – review & editing. HL: Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the following grants: The National Key Research and Development Program of China (no.2022YFC2502700), the National Natural Science Foundation of China (NSFC, 82170218, 82470221) to Shaoyan Hu, NSFC 82100229 to Yuanyuan Tian, NSFC 82200177 to Li Gao, NSFC 82470127 to Yixin Hu, NSFC 82300244 to Bohan Li, NSFC 82400264 to Yongping Zhang, NSFC 82300182 to Shuiyan Wu, NSFC 82470160 to Yizhen Li, Suzhou Projects (DZXYJ202305, GSWS2023048, 2020ZKPB02) to Shaoyan Hu, and the Suzhou Municipal Key Laboratory (SZS201615, SKY2022012, SZS2023014)to Shaoyan Hu. Soochow University of Medical School, ML13101223 to Shaoyan Hu.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1666527/full#supplementary-material
References
1. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. (2002) 1:75–87. doi: 10.1016/S1535-6108(02)00018-1
2. Pui CH, Robison LL, and Look AT. Acute lymphoblastic leukaemia. Lancet. (2008) 371:1030–43. doi: 10.1016/S0140-6736(08)60457-2
3. Zhang X, Wang Y, Tian X, Sun L, Jiang H, Chu J, et al. Delineation of features, responses, outcomes, and prognostic factors in pediatric PDGFRB-positive acute lymphoblastic leukemia patients: A retrospective multicenter study. Cancer. (2024) 130:3902–12. doi: 10.1002/cncr.35481
4. Teachey DT, Devidas M, Wood BL, Chen Z, Hayashi RJ, and Hermiston ML. Children’s oncology group trial AALL1231: A phase III clinical trial testing bortezomib in newly diagnosed T-cell acute lymphoblastic leukemia and lymphoma. J Clin Oncol. (2022) 40:2106–18. doi: 10.1200/JCO.21.02678
5. Burns MA, Place AE, Stevenson KE, Gutiérrez A, Forrest S, Pikman Y, et al. Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05–001 and 11-001. Pediatr Blood Cancer. (2021) 68:e28719. doi: 10.1002/pbc.28719
6. Chang YH, Yu CH, Jou ST, Lin CY, Lin KH, Lu MY, et al. Targeted sequencing to identify genetic alterations and prognostic markers in pediatric T-cell acute lymphoblastic leukemia. Sci Rep. (2021) 11:769. doi: 10.1038/s41598-020-80613-6
7. Yuan L, Lu L, Yang Y, Sun H, Chen X, Huang Y, et al. Genetic mutational profiling analysis of T cell acute lymphoblastic leukemia reveal mutant FBXW7 as a prognostic indicator for inferior survival. Ann Hematol. (2015) 94:1817–28. doi: 10.1007/s00277-015-2474-0
8. Yuan Y, Li J, Xue TL, Hu HR, Lin W, Liu SGLi J, et al. Prognostic significance of NOTCH1/FBXW7 mutations in pediatric T cell acute lymphoblastic leukemia: a study of minimal residual disease risk-directed CCLG-ALL 2008 treatment protocol. Leuk Lymphoma. (2022) 63:1624–33. doi: 10.1080/10428194.2022.2032033
9. Natarajan V, Bandapalli OR, Rajkumar T, Sagar TG, and Karunakaran N. NOTCH1 and FBXW7 mutations favor better outcome in pediatric South Indian T-cell acute lymphoblastic leukemia. J Pediatr Hematol Oncol. (2015) 37:e23–30. doi: 10.1097/MPH.0000000000000290
10. Fogelstrand L, Staffas A, Wasslavik C, Sjögren H, Söderhäll S, Frost BM, et al. Prognostic implications of mutations in NOTCH1 and FBXW7 in childhood T-ALL treated according to the NOPHO ALL-1992 and ALL-2000 protocols. Pediatr Blood Cancer. (2014) 61:424–30. doi: 10.1002/pbc.24803
11. Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. (2010) 24:2014–22. doi: 10.1038/leu.2010.204
12. Yeh TC, Liang DC, Liu HC, Jaing TH, Chen SH, Hou JY, et al. Clinical and biological relevance of genetic alterations in pediatric T-cell acute lymphoblastic leukemia in Taiwan. Pediatr Blood Cancer. (2019) 66:e27496. doi: 10.1002/pbc.27496
13. Pölönen P, Mullighan CG, and Teachey DT. Classification and risk stratification in T-lineage acute lymphoblastic leukemia. Blood. (2025) 145:1464–74. doi: 10.1182/blood.2023022920
14. Cui L, Li ZG, Chai YH, Yu J, Gao J, Zhu XF, et al. Outcome of children with newly diagnosed acute lymphoblastic leukemia treated with CCLG-ALL 2008: The first nation-wide prospective multicenter study in China. Am J Hematol. (2018) 93:913–20. doi: 10.1002/ajh.25124
15. Yang W, Cai J, Shen S, Gao J, Yu J, Hu S, et al. Pulse therapy with vincristine and dexamethasone for childhood acute lymphoblastic leukaemia (CCCG-ALL-2015): an open-label, multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol. (2021) 22:1322–32. doi: 10.1016/S1470-2045(21)00328-4
16. Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. (2009) 113:3918–24. doi: 10.1182/blood-2008-10-184069
17. Inaba H and Pui CH. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. (2010) 11:1096–106. doi: 10.1016/S1470-2045(10)70114-5
