 Prashant Kumar Verma
Prashant Kumar Verma Ashraf A. El-Harouni
Ashraf A. El-Harouni- 1Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 2Department of Clinical Genetics, National Research Center, Cairo, Egypt
Background: Postaxial polydactyly (PAP) is one of the commonest congenital malformations and usually is associated to several syndromes. There is no primary investigational strategy for PAP cases with single gene disorder in literature. PAP cases with single gene disorder can be classified according to common pathways and molecular basis. Molecular classification may help in diagnostic approach.
Materials and Methods: All single gene disorders associated with PAP reported on PubMed and OMIM are analyzed and classified according to molecular basis.
Results: Majority of genes related to cilia structure and functions are associated with PAP, so we classified them as ciliopathies and non-ciliopathies groups. Genes related to Shh–Gli3 pathway was the commonest group in non-ciliopathies.
Conclusion: Genes related to cilia are most commonly related to PAP due to their indirect relationship to Shh–Gli3 signaling pathway. Initially, PAP may be the only clinical finding with ciliopathies so those cases need follow up. Proper diagnosis is helpful for management and genetic counseling. Molecular approach may help to define pleiotropy.
Introduction
Postaxial polydactyly (PAP) is defined as an extra digit or a part of digit on the ulnar or fibular side of hand or foot. A small projection of tissue or scar mark just below the proximal interphalangeal crease can also be the only clinical finding. Prevalence of PAP is 1–2/1000 live births with some difference in ethnic groups (1, 2). PAP is more common (75%) than preaxial polydactyly (25%). About 8% of cases with bilateral PAP in upper and lower limbs are frequently associated with multiple congenital anomalies. Distribution of PAP is shown in Figure 1 (3). PAP is clinically classified into type A with fully developed extra digit and type B with incompletely developed digit (4). Type B PAP is commonly associated with isolated familial PAP (5).
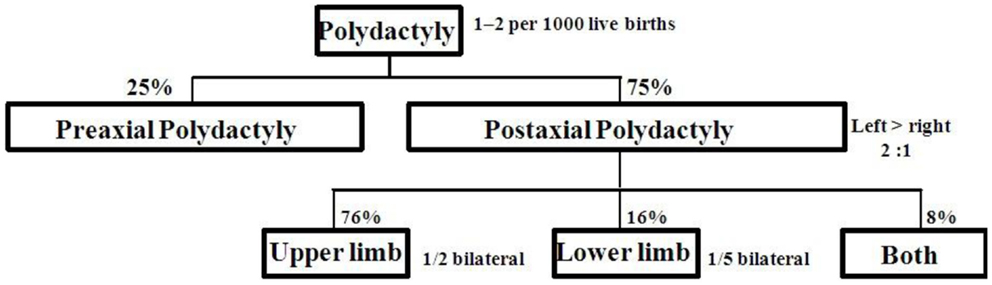
Figure 1. Limb involvement in postaxial polydactyly.
Many congenital malformations associated to PAP are reported in literature (6–12). There are no reported guidelines or protocols to investigate these malformation syndromes.
Limb growth is controlled by a set of genes. The limb buds grow in three directions. The axis of growth is proximal to distal, dorsal to ventral, and anterior to posterior (first to fifth digit). Although a set of genes for limb growth are interacted to each other but the genes more specifically related to anterior to posterior axis shown in Figure 2 (13–27) are strongly related to molecular basis of PAP. Shh–Gli pathway is the well known pathway related to anterior to posterior growth pattern. Single gene disorders associated with PAP may be directly or indirectly related to Shh–Gli pathway. Classification of all reported single gene disorders associated with PAP on the basis of molecular association may help in making a common approach for investigation and genetic counseling of PAP.
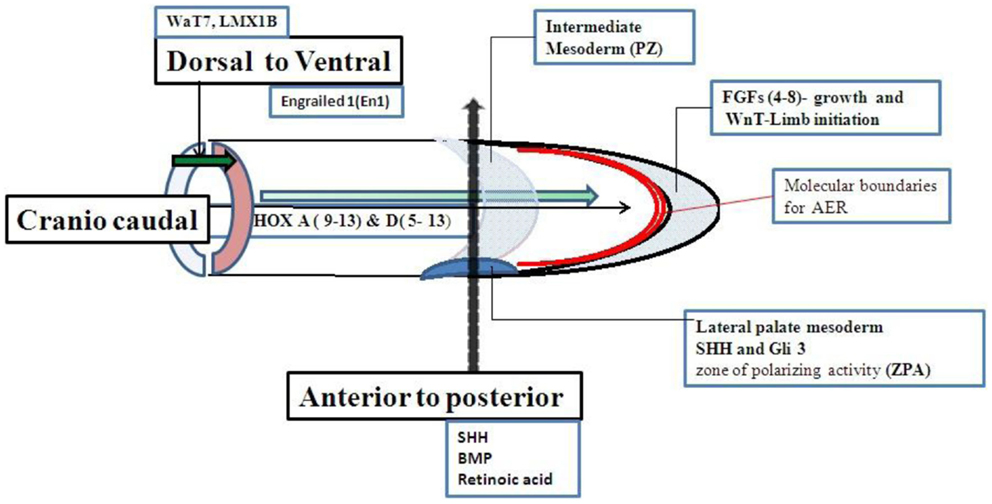
Figure 2. Genes related to limb growth.
