 Henrik Horndalsveen1,2,3*
Henrik Horndalsveen1,2,3* Vilde Drageset Haakensen1,2
Vilde Drageset Haakensen1,2 Tesfaye Madebo4,5
Tesfaye Madebo4,5 Bjørn Henning Grønberg6,7Tarje Onsøien Halvorsen6,7Jussi Koivunen8,9Kersti Oselin10Saulius Cicenas11Nina Helbekkmo12Marianne Aanerud5,13Jarkko Ahvonen14Maria Silvoniemi15Maria Moksnes Bjaanæs2
Bjørn Henning Grønberg6,7Tarje Onsøien Halvorsen6,7Jussi Koivunen8,9Kersti Oselin10Saulius Cicenas11Nina Helbekkmo12Marianne Aanerud5,13Jarkko Ahvonen14Maria Silvoniemi15Maria Moksnes Bjaanæs2 Saima Farooqi1,2
Saima Farooqi1,2 Daniel Nebdal1Astrid Marie Dalsgaard1Britina Kjuul Danielsen1Mari Børve1Tonje Sofie Dalen1Åsa Kristina Öjlert1,2
Daniel Nebdal1Astrid Marie Dalsgaard1Britina Kjuul Danielsen1Mari Børve1Tonje Sofie Dalen1Åsa Kristina Öjlert1,2 Åslaug Helland1,2,3
Åslaug Helland1,2,3- 1Institute for Cancer Research, Department of Cancer Genetics, Oslo University Hospital, Oslo, Norway
- 2Department of Oncology, Oslo University Hospital, Oslo, Norway
- 3Department of Clinical Medicine, University of Oslo, Oslo, Norway
- 4Department of Pulmonology, Stavanger University Hospital, Stavanger, Norway
- 5Department of Clinical Science, University of Bergen, Bergen, Norway
- 6Department of Clinical and Molecular Medicine, NTNU, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
- 7Department of Oncology, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
- 8Department of Oncology and Radiotherapy, Oulu University Hospital, Oulu, Finland
- 9Cancer Center, Medical Research Center Oulu, Oulu University Hospital, Oulu, Finland
- 10Oncology and Haematology Clinic, North Estonia Medical Centre, Tallinn, Estonia
- 11Department of Thoracic Surgery and Oncology, National Cancer Center, Affiliate of Vilnius University Hospital Santaros Klinikos, Vilnius, Lithuania
- 12Department of Pulmonology, University Hospital of North Norway, Tromsø, Norway
- 13Department of Thoracic Medicine, Haukeland University Hospital, Bergen, Norway
- 14Tays Cancer Center, Department of Oncology, Tampere University Hospital, Tampere, Finland
- 15Department of Pulmonary Medicine, Turku University Hospital, Turku, Finland
Introduction: Chemoradiotherapy followed by durvalumab is a potentially curative treatment for unresectable, locally advanced non-small cell lung cancer (NSCLC), but clinical outcomes remain highly variable. Identifying robust biomarkers is essential to refine treatment selection and enable risk-adapted strategies.
Methods: In this multicenter, prospective cohort study, 86 patients with unresectable stage III NSCLC were treated with chemoradiotherapy followed by durvalumab. Baseline plasma samples underwent genomic profiling and blood tumor mutational burden (bTMB) assessment using targeted next-generation sequencing. Associations between bTMB, circulating tumor DNA (ctDNA) alterations, PD-L1 expression, and progression-free survival (PFS) were evaluated using a one-sided significance threshold of p < 0.10.
Results: Median PFS was 18.9 months (95% CI: 14.7–not reached), and median bTMB was 6.6 mutations/megabase. In univariable analysis, high bTMB was associated with longer PFS using both the prespecified 8.5 mut/Mb cut-off (HR: 0.65; p = 0.088) and the median 6.6 mut/Mb cut-off (HR: 0.52; p = 0.016). PD-L1 ≥ 1% was associated with longer PFS (HR: 0.38; p = 0.0003), while STK11, KEAP1, or NFE2L2 mutations in ctDNA were linked to shorter PFS (HR: 1.84; p = 0.040). In multivariable analysis, PD-L1 remained significantly associated with PFS in both models, while bTMB and STK11/KEAP1/NFE2L2 mutations were significant using the 6.6 mut/Mb cut-off.
Conclusion: High bTMB, PD-L1 expression ≥ 1%, and absence of STK11/KEAP1/NFE2L2 mutations were associated with longer PFS. These findings support integrating multiple biomarkers to improve risk stratification and personalize treatment in unresectable stage III NSCLC.
Clinical Trial Registration: The study is registered on www.clinicaltrials.gov (ClinicalTrials.gov identifier: NCT04392505).
1 Introduction
Approximately 20-30% of non-small cell lung cancer (NSCLC) patients are diagnosed with stage III disease (1, 2). Patients with unresectable stage III disease and a good performance status may undergo radical therapy using radiotherapy (60–66 Gy) with concurrent platinum-based doublet chemotherapy (3). The PACIFIC trial, supported by real-world data, demonstrated superior outcomes when chemoradiotherapy (CRT) is consolidated with one year of the anti-programmed death-ligand 1 (PD-L1) inhibitor durvalumab (4–6). However, many patients relapse despite durvalumab treatment, while up to 20% of patients not receiving durvalumab achieve long-term disease-free survival. These observations highlight the need for new biomarkers to better predict treatment responses and enable personalized, risk-adaptive treatment.
Currently, PD-L1 expression is the most clinically useful yet imperfect biomarker for predicting the efficacy of immune checkpoint inhibitors (ICIs) in NSCLC (7). Tumor mutational burden (TMB), defined as the number of somatic non-synonymous mutations per coding area of the tumor genome, has emerged as another promising biomarker (8, 9). Theoretically, a high TMB may increase tumor neoantigen formation to enhance neoantigen-specific T-cell responses and improve sensitivity to ICIs (10–12). While TMB was originally analyzed in tumor tissue samples by whole exome sequencing (WES), mounting evidence suggests that targeted gene panels may offer comparable precision, provided that the panel size is sufficient (≥1 Mb) (8, 13). Recently, methods for determining TMB in circulating tumor DNA (ctDNA) have emerged. Blood-based TMB (bTMB) analysis involves less invasive sampling and is the only viable option in cases where tumor tissue is difficult to obtain. Furthermore, bTMB may be less susceptible to tumor heterogeneity and allows for repeated assessments during treatment (13). However, the consistency between liquid- and tissue-based TMB analyses and the optimal approach remain to be defined (8, 11).
In advanced NSCLC, studies have demonstrated that high TMB correlates with greater benefit from ICIs, particularly when immunotherapy is administered alone and not in combination with chemotherapy (14–23). Further, TMB in this context appears to be independent of PD-L1 expression (7, 8). In early-stage resected NSCLC, high tissue TMB (tTMB) has been reported to predict improved locoregional control after post-operative radiotherapy, suggesting that TMB may serve as a biomarker of radiosensitivity (24). Still, the role of TMB in locally advanced NSCLC treated with chemoradiation and durvalumab remains underexplored (25–28).
Detection of specific mutations offers an alternative approach to examine the mutational landscape of NSCLC for prognostic and predictive biomarkers. STK11 mutations impair DNA damage repair, while KEAP1/NFE2L2 mutations enhance the ability of cancer cells to tolerate oxidative stress, both contributing to radiotherapy resistance (24, 29). Additionally, alterations in STK11, KEAP1, and NFE2L2 are linked to immunologically cold tumor microenvironments, potentially serving as negative predictive biomarkers for immunotherapy (29, 30).
The Durvalumab After ChemoRadiotherapy (DART) study enrolled patients with unresectable stage III NSCLC eligible for CRT followed by durvalumab. The aim was to explore the biology underlying treatment response and resistance. Here, we evaluate TMB, PD-L1 expression, and ctDNA-based pathogenic gene alterations as biomarkers in this setting, with a primary focus on associations between bTMB and progression-free survival (PFS).
2 Materials and methods
2.1 Patients, study design and treatment
The DART study is a multicenter phase II translational and biomarker study conducted at ten hospitals in Norway, Finland, Lithuania, and Estonia. Patients with unresectable stage III NSCLC were enrolled and treated with curatively intended CRT, consisting of two cycles of platinum-based doublet chemotherapy every three weeks and radiotherapy at 2 Gy per fraction to a total dose of 60–66 Gy. Patients without disease progression following CRT received durvalumab 1500 mg every four weeks, preferably starting within five weeks of CRT completion, and continued until progression, intolerable toxicity, or a maximum duration of 12 months. Participants not starting durvalumab were excluded from the analyses.
2.2 Ethics statement
The study adhered to the Declaration of Helsinki (31), Good Clinical Practice, and all applicable laws and institutional guidelines. Approval was granted by the Regional Committee for Medical and Health Research Ethics (reference 48665, November 28, 2019). All participants provided informed consent. The trial is registered at ClinicalTrials.gov (NCT04392505).
2.3 Clinical assessments
Baseline imaging included CT of the chest/upper abdomen, MRI or CT of the brain, and whole body 18F-FDG PET/CT. Tumor evaluation by CT was performed between completion of CRT and the first durvalumab infusion, every 12 weeks during durvalumab therapy, and for the next two years, then every 26 weeks for an additional three years until progression or death. Supplemental MRI and PET/CT were conducted if clinically indicated. Tumor response was assessed per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (32). Lesions receiving radiotherapy as part of CRT were considered measurable, since radiotherapy was part of the study protocol and applied to all baseline lesions. Disease progression required radiologic progression by RECIST 1.1, supported by one of the following: 1) a biopsy or PET showing clear progression, 2) clinical deterioration, or 3) a confirmatory CT scan performed at least four weeks after the initial scan. If progression was confirmed, the date of the first scan indicating progression was recorded. The primary endpoint was PFS, defined as time from the start of durvalumab to disease progression or death from any cause. Overall survival (OS) was calculated from durvalumab initiation to death.
