 Michael E. Fant
Michael E. Fant Juan Fuentes1
Juan Fuentes1- 1Department of Pediatrics, Morsani College of Medicine, University of South Florida, Tampa, FL, USA
- 2Department of Obstetrics and Gynecology, Morsani College of Medicine, University of South Florida, Tampa, FL, USA
- 3Department of Pathology and Cell Biology, Morsani College of Medicine, University of South Florida, Tampa, FL, USA
Epidemiological studies have demonstrated an increased prevalence of birth defects and intrauterine growth restriction (IUGR) among infants born prematurely suggesting they share common biological determinants. The identification of key regulatory pathways contributing to this nexus is essential to ongoing efforts to develop effective intervention strategies. Plac1 is a paternally imprinted and X-linked gene that conforms to this paradigm. Examination of a mutant mouse model has confirmed that Plac1 is essential for normal placental development and function. Moreover, it is expressed throughout the developing embryo indicating that it also has broad relevance to embryogenesis. Most notably, its absence in the developing embryo is associated with abnormal brain development and an increased risk of lethal, postnatal hydrocephalus identifying it as a novel, X-linked determinant of brain development. The essential and non-redundant roles of Plac1 in placental and neurological development represent a novel regulatory paradigm for embryonic growth and pregnancy maintenance. Regulatory pathways influenced, in part, by Plac1 are likely to contribute to the observed nexus of IUGR, prematurity, and birth defects.
Introduction
In the United States, birth defects and prematurity are the major causes of infant mortality and long-term disability in children (1). Globally, intrauterine growth restriction (IUGR) also emerges as a major determinant of infant mortality and morbidity (2). Together they impose heavy medical, social, and economic burdens on individuals, families, communities, and national development. Often considered as separate entities, increasing evidence suggests they are rooted in common biological pathways. Innovative approaches aimed at reducing their prevalence and associated morbidities will require fundamentally new knowledge regarding the regulatory pathways they share. We will review the evidence supporting their common origins. We will also review a recently identified X-linked gene, Plac1, which is essential to both placental and embryonic development and likely contributes to regulatory pathways that link these three outcomes.
Epidemiological Evidence Linking Prematurity and Structural Birth Defects
Epidemiological studies have demonstrated an unambiguous association between being born prematurely and having a BD (3, 4). In fact, the prevalence of BDs increases progressively as gestational age decreases. The defects most strongly associated with prematurity involve the central nervous system (CNS) (Prevalence Ratio = 16.23), followed by respiratory tract defects (PR = 11.51), cardiovascular defects (PR = 9.29), gastrointestinal defects (PR = 4.98), and genitourinary defects (PR = 4.09). This epidemiological linkage is not surprising. Regulatory interactions required for placental morphogenesis, a fetal tissue uniquely required to establish and maintain pregnancy, are also required for the development of other fetal organs. Signaling mechanisms known to be relevant to both embryonic and extra-embryonic development include HuR, Wnt, integrin, RxR, and PPAR-dependent pathways (5–9). Additionally, disruptions in placental function, irrespective of the underlying cause, can secondarily affect embryonic organ development (10, 11). Consequently, disruptions in the placental developmental program are not only likely to increase the probability for pregnancy failure but also likely to influence the development of other fetal organs. While progress has been made in preventing specific BDs and improving pregnancy outcomes complicated by specific maternal conditions, i.e., folate deficiency and diabetes, further progress requires identification of critical pathways that contribute to the “developmental program” shared by both processes. This intuitively implicates mechanisms underlying the complex spatial and temporal organization that define tissue morphogenesis in both the placenta and embryo.
Prematurity and Intrauterine Growth Restriction
Compelling evidence has emerged over the last three decades demonstrating that infants delivered prematurely are also disproportionately affected by IUGR. By inference, prematurity and birth defects are part of a continuum that also includes IUGR. Heinonen et al. (12), described a cohort of 120 preterm infants (≤36 weeks) born in the Kuopio province in Finland over a 2-year period, representing 96% of all the preterm births in that region. Birth weight (BW), length (L), and ponderal index (PI) were recorded and IUGR was defined as 2SD below the mean for gestational age for either parameter. IUGR was observed in 41% of the infants when considering at least one growth parameter and a low PI was the most common parameter affected (86%). Additionally, 33% of the infants had more than one growth parameter affected. These observations helped to change the existing pathophysiological paradigm by clinically linking these two entities. The high prevalence of IUGR that was associated with their preterm infants suggested that the sequence of events resulting in their birth actually started weeks earlier, in utero, leading to a diminution in growth rate and culminating in early delivery. In other words, the clinical presentation of preterm labor, premature rupture of membranes, or fetal distress represent late or end-terminal stages of a more chronic process.