Materials and Methods
By using Mesh term “postaxial AND polydactyly” in searching PubMed and OMIM, we got total 667 entries. We included genetically well known syndromes with PAP and classified on the basis of common pathways and molecular association. We did not include single case reports and cases without molecular study. We also did not describe all phenotypic series of particular syndrome if genes are functionally related as Meckel, Bardet–Biedl syndrome (BBS), etc. As for clinical and molecular references for syndromes with PAP, we used NCBI resources like OMIM number, Gene ID, and relevant references related to gene function. We tried to define interactions between these genes for understanding the molecular mechanism how to PAP as related phenotype with particular gene.
Results
Total of 36 genetically well known syndromes and entries were identified in which 16 (44%) related to ciliopathies group (Table 1) and 20 (56%) were unrelated groups and we classified them as non-ciliopathies group (Table 2). Most of the genes related to formation and development of embryo. Shh–Gli3 signaling pathway was the commonest pathway involved with PAP. PAP is more frequently associated with ciliopathy.
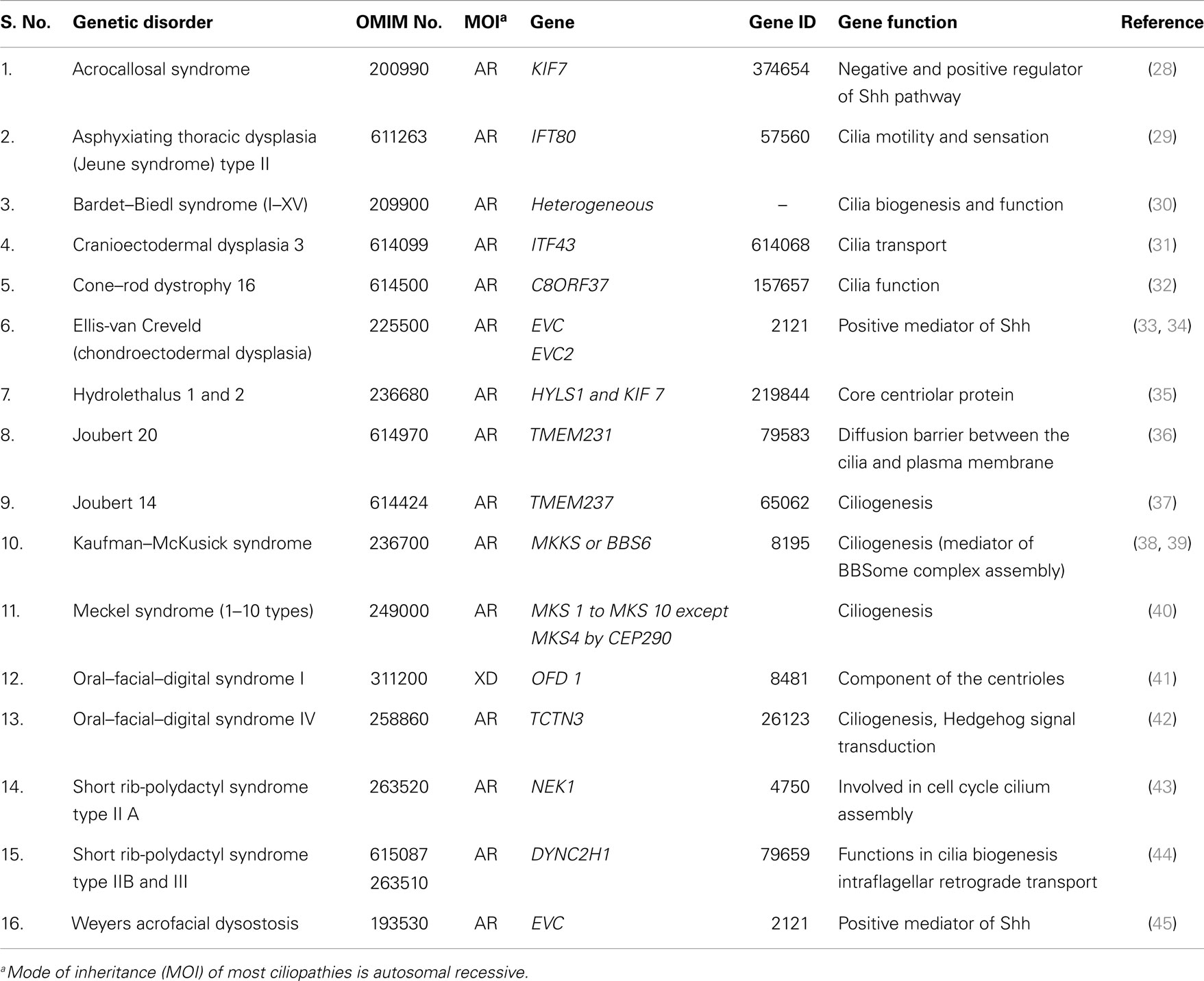
Table 1. Ciliopathies (genes related to cilia biogenesis, structure, and functions).