2.4 Tumor tissue collection, sequencing and tTMB calculation
Formalin-fixed paraffin-embedded (FFPE) tumor tissue and matched buffy coat samples for germline variant filtering were obtained at baseline. HE-stained FFPE sections were reviewed by a pathologist to confirm tumor content. DNA was extracted using the AllPrep DNA/RNA FFPE Kit (Qiagen) for tumor and QIAamp DNA Blood Mini Kit (Qiagen) for buffy coats. DNA concentration and quality were assessed using Qubit (ThermoFisher Scientific), Nanodrop (ThermoFisher Scientific), and Genomic DNA ScreenTape (Agilent). Samples with tumor DNA concentration > 3 ng/µl and matched buffy coats were submitted for sequencing. WES was performed at the OUH Genomics Core Facility using the Twist Biosciences Library Preparation Kit and the Twist Human Comprehensive Exome Enrichment Kit (Illumina). Sequencing (2 × 150 bp) was performed on a NovaSeq6000 system at average coverages of 150× (tumor) and 50× (buffy coat). Sequencing reads were aligned to the human reference genome GRCh38 using the Burrows–Wheeler Aligner (BWA_MEM2). Somatic variants were identified with GATK Mutect2 (v4.2.6.1) and Strelka (v2.9.10), and annotated using the Personal Cancer Genome Reporter (33). Variants with a variant allele frequency (VAF) ≥ 5% and tumor read depth ≥ 100× were included in tTMB calculation, defined as the number of non-synonymous SNVs and indels per megabase of targeted exome. Additional details are provided in the Supplementary Methods.
2.5 Plasma sample collection, sequencing and bTMB calculation
Peripheral blood was collected in three 10 ml cfDNA BCT tubes (Streck) at baseline. Plasma was separated via two-step centrifugation before storage at –80 °C. cfDNA was extracted from 8 ml plasma using the Mag-Bind cfDNA Kit (Omega Bio Tek) on an automated platform (Opentrons OT-2, KingFisher Flex). DNA library preparation followed established protocols (34) including dA-tailing, adaptor ligation, and indexing PCR, with intermediate quality control using the Agilent 4150 TapeStation. Target regions were captured by hybridization using TACS (target capture sequences). The NeoThetis Pan Cancer Plus assay (MEDICOVER Genetics), targeting 222 cancer-related genes and a total of 1.25 Mb, was used to identify single nucleotide variants (SNVs), small insertions and deletions (indels), copy number amplifications (CNAs) and structural rearrangements (Supplementary Table 1). Captured libraries were sequenced on a NovaSeq6000 platform (Illumina). Reads were demultiplexed using bcl-convert (v4.2), with poor-quality reads and adaptor sequences removed before alignment to GRCh37 using the Burrows-Wheeler algorithm (35). Duplicate reads were grouped by unique adaptor families to generate consensus reads. To further refine the set of positive variant calls, a statistical error correction model (at base-pair resolution), followed by a filtering bioinformatics pipeline, was applied. ctDNA variant calling was performed de novo (not tumor-guided) and variants were classified per AMP guidelines using automated tiering (VarSomeClinical), followed by manual curation by at least two variant analysts. Variants were excluded if they had VAF < 0.25%, population frequency > 1% (gnomAD), were synonymous, or were deemed low-confidence. For bTMB calculation, only SNVs and indels in targeted regions with ≥ 1000× coverage were counted. Additional details are provided in the Supplementary Methods.
2.6 Statistical analysis
PFS and OS were estimated using the Kaplan-Meier method. Follow-up time was calculated using reverse Kaplan-Meier. TMB was analyzed as a categorical variable (low vs. high) using several cut-offs, including 8.5 mut/Mb (protocol-prespecified primary) and the cohort median. The 8.5 mut/Mb cut-off was set when the protocol was planned in 2019, informed by metastatic NSCLC medians (7–10 mut/Mb) and the then-common use of 10 mut/Mb. Given limited data in stage III NSCLC and the expectation of slightly lower TMB, 8.5 mut/Mb was chosen to balance biological plausibility and statistical power. STK11, KEAP1 and NFE2L2 mutations were analyzed as a grouped variable, reflecting shared biology linked to treatment resistance and the low individual frequencies of these alterations. Associations between patient characteristics and genomic variables at baseline were assessed using Fisher’s exact test or Chi-square test for categorical variables. For categorical vs. continuous variables, Wilcoxon rank-sum (two groups) or Kruskal-Wallis (more than two groups) tests were applied. Correlations were examined using Spearman’s method. Associations between clinical/genomic characteristics and PFS were assessed using log-rank tests and Cox proportional hazards models. Key variables significantly associated with outcome in univariable analysis were further evaluated in multivariable Cox regression models, adjusted for age and performance status, as established prognostic factors. As prespecified in the study protocol, the significance threshold (alpha) was set at 0.10, with one-sided p-values to test effects in the expected direction. Statistical analyses were performed in R(v4.1.1)
3 Results
3.1 Clinical and treatment characteristics
Between May 5, 2020, and September 7, 2023, 123 patients were screened, of whom 90 met all eligibility criteria and completed CRT. Of these, 87 initiated durvalumab (Figure 1). One patient was excluded after a re-examination of the lung tumor biopsy concluded that it was a metastasis from rectal cancer. Another patient was found to have stage IIB disease upon later radiological review but was included in the primary analysis since the patient had unresectable NSCLC and was treated according to protocol. Baseline clinical characteristics of the 86 patients are shown in Table 1. The median age was 69 years (range 36-85), 60% (n=52) were male and 95% (n=82) had a history of smoking. Histologically, 57% (n=49) of tumors were squamous cell carcinoma and 41% (n=35) had PD-L1 expression <1%. The median time from end of CRT to durvalumab initiation was 24 days (range 6-45) with a median of 11 durvalumab infusions administered (range 1–13).
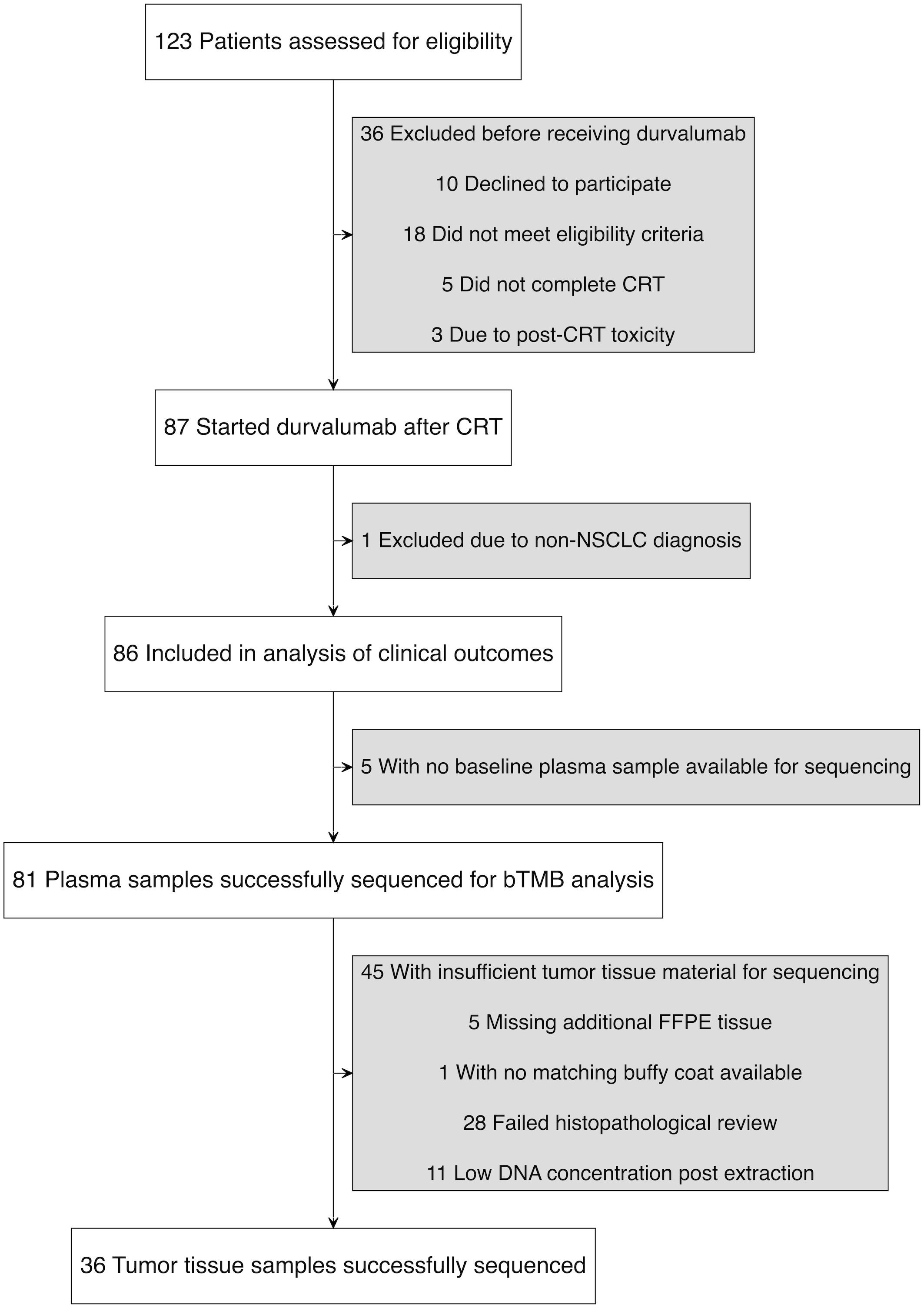
Figure 1. Study flow diagram illustrating patient enrollment, exclusions, and sample availability.