Subsequent studies in different populations supported these findings. In 2000, Zeitlin et al. (13) reported the results of a case–control study examining the determinants of preterm birth using data from 16 European countries derived from the European Program of Occupational Risks and Pregnancy Outcome (EUROPOP) between 1994 and 1997. A total of 4700 preterm infants born between 22 and 36 weeks of gestation and 6460 control infants born between 37 and 40 weeks were analyzed. Small-for-gestational age (SGA) was defined as a BW below the 10th percentile. Being SGA was found to be significantly associated with preterm birth. Over 40% of preterm births induced for reasons other than the premature rupture of membranes were SGA compared with 10.7% of term infants (OR 6.41). The association was smaller but still significant for preterm births associated with the spontaneous or premature rupture of membranes (OR 1.51). The relationship between growth restriction and prematurity was strongest before 34 weeks gestation. Subsequent clinical studies were consistent with these findings. Doubilet et al. (14) obtained estimates of fetal growth by ultrasound at 24–29.9 weeks gestation and followed the pregnancies until delivery. The measurements of infants delivered very prematurely (24–29.9 weeks) were compared to those of infants delivering at ≥37 weeks. All growth parameters were significantly lower in the group that delivered prematurely compared to those delivering at term. A subsequent study recapitulated these findings. Gardosi (15) obtained ultrasound-derived estimates of fetal weight in a cohort of pregnant women later in gestation (32 weeks). The weights of infants delivering prematurely were compared to those that eventually delivered at term. The weights of term infants displayed a normal distribution whereas the distribution of weights associated with preterm infants were skewed to the left and had a lower median, reflecting a greater proportion of IUGR infants.
The presence of IUGR is underappreciated by most clinicians caring for premature infants unless the infant has been severely affected and is at the extreme end of his/her growth curve. It is important to remain cognizant, however, of the fact that the overall growth (BW, PI, length) trajectory of premature infants is shifted lower, compared to infants delivering at term, even though many have not yet reached the extreme or SGA threshold by the time they deliver. It is clear that a significant portion of preterm infants endure days or weeks of an adverse intrauterine environment that negatively affects their growth and increases their risk for long-term morbidities, independent of being premature.
Prematurity and the Risk for Cognitive and Psychiatric Impairment
Many studies define birth defects in terms of structurally identifiable anomalies. The March of Dimes, however, has extended this definition to include functional neurological impairments not apparent at birth or associated with identifiable, structural abnormalities (16). This more comprehensive definition of birth defects has been validated by the cumulative weight of several important studies demonstrating an association between prematurity and a variety of functional neurological disorders.
Psychiatric Disorders
Nosarti et al. (17) performed an historical population-based cohort study of all live-born individuals (N = 1,301,522) who were registered in the Swedish Medical Birth Register between 1973 and 1985 and living in Sweden at age 16 by 2002. Infants born at 32–36 weeks gestation had a 1.6-fold increase in risk of non-affective psychosis, 1.3× the risk of depressive disorder, and 2.7× the risk of bipolar affective disorder, compared to infants born at term. These risks were higher for infants born earlier than 32 weeks, 2.5×, 2.9×, and 7.9× respectively.
Attention-Deficit Hyperactivity Disorder
Similarly, Lindström et al. (18) demonstrated an association between preterm birth and an elevated risk for ADHD in Swedish schoolchildren. The children born between 1987 and 2000 (N = 1,180,616) who were prescribed ADHD medication between 6 and 19 years of age were identified and linked to their gestational age at birth. There was a significant increase in the use of ADHD medication in children who were born prematurely. Infants born at 23–28 weeks gestation were 2.1× more likely to be taking these medications than children delivered at term. This was followed by 1.6× at 29–32 weeks, 1.4× at 33–34 weeks, 1.3× at 35–36 weeks, and 1.1× at 37–38 weeks. These findings were not affected by genetic, perinatal, or socioeconomic status.
Autism Spectrum Disorders
Finally, Johnson et al. (19) conducted a prospective study of all births <26 weeks gestation in the United Kingdom and Ireland in 1995 and found an increased risk for autism spectrum symptoms and autism spectrum disorders (ASD) by middle childhood. Subsequently, by analyzing the medical records of all children ages 0–17 years who resided in Stockholm County from 2001 to 2007 (N = 589,114), Abel et al. (20) confirmed the association between ASD and prematurity and further demonstrated an independent effect of fetal growth.