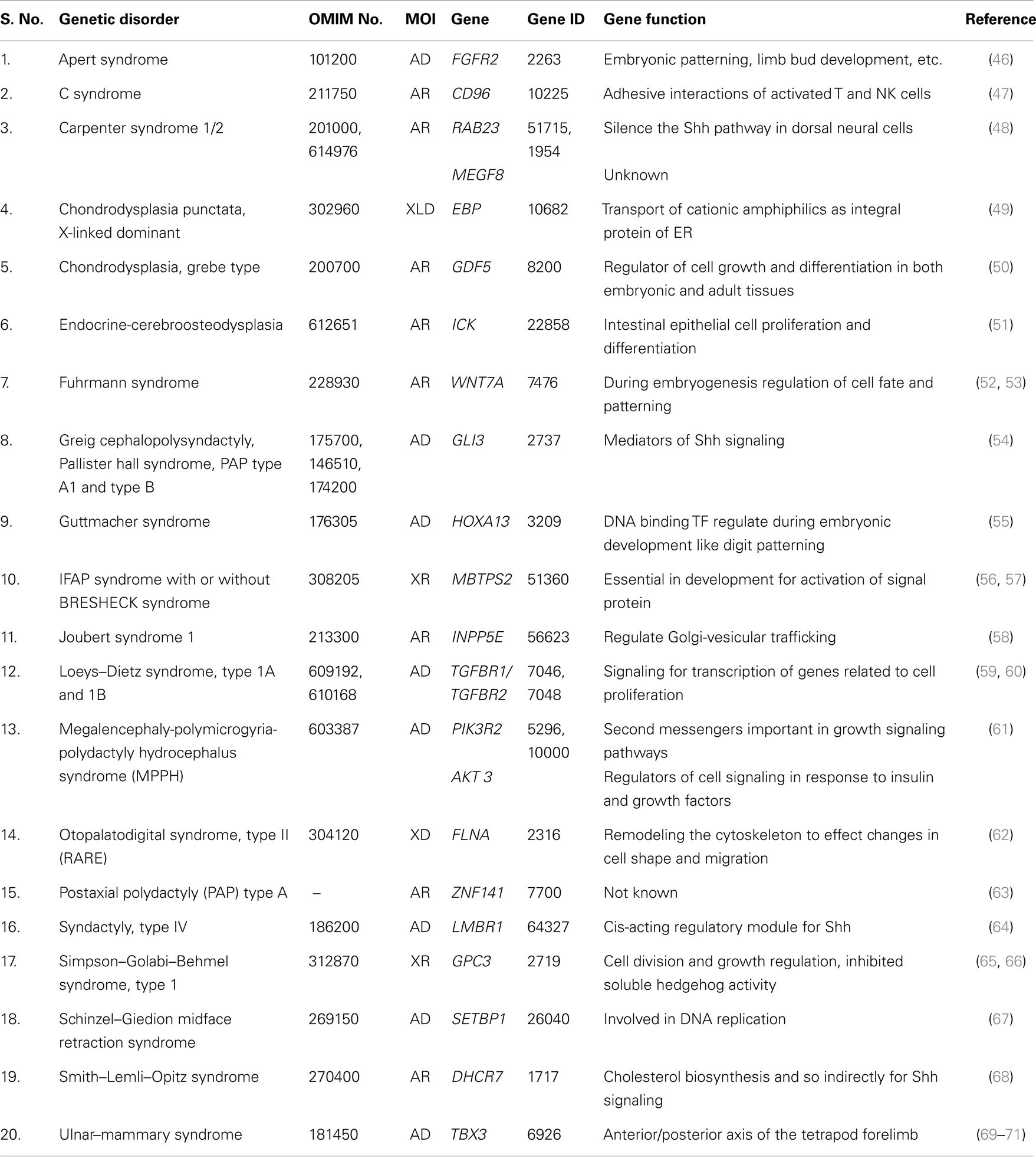
Table 2. Non-ciliopathies (genes not related to cilia biogenesis, structure, and functions).
