 Hang Dong1
Hang Dong1 Boyin Zhang
Boyin Zhang Xiaowen Wu
Xiaowen Wu Guangyao Liu
Guangyao Liu- 1Department of Traumatic Orthopedics, Jilin University China-Japan Union Hospital, Changchun, China
- 2Department of Breast Surgery, Jilin University China-Japan Union Hospital, Changchun, China
- 3Department of Spine Surgery, Jilin University China-Japan Union Hospital, Changchun, Changchun, China
- 4Department of Genitourinary Oncology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Peking University Cancer, Beijing, China
Chromatin domain-binding protein 4 (CHD4), the ATPase core component of the NuRD complex, exerts dual roles in epigenetic regulation—mediating both gene silencing and activation. We report a case of occult breast cancer with extensive bone metastasis harboring a novel somatic truncation mutation in the catalytic SNF2 domain (CHD4 p.Trp736Ter). This mutation, unreported in other cancers to date, abolishes ATPase activity and disrupts NuRD complex assembly. Multimodal analysis (PET/CT, biomarkers, pathology) confirmed the case as luminal-A subtype and revealed a significant response to endocrine therapy. We hypothesize that this CHD4 mutation may alter the protein’s dual regulatory balance, particularly enhancing its potential gene-activating functions, and propose that impaired chromatin remodeling driven by such mutations is associated with metastatic progression of breast cancer.
1 Introduction
CHD4, a dominant ATP-dependent chromatin remodeler within the NuRD complex, utilizes ATP hydrolysis to restructure nucleosomes (1, 2). As a core NuRD component, it recruits DNMTs to induce DNA methylation and partners with HDAC1/2, EZH2, and G9a to deposit repressive histone marks (H3K27me3/H3K9me2), silencing gene expression (3). Further evidence has emerged that the N-terminal region of CHD4 has gene-activating functions. These functions involve direct interaction with the ATPase domain of BRG1, which enhances transcriptional activity. This highlights the multifaceted role of CHD4 in epigenetic regulation (4). Beyond gene regulation, CHD4 is also essential for DNA damage repair and epigenetic reprogramming. Dysregulation profoundly impacts gene transcription and cellular processes.
CHD4 function relies on coordinated domain interactions. Pathogenic mutations disrupting these domains impair its activity, leading to defective DNA repair, aberrant gene expression, and tumorigenesis (5–7). Supporting this, a novel SNF2 domain nonsense mutation was identified in a patient with metastatic breast cancer (primary unknown, diagnosed via metastatic biopsies) who remains stable on endocrine therapy. Computational analysis suggests that this mutation has the potential to disrupt CHD4’s DNA-binding ability, which could in turn impair its gene-silencing function. Additionally, the mutation may also enhance its capacity for transcriptional activation. The integration of this case with existing research endeavors to elucidate the manner in which CHD4 mutations perturb its dual regulatory role and drive oncogenesis.
2 Clinical case
A 66-year-old postmenopausal woman with an unremarkable medical history presented with a diagnostically challenging metastatic tumor. Her clinical timeline began 35 years prior with a left acute mastitis episode requiring surgical incision and drainage, which evolved into chronic mastitis. In May 2022, she underwent ultrasound-guided closed drainage for a liver abscess, achieving full resolution. Seven months later in December 2022, acute-onset chest and back pain prompted abdominal CT imaging to evaluate potential liver abscess recurrence. While hepatic architecture appeared normal, the scan incidentally detected a T10 vertebral compression fracture. Subsequent spinal MRI revealed multiple osteolytic-osteoblastic mixed vertebral metastases, predominantly involving pedicles and posterior elements with characteristic rounded morphology. Serum tumor marker profiling was conducted to identify a primary malignancy. Carcinoembryonic antigen levels were notably elevated above reference ranges, whereas other markers including CA 15-3, CA 125, and alpha-fetoprotein remained within normal parameters. Despite extensive diagnostic investigations encompassing biochemical analyses and advanced imaging modalities, the origin of the primary tumor remains undetermined at this stage of evaluation.