The Placenta–Brain Axis in Neurological Development
Recently, Bonnin et al. (21) provided compelling evidence for the functional interdependence of the placenta and brain during development that may have relevance to these observations and supports the concept of “premature placental separation” as an independent determinant of brain development. Forebrain circuits are important in ASD, schizophrenia, bipolar disorder, anxiety, and depression. By creating a novel and innovative ex vivo mouse model, Bonnin and colleagues were able to demonstrate that the serotonin required for forebrain development was derived exclusively from the placenta between E10.5 and E15.5 after conversion from tryptophan. The hindbrain was not a significant source of forebrain serotonin until after E15.5. Thus, premature separation from the placenta during this time would deprive the developing brain of an essential neurotransmitter during a critical period of development.
The Placenta as an Experimental Model
Cumulative evidence derived from epidemiological, clinical, and animal studies provide compelling evidence that prematurity, structural birth defects, and cognitive/psychiatric disorders have roots in shared biological pathways (Figure 1). Identifying and characterizing these pathways and delineating loci that are susceptible to genetic and environmental perturbations are essential for efforts to develop effective intervention strategies aimed at improving outcomes. The placenta, in its unique role of providing an optimal milieu for fetal development, plays an essential role in this regulatory context. Plac1 (Placenta-specific 1), an X-linked, imprinted gene was recently identified that conforms to this paradigm.
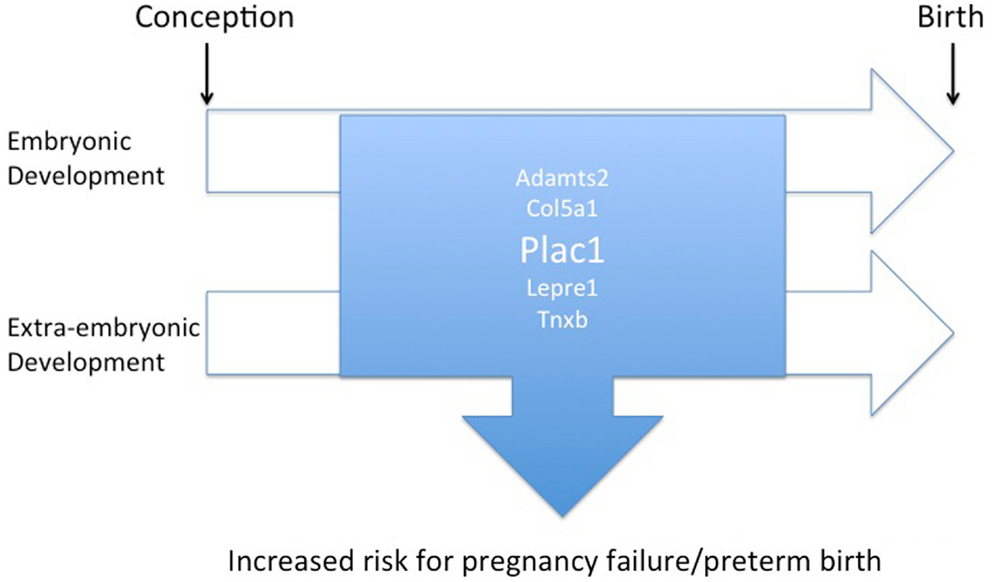
Figure 1. Diagram illustrating the overlap of regulatory pathways involved with placental and embryonic development that, when disrupted, may contribute to an increased risk of preterm birth or miscarriage as well as birth defects.
Identification of the Plac1 Gene
The Plac1 gene (placenta-specific 1) was first reported by Cocchia et al. (22). It maps to a region of the X chromosome known to be important for fetal development. Large deletions of the mouse X chromosome, spanning 200–700 kb near the Plac1 locus, result in fetal growth retardation and neonatal death (23). Further evidence supporting the importance of this locus was provided by Zechner et al. (24) and Hemberger et al. (25). The Ihpd (interspecific hybrid placental dysplasia) locus maps in proximity to the Plac1 locus and is associated with abnormal placental development where several X-linked genes may be involved.
Northern analysis and in situ hybridization indicated Plac1 expression is restricted primarily to cells of trophoblastic lineage (22, 26) hence its name Placenta-specific 1. However, recent studies (using qRT-PCR) have demonstrated expression in multiple embryonic tissues (1–10% of placental expression). Human PLAC1 expression is tightly linked to trophoblast differentiation and localizes to the apical region of the cytosolic compartment of the syncytium in proximity to the maternal-facing, microvillus brush border membrane, consistent with function at or near the maternal–fetal interface (27, 28).
Plac1 in Placental Development
Using a mutant mouse model, we have confirmed that Plac1 is essential for normal placental development (29). Plac1 ablation results in placentomegaly and IUGR. At E16.5, knockout (KO) and heterozygous (Het) placentae deriving the Plac1-null allele from the mother (Xm−X) weigh approximately 100% more than WT placentae whereas the corresponding embryos weigh 7–12% less. Histologically, the Plac1 mutants exhibit an expanded junctional zone that migrates into the labyrinth where maternal–fetal transport takes place (Figure 2A). By contrast, Het placentae that derive the null allele from the father (XXp−) exhibit normal growth and are histologically indistinguishable from WT placentae consistent with paternal imprinting. While Plac1 ablation does not result in an obligatory lethal phenotype it does reduce postnatal viability of nullizygous offspring.