Discussion
Postaxial polydactyly is one of the most common congenital malformations and a key feature for dysmorphic syndromes. Genetic syndromes related to cilia dysfunction are called ciliopathies, and the majority of this group is associated with PAP. Most of ciliopathies related genes work together as common unit and any defect in one component leads to dysfunction of overall cilia function, either directly or indirectly. This is the cause for overlapping clinical phenotypes of different ciliopathies. We were not discussing the complex genetics of human ciliopathies but focusing more on the molecular mechanism for PAP association with ciliopathies.
Genes associated with anterior to posterior patterning may be responsible for molecular etiology of PAP (Figure 2). Cilia should be involved with the genes associated with anterior to posterior patterning of the limb. The Shh–Gli3-activated Ptch transcription pathway is the most important pathway related to control anterior to posterior patterning and associated with PAP. Shh, Ptch 1, Smo, and Gli3 are the main genes in Shh–Gli3 pathway. Bone morphogenic protein (BMP) and retinoic acid are also needed for anterior to posterior patterning but their association with PAP is not reported in the literature.
Sonic hedgehog mutations are usually not reported with PAP in humans because most of these mutations are heterozygous. Haploinsufficiency of Shh gene does not affect the long range process of patterning (72).
Ptch 1 and Smo are the other intermediate genes in this pathway and both of them have an inhibitory function in Shh–Gli3 pathway. Mutations in these genes were not reported with PAP. Homozygous mutations in Ptch 1 and Smo are lethal during embryonic development and haploinsufficiency do not affect long range process of patterning (73, 74).
Gli3 gene is the most important gene in this pathway and mutations in this gene are reported with PAP. Gli3R is a repressor form without Shh signaling. Smo activated Gli3R to an activated Gli3A form after Shh–Ptch interaction due to loss of inhibitory effect of Ptch on Smo (Figure 3). Gli3 works as a dual function transcription factor. These two forms of Gli3R and Gli3A and their proportion of Gli3R/Gli3A forms directly are related to digit types and number (75, 76). Complete regulatory mechanism of the Gli3R/Gli3A ratio is still unclear. There is no exact genotype and phenotype correlation with Gli3 mutations due to complex interaction to other genes and bifunctional transcriptional switch (77, 78).
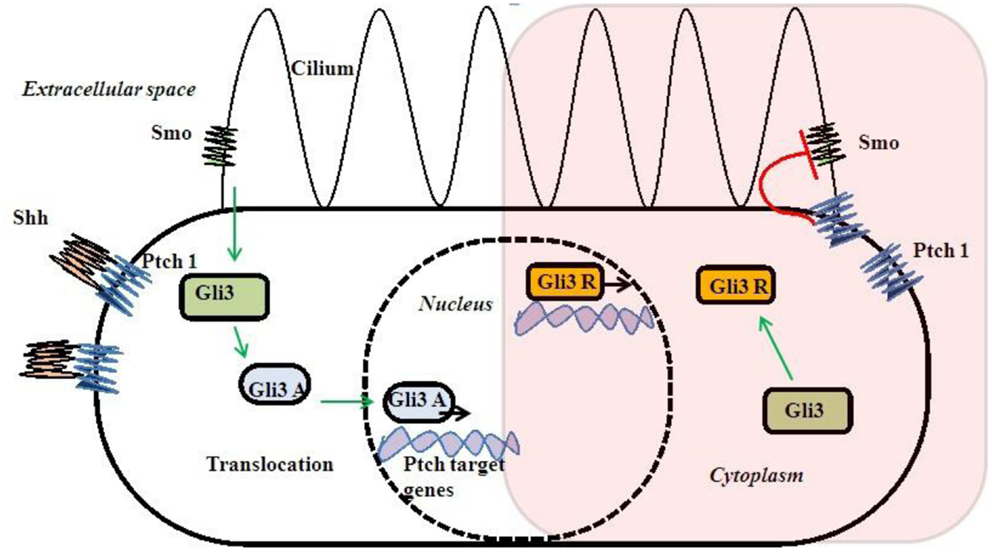
Figure 3. Shh–Gli3 signaling and Ptch–Smo interaction and dual transcription.