2.1 Baseline 18F-FDG PET/CT imaging
18F-FDG PET/CT (Figure 1D) demonstrated a highly metabolic right thyroid nodule (13.0 × 6.7mm, SUVmax 9.41; Figure 1A) alongside FDG-avid lymphadenopathy in submandibular/cervical/supraclavicular (SUVmax 2.58) and mediastinal regions (SUVmax 4.19). Extensive hypermetabolic bone metastases were evident, including a pathological T10 fracture (SUVmax 11.64, CT HU 876; Figure 1B), right iliac lesion (SUVmax 6.32; Figure 1C), and left proximal humeral lesion (SUVmax 11.47; Figure 1E). A left breast nodule showed only borderline FDG uptake (SUVmax 2.19; Figure 1F), subsequently confirmed as benign (BI-RADS II) with periareolar fibrosis on ultrasound, consistent with chronic mastitis.
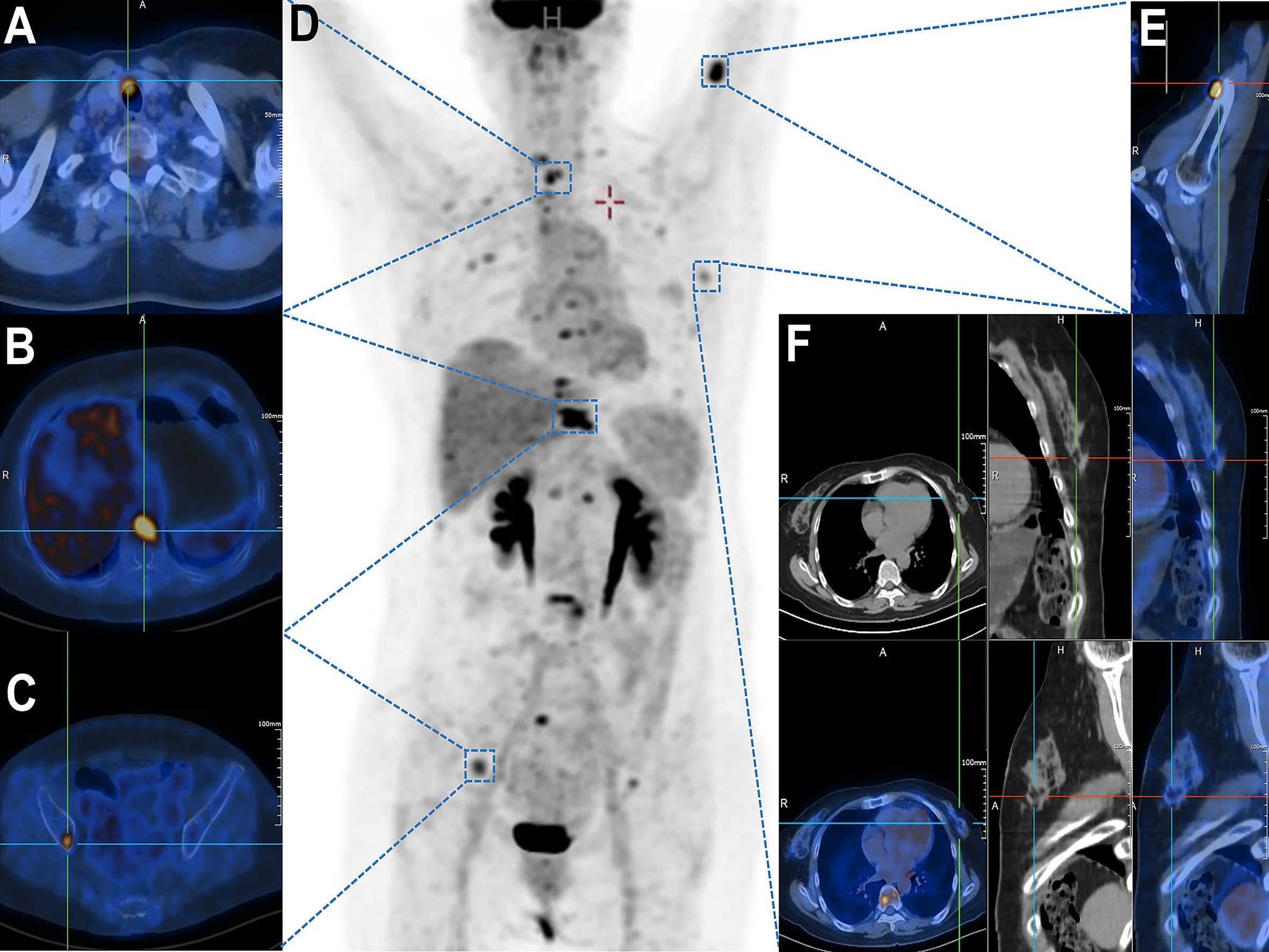
Figure 1. Whole-body 18F-FDG PET/CT demonstrating multifocal hypermetabolic lesions. (A) Thyroid isthmus lesion with focally increased FDG uptake (SUVmax = 9.41). (B) Vertebral metastasis at T10 level (SUVmax = 11.64). (C) Right iliac bone lesion showing elevated metabolic activity (SUVmax = [value]). (D) Maximum intensity projection (MIP) image revealing systemic dissemination of metabolically active metastases. (E) Left proximal humeral lesion with abnormal FDG avidity (SUVmax = [value]). (F) A suspicious mass in the left breast upper-outer quadrant demonstrated focal FDG avidity (SUVmax = 2.19) on axial, coronal, and sagittal fused PET/CT views.