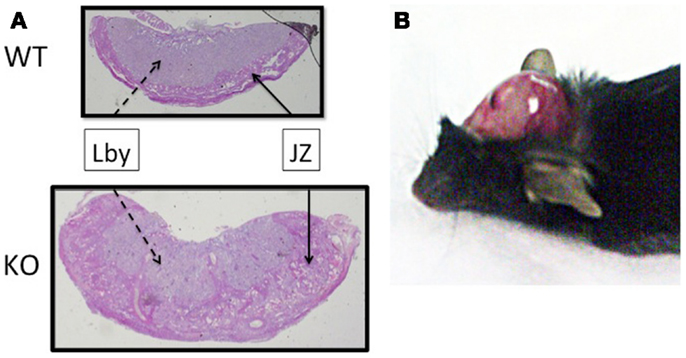
Figure 2. Effect of Plac1 ablation on placental and brain development. (A) H&E stain of E16.5 mouse placentae associated with KO and WT males. JZ, junctional zone; Lby, labyrinth. (B) Postmortem, 6-week-old Xm−X Het female with hydrocephalus. The cranium was retracted to expose the brain tissue.
Observations from human studies have provided evidence that PLAC1 is relevant to the establishment and maintenance of pregnancy by identifying it as a potential biomarker for gestational pathologies. We reported that women can become sensitized to the PLAC1 antigen during pregnancy and the presence of anti-PLAC1 antibodies may be associated with infertility and/or recurrent pregnancy loss (30). This observation was confirmed by Matteo et al. (31) who demonstrated increased anti-PLAC1 antibody titers in women with a history of infertility. Additionally, Farina et al. (32) and Concu et al. (33) demonstrated that PLAC1 mRNA can be detected in maternal serum as early as 8 weeks gestation and throughout pregnancy. Circulating PLAC1 mRNA is markedly diminished in pregnancies associated with vaginal bleeding prior to 20 weeks gestation. Subsequent studies demonstrated elevated levels of circulating PLAC1 mRNA in pre-eclampsia that were directly related to disease severity (34, 35). These findings are consistent with a role for PLAC1 in pregnancy maintenance and possibly as a useful biomarker of gestational pathologies.
Although there is no experimental evidence directly linking PLAC1 to preterm delivery, it does appear to disrupt placental development in a manner likely to interfere with its ability to adapt to the physiological and environmental challenges of pregnancy, thereby predisposing the pregnancy to early failure. There is some evidence linking genes expressed at the maternal–fetal interface with the maintenance of pregnancy. In a recent review, Anum et al. (36) identified mutations in several genes (COL5A1, COL5A2, COL3A1, COL1A1, COL1A2, TNXB, PLOD1, ADAMTS2, CRTAP, LEPRE1, and ZMPSTE24) associated with connective tissue disorders involving multiple organs, i.e., Ehlers–Danlos syndrome, osteogenesis imperfecta, and restrictive dermopathy that were also associated with prematurity, presumably resulting from the premature rupture of membranes.
Plac1 in Brain Development
While characterizing the mutant mouse model we unexpectedly discovered that Plac1 is also expressed throughout the developing embryo where it plays a major role in brain development (37). Plac1 ablation is associated with a 22 and 11% increased risk for lethal hydrocephalus (HC) and other brain abnormalities in KO males (Xm−Y) and Het females inheriting the null allele from the mother (Xm−X), respectively (Figure 2B). By contrast, Het females inheriting the null allele from the father (XXp−) do not develop HC, consistent with paternal imprinting. These observations established Plac1 as a novel, X-linked determinant of brain development.
The X chromosome is enriched for genes involved in brain development and cognitive function and several are clustered around Xq26.3 (38, 39), where human PLAC1 localizes. Families carrying microdeletions or duplications in this region (encompassing PLAC1) report family members with a variety of CNS defects including reduced cognitive function, microcephaly, and neural tube defects (40–43). Our experimental findings indicate PLAC1 is one of the genes at this locus (Xq26.3) that is essential for normal brain development and likely contributes to the clinical abnormalities observed in the reported families.