Sonic hedgehog pathway needs cilia for signaling (79, 80) (Figure 3). So, Shh–Gli3 signaling pathway is affected with most of the ciliopathies with PAP association and that may be due to altered Gli3R/Gli3A ratio. Cell lacking cilia or alteration of intraflagellar transport (IFT) cause changes in Gli3 processing and unable them to proceed Shh signaling (81, 82). Those single gene disorders associated with PAP directly or indirectly alter Gli3 signaling causing them to have some rational pleiotropy for PAP association.
Besides PAP, there were few cases reported overlapping in their clinical features with Gli3 and ciliopathies (83). For example, acrocallosal syndrome has some overlapping features with GCPS (Greig cephalopolysyndactyly syndrome). This may be due to KIF7 gene, which has negative or positive regulator mechanism in Shh pathway and needs molecular testing to confirm the diagnosis (84). PAP may be the only external malformation appreciated at birth, while other features may develop later in many ciliopathies. So, we made an investigation approach chart (Figure 4) during the first visit of any patient with PAP to the genetic clinic. Any patient with non-familial symmetrical PAP, even without congenital malformations, should be thoroughly investigated to rule out associated complications of ciliopathies (85) (Table 3). The recurrence risk for all ciliopathies is 25% per each pregnancy except OFD 1, which inherited as an X liked dominant trait.
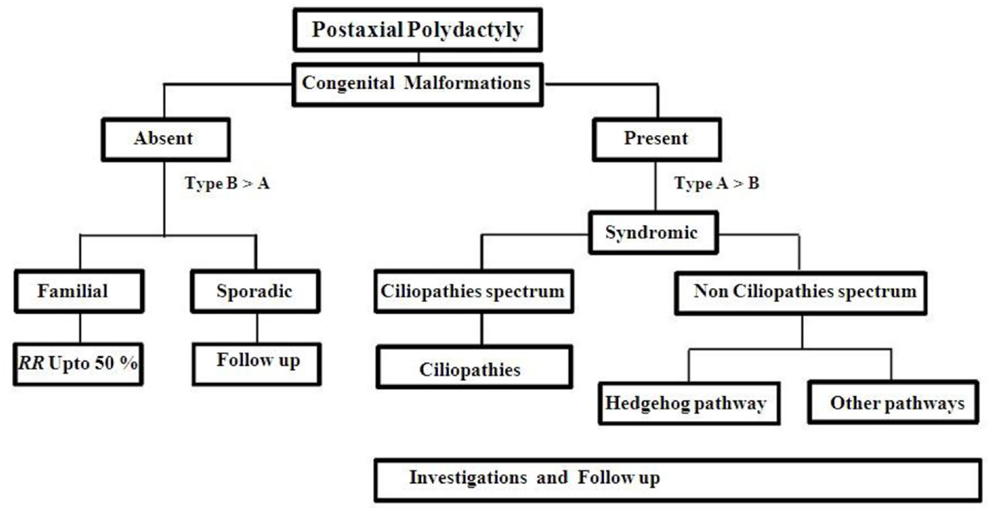
Figure 4. Approach to a PAP case.

Table 3. Primary investigation protocol for a case of post axial polydactyly with malformations.
Single gene disorders other than ciliopathies associated with PAP were classified as non-ciliopathies. In this group, functionally related genes to Shh–Gli3 pathways are Gli3, LMBR1, and DHCR7. GDF5 and TGFBR1, 2 genes are belonging to TGF-β signaling pathways. While WNT7A and FGFR2 genes are belonging to Wnt and FGF signaling pathways. Although WNT7A and FGFR2 genes interact with Shh pathway during limb development, but the exact molecular mechanism for PAP is still unclear. Most of other genes in this group belongs to gene families, which were not yet included in a specific pathway.
We also tried to find out the type of pleiotropy for PAP association. Pleiotropy is defined as multiple distinct phenotypic variants caused by a single gene. Most of these genes with PAP association are related to embryonic patterning and development. Rational and mosaic are the two most common types of pleiotropy. Genes, which have a molecular mechanism for explaining particular trait, are called rational pleiotropy whereas those not having it are called mosaic pleiotropy (86). In our study, we found out rational pleiotropy for PAP association only with syndromes is related to Shh–Gli3 pathway. Other syndromes may be having mosaic pleiotropy for PAP association.
Recurrence risk for familial autosomal dominant (AD) PAP is up to 50% per each pregnancy with variable expressivity. Non-familial case should be kept in follow up (Figure 4). Cytogenetic studies should be done for multiple congenital anomalies associated with PAP without specific dysmorphology. Chromosomes abnormalities in 2, 3, 4, 7, 13, 14, and 18 were reported with PAP (87–97). Single gene testing is not acceptable to most of ciliopathies disorders because of genetic heterogeneity, oligogenic inheritance, and age dependent penetrance. So, initially most of the cases are classified upon the clinical basis but further more investigations are necessary for proper diagnosis and genetic counseling.