Breast cancer bone metastasis imaging: 18F-sodium fluoride PET/CT excels for osteoblastic metastases (alters management in 24–60% cases), while 18F-FDG PET/CT targets osteolytic disease. PET/CT is critical for advanced/high-risk staging, metastasis detection, NAC response, and recurrence surveillance (8). In suspected recurrence, PET/CT outperformed CECT/BS with perfect NPV, near-perfect AUC (0.99), and superior specificity (98.9%) for distant metastases, establishing it as a definitive standalone modality (9).
2.2 Longitudinal imaging progression of spinal metastases
Serial imaging during the patient’s liver abscess hospitalization (initial abdominal CT: Figure 2A; follow-up CECT: Figure 2B) revealed incidental T9 vertebral body sclerosis (Red Arrow) and subtle T10 anterior margin moth-eaten destruction (Yellow Arrow), with T11 appearing unremarkable. These early metastatic features were only recognized retrospectively. Subsequent MRI during back pain episodes confirmed progressive T10 destruction (Figure 2C). Eight months post-metastasis, thoracic CT (Figure 2D) demonstrated T9 osteoblastic progression, T10 osteolysis with peripheral sclerosis, and new T11 sclerotic lesions. Follow-up CT (Figures 2E, F) showed expanding osteoblastic lesions at T9/T11 (Red Arrows), advancing T10 osteolysis (Yellow Arrows), and a visible biopsy tract (Green box). Concurrent MRI (Figures 2G, H) revealed T10 vertebral collapse and T11 involvement. Post-endocrine therapy imaging (Figure 2I) documented stabilized T9/T11 lesions, regressing T10 osteolysis, no new fractures, and persistent biopsy changes, demonstrating therapeutic response. This progression highlights breast cancer metastases’ heterogeneous radiographic presentation (osteolytic/osteoblastic/mixed), their subtle early imaging profile, and distinct growth patterns: osteoblastic foci exhibiting rapid vertebral dissemination within 6–8 months versus typically localized osteolytic lesions (10).
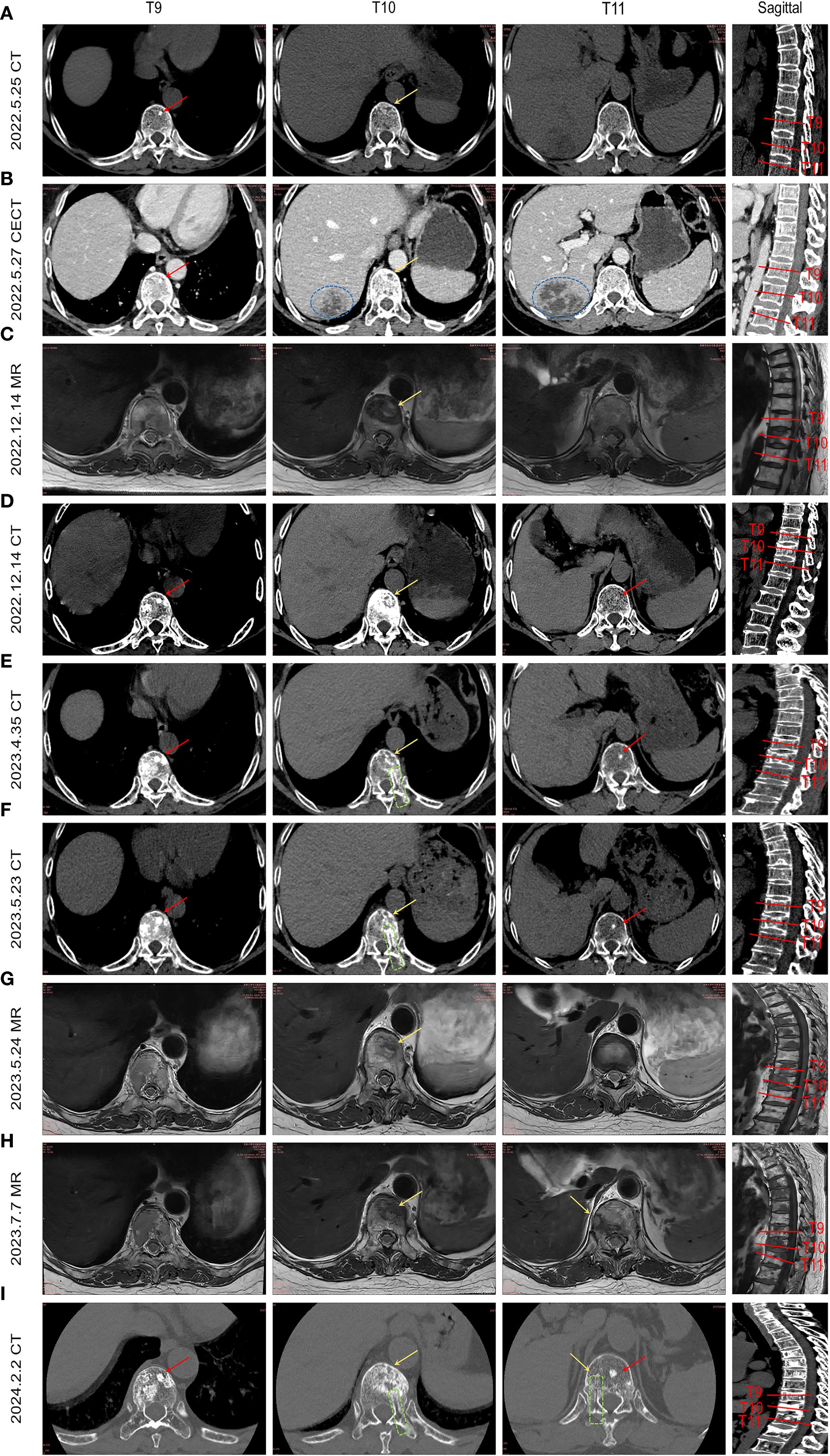
Figure 2. illustrates the temporal evolution of spinal metastasis in the patient. By integrating abdominal CT scans obtained during hospitalization for liver abscess and collating prior MRI/CT images, the imaging progression of breast cancer spinal metastases from onset to final outcome is presented sequentially. (A) Abdominal CT during liver abscess hospitalization shows a high-density shadow in the T9 