The development of HC was the first clue that Plac1 was involved in mouse brain development. At least 43 genetic loci linked to hereditary HC have been identified in animal models and humans, irrespective of the presence of other associated defects (44, 45). In fact, X-linked HC is the most common form of hereditary HC accounting for 15–25% of primary idiopathic HC in newborn males (46–49). A variety of CNS malformations and clinical symptoms have been reported in humans with X-linked HC including ataxia, reduced cognitive function, and gait abnormalities. Linkage and pedigree analysis led to the discovery of the neural cell adhesion molecule, L1CAM, as the genetic basis for most of the cases of this disorder (50). Mouse models of L1cam mutations recapitulated the phenotypic variability observed in humans, including the absence of overt HC in some cases, pointing to interactions with other genetic or environmental factors (51–53). Cases of X-linked HC, not associated with mutations in L1CAM, have also been reported. A family with adult onset X-linked HC has also been described (54), characterized by normal pressure hydrocephalus (NPH), seizures, mental disorganization, behavior problems, and urinary incontinence. Although no gene locus has been identified, its transmission is consistent with X chromosome linkage. Many of the genetic determinants of developmental brain disorders have yet to be identified. These studies underscore the importance of the X chromosome, particularly the PLAC1 locus, to brain development and neurological function.
Plac1 Protein Function
While nothing is known regarding the functional properties of Plac1, inferences relevant to tissue morphogenesis can be made based on its sequence. The human ORF encodes a putative protein of 212 amino acids whereas the mouse encodes a 173 amino acid product. Both contain a cleavable signal peptide and are predicted to exist as extracellular peptides. Significant sequence homology (approximately 30%) exists between Plac1 and the zona pellucida 3 (ZP3) protein, a sperm-binding glycoprotein important for fertilization. The zona pellucida domain (ZPD) defines a conserved family of membrane-anchored matrix proteins having important roles in the localized organization of epidermal cell membranes to influence organ development and plasticity (55–61). Therefore, a key feature of Plac1’s function is likely the modulation and integration of signaling pathways linking the cell membrane to the extracellular environment to alter cell shape, motility, and plasticity during morphogenesis.
Summary
Recognition of the paradigm linking IUGR, prematurity, and birth defects to common biological substrates will facilitate efforts to understand their root causes. The identification of key regulatory pathways contributing to this nexus will provide important knowledge necessary to enhance efforts to prevent, diagnose, and intervene in troubled pregnancies and affected offspring. Plac1, an X-linked, imprinted gene with unique relevance to placental and embryonic development, is a novel candidate gene conforming to this paradigm. Defining its developmental role will provide important new insights into embryonic development and pregnancy maintenance.
Author Contributions
Michael E. Fant conceived the work and contributed to its design, data analysis, and interpretation and wrote the initial draft of the manuscript. Juan Fuentes contributed to the acquisition, analysis, and interpretation of data and was involved in critically reviewing the manuscript for intellectual content. Xiaoyuan Kong contributed to the acquisition, analysis, and interpretation of data as well as critically reviewing the manuscript. Suzanne Jackman contributed to the acquisition, analysis, and interpretation of data and critically reviewed and revised the manuscript. All authors approved the final version submitted for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The studies presented in this manuscript were funded in part by grants from the National Institutes of Health, HD 048862 and the March of Dimes, #09-503 (Michael E. Fant).
References
1. Kochanek KD, Kirmeyer SE, Martin JA, Strobino DM, Guyer B. Annual summary of vital statistics: 2009. Pediatrics (2012) 129:338. doi: 10.1542/peds.2011-3435
2. de Onis M, Blössner M, Villar J. Levels and patterns of intrauterine growth retardation in developing countries. Eur J Clin Nutr (1998) 52(Suppl 1):S5–15.
3. Honein MA, Kirby RS, Meyer RE, Xing J, Skerrette NI, Yuskiv N, et al. The association between major birth defects and preterm birth. Matern Child Health J (2009) 13:164–75. doi:10.1007/s10995-008-0348-y
4. Rasmussen SA, Erickson JD, Reef SE, Ross DS. Teratology: from science to birth defects prevention. Birth Defects Res A Clin Mol Teratol (2009) 85:82–92. doi:10.1002/bdra.20506
5. Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou A, et al. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol (2009) 29(10):2762–76. doi:10.1128/MCB.01393-08
6. Barak Y, Sadovsky Y, Shalom-Barak T. PPAR signaling in placental development and function. PPAR Res (2008) 2008:142082. doi:10.1155/2008/142082
7. Hemberger M, Cross JC. Genes governing placental development. Trends Endocrinol Metab (2001) 12(4):162–8. doi:10.1016/S1043-2760(01)00375-7
8. Hynes RO, Hodivala-Dilke KM. Insights and questions arising from studies of a mouse model of Glanzmann thrombasthenia. Thromb Haemost (1999) 82(2):481–5.