18. Pieters R, Huismans DR, Loonen AH, Hählen K, van der Does-van den Berg A, van Wering ER, et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet. (1991) 338:399–403. doi: 10.1016/0140-6736(91)91029-T
19. Li Y, Buijs-Gladdines JG, Canté-Barrett K, Stubbs AP, Vroegindeweij EM, Smits WK, et al. IL-7 receptor mutations and steroid resistance in pediatric T cell acute lymphoblastic leukemia: A genome sequencing study. PloS Med. (2016) 13:e1002200. doi: 10.1371/journal.pmed.1002200
20. Sanchez-Martin M and Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. (2017) 129:1124–33. doi: 10.1182/blood-2016-09-692582
21. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49:1211–8. doi: 10.1038/ng.3909
22. Dai YT, Zhang F, Fang H, Li JF, Lu G, Jiang L, et al. Transcriptome-wide subtyping of pediatric and adult T cell acute lymphoblastic leukemia in an international study of 707 cases. Proc Natl Acad Sci U.S.A. (2022) 119:e2120787119. doi: 10.1073/pnas.2120787119
23. Jenkinson S, Koo K, Mansour MR, Goulden N, Vora A, Mitchell C, et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. (2013) 27:41–7. doi: 10.1038/leu.2012.176
24. Pölönen P, Di Giacomo D, Seffernick AE, Elsayed A, Kimura S, Benini F, et al. The genomic basis of childhood T-lineage acute lymphoblastic leukaemia. Nature. (2024) 632:1082–91. doi: 10.1038/s41586-024-07807-0
25. Liu X, Zou Y, Zhang L, Guo Y, Chen Y, Yang W, et al. A novel risk defining system for pediatric T-cell acute lymphoblastic leukemia from CCCG-ALL-2015 group. Front Oncol. (2022) 12:841179. doi: 10.3389/fonc.2022.841179
26. Álvarez-Garcia V, Tawil Y, Wise HM, and Leslie NR. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin Cancer Biol. (2019) 59:66–79. doi: 10.1016/j.semcancer
27. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, et al. The PI3K pathway in human disease. Cell. (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
28. Martelli AM, Paganelli F, Fazio A, Bazzichetto C, Conciatori F, McCubrey JA, et al. The key roles of PTEN in T-cell acute lymphoblastic leukemia development, progression, and therapeutic rSesponse. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11050629
29. Jenkinson S, Kirkwood AA, Goulden N, Vora A, Linch DC, Gale RE, et al. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia. (2016) 30:39–47. doi: 10.1038/leu.2015.206
30. Szarzyńska-Zawadzka B, Kunz JB, Sędek Ł, Kosmalska M, Zdon K, Biecek P, et al. PTEN abnormalities predict poor outcome in children with T-cell acute lymphoblastic leukemia treated according to ALL IC-BFM protocols. Am J Hematol. (2019) 94:E93–e96. doi: 10.1002/ajh.25396
31. Li S, Balmain A, and Counter CM. A model for RAS mutation patterns in cancers: finding the sweet. spot. Nat Rev Cancer. (2018) 18:767–77. doi: 10.1038/s41568-018-0076-6
32. Perentesis JP, Bhatia S, Boyle E, Shao Y, Shu XO, Steinbuch M, et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia. (2004) 18:685–92. doi: 10.1038/sj.leu.2403272
33. Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. (2010) 42:338–42. doi: 10.1038/ng.542
34. Uy GL, Duncavage EJ, Chang GS, Jacoby MA, Miller CA, Shao J, et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia. (2017) 31:872–81. doi: 10.1038/leu.2016.282
35. Zhang X, Huang A, Liu L, Qin J, Wang C, Yang M, et al. The clinical impact of IKZF1 mutation in acute myeloid leukemia. Exp Hematol Oncol. (2023) 12:33. doi: 10.1186/s40164-023-00398-y
Keywords: T-cell acute lymphoblastic leukaemia, gene mutations, paediatric, next-generation sequencing, prognosis
Citation: Zhang S, Chu X, Li Z, Ji Q, Meng N, Gao W, Zhang X, Hu Y, Gao L, Li B, Chen P, Wan H, Liu Y, Zhang Y, Tian Y, Wu S, Li Y, Hu S and Liu H (2025) Next-generation sequencing reveals clinical features and prognosis of gene mutations in Chinese children with T-cell acute lymphoblastic leukaemia. Front. Oncol. 15:1666527. doi: 10.3389/fonc.2025.1666527
Received: 15 July 2025; Accepted: 25 August 2025;
Published: 26 September 2025.
Edited by:
Joanna Zawitkowska, Medical University of Lublin, PolandReviewed by:
Hema Dave, Merck, United StatesArchana Sasi, All India Institute of Medical Sciences, India
Copyright © 2025 Zhang, Chu, Li, Ji, Meng, Gao, Zhang, Hu, Gao, Li, Chen, Wan, Liu, Zhang, Tian, Wu, Li, Hu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hu Liu, NDU1NDM0MTUyQHFxLmNvbQ==; Shaoyan Hu, aHVzaGFveWFuQHN1ZGEuZWR1LmNu; Yizhen Li, bGl5ekBzdWRhLmVkdS5jbg==
†These authors have contributed equally to this work