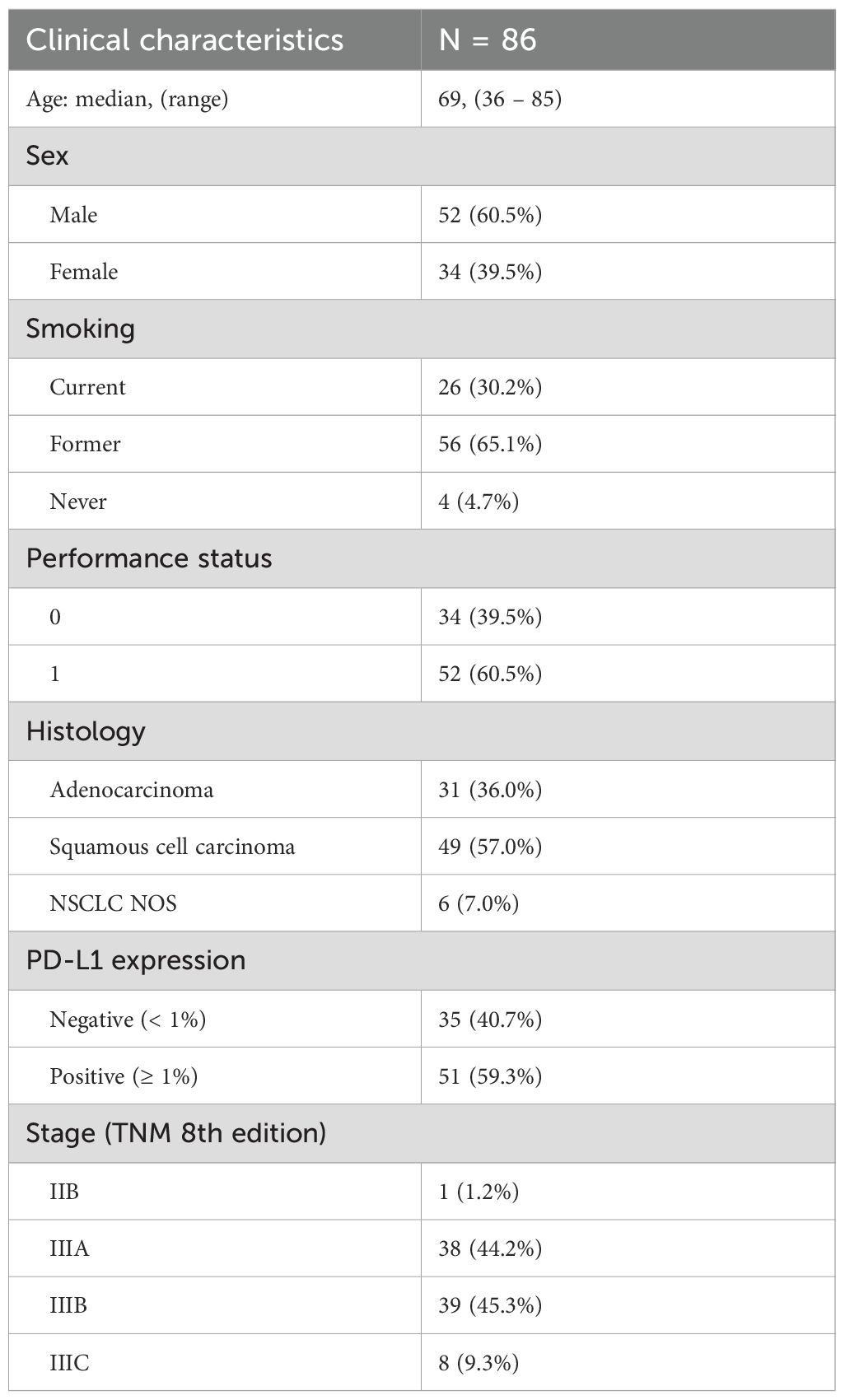
Table 1. Patient characteristics.
3.2 Genomic characteristics
Baseline plasma samples for sequencing were available from 81 of 86 patients, all of which passed quality control. The median bTMB was 6.6 mut/Mb (range: 0–41.9 mut/Mb, Figure 2). Using the prespecified cutoff of 8.5 mut/Mb, 49 patients were categorized as bTMB low, and 32 as bTMB high. No significant associations were found between bTMB and baseline patient characteristics. The oncoprint summarizes functionally relevant genomic alterations detected in plasma ctDNA (Figure 2). TP53 was the most frequently altered gene in plasma (68% of patients) followed by KRAS (17%). Alterations in STK11, KEAP1 or NFE2L2 were observed in 21% of patients. Patients with TP53 mutations in plasma ctDNA had higher bTMB (p < 0.001). No other individual mutations showed significant associations with bTMB, but combined STK11/KEAP1/NFE2L2 alterations were linked to higher bTMB (p = 0.046) and PD-L1 negativity (p = 0.035).
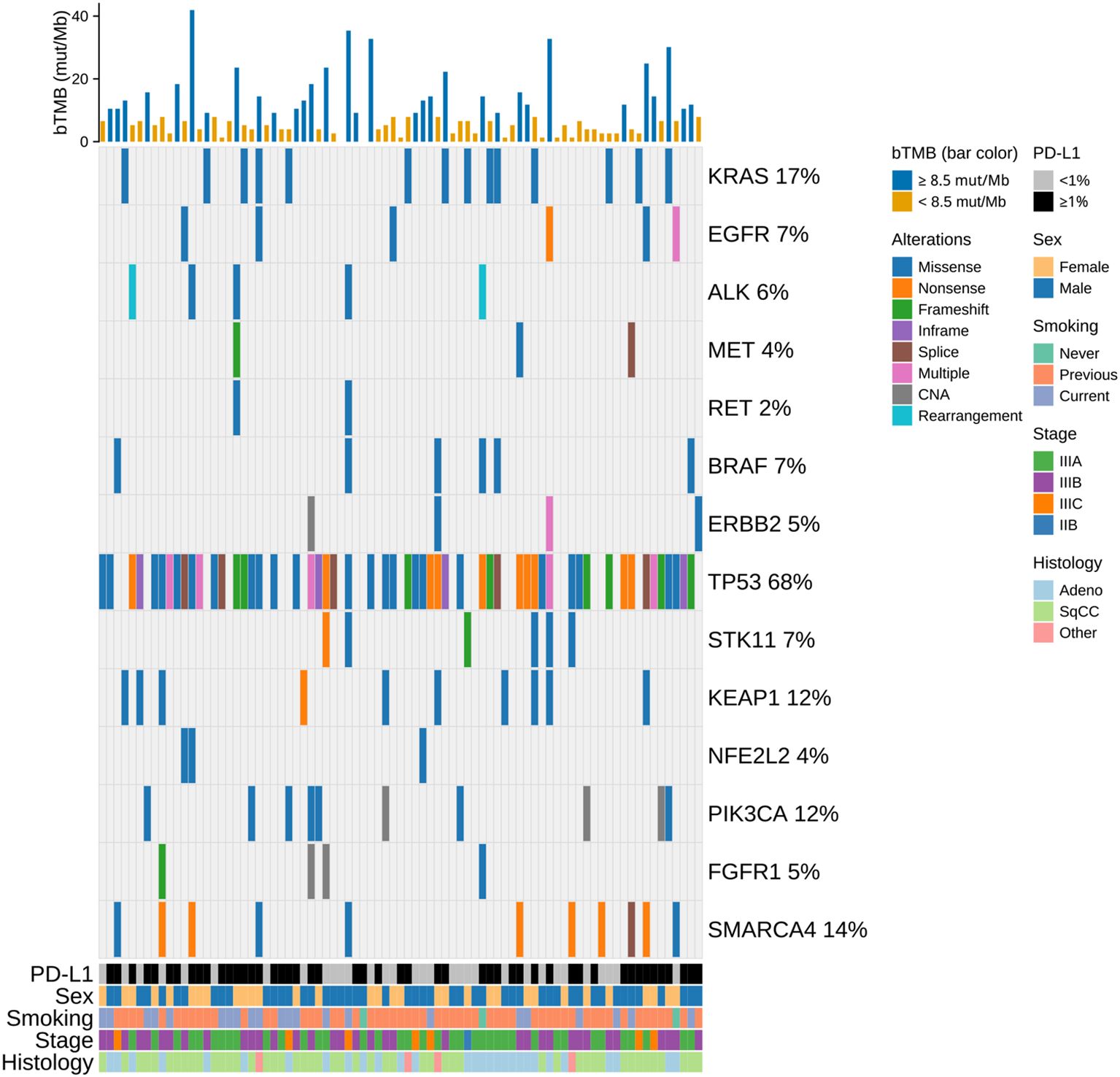
Figure 2. Oncoprint of the study population showing functionally relevant genomic alterations detected in plasma ctDNA. Clinical annotations (PD-L1, sex, smoking status, stage, histology) and bTMB are displayed as overlays; alteration types are color-coded, and each column represents one patient. Bars above indicate bTMB scores, color-coded by bTMB category using a cut-off of 8.5 mut/Mb. bTMB, blood tumor mutational burden; CAN, copy number amplification; mut/Mb, mutations per megabase; SqCC, squamous cell carcinoma.