Conclusion
Genes related to anterior to posterior patterning are responsible for PAP. Dysregulation or mutations of the Gli3 gene was associated with PAP. Genes related to cilia are most commonly related to PAP due to their indirect relationship to Shh–Gli3 signaling pathway. Initially, PAP may be the only clinical findings with ciliopathies so these cases need continuous follow up.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AD, autosomal dominant; AR, autosomal recessive; BMP, bone morphogenetic protein; DHCR7, 7-dehydrocholesterol reductase; FGFR2, fibroblast growth factor receptor 2; GDF5, growth/differentiation factor 5; GLI3, GLI-Kruppel family member 3; GLIR and GLI3A, GLI-Kruppel family member 3 activators and GLI-Kruppel family member 3 repressors; LMBR1, limb region 1, mouse, homolog of; MOI, mode of inheritance; OFD 1, orofaciodigital syndrome I; PAP, post axial polydactyly; PTCH 1, patched, Drosophila, homolog of, 1; Shh, sonic hedgehog; SMO, smoothened, Drosophila, homolog of; TGFBR1, 2, transforming growth factor, beta type 1,2 receptor; WNT7A, wingless-type MMTV integration site family, member 7A; XD, X-linked dominant; XR, X-linked recessive.
References
1. Zhou GX, Dai L, Zhu J, Miao L, Wang YP, Liang J, et al. [Epidemiological analysis of polydactylies in Chinese perinatals]. Sichuan Da Xue Xue Bao Yi Xue Ban (2004) 35(5):708–10.
2. Boeing M, Paiva Lde C, Garcias Gde L, Roth Mda G, Santos IS. [Epidemiology of polydactylies: a case-control study in the population of Pelotas-RS]. J Pediatr (Rio J) (2001) 77(2):148–52. doi:10.2223/JPED.196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Castilla EE, Lugarinho da Fonseca R, da Graca Dutra M, Bermejo E, Cuevas L, Martinez-Frias ML. Epidemiological analysis of rare polydactylies. Am J Med Genet (1996) 65(4):295–303. doi:10.1002/(SICI)1096-8628(19961111)65:4<295::AID-AJMG10>3.0.CO;2-P
4. Temtamy SA, McKusick VA. The genetics of hand malformations. Birth Defects Orig Artic Ser (1978) 14(3):1–619.
5. Watson BT, Hennrikus WL. Postaxial type-B polydactyly. Prevalence and treatment. J Bone Joint Surg Am (1997) 79(1):65–8.
6. Planas S, Peiro R, Rubio R, Villanueva R, Seres A, Carreras R. A new report of mesomelic camptomelia, polydactyly and Dandy-Walker complex in siblings. Prenat Diagn (2003) 23(5):372–4. doi:10.1002/pd.601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Roman Corona-Rivera J, Corona-Rivera E, Fragoso-Herrera R, Nuno-Arana I, Loera-Castaneda V. Probable new syndrome in a Mexican family with congenital palmar polyonychia and postaxial limb defects. Am J Med Genet A (2004) 125A(2):205–9. doi:10.1002/ajmg.a.20443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Schaefer GB, Olney AH. Hypothalamic dysfunction with polydactyly and hypoplastic nails. Semin Pediatr Neurol (1999) 6(3):238–42. doi:10.1016/S1071-9091(99)80021-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Sonoda T. [Scalp defects-postaxial polydactyly]. Ryoikibetsu Shokogun Shirizu (2001) (34 Pt 2):604–5.
11. Sugiura Y, Lenz W. New type of synpolydactyly of hands and feet in two unrelated males. Am J Med Genet (1999) 83(5):353–5. doi:10.1002/(SICI)1096-8628(19990423)83:5<353::AID-AJMG1>3.0.CO;2-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Upton J. Heterotopic finger transfer in ulnar ray deficiency associated with contralateral postaxial polydactyly: a case report. J Reconstr Microsurg (1993) 9(2):109–10.
13. Laufer E, Nelson CE, Johnson RL, Morgan BA, Tabin C. Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell (1994) 79(6):993–1003. doi:10.1016/0092-8674(94)90030-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Niswander L, Jeffrey S, Martin GR, Tickle C. A positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature (1994) 371(6498):609–12. doi:10.1038/371609a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Yang Y, Niswander L. Interaction between the signaling molecules WNT7a and SHH during vertebrate limb development: dorsal signals regulate anteroposterior patterning. Cell (1995) 80(6):939–47. doi:10.1016/0092-8674(95)90297-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Zuniga A, Haramis AP, McMahon AP, Zeller R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature (1999) 401(6753):598–602. doi:10.1038/44157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Martin GR. The roles of FGFs in the early development of vertebrate limbs. Genes Dev (1998) 12(11):1571–86. doi:10.1101/gad.12.11.1571
18. Summerbell D, Lewis JH, Wolpert L. Positional information in chick limb morphogenesis. Nature (1973) 244(5417):492–6. doi:10.1038/244228a0
19. Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell (1993) 75(7):1401–16. doi:10.1016/0092-8674(93)90626-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Chang DT, Lopez A, von Kessler DP, Chiang C, Simandl BK, Zhao R, et al. Products, genetic linkage and limb patterning activity of a murine hedgehog gene. Development (1994) 120(11):3339–53.