vertebral body (red arrow) and worm-eaten changes at the anterior margin of T10 (yellow arrow). (B) Contrast-enhanced abdominal CT highlights the liver abscess (blue circle), T9 high-density shadow, and more pronounced worm-eaten changes at T10 anterior margin (yellow arrow). (C) Incidental MRI for low back pain reveals a metastatic lesion at T10 anterior margin (yellow arrow) and mixed signal intensity changes in T9. (D) Thoracic CT after 8 months shows multiple osteoblastic changes in T9 (red arrow), resorptive changes at T10 anterior margin, osteoblastic rim around osteolytic lesions, and new high-density shadows in T11. (E, F) Lesions progress with expanding osteoblastic foci in T9 and T11 (red arrows), enlarged worm-eaten changes at T10 anterior margin (yellow arrow), and a visible biopsy tract (green box). (G, H) Sagittal views show expanded T10 anterior lesions with compression fracture, metastatic involvement at T11 edge. (I) Eight-month follow-up thoracic CT after endocrine therapy demonstrates stabilized T9/T11 lesions (red arrows), improved T11 anterior worm-eaten changes (yellow arrow), no new compression fractures, and persistent biopsy tract (green box).
2.3 Diagnosis and endocrine therapy
Histopathological evaluation of the third biopsy specimen from the T10 vertebral lesion demonstrated poorly differentiated adenocarcinoma cells exhibiting cytokeratin positivity. Immunohistochemical profiling robustly supported a mammary origin, characterized by strong nuclear expression of estrogen receptor with 3+ intensity and 70% positivity, progesterone receptor with 3+ intensity and 70% positivity, HER-2 negativity at 1+ intensity, and a low Ki-67 proliferation index of 5%. While the low Ki-67 value suggested limited proliferative activity, potential sampling bias due to minimal tissue availability was acknowledged as a confounding factor. Further immunophenotypic analysis revealed CK5/6 negativity, excluding adenoepithelial differentiation, whereas CK7, mammaglobin, and GATA-3 positivity reinforced breast origin. Critical exclusion of alternative primary sites was achieved through PAX-8 negativity to rule out urothelial and renal carcinomas, CK20 and villin negativity to exclude gastrointestinal, pancreatic, biliary, and ovarian mucinous tumors, thyroglobulin and TTF-1 negativity to discard thyroid carcinoma, and vimentin negativity to eliminate mesenchymal neoplasms.