9. Wendling O, Chambon P, Mark M. Retinoid X receptors are essential for early mouse development and placentogenesis. Proc Natl Acad Sci U S A (1999) 96(2):547–51. doi:10.1073/pnas.96.2.547
10. Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL, McMillen IC. Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol (2007) 293(1):R306–13. doi:10.1152/ajpregu.00798.2006
11. Sparrow DB, Boyle SC, Sams RC, Mazuruk B, Zhang L, Moeckel G, et al. Placental insufficiency associated with loss of Cited1 causes renal medullary dysplasia. J Am Soc Nephrol (2009) 20(4):777–86. doi:10.1681/ASN.2008050547
12. Heinonen K, Matilainen R, Koski H, Launiala K. Intrauterine growth retardation (IUGR) in pre-term infants. J Perinat Med (1985) 13(4):171–8. doi:10.1515/jpme.1985.13.4.171
13. Zeitlin J, Ancel PY, Saurel-Cubizolles MJ, Papiernik E. The relationship between intrauterine growth restriction and preterm delivery: an empirical approach using data from a European case-control study. BJOG (2000) 107(6):750–8. doi:10.1111/j.1471-0528.2000.tb13336.x
14. Doubilet PM, Benson CB, Wilkins-Haug L, Ringer S. Fetuses subsequently born premature are smaller than gestational age-matched fetuses not born premature. J Ultrasound Med (2003) 22(4):359–63.
15. Gardosi JO. Prematurity and fetal growth restriction. Early Hum Dev (2005) 81(1):43–9. doi:10.1016/j.earlhumdev.2004.10.015
16. Christianson A, Howson CP, Modell B. March of Dimes: Global Report on Birth Defects – The Hidden Toll of Dying and Disabled Children March of Dimes Birth Defects Foundation. White Plains, NY: (2006). Available from: www.marchofdimes.com/glue/files/Birth_Defects_Report-PF.pdf
17. Nosarti C, Reichenberg A, Murray RM, Cnattingius S, Lambe MP, Yin L, et al. Preterm birth and psychiatric disorders in young adult life. Arch Gen Psychiatry (2012) 69(6):E1–8. doi:10.1001/archgenpsychiatry.2011.1374
18. Lindström K, Lindblad F, Hjern A. Preterm birth and attention-deficit/hyperactivity disorder in schoolchildren. Pediatrics (2011) 127(5):858–65. doi:10.1542/peds.2010-1279
19. Johnson S, Hollis C, Kochhar P, Hennessy E, Wolke D, Marlow N. Autism spectrum disorders in extremely preterm children. J Pediatr (2010) 156(4):525–31.e2. doi:10.1016/j.jpeds.2009.10.041
20. Abel KM, Dalman C, Svensson AC, Susser E, Dal H, Idring S, et al. Deviance in fetal growth and risk of autism spectrum disorder. Am J Psychiatry (2013) 170(4):391–8. doi:10.1176/appi.ajp.2012.12040543
21. Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, et al. A transient placental source of serotonin for the fetal forebrain. Nature (2011) 472(7343):347–50. doi:10.1038/nature09972
22. Cocchia M, Huber R, Pantano S, Chen EY, Ma P, Forabosco A, et al. PLAC1, an Xq26 gene with placenta-specific expression. Genomics (2000) 68:305–12. doi:10.1006/geno.2000.6302
23. Kushi A, Edamura K, Noguchi M, Akiyama K, Nishi Y, Sasai H. Generation of mutant mice with large chromosomal deletion by use of irradiated ES cells – analysis of large deletion around HPRT locus of ES cell. Mamm Genome (1998) 9:269–73. doi:10.1007/s003359900747
24. Zechner U, Reule M, Orth A, Bonhomme F, Strack B, Guenet, et al. An X-chromosome linked locus contributes to abnormal placental development in mouse interspecific hybrid. Nat Genet (1996) 12:398–403. doi:10.1038/ng0496-398
25. Hemberger MC, Pearsall RS, Zechner U, Orth A, Otto S, Ruschendorf F, et al. Genetic dissection of X-linked interspecific hybrid placental dysplasia in congenic mouse strains. Genetics (1999) 153:383–90.
26. Fant M, Weisoly DL, Cocchia M, Huber R, Khan S, Lunt T, et al. Plac1, a trophoblast-specific gene, is expressed throughout pregnancy in the human placenta and modulated by keratinocyte growth factor (KGF). Mol Reprod Dev (2002) 4:430–6. doi:10.1002/mrd.10200
27. Massabbal E, Parveen S, Weisoly DL, Nelson DM, Smith SD, Fant M. PLAC1 expression increases during trophoblast differentiation: evidence for regulatory interactions with the fibroblast growth factor-7 (FGF-7) axis. Mol Reprod Dev (2005) 71:299–304. doi:10.1002/mrd.20272
28. Fant M, Barerra-Saldana H, Dubinsky W, Poindexter B, Bick R. The PLAC1 protein localizes to membranous compartments in the apical region of the syncytiotrophoblast. Mol Reprod Dev (2007) 74:922–9. doi:10.1002/mrd.20673
29. Jackman S, Kong X, Fant ME. Plac1 (Placenta-specific 1) is essential for normal placental and embryonic development. Mol Reprod Dev (2012) 79:564–72. doi:10.1002/mrd.22062
30. Kotto-Kome A, Silva C, Whiteman V, Kong X, Fant ME. Circulating anti-PLAC antibodies during pregnancy and in women with reproductive failure: a preliminary analysis. ISRN Immunology (2011) 2011:5.