21. Lopez-Martinez A, Chang DT, Chiang C, Porter JA, Ros MA, Simandl BK, et al. Limb-patterning activity and restricted posterior localization of the amino-terminal product of Sonic hedgehog cleavage. Curr Biol (1995) 5(7):791–6. doi:10.1016/S0960-9822(95)00156-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Parr BA, McMahon AP. Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature (1995) 374(6520):350–3. doi:10.1038/374350a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Riddle RD, Ensini M, Nelson C, Tsuchida T, Jessell TM, Tabin C. Induction of the LIM homeobox gene Lmx1 by WNT7a establishes dorsoventral pattern in the vertebrate limb. Cell (1995) 83(4):631–40. doi:10.1016/0092-8674(95)90103-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Vogel A, Rodriguez C, Warnken W, Izpisua Belmonte JC. Dorsal cell fate specified by chick Lmx1 during vertebrate limb development. Nature (1995) 378(6558):716–20. doi:10.1038/378716a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Logan C, Hornbruch A, Campbell I, Lumsden A. The role of engrailed in establishing the dorsoventral axis of the chick limb. Development (1997) 124(12):2317–24.
26. Loomis CA, Harris E, Michaud J, Wurst W, Hanks M, Joyner AL. The mouse engrailed-1 gene and ventral limb patterning. Nature (1996) 382(6589):360–3. doi:10.1038/382360a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Loomis CA, Kimmel RA, Tong CX, Michaud J, Joyner AL. Analysis of the genetic pathway leading to formation of ectopic apical ectodermal ridges in mouse engrailed-1 mutant limbs. Development (1998) 125(6):1137–48.
28. Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet (2011) 43(6):601–6. doi:10.1038/ng.826
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in jeune asphyxiating thoracic dystrophy. Nat Genet (2007) 39(6):727–9. doi:10.1038/ng2038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest (2009) 119(3):428–37. doi:10.1172/JCI37041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet (2011) 48(6):390–5. doi:10.1136/jmg.2011.088864
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Estrada-Cuzcano A, Neveling K, Kohl S, Banin E, Rotenstreich Y, Sharon D, et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet (2012) 90(1):102–9. doi:10.1016/j.ajhg.2011.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet (2000) 24(3):283–6. doi:10.1038/73508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet (2003) 72(3):728–32. doi:10.1086/368063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Dammermann A, Pemble H, Mitchell BJ, McLeod I, Yates JR III, Kintner C, et al. The hydrolethalus syndrome protein HYLS-1 links core centriole structure to cilia formation. Genes Dev (2009) 23(17):2046–59. doi:10.1101/gad.1810409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol (2012) 14(1):61–72. doi:10.1038/ncb2410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet (2011) 89(6):713–30. doi:10.1016/j.ajhg.2011.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Katsanis N, Beales PL, Woods MO, Lewis RA, Green JS, Parfrey PS, et al. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat Genet (2000) 26(1):67–70. doi:10.1038/79201
39. Slavotinek AM, Stone EM, Mykytyn K, Heckenlively JR, Green JS, Heon E, et al. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet (2000) 26(1):15–6. doi:10.1038/79116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol (2012) 22(4):201–10. doi:10.1016/j.tcb.2012.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Emes RD, Ponting CP. A new sequence motif linking lissencephaly, Treacher Collins and oral-facial-digital type 1 syndromes, microtubule dynamics and cell migration. Hum Mol Genet (2001) 10(24):2813–20. doi:10.1093/hmg/10.24.2813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Reiter JF, Skarnes WC. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev (2006) 20(1):22–7. doi:10.1101/gad.1363606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet (2011) 88(1):106–14. doi:10.1016/j.ajhg.2010.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Vaughan KT, Mikami A, Paschal BM, Holzbaur EL, Hughes SM, Echeverri CJ, et al. Multiple mouse chromosomal loci for dynein-based motility. Genomics (1996) 36(1):29–38. doi:10.1006/geno.1996.0422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Howard TD, Guttmacher AE, McKinnon W, Sharma M, McKusick VA, Jabs EW. Autosomal dominant postaxial polydactyly, nail dystrophy, and dental abnormalities map to chromosome 4p16, in the region containing the Ellis-van Creveld syndrome locus. Am J Hum Genet (1997) 61(6):1405–12. doi:10.1086/301643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet (1995) 9(2):165–72. doi:10.1038/ng0295-165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kaname T, Yanagi K, Chinen Y, Makita Y, Okamoto N, Maehara H, et al. Mutations in CD96, a member of the immunoglobulin superfamily, cause a form of the C (Opitz trigonocephaly) syndrome. Am J Hum Genet (2007) 81(4):835–41. doi:10.1086/522014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Eggenschwiler JT, Espinoza E, Anderson KV. Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature (2001) 412(6843):194–8. doi:10.1038/35084089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Labit-Le Bouteiller C, Jamme MF, David M, Silve S, Lanau C, Dhers C, et al. Antiproliferative effects of SR31747A in animal cell lines are mediated by inhibition of cholesterol biosynthesis at the sterol isomerase step. Eur J Biochem (1998) 256(2):342–9. doi:10.1046/j.1432-1327.1998.2560342.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Settle SH Jr, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol (2003) 254(1):116–30. doi:10.1016/S0012-1606(02)00022-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Togawa K, Yan YX, Inomoto T, Slaugenhaupt S, Rustgi AK. Intestinal cell kinase (ICK) localizes to the crypt region and requires a dual phosphorylation site found in map kinases. J Cell Physiol (2000) 183(1):129–39. doi:10.1002/(SICI)1097-4652(200004)183:1<129::AID-JCP15>3.0.CO;2-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Johnson RL, Tabin CJ. Molecular models for vertebrate limb development. Cell (1997) 90(6):979–90. doi:10.1016/S0092-8674(00)80364-5
53. Woods CG, Stricker S, Seemann P, Stern R, Cox J, Sherridan E, et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet (2006) 79(2):402–8. doi:10.1086/506332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell (2000) 100(4):423–34. doi:10.1016/S0092-8674(00)80678-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Sheth R, Marcon L, Bastida MF, Junco M, Quintana L, Dahn R, et al. Hox genes regulate digit patterning by controlling the wavelength of a Turing-type mechanism. Science (2012) 338(6113):1476–80. doi:10.1126/science.1226804
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Zelenski NG, Rawson RB, Brown MS, Goldstein JL. Membrane topology of S2P, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J Biol Chem (1999) 274(31):21973–80. doi:10.1074/jbc.274.31.21973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell (2000) 6(6):1355–64. doi:10.1016/S1097-2765(00)00133-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Kisseleva MV, Wilson MP, Majerus PW. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J Biol Chem (2000) 275(26):20110–6. doi:10.1074/jbc.M910119199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Lin HY, Wang XF, Ng-Eaton E, Weinberg RA, Lodish HF. Expression cloning of the TGF-beta type II receptor, a functional transmembrane serine/threonine kinase. Cell (1992) 68(4):775–85. doi:10.1016/0092-8674(92)90152-3
60. Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science (2005) 307(5715):1621–5. doi:10.1126/science.1105776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Riviere JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet (2012) 44(8):934–40. doi:10.1038/ng.2331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Ehrlicher AJ, Nakamura F, Hartwig JH, Weitz DA, Stossel TP. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature (2011) 478(7368):260–3. doi:10.1038/nature10430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kalsoom UE, Klopocki E, Wasif N, Tariq M, Khan S, Hecht J, et al. Whole exome sequencing identified a novel zinc-finger gene ZNF141 associated with autosomal recessive postaxial polydactyly type A. J Med Genet (2013) 50(1):47–53. doi:10.1136/jmedgenet-2012-101219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet (2003) 12(14):1725–35. doi:10.1093/hmg/ddg180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acad Sci USA (2006) 103(14):5478–83. doi:10.1073/pnas.0508035103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Capurro MI, Xu P, Shi W, Li F, Jia A, Filmus J. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev Cell (2008) 14(5):700–11. doi:10.1016/j.devcel.2008.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Minakuchi M, Kakazu N, Gorrin-Rivas MJ, Abe T, Copeland TD, Ueda K, et al. Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia-associated protein SET. Eur J Biochem (2001) 268(5):1340–51. doi:10.1046/j.1432-1327.2001.02000.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science (1996) 274(5285):255–9. doi:10.1126/science.274.5285.255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Bamshad M, Le T, Watkins WS, Dixon ME, Kramer BE, Roeder AD, et al. The spectrum of mutations in TBX3: genotype/phenotype relationship in ulnar-mammary syndrome. Am J Hum Genet (1999) 64(6):1550–62. doi:10.1