Synthesizing imaging, histopathology, and biomarker data, the metastatic lesions were conclusively attributed to advanced Luminal A breast cancer, defined by ER positivity, PR positivity, HER2 negativity, disseminated skeletal involvement, and absent visceral metastases. Given the incurable nature of widespread bone metastases and the endocrine sensitivity intrinsic to Luminal A subtypes, therapy was aligned with National Comprehensive Cancer Network (NCCN) guidelines. The regimen comprised fulvestrant, an estrogen receptor antagonist, combined with a CDK4/6 inhibitor such as palbociclib to target hormonal pathways and suppress cell cycle progression. Additionally, denosumab was administered to mitigate osteoclast-mediated bone destruction and reduce fracture risk, addressing the patient’s pathologic vertebral fracture.
2.4 Novel mutation in CHD4
Plasma NGS (580 genes) revealed a novel somatic CHD4 truncating mutation (c.2208G>A, p.Trp736Ter; VAF=4%), confirmed as pathogenic and previously undocumented. No pathogenic alterations were detected in key breast cancer drivers (PIK3CA/BRCA1/BRCA2/ERBB2/TP53), structural rearrangements, or CNVs. The p.Trp736Ter mutation causes truncation of the residues encoded by exon 15 (708–771), thereby preventing nucleosome disassembly and NuRD complex formation. TCGA analysis identified similar pathogenic truncating mutations (Table 1): adjacent variants associated with uterine malignancies (p.W728*/p.W741*), and upstream hinge region truncating mutations (p.684*/p.685*, etc.) associated with colorectal cancer/bladder cancer/endometrial cancer and melanoma. Additionally, similar truncating mutations in the subfamily homologous proteins CHD3 and CHD5 were summarized.
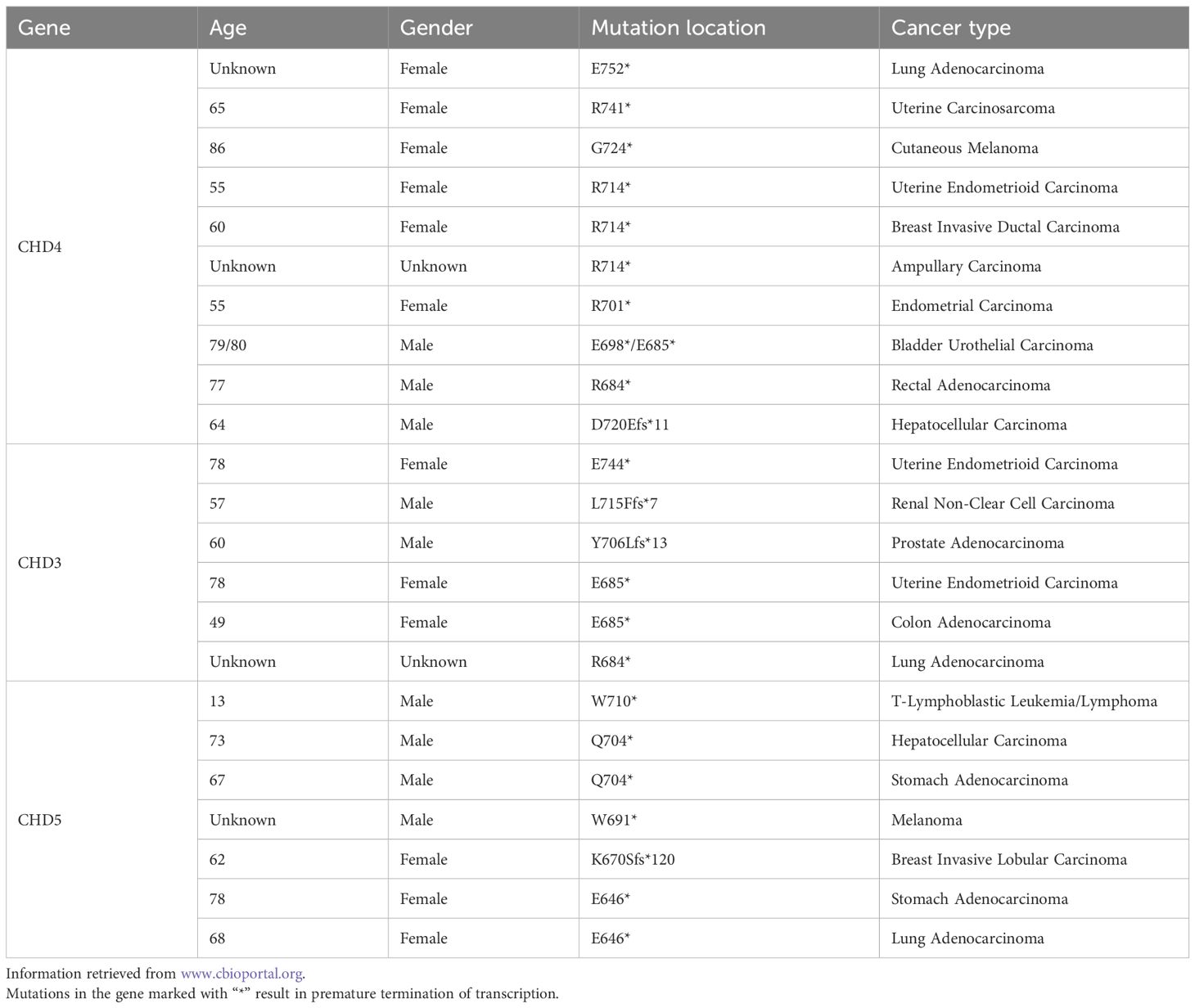
Table 1. Similar mutations in CHD3/4/5 across malignancies.