31. Matteo M, Greco P, Levi Setti PE, Morenghi E, De Rosario F, Massenzio F, et al. Preliminary evidence for high anti-PLAC1 antibody levels in infertile patients with repeated unexplained implantation failure. Placenta (2013) 34(4):335–9. doi:10.1016/j.placenta.2013.01.006
32. Farina A, Rizzo N, Concu M, Banzola I, Sekizawa A, Grotti S, et al. Lower maternal PLAC1 mRNA in pregnancies complicated with vaginal bleeding (threatened abortion <20 weeks) and a surviving fetus. Clin Chem (2005) 51:224–7. doi:10.1373/clinchem.2004.041228
33. Concu M, Banzola I, Farina A, Sekizawa A, Rizzo N, Marini M, et al. Rapid clearance of mRNA for PLAC1 gene in maternal blood after delivery. Fetal Diagn Ther (2005) 20(1):27–30. doi:10.1159/000081365
34. Fujito N, Samura O, Miharu N, Tanigawa M, Hyodo M, Kudo Y. Increased plasma mRNAs of placenta-specific 1 (PLAC1) and glial cells-missing 1 (GCM1) in mothers with pre-eclampsia. Hiroshima J Med Sci (2006) 55:9–15.
35. Purwosunu Y, Sekizawa A, Farina A, Wibowo N, Okazaki S, Nakamura M, et al. Cell-free mRNA concentrations of CRH, PLAC1, and selectin-P are increased in the plasma of pregnant women with preeclampsia. Prenat Diagn (2007) 27(8):772–7. doi:10.1002/pd.1780
36. Anum EA, Hill LD, Pandya A, Strauss JF III. Connective tissue and related disorders and preterm birth: clues to genes contributing to prematurity. Placenta (2009) 30(3):207–15. doi:10.1016/j.placenta.2008.12.007
37. Kong X, Jackman S, Fant ME. Plac1 (Placenta-specific 1) is widely expressed during fetal development and is and associated with a lethal form of hydrocephalus. Birth Defects Res A Clin Mol Teratol (2013) 97:571–7. doi:10.1002/bdra.23171
38. Nguyen DK, Disteche CM. High expression of the mammalian X-chromosome in brain. Brain Res (2006) 1126(1):46–9. doi:10.1016/j.brainres.2006.08.053
39. Wolff DJ, Gustashaw KM, Zurcher V, Ko L, White W, Weiss L, et al. Deletions in Xq26.3-q27.3 including FMR1 result in a severe phenotype in a male and variable phenotypes in females depending upon the X inactivation pattern. Hum Genet (1997) 100:256–61. doi:10.1007/s004390050501
40. Madrigal I, Fernandez-Burriel M, Rodriguez-Revenga L, Cabrera JC, Merti M, Mur A, et al. Xq26.2-q26.3 microduplication in two brothers with intellectual disabilities: clinical and molecular characterization. J Hum Genet (2010) 55(12):822–6. doi:10.1038/jhg.2010.119
41. Hol FA, Schepens MT, van Beersum SE, Redolfi E, Affer M, Vezzoni P, et al. Identification and characterization of an Xq25-q27 duplication in a family with spina bifida and panhypopituitarism suggests the involvement of two distinct genes. Genomics (2000) 69:174–81. doi:10.1006/geno.2000.6327
42. Vasquez AI, Rivera H, Bobadilla L, Crolla JA. A familial Xp+, dup (Xq26.3–>qter). J Med Genet (1995) 32:891–3. doi:10.1136/jmg.32.11.891
43. Akiyama M, Kawame H, Ohashi H, Tohma T, Ohta H, Shishikura A, et al. Functional disomy for Xq26.3-qter in a boy with unbalanced t(X;21)(q26.3;p11.2) translocation. Am J Med Genet (2001) 99:111–4. doi:10.1002/1096-8628(2001)9999:9999<::AID-AJMG1150>3.0.CO;2-C
44. Haverkamp F, Wolfle J, Aretz M, Kramer A, Hohmann B, Fahnenstich H, et al. Congenital hydrocephalus internus and aqueduct stenosis: aetiology and implications for genetic counseling. Eur J Pediatr (1999) 158:474–8. doi:10.1007/s004310051123
45. Zhang J, Williams MA, Rigamonti D. Genetics of human hydrocephalus. J Neurol (2006) 253:1255–66. doi:10.1007/s00415-006-0245-5
46. Burton BK. Recurrence risk for congenital hydrocephalus. Clin Genet (1979) 16:47–53. doi:10.1111/j.1399-0004.1979.tb00848.x