Of the 81 patients with sequenced plasma samples, 36 had tumor tissue with sufficient tumor content for WES (Figure 1). The median tTMB was 11.6 mut/Mb (range: 2.6–49.5 mut/Mb). The correlation between bTMB and tTMB was moderate (Spearman’s ρ = 0.50, p = 0.002), with tTMB values being significantly higher (p = 0.012). When using the median to categorize TMB as high or low, 75% (27/36) of patients were concordantly classified by bTMB and tTMB. Neither bTMB nor tTMB significantly correlated with PD-L1 expression, although a weak trend toward higher bTMB was seen in patients with PD-L1 ≥ 1% (bTMB: Spearman’s ρ = 0.15, p = 0.172; tTMB: Spearman’s ρ = 0.01, p = 0.937; Supplementary Figure 1). Among patients with matched plasma and tissue samples (gene-level presence/absence comparison), 60% of mutations (SNVs and indels) in key genes were detected in both plasma and tumor tissue, while 25% were exclusive to tumor and 15% to plasma (Supplementary Figure 2).
3.3 Clinical outcomes
At the cut-off date (December 1, 2024), median follow-up was 33.1 months (IQR 22.3–35.6). The median PFS was 18.9 months (95% CI: 14.7–not reached, NR). A total of 47 patients (54.7%) had experienced a progression event at a median of 8.3 months. Of these, 20 had local recurrences, 19 had distant metastases, two had both local recurrence and distant metastasis, and six died without documented progression. The median OS was not reached. The 12- and 24-month OS rates were 87.2% (95% CI: 80.4–94.5) and 71.5% (95% CI: 62.1–82.2), respectively. In univariable analyses of baseline clinical characteristics and PFS, age ≥75 years (HR: 2.02; 95% CI: 0.80–5.13; p = 0.07) and male sex (HR: 1.63; 95% CI: 0.88–3.02; p = 0.06) were associated with shorter PFS (Supplementary Figure 3). No significant associations with PFS were found for smoking status, ECOG performance status, disease stage, histology, or time from CRT to durvalumab (<28 vs. ≥28 days).
3.4 Association between TMB and PFS
Using the prespecified cut-off of 8.5 mut/Mb, patients with high bTMB had improved PFS compared to those with low bTMB (HR: 0.65; 95% CI: 0.35–1.21; p = 0.088; Figure 3A). The median PFS was NR for high bTMB (95% CI: 16.2–NR) and 16.7 months for low bTMB (95% CI: 11.8–NR). Applying the median value of 6.6 mut/Mb as an alternative cut-off, high bTMB was significantly associated with longer PFS (HR: 0.52; 95% CI: 0.28–0.96; p = 0.016; Figure 3B). The median PFS was NR (95% CI: 16.3–NR) in high bTMB vs. 14.8 months (95% CI: 10.9–24.8) in low bTMB. Higher thresholds (10, 16, and 20 mut/Mb) did not yield significant associations with PFS (p = 0.181, p = 0.369 and p = 0.241, respectively; Supplementary Figure 4). Notably, very few patients were classified as having high bTMB when applying these higher thresholds.
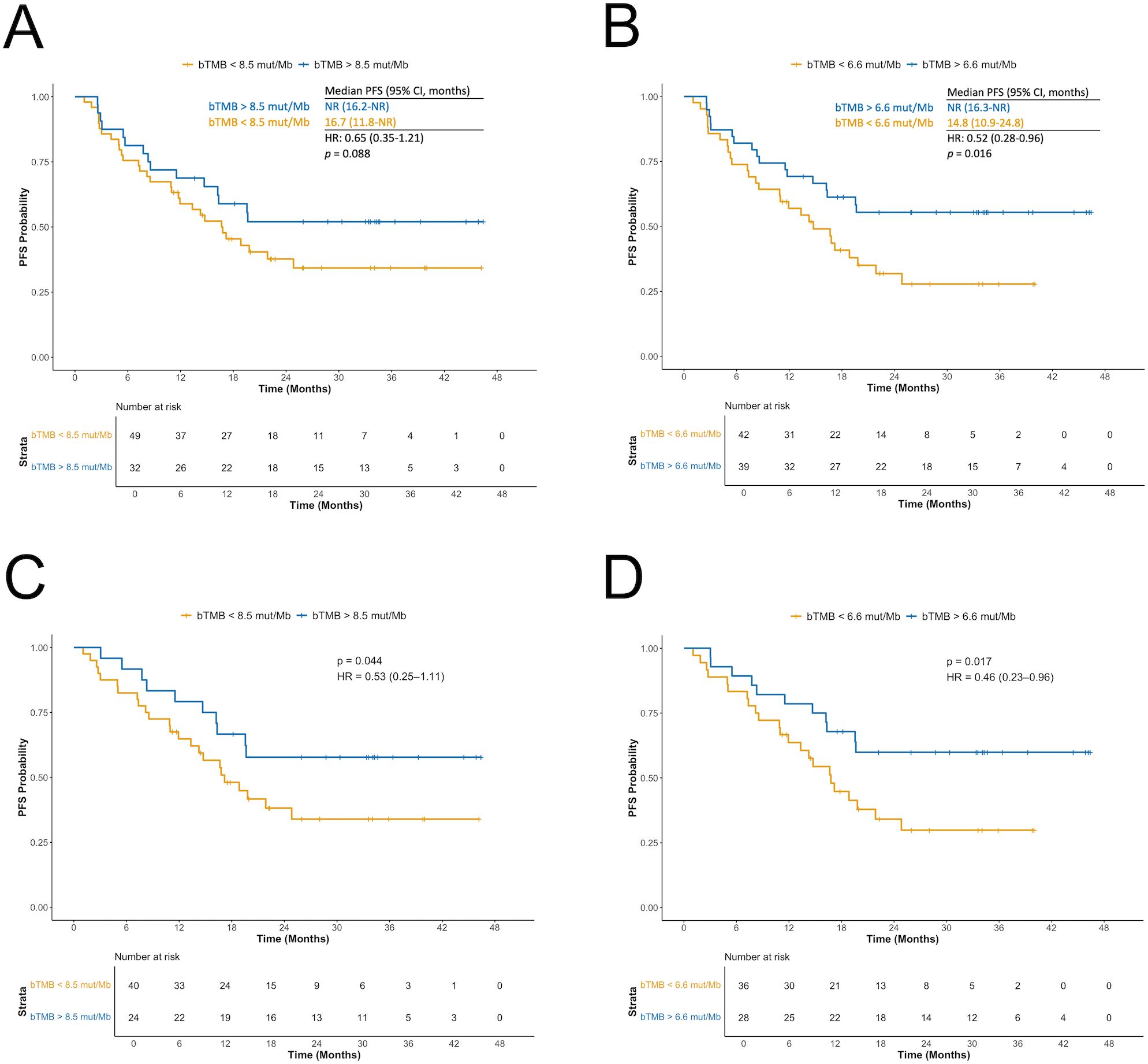
Figure 3. Association between blood tumor mutational burden (bTMB) and progression-free survival (PFS) based on two different cut-offs. (A) bTMB </> 8.5 mutations per megabase (mut/Mb) in the full cohort. (B) bTMB </> median value of 6.6 mut/Mb in the full cohort. (C) bTMB </> 8.5 mut/Mb after excluding patients with STK11, KEAP1, or NFE2L2 mutations detected in blood. (D) bTMB </> 6.6 mut/Mb after excluding patients with STK11, KEAP1, or NFE2L2 mutations detected in blood. bTMB, blood tumor mutational burden; mut/Mb, mutations per megabase; PFS, progression-free survival.
Excluding patients with STK11/KEAP1/NFE2L2 mutations strengthened the association between high bTMB and longer PFS for both the 8.5 mut/Mb cut-off (HR: 0.53; 95% CI: 0.25–1.11; p = 0.044; Figure 3C) and 6.6 mut/Mb (HR: 0.46; 95% CI: 0.23–0.96; p = 0.017; Figure 3D). The 10 mut/Mb cut-off also reached significance (HR: 0.59; 95% CI: 0.27–1.30; p = 0.096).
Among the 36 patients with tTMB data, no significant PFS difference was observed between high and low tTMB groups.
3.5 Association between PD-L1 expression and PFS
Patients with PD-L1 tumor expression ≥ 1% had improved PFS compared to those with PD-L1 < 1% (HR: 0.38; 95% CI: 0.21–0.67; p = 0.0003; Figure 4A). When combining bTMB and PD-L1 status, the longest PFS was observed in patients with both PD-L1 ≥ 1% and high bTMB, using either the 8.5 mut/Mb (Supplementary Figure 5A) or 6.6 mut/Mb (Supplementary Figure 5B) cut-offs. Compared to the reference group (PD-L1 < 1% and low bTMB), those with PD-L1 ≥ 1% and bTMB ≥ 8.5 mut/Mb had a significantly reduced risk of progression or death (HR: 0.29; 95% CI: 0.13–0.65; p = 0.001). The association was even stronger when using the 6.6 mut/Mb cut-off value (HR: 0.22; 95% CI: 0.10-0.50, p < 0.001).