The CHD4 protein contains sequentially arranged domains: CHDNT, two PHDs, two chromodomains, SNF2 ATPase, Helicase domain, two DUFs, and CHDCT (11). Its N-terminal region (CHD4-N) is basic and binds both DNA and poly(ADP-ribose) (PAR), with significantly higher affinity for PAR. This specific PAR binding is essential for recruiting CHD4 to DNA damage site (12). Additionally, CHD4-N acts as a transcriptional activator, interacting directly with the BRG1 ATPase domain to promote gene expression and autoregulating its function through competitive binding to BRG1 (4).
3 Discussion
The oncogenic role of CHD4 mutations is validated across cancers—suppressing WNT antagonists through DNMT cooperation in colorectal cancer, governing repair-associated methylation dynamics in ovarian cancer, and enabling TERT promoter epigenetic remodeling in thyroid cancer—demonstrating broad tumorigenic influence through direct DNMT modulation and NuRD-dependent methylation/deacetylation (13–15). CHD4-NuRD drives breast cancer metastasis initiation by orchestrating EMT through MTA1/3-mediated E-cadherin suppression and β-catenin mislocalization, while antagonizing GATA3-dependent MET via chromatin occlusion, enabling early dissemination from small primaries (16–18). Beyond EMT, CHD4 amplifies metastasis through Wnt/β-catenin co-activation, HIF-enhanced hypoxic responses, RhoA/ROCK-driven migration, and immune evasion via EZH2-coupled H3K27me3 silencing of chemokines, while concurrently promoting chemoresistance via MEK/ERK overactivation—establishing CHD4 as a multi-mechanistic therapeutic target across cancers (14, 19, 20).
Despite unclear primary origin, widespread bone metastases occurred, warranting investigation into links with breast cancer molecular subtypes (especially high-risk Luminal tumors at 29.9%) and potential novel CHD4 mutations (21). Bone tropism is driven by specific genetic programs (e.g., RUNX2, IL11/CTGF/CXCR4) and the “seed and soil” hypothesis involving RANKL/TGF-β cascades (22). CHD4 drives breast cancer epigenetic dysregulation by recruiting HDAC1/2 and DNMTs via the NuRD complex to silence tumor suppressor genes, with its global impact manifested in breast cancer CpG island methylator phenotype (B-CIMP) linked to metastatic transcriptomes, ERα/immune dual-cluster regulatory networks, and replication-linked methylation clocks (23–25).