47. Halliday J, Chow CW, Wallace D, Danks DM. X-linked hydrocephalus: a survey of a 20 year period in Victoria, Australia. J Med Genet (1986) 23:23–31. doi:10.1136/jmg.23.1.23
48. Finckh U, Schroder J, Ressler B, Veske A, Gal A. Spectrum and detection rate of L1CAM mutations in isolated and familial cases with clinically suspected L1 disease. Am J Med Genet (2000) 92:40–6. doi:10.1002/(SICI)1096-8628(20000501)92:1<40::AID-AJMG7>3.0.CO;2-R
49. Weller S, Gartner J. Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): mutations in the L1CAM gene. Hum Mutat (2001) 18:1–12. doi:10.1002/humu.1144
50. Jouet M, Rosenthal A, McFarlane J, Kennrick S, Donnai D. A missense mutation confirms the L1 defect in X-linked hydrocephalus (HSAS). Nat Genet (1993) 4:331. doi:10.1038/ng0893-331
51. Cohen NR, Taylor JSH, Scott LB, Guillery LW, Soriano P, Furley AJ. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol (1997) 8:26–33. doi:10.1016/S0960-9822(98)70017-X
52. Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the gene coding for the cell adhesion molecule L1 leads to malformations of the nervous system in mice. Nat Genet (1997) 17:346–9. doi:10.1038/ng1197-346
53. Fransen E, D’Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E, et al. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum Mol Genet (1998) 7(6):999–1009. doi:10.1093/hmg/7.6.999
54. Katsuragi S, Teraoka K, Ikegami K, Amano K, Yamashita K, Ishizuka K, et al. Late onset hydrocephalus with normal cerebrospinal fluid pressure. Psychiatry Clin Neurosci (2000) 54:487–92. doi:10.1046/j.1440-1819.2000.00740.x
55. Jovine L, Qi H, Williams Z, Litscher E, Wassarman PM. The ZP domain is a conserved module for polymerization of extracellular proteins. Nat Cell Biol (2002) 4(6):457–61. doi:10.1038/ncb802
56. Jovine L, Qi H, Williams Z, Litscher ES, Wassarman PM. A duplicated motif controls assembly of zona pellucida domain proteins. Proc Natl Acad Sci U S A (2004) 101:5922–7. doi:10.1073/pnas.0401600101
57. Jovine L, Janssen WG, Litscher ES, Wassarman PM. The PLAC1-homology region of the ZP domain is sufficient for protein polymerisation. BMC Biochem (2006) 7:11. doi:10.1186/1471-2091-7-11
58. Diestel U, Resch M, Meinhardt K, Weiler S, Hellmann TV, Mueller TD, et al. Identification of a novel TGF-β-binding site in the zona pellucida C-terminal (ZP-C) domain of TGF-β-receptor-3 (TGFR-3). PLoS One (2013) 8(6):e67214. doi:10.1371/journal.pone.0067214
59. Bökel C, Prokop A, Brown NH. Papillote and piopio: Drosophila ZP-domain proteins required for cell adhesion to the apical extracellular matrix and microtubule organization. J Cell Sci (2005) 118(Pt 3):633–42. doi:10.1242/jcs.01619
60. Jazwinska A, Ribeiro C, Affolter M. Epithelial tube morphogenesis during Drosophila tracheal development requires piopio, a luminal ZP protein. Nat Cell Biol (2003) 5(10):895–901. doi:10.1038/ncb1049
Keywords: prematurity, Plac1, placenta, birth defects, IUGR, fetal growth
Citation: Fant ME, Fuentes J, Kong X and Jackman S (2014) The nexus of prematurity, birth defects, and intrauterine growth restriction: a role for Plac1-regulated pathways. Front. Pediatr. 2:8. doi: 10.3389/fped.2014.00008
Received: 09 November 2013; Paper pending published: 02 December 2013;
Accepted: 24 January 2014; Published online: 21 February 2014.
Edited by:
Heber C. Nielsen, Tufts School of Medicine, USACopyright: © 2014 Fant, Fuentes, Kong and Jackman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael E. Fant, Department of Pediatrics, Morsani College of Medicine, University of South Florida, 140 7th Avenue South, CRI 2008, Tampa, FL 33701, USA e-mail:bWZhbnRAaGVhbHRoLnVzZi5lZHU=