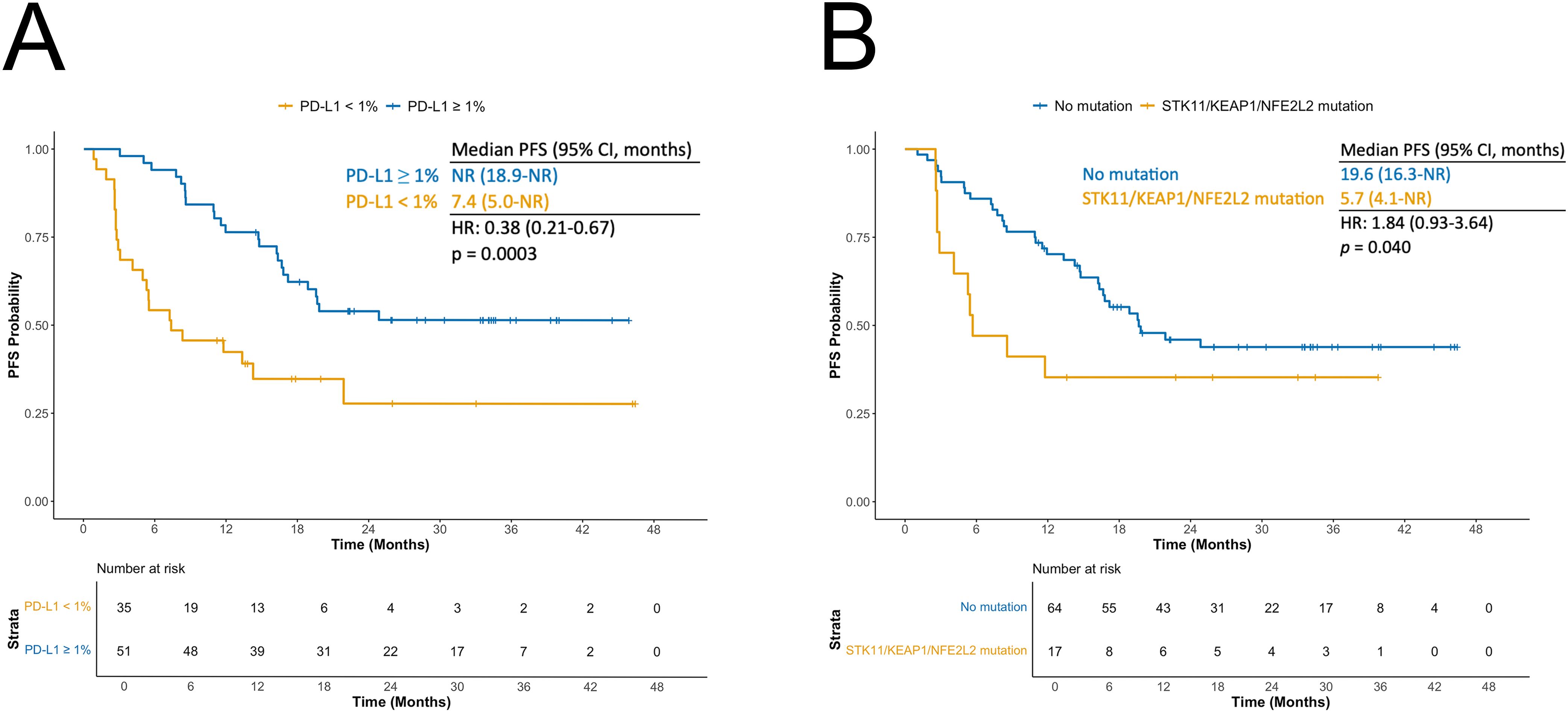
Figure 4. (A) Kaplan-Meier curves for progression-free survival (PFS) according to PD-L1 status. (B) Kaplan-Meier curves for PFS according to STK11/KEAP1/NFE2L2 mutation status.
3.6 Association between genomic alterations in blood and PFS
In univariable analysis, the presence of mutations in STK11, KEAP1, or NFE2L2 in plasma was associated with shorter PFS (HR 1.84, 95% CI 0.93–3.64; p = 0.040). Median PFS was 5.7 months (95% CI 4.1–NR) in patients with ≥1 of these mutations versus 19.6 months (95% CI 16.3–NR) in wild-type patients (Figure 4B). Combining STK11/KEAP1/NFE2L2 status with bTMB identified a particularly favorable cohort: patients with wild-type STK11/KEAP1/NFE2L2 and high bTMB (>8.5 mut/Mb) had an HR of 0.37 (95% CI 0.14–1.03; p = 0.029) compared with those with STK11/KEAP1/NFE2L2 alterations and low bTMB. Associations with PFS for other ctDNA-detected alterations present in ≥10 patients are shown in Supplementary Figure 6. KRAS mutations were linked to longer PFS (HR 0.52, 95% CI 0.21–1.33; p = 0.087). Prognosis improved further in KRAS-mutated patients after excluding those with STK11 or KEAP1 co-mutations (HR 0.35, 95% CI 0.11–1.12; p = 0.034).
3.7 Multivariable analysis of factors associated with PFS
Since high bTMB was associated with longer PFS in univariable analyses using both the prespecified 8.5 mut/Mb cut-off and the median value of 6.6 mut/Mb, we performed two separate multivariable analyses for these cut-offs. In the 8.5 mut/Mb model, only PD-L1 expression ≥ 1% was significantly associated with longer PFS (HR: 0.41; 95% CI: 0.22–0.76; p = 0.002), while STK11/KEAP1/NFE2L2 mutations showed a trend toward shorter PFS (HR: 1.58; 95% CI: 0.78–3.24; p = 0.104; Figure 5A). In the 6.6 mut/Mb model, high bTMB (HR: 0.48; 95% CI: 0.25–0.91; p = 0.012), PD-L1 ≥ 1% (HR: 0.43; 95% CI: 0.23–0.79; p = 0.003), and STK11/KEAP1/NFE2L2 mutations (HR: 1.86; 95% CI: 0.87–3.96; p = 0.055) were all significantly associated with PFS, with high bTMB and PD-L1 ≥ 1% linked to longer PFS and STK11/KEAP1/NFE2L2 mutations linked to shorter PFS (Figure 5B).
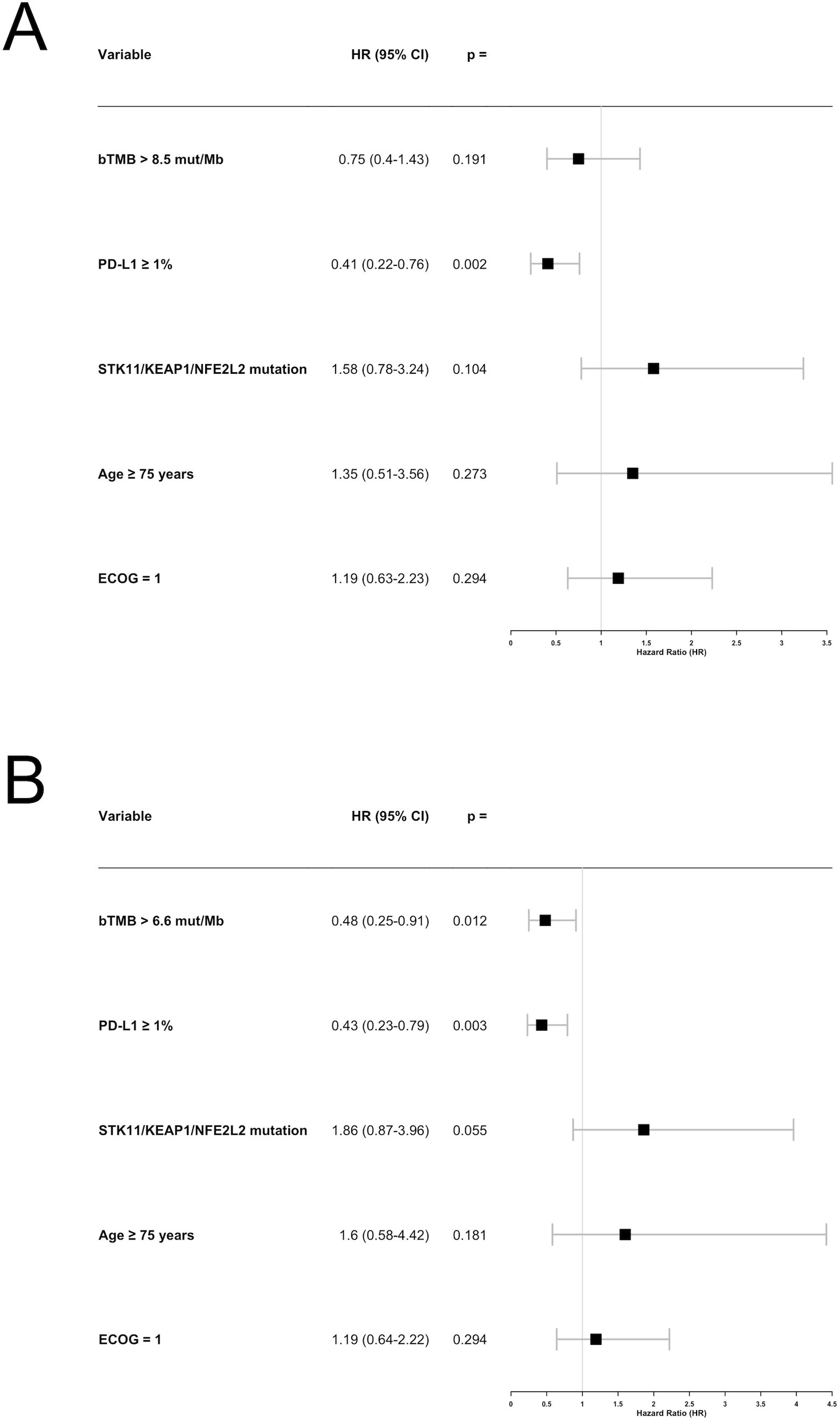
Figure 5. Forest plot for the multivariable analysis of factors associated with progression-free survival (PFS). (A) Using the blood tumor mutational burden (bTMB) cut-off of 8.5 mutations per megabase (mut/Mb). (B) Using the bTMB cut-off of 6.6 mut/Mb. bTMB, blood tumor mutational burden; mut/Mb, mutations per megabase.