The role of CHD4 in gene silencing provides a key framework for explaining the carcinogenic effect of its mutations. Although the classic CHD4 mutation is believed to lead to loss of function, especially impounding its NURD-dependent gene silencing ability, thereby disrupting the inhibition of tumor suppressor genes, the research results may suggest a unique mechanism: The novel SNF2 domain truncation mutation (p.RP736TER) may lead to gene activation rather than simply eliminating the activity of CHD4. The molecular model of the mutant CHD4 confirmed that its N-terminal region retains a specific binding to the bromine domain of BRG1, and that this interaction mediates the gene activation function of CHD4 (4). This combination has the potential to disrupt the normal interaction between BRG1 and chromatin, thereby tilting CHD4 towards excessive transcriptional activation and simultaneously damaging its ATPase-dependent gene silencing function (due to SNF2 domain truncation). This functional imbalance, characterized by enhanced activation in conjunction with reduced silencing, has the potential to drive abnormal expression of oncogenes or metastasis-related genes. This, in turn, can promote the occurrence and progression of occult breast cancer with bone metastases. Consequently, it is imperative to ascertain the epigenetic consequences of this mutation and its impact on BRG1 binding.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
This study was approved by the Ethical Review Committee of Jilin University China-Japan Union Hospital (Approval No.: [2024110706]). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
HD: Investigation, Data curation, Writing – original draft, Formal Analysis. JZ: Writing – original draft, Resources, Software, Methodology. BZ: Data curation, Methodology, Supervision, Writing – review & editing. XW: Writing – review & editing. GL: Funding acquisition, Visualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the the Project of the National Science Foundation of China (Grant No. 82202668) and Jilin Province Health Talent Special Project (Grant No. 2018SCZ018).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1682794/full#supplementary-material
References
1. Zhong Y, Paudel BP, Ryan DP, Low JKK, Franck C, Patel K, et al. CHD4 slides nucleosomes by decoupling entry- and exit-side DNA translocation. Nat Commun. (2020) 11. doi: 10.1038/s41467-020-15183-2
2. Wang Y, Chen Y, Bao L, Zhang B, Wang JE, Kumar A, et al. CHD4 promotes breast cancer progression as a coactivator of hypoxia-inducible factors. Cancer Res. (2020) 80:3880–91. doi: 10.1158/0008-5472.CAN-20-1049
3. Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell. (2017) 31:653–68.e7. doi: 10.1016/j.ccell.2017.04.005
4. Shimono Y, Murakami H, Kawai K, Wade PA, Shimokata K, and Takahashi M. Mi-2β Associates with BRG1 and RET finger protein at the distinct regions with transcriptional activating and repressing abilities. J Biol Chem. (2003) 278:51638–45. doi: 10.1074/jbc.M309198200
5. Morra R, Lee BM, Shaw H, Tuma R, and Mancini EJ. Concerted action of the PHD, chromo and motor domains regulates the human chromatin remodelling ATPase CHD4. FEBS Lett. (2012) 586:2513–21. doi: 10.1016/j.febslet.2012.06.017
6. Watson AA, Mahajan P, Mertens HDT, Deery MJ, Zhang W, Pham P, et al. The PHD and chromo domains regulate the ATPase activity of the human chromatin remodeler CHD4. J Mol Biol. (2012) 422:3–17. doi: 10.1016/j.jmb.2012.04.031
7. Zhang Q, Zhu F, Tong Y, Shi D, and Zhang J. CHD4 R975H mutant activates tumorigenic pathways and promotes stemness and M2-like macrophage polarization in endometrial cancer. Sci Rep. (2024) 14. doi: 10.1038/s41598-024-69233-6
8. Paydary K, Seraj SM, Zadeh MZ, Emamzadehfard S, Shamchi SP, Gholami S, et al. The evolving role of FDG-PET/CT in the diagnosis, staging, and treatment of breast cancer. Mol Imaging Biol. (2018) 21:1–10. doi: 10.1007/s11307-018-1181-3
9. Hildebrandt MG, Gerke O, Baun C, Falch K, Hansen JA, Farahani ZA, et al. 18F]Fluorodeoxyglucose (FDG)-positron emission tomography (PET)/computed tomography (CT) in suspected recurrent breast cancer: A prospective comparative study of dual-time-point FDG-PET/CT, contrast-enhanced CT, and bone scintigraphy. J Clin Oncol. (2016) 34:1889–97. doi: 10.1200/JCO.2015.63.5185