4 Discussion
In this prospective cohort study of patients with unresectable stage III NSCLC treated with CRT and durvalumab, high bTMB and PD-L1 ≥ 1% were associated with longer PFS, while ctDNA-detected mutations in STK11, KEAP1, or NFE2L2 were linked to shorter PFS. These findings provide insight into treatment response and resistance and reveal potential biomarkers to guide clinical decision-making in locally advanced NSCLC.
While TMB is a known predictor of immunotherapy benefit in stage IV NSCLC, its role in locally-advanced disease treated with multimodal therapy remains less established. Recently, retrospective analyses have reported high tTMB to be associated with longer disease control after CRT and consolidative durvalumab (25, 26). However, in locally advanced NSCLC, obtaining sufficient tumor tissue for routine diagnostics can be challenging, often leaving too little material for tTMB and additional biomarker analysis (9, 36). In our trial, only 36 of 81 patients had baseline tumor tissue samples with enough tumor content for tTMB determination. ctDNA-based genomic profiling and bTMB assessment offer a practical alternative when tissue is limited, with several advantages: less invasiveness, reduced susceptibility to intra- and intertumoral heterogeneity, and greater feasibility for repeated measurements throughout treatment for dynamic bTMB monitoring (13, 28).
TMB may influence responses to CRT and durvalumab through multiple mechanisms. High TMB is a predictor of immunotherapy benefit. While its predictive value appears diminished when immunotherapy is paired with chemotherapy (37), this might not apply when combined with radiotherapy (25, 26). Tumors with high TMB offer a more immunogenic tumor microenvironment with more tumor neoantigens and increased CD8-positive and PD-1-positive T-cell infiltration, which may increase the vulnerability of tumor cells to the immune-related effects of radiotherapy (21, 24). Additionally, high TMB correlates with alterations in DNA damage response and repair (DDR) genes, which play key roles in radiation repair. In theory, pathogenic mutations in these genes could further increase radiosensitivity (38, 39).
There is currently no consensus on the optimal threshold to define high versus low TMB (11). In our study, both the 6.6 mut/Mb (median) and the protocol-prespecified 8.5 mut/Mb cut-offs were significantly associated with PFS in univariable analyses. However, only when using the 6.6 mut/Mb cut-off value, did the association remain significant in the multivariable analysis. Applying higher cut-off values yielded no significant association between high bTMB and longer PFS, possibly due to the small number of patients classified as high bTMB and limited statistical power. While the FDA approved pembrolizumab for solid tumors with high TMB using a 10 mut/Mb cut-off (40), some trials suggest higher thresholds, in the 80th-90th percentiles, to better predict immunotherapy efficacy (41, 42). Conversely, a meta-analysis by Meng et al. indicated that lower cut-offs may more effectively identify patients likely to benefit from immunotherapy (11). Ultimately, the optimal threshold likely depends on tumor type, disease stage, methodology, and assay, making it difficult to define a universal standard (13).
Consistent with findings from the PACIFIC and PACIFIC-R studies, we found that patients with PD-L1 tumor expression ≥ 1% had better PFS after CRT and durvalumab compared to PD-L1-negative patients (5, 43). In our cohort, PD-L1, treated as a dichotomous variable with a 1% cut-off value, was the biomarker most strongly associated with PFS, both in univariable and multivariable analyses, reinforcing its clinical relevance in this setting. However, some trials categorizing PD-L1 expression into multiple levels have reported similar outcomes in PD-L1 negative and PD-L1 low (1-49%) disease (26, 44), suggesting that PD-L1 might be better evaluated as a continuous variable and in combination with other biomarkers. Our data support bTMB and PD-L1 as independent markers with the longest PFS observed in patients with both high bTMB and PD-L1 ≥ 1%. While exploratory rather than practice-changing, these results support a multi-biomarker approach integrating PD-L1, TMB, and additional tumor features to refine prognosis and guide treatment selection for this patient group.
Our data indicate that pathogenic mutations in STK11, KEAP1, and NFE2L2, as detected by ctDNA analysis, are associated with inferior PFS in patients undergoing CRT and durvalumab. These mutations have been linked to increased resistance to radiotherapy (24, 29, 30). Increasing evidence also supports that tumors with STK11 or KEAP1 mutations are less responsive to chemotherapy and PD-L1-targeted immunotherapy, suggesting their role as negative prognostic biomarkers (45, 46). If our findings and a median PFS of six months reflect the expected benefit of CRT and durvalumab in this subgroup, risk-adaptive strategies could be warranted. Subgroup analyses from POSEIDON and CheckMate 227 suggest that adding a CTLA-4 inhibitor may improve outcomes in metastatic NSCLC with STK11 and KEAP1 mutations (47, 48). However, given the already intensive combination of CRT and durvalumab, further escalation of treatment for a minor improvement in outcome should be considered with caution. Novel therapeutics targeting STK11- and KEAP1/NRF2 pathways are being investigated and could play a role in the future (45). Importantly, not all STK11/KEAP1/NFE2L2-mutations are equally deleterious. Mutation subtype, clonality, the broader genomic landscape and co-mutations (particularly KRAS) should be factored in when assessing the clinical impact of these alterations.
Although high bTMB was associated with longer PFS, our multivariable analyses suggest it may not be sufficiently robust as a stand-alone biomarker in unresectable, locally-advanced NSCLC. Combining bTMB with additional molecular markers, such as pathogenic gene alterations, could better capture the tumor’s molecular characteristics and improve outcome prediction (8, 9). In our trial, three patients with bTMB > 20 mut/Mb experienced disease progression within 12 months, all of whom had deleterious mutations in STK11, KEAP1, or NFE2L2. Furthermore, the association between high bTMB and longer PFS was strengthened when patients with these mutations were excluded. A similar finding was reported by Shaverdian et al., where high tTMB predicted improved locoregional control following post-operative radiotherapy, primarily in NSCLC patients without mutations in genes associated with radioresistance (24). In our study, the combination of high bTMB and STK11/KEAP1/NFE2L2 wild-type status identified a subgroup with a particularly favorable prognosis. If validated in future trials, this cohort may be considered for treatment de-intensification, such as reduced duration of durvalumab therapy.
A combinatorial strategy could incorporate not only bTMB, PD-L1 status, and pathogenic mutations in key genes, but potentially also factors such as cytokines, immune cell composition, and tumor microenvironment features (13). A multi-biomarker model may provide a stronger foundation for personalized treatment. However, for such an approach to be clinically applicable, it must be practical, time-efficient, and cost-effective. Most importantly, further prospective validation is needed before implementing these biomarker-guided strategies in routine clinical practice.
Some limitations of this study should be acknowledged. Its exploratory nature is reflected in the small size of certain genetic subgroups and the use of a significance level of 0.10. Survival data are still immature, and it remains to be seen whether differences in PFS between biomarker-related subgroups will translate into OS differences. As all patients received the same treatment, it is also difficult to determine whether the investigated biomarkers are predictive or merely prognostic. For comparisons of bTMB vs. tTMB, and plasma vs. tissue-based mutation detection, different assays (targeted panel vs. WES) and reference genomes (GRCh37 vs. GRCh38) were used, which limits strict variant-level matching without liftover/re-validation. Thus, there are potentially several technical reasons in addition to biological reasons for the moderate concordance previously reported (49). Finally, only 36 patients had baseline tissue samples with sufficient tumor content for sequencing, and in some cases, DNA concentrations were below the recommended threshold (10 ng/µl), increasing the uncertainty of the tTMB results.
In conclusion, high bTMB and PD-L1 expression ≥ 1% were associated with longer PFS in patients with stage III NSCLC undergoing CRT and consolidative durvalumab, while ctDNA-detected pathogenic mutations in STK11, KEAP1, or NFE2L2 were linked to shorter PFS. Future studies are needed to validate these as complementary biomarkers and to explore personalized treatment strategies, including risk-adapted escalation or de-escalation of therapy.
Data availability statement
The datasets presented in this article are not readily available because they contain information that could compromise participant privacy. Requests to access the datasets should be directed to the corresponding author, Henrik Horndalsveen (aGVucmlrLmhvcm5kYWxzdmVlbkBnbWFpbC5jb20=). Summary-level data supporting the conclusions of this article are included in the article and its Supplementary Material.
Ethics statement
The studies involving humans were approved by The Regional Committee for Medical and Health Research Ethics South-East Norway (REK sør-øst), Norway. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
HH: Data curation, Formal Analysis, Investigation, Visualization, Writing – original draft. VH: Data curation, Investigation, Methodology, Supervision, Writing – review & editing. TM: Investigation, Writing – review & editing. BG: Investigation, Writing – review & editing. TH: Investigation, Writing – review & editing. JK: Investigation, Writing – review & editing. KO: Investigation, Writing – review & editing. SC: Investigation, Writing – review & editing. NH: Investigation, Writing – review & editing. MA: Investigation, Writing – review & editing. JA: Investigation, Writing – review & editing. MS: Investigation, Writing – review & editing. MB: Investigation, Writing – review & editing. SF: Investigation, Writing – review & editing. DN: Formal Analysis, Software, Writing – review & editing. AD: Investigation, Writing – review & editing. BD: Investigation, Writing – review & editing. MB: Investigation, Writing – review & editing. TD: Investigation, Writing – review & editing. ÅÖ: Investigation, Writing – review & editing. ÅH: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was researcher-initiated and funded by grants from the South-Eastern Norway Regional Health Authority (grant no: 2019119 and 26011) and AstraZeneca. The authors declare that this study received funding from AstraZeneca. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
The authors thank all participating patients and their families, study nurses, engineers and lab personnel in all hospitals for their valuable contribution to the trial. We thank the South-Eastern Norway Regional Health Authority and AstraZeneca for their funding and MEDICOVER Genetics for their contribution to conducting the ctDNA analysis in the study.