10. Hamaoka T, Madewell JE, Podoloff DA, Hortobagyi GN, and Ueno NT. Bone imaging in metastatic breast cancer. J Clin Oncol. (2004) 22:2942–53. doi: 10.1200/JCO.2004.08.181
11. Goswami K, Venkatachalam K, Singh SP, Rao CV, and Madka V. Chromatin remodulator CHD4: A potential target for cancer interception. Genes. (2025) 16. doi: 10.3390/genes16020225
12. Silva APG, Ryan DP, Galanty Y, Low JKK, Vandevenne M, Jackson SP, et al. The N-terminal region of chromodomain helicase DNA-binding protein 4 (CHD4) is essential for activity and contains a high mobility group (HMG) box-like-domain that can bind poly(ADP-ribose). J Biol Chem. (2016) 291:924–38. doi: 10.1074/jbc.M115.683227
13. Cai Y, Geutjes EJ, de Lint K, Roepman P, Bruurs L, Yu LR, et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene. (2013) 33:2157–68. doi: 10.1038/onc.2013.178
14. Wang J, Zhong F, Li J, Yue H, Li W, and Lu X. The epigenetic factor CHD4 contributes to metastasis by regulating the EZH2/β-catenin axis and acts as a therapeutic target in ovarian cancer. J Trans Med. (2022) 21:2–15. doi: 10.21203/rs.3.rs-2021260/v1
15. Li S, Hu G, Chen Y, Sang Y, Tang Q, and Liu R. TERT upstream promoter methylation regulates TERT expression and acts as a therapeutic target in TERT promoter mutation-negative thyroid cancer. Cancer Cell Int. (2024) 24. doi: 10.1186/s12935-024-03459-2
16. Li D-Q, Pakala SB, Nair SS, Eswaran J, and Kumar R. Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer Res. (2012) 72:387–94. doi: 10.1158/0008-5472.CAN-11-2345
17. Mahmood N, Arakelian A, Szyf M, and Rabbani SA. Methyl-CpG binding domain protein 2 (Mbd2) drives breast cancer progression through the modulation of epithelial-to-mesenchymal transition. Exp Mol Med. (2024) 56:959–74. doi: 10.1038/s12276-024-01205-2
18. Saotome M, Grimm SA, Nagornyuk A, Gunarathna S, Shimbo T, Wade PA, et al. Genomic transcription factor binding site selection is edited by the chromatin remodeling factor CHD4. Nucleic Acids Res. (2024) 52:3607–22. doi: 10.1093/nar/gkae025
19. Xu N, Liu F, Wu S, Ye M, Ge H, Zhang M, et al. CHD4 mediates proliferation and migration of non-small cell lung cancer via the RhoA/ROCK pathway by regulating PHF5A. BMC Cancer. (2020) 20. doi: 10.1186/s12885-020-06762-z
20. Li C, Wang Z, Yao L, Lin X, Jian Y, Li Y, et al. Mi-2β promotes immune evasion in melanoma by activating EZH2 methylation. Nat Commun. (2024) 15. doi: 10.1038/s41467-024-46422-5
21. Diessner J, Wischnewsky M, Stüber T, Stein R, Krockenberger M, Häusler S, et al. Evaluation of clinical parameters influencing the development of bone metastasis in breast cancer. BMC Cancer. (2016) 16. doi: 10.1186/s12885-016-2345-7
22. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. (2005) 115:44–55. doi: 10.1172/JCI22320
23. Batra RN, Lifshitz A, Vidakovic AT, Chin S-F, Sati-Batra A, Sammut S-J, et al. DNA methylation landscapes of 1538 breast cancers reveal a replication-linked clock, epigenomic instability and cis-regulation. Nat Commun. (2021) 12. doi: 10.1038/s41467-021-25661-w
24. Fleischer T, Tekpli X, Mathelier A, Wang S, Nebdal D, Dhakal HP, et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat Commun. (2017) 8. doi: 10.1038/s41467-017-00510-x
Keywords: CHD4 mutation, occult breast cancer, bone metastases, NuRD complex, epigenetic modification
Citation: Dong H, Zhao J, Zhang B, Wu X and Liu G (2025) Case Report: Novel mutation in CHD4 triggers occult breast cancer with bone metastases. Front. Oncol. 15:1682794. doi: 10.3389/fonc.2025.1682794
Received: 09 August 2025; Accepted: 16 September 2025;
Published: 29 September 2025.
Edited by:
Maha Mohamed Saber-Ayad, University of Sharjah, United Arab EmiratesReviewed by:
Krishnendu Goswami, University of Oklahoma Health Sciences, United StatesQinglin Zhang, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2025 Dong, Zhao, Zhang, Wu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangyao Liu, Z3lsaXVAamx1LmVkdS5jbg==