Conflict of interest
HH: Advisory board: Johnson and Johnson. Honoraria: AstraZeneca, Pfizer, Roche. VH: Advisory board: AstraZeneca, Merck Sharp & Dome, Johnson & Johnson, Novartis, Bristol-Myers Squibb. Honoraria: AstraZeneca, Merck Sharp & Dome, Johnson & Johnson, Novartis, Bristol-Myers Squibb, Pfizer, Takeda. TM: Advisory board: Johnson and Johnson. Honoraria: AstraZeneca, GlaxoSmithKline, Takeda AS, Merck Sharp & Dome. BG: Advisory board: Janssen, Accord, Merck Sharp & Dome, AstraZeneca, Pharmacosmos. Honoraria: AstraZeneca, Pfizer, Accord, Eli Lilly, Merck Sharp & Dome, Gilead, Bristol-Myers Squibb. Research funding: Roche, AstraZeneca. TH: Advisory board: AstraZeneca, Sanofi, Immedica. Honoraria: AstraZeneca, Takeda, Merck Sharp & Dome, Pfizer. Research funding: Roche, AstraZeneca. JK: Honoraria: Roche, AstraZeneca, Johnson and Johnson, Bristol-Myers Squibb, Merck Sharp & Dome, Amgen, Merck KGaA, Novartis, Sanofi and Pfizer. Research Funding: Institutional grants from AstraZeneca and Roche outside of current study. Lecturing: Siemens Healthineers. Employment: Former employee of Faron Pharmaceuticals. KO: Advisory board: Merck Sharp & Dome, AstraZeneca, Roche. Research Funding: Optellum. MA: Honoraria: Bristol-Myers Squibb, Astra Zeneca. JA: Advisory board: AstraZeneca. MS: Advisory board: AstraZeneca, Merck Sharp & Dome, Johnson and Johnson, Bristol-Myers Squibb, Pfizer, Roche. Honoraria: AstraZeneca, Merck Sharp & Dome, Johnson and Johnson, Bristol-Myers Squibb, Pfizer, Roche, Boehringer-Ingelheim. SF: Honoraria: Bristol-Myers Squibb. ÅÖ: Advisory board: Sanofi. ÅH: Research Funding: Roche, AstraZeneca, Novartis, Incyte, Eli Lilly, Bristol-Myers Squibb, Ultimovacs, Merck, GlaxoSmithKline, Illumina, Nanopore, Johnson and Johnson. Advisory boards and Honoraria: ABBVIE, Takeda, AstraZeneca, Roche, Pfizer, Janssen, EliLilly, Bristol-Myers Squibb, PierreFabre, Bayer, Merck Sharp & Dome, Novartis, Merck, Sanofi, Medicover. All funds go to Oslo University Hospital.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1681420/full#supplementary-material
Abbreviations
bTMB, blood tumor mutational burden; cfDNA, cell-free DNA; CNA, copy number amplifications; CRT, chemoradiotherapy; ctDNA, circulating tumor DNA; DDR, DNA damage response and repair; FFPE, formalin-fixed paraffin-embedded; HR, hazard ratio; ICI, immune checkpoint inhibitor; indels, small insertions and deletions; mut/Mb, mutations per megabase; NR, not reached; NSCLC, non-small cell lung cancer; OS, overall survival; QC, quality control; PD-1, programmed cell death protein-1; PD-L1, Programmed death-ligand 1; PFS, progression-free survival; SNVs, single nucleotide variants; TACS, target capture sequences; tTMB, tissue tumor mutational burden; TMB, tumor mutational burden; VAF, variant allele frequency; WES, whole exomeSequencing.
References
1. Ganti AK, Klein AB, Cotarla I, Seal B, and Chou E. Update of incidence, prevalence, survival, and initial treatment in patients with non–small cell lung cancer in the US. JAMA Oncol. (2021) 7:1824–32. doi: 10.1001/JAMAONCOL.2021.4932
2. Varlotto JM, Sun Z, Ky B, Upshaw J, Fitzgerald TJ, Diehn M, et al. A review of concurrent chemo/radiation, immunotherapy, radiation planning, and biomarkers for locally advanced non-small cell lung cancer and their role in the development of ECOG-ACRIN EA5181. Clin Lung Cancer. (2022) 23:547–60. doi: 10.1016/j.cllc.2022.06.005
3. Crvenkova S and Pesevska M. Important prognostic factors for the long-term survival in non-small cell lung cancer patients treated with combination of chemotherapy and conformal radiotherapy. J BUON. (2015) 20:775–81.
4. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. New Engl J Med. (2017) 377:1919–29. doi: 10.1056/NEJMOA1709937/SUPPL_FILE/NEJMOA1709937_DISCLOSURES.PDF
5. Spigel DR, Faivre-Finn C, Gray JE, Vicente D, Planchard D, Paz-Ares L, et al. Five-year survival outcomes from the PACIFIC trial: durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. J Clin Oncol. (2022) 40:1301–11. doi: 10.1200/JCO.21.01308
6. Wang Y, Zhang T, Huang Y, Li W, Zhao J, Yang Y, et al. Real-world safety and efficacy of consolidation durvalumab after chemoradiation therapy for stage III non-small cell lung cancer: A systematic review and meta-analysis. Int J Radiat Oncol Biol Phys. (2022) 112:1154–64. doi: 10.1016/J.IJROBP.2021.12.150
7. Mino-Kenudson M, Schalper K, Cooper W, Dacic S, Hirsch FR, Jain D, et al. Predictive biomarkers for immunotherapy in lung cancer: perspective from the international association for the study of lung cancer pathology committee. J Thorac Oncol. (2022) 17:1335–54. doi: 10.1016/j.jtho.2022.09.109
8. Meri-Abad M, Moreno-Manuel A, García SG, Calabuig-Fariñas S, Pérez RS, Herrero CC, et al. Clinical and technical insights of tumour mutational burden in non-small cell lung cancer. Crit Rev Oncol Hematol. (2023) 182:103891. doi: 10.1016/J.CRITREVONC.2022.103891
9. Sholl LM. Biomarkers of response to checkpoint inhibitors beyond PD-L1 in lung cancer. Modern Pathol. (2022) 35:66–74. doi: 10.1038/s41379-021-00932-5
10. Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria JC, and Postel-Vinay S. Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res. (2016) 22:4309–21. doi: 10.1158/1078-0432.CCR-16-0903/128815/AM/MUTATIONAL-LANDSCAPE-AND-SENSITIVITY-TO-IMMUNE
11. Meng G, Liu X, Ma T, Lv D, and Id S. Predictive value of tumor mutational burden for immunotherapy in non-small cell lung cancer: A systematic review and meta-analysis. PloS One. (2022) 17:e0263629. doi: 10.1371/journal.pone.0263629
12. Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. (2018) 33:843–852.e4. doi: 10.1016/j.ccell.2018.03.018
13. Budczies J, Kazdal D, Menzel M, Beck S, Kluck K, Altbürger C, et al. Tumour mutational burden: clinical utility, challenges and emerging improvements. Nat Rev Clin Oncol. (2024) 21:725–42. doi: 10.1038/s41571-024-00932-9
14. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Sci (1979). (2015) 348:124–8. doi: 10.1126/science.aaa1348
15. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. New Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMOA1613493/SUPPL_FILE/NEJMOA1613493_DISCLOSURES.PDF
16. Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. New Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMOA1801946/SUPPL_FILE/NEJMOA1801946_DISCLOSURES.PDF
17. Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1–selected patients with NSCLC. New Engl J Med. (2020) 383:1328–39. doi: 10.1056/NEJMOA1917346/SUPPL_FILE/NEJMOA1917346_DATA-SHARING.PDF
18. Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. (2018) 24:1441–8. doi: 10.1038/s41591-018-0134-3
19. Kim ES, Velcheti V, Mekhail T, Leal TA, Dowell JE, Tsai ML, et al. Primary efficacy results from B-F1RST, a prospective phase II trial evaluating blood-based tumour mutational burden (bTMB) as a predictive biomarker for atezolizumab (atezo) in 1L non-small cell lung cancer (NSCLC). Ann Oncol. (2018) 29:viii744. doi: 10.1093/annonc/mdy424.067
20. Peters S, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M-J, et al. Abstract CT074: Tumor mutational burden (TMB) as a biomarker of survival in metastatic non-small cell lung cancer (mNSCLC): Blood and tissue TMB analysis from MYSTIC, a Phase III study of first-line durvalumab ± tremelimumab vs chemotherapy. Cancer Res. (2019) 79:CT074. doi: 10.1158/1538-7445.AM2019-CT074
21. Ricciuti B, Wang X, Alessi JV, Rizvi H, Mahadevan NR, Li YY, et al. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. (2022) 8:1160–8. doi: 10.1001/JAMAONCOL.2022.1981
22. Di Federico A, De Giglio A, Gelsomino F, Sperandi F, Melotti B, and Ardizzoni A. Predictors of survival to immunotherapy and chemoimmunotherapy in non-small cell lung cancer: A meta-analysis. J Natl Cancer Inst. (2023) 115:29–42. doi: 10.1093/jnci/djac205
23. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti–programmed cell death (PD)-1 and anti–programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. (2018) 36:633. doi: 10.1200/JCO.2017.75.3384
24. Shaverdian N, Shepherd AF, Li X, Offin M, Lengel HB, Gelblum DY, et al. Impact of tumor mutational burden and gene alterations associated with radiation-response on outcomes of post-operative radiation therapy in non-small cell lung cancer. Int J Radiat Oncol Biol Phys. (2022) 113:335–44. doi: 10.1016/J.IJROBP.2022.02.014
25. Lebow ES, Shepherd A, Eichholz JE, Offin M, Gelblum DY, Wu AJ, et al. Analysis of tumor mutational burden, progression-free survival, and local-regional control in patients with locally advanced non-small cell lung cancer treated with chemoradiation and durvalumab. JAMA Netw Open. (2023) 6:e2249591. doi: 10.1001/jamanetworkopen.2022.49591
26. Alessi JV, Ricciuti B, Wang X, Pecci F, Di Federico A, Lamberti G, et al. Impact of TMB/PD-L1 expression and pneumonitis on chemoradiation and durvalumab response in stage III NSCLC. Nat Commun. (2023) 14:4238. doi: 10.1038/s41467-023-39874-8
27. Ye L, Chu X, Ni J, Chu L, Yang X, and Zhu Z. NGS-based tissue-blood TMB comparison and blood-TMB monitoring in stage-III non-small cell lung cancer treated with concurrent chemoradiotherapy. Cancer Invest. (2024) 42:165–75. doi: 10.1080/07357907.2024.2316297
28. Wang Y, Wang W, Zhang T, Yang Y, Wang J, Li C, et al. Dynamic bTMB combined with residual ctDNA improves survival prediction in locally advanced NSCLC patients with chemoradiotherapy and consolidation immunotherapy. J Natl Cancer Center. (2024) 4:177–87. doi: 10.1016/j.jncc.2024.01.008
29. Scalera S, Mazzotta M, Cortile C, Krasniqi E, De Maria R, Cappuzzo F, et al. KEAP1-mutant NSCLC: the catastrophic failure of a cell-protecting hub. J Thorac Oncol. (2022) 17:751–7. doi: 10.1016/j.jtho.2022.03.011
30. Workman S, Jabbour SK, and Deek MP. A narrative review of genetic biomarkers in non-small cell lung cancer: an update and future perspectives. AME Med J. (2023) 8:6. doi: 10.21037/amj-2022-01
31. World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. (2013) 310:2191–4. doi: 10.1001/JAMA.2013.281053
32. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. (2009) 45:228–47. doi: 10.1016/j.ejca.2008.10.026
33. Nakken S, Fournous G, Vodák D, Aasheim LB, Myklebost O, and Hovig E. Personal Cancer Genome Reporter: variant interpretation report for precision oncology. Bioinformatics. (2018) 34:1778–80. doi: 10.1093/BIOINFORMATICS/BTX817
34. Kyrochristos ID, Glantzounis GK, Goussia A, Eliades A, Achilleos A, Tsangaras K, et al. Proof-of-concept pilot study on comprehensive spatiotemporal intra-patient heterogeneity for colorectal cancer with liver metastasis. Front Oncol. (2022) 12:855463. doi: 10.3389/fonc.2022.855463
35. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. eprint arXiv. (2013).
36. Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non–small cell lung cancer. JAMA Oncol. (2018) 5:173. doi: 10.1001/JAMAONCOL.2018.4305
37. Paz-Ares L, Langer CJ, Novello S, Halmos B, Cheng Y, Gadgeel SM, et al. Pembrolizumab (pembro) plus platinum-based chemotherapy (chemo) for metastatic NSCLC: Tissue TMB (tTMB) and outcomes in KEYNOTE-021, 189, and 407. Ann Oncol. (2019) 30:v917–8. doi: 10.1093/annonc/mdz394.078
38. Gu W, Zhuang W, Zhuang M, He M, and Li Z. DNA damage response and repair gene mutations are associated with tumor mutational burden and outcomes to platinum-based chemotherapy/immunotherapy in advanced NSCLC patients. Diagn Pathol. (2023) 18:119. doi: 10.1186/S13000-023-01401-0
39. Li Z, Niu Y, Ma T, and Yuan H. MA13.03 DNA damage response gene alterations and their association with tumor mutation burden and response to immunotherapy in NSCLC and SCLC. J Thorac Oncol. (2021) 16:S182. doi: 10.1016/j.jtho.2021.01.265
40. Marcus L, Fashoyin-Aje LA, Donoghue M, Yuan M, Rodriguez L, Gallagher PS, et al. FDA approval summary: Pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin Cancer Res. (2021) 27:4685–9. doi: 10.1158/1078-0432.CCR-21-0327/672264/AM/FDA-APPROVAL-SUMMARY-PEMBROLIZUMAB-FOR-THE
41. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
42. Vokes NI, Liu D, Ricciuti B, Jimenez-Aguilar E, Rizvi H, Dietlein F, et al. Harmonization of tumor mutational burden quantification and association with response to immune checkpoint blockade in non–small-cell lung cancer. JCO Precis Oncol. (2019) 3:PO.19.00171. doi: 10.1200/PO.19.00171
43. Girard N, Bar J, Garrido P, Garassino MC, McDonald F, Mornex F, et al. Treatment characteristics and real-world progression-free survival in patients with unresectable stage III NSCLC who received durvalumab after chemoradiotherapy: findings from the PACIFIC-R study. J Thorac Oncol. (2023) 18:181–93. doi: 10.1016/j.jtho.2022.10.003
44. Desilets A, Blanc-Durand F, Lau S, Hakozaki T, Kitadai R, Malo J, et al. Durvalumab therapy following chemoradiation compared with a historical cohort treated with chemoradiation alone in patients with stage III non–small cell lung cancer: A real-world multicentre study. Eur J Cancer. (2021) 142:83–91. doi: 10.1016/j.ejca.2020.10.008
45. Ricciuti B and Garassino MC. Precision immunotherapy for STK11/KEAP1-mutant NSCLC. J Thorac Oncol. (2024) 19:877–82. doi: 10.1016/j.jtho.2024.03.002
46. Rizvi N, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M, et al. OA04.07 mutations associated with sensitivity or resistance to immunotherapy in mNSCLC: analysis from the MYSTIC trial. J Thorac Oncol. (2019) 14:S217. doi: 10.1016/j.jtho.2019.08.428
47. Peters S, Cho BC, Luft A, Alatorre-Alexander J, Geater SL, Kim S-W, et al. OA15.04 association between KRAS/STK11/KEAP1 mutations and outcomes in POSEIDON: durvalumab ± Tremelimumab + Chemotherapy in mNSCLC. J Thorac Oncol. (2022) 17:S39–41. doi: 10.1016/J.JTHO.2022.07.073
48. Ramalingam SS, Balli D, Ciuleanu T-E, Pluzanski A, Lee J-S, Schenker M, et al. 4O Nivolumab (NIVO) + ipilimumab (IPI) versus chemotherapy (chemo) as first-line (1L) treatment for advanced NSCLC (aNSCLC) in CheckMate 227 part 1: Efficacy by KRAS, STK11, and KEAP1 mutation status. Ann Oncol. (2021) 32:S1375–6. doi: 10.1016/J.ANNONC.2021.10.020
49. Si H, Kuziora M, Quinn KJ, Helman E, Ye J, Liu F, et al. A blood-based assay for assessment of tumor mutational burden in first-line metastatic NSCLC treatment: Results from the MYSTIC study A C. Clin Cancer Res. (2021) 27:1631–40. doi: 10.1158/1078-0432.CCR-20-3771/78940/AM/A-BLOOD-BASED-ASSAY-FOR-ASSESSMENT-OF-TUMOR
Keywords: locally advanced NSCLC, immunotherapy, biomarker, TMB, circulating tumorDNA
Citation: Horndalsveen H, Haakensen VD, Madebo T, Grønberg BH, Halvorsen TO, Koivunen J, Oselin K, Cicenas S, Helbekkmo N, Aanerud M, Ahvonen J, Silvoniemi M, Bjaanæs MM, Farooqi S, Nebdal D, Dalsgaard AM, Danielsen BK, Børve M, Dalen TS, Öjlert ÅK and Helland Å (2025) Blood-based tumor mutational burden as a biomarker in unresectable non-small cell lung cancer treated with chemoradiotherapy and durvalumab. Front. Oncol. 15:1681420. doi: 10.3389/fonc.2025.1681420
Received: 07 August 2025; Accepted: 08 October 2025;
Published: 22 October 2025.
Edited by:
John Varlotto, Marshall University Chief of Radiation Oncology, United StatesReviewed by:
Stephen J Kuperberg, New York University, United StatesKarthigayini Sivaprakasam, Memorial Sloan Kettering Cancer Center, United States
Copyright © 2025 Horndalsveen, Haakensen, Madebo, Grønberg, Halvorsen, Koivunen, Oselin, Cicenas, Helbekkmo, Aanerud, Ahvonen, Silvoniemi, Bjaanæs, Farooqi, Nebdal, Dalsgaard, Danielsen, Børve, Dalen, Öjlert and Helland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henrik Horndalsveen, aGVucmlrLmhvcm5kYWxzdmVlbkBnbWFpbC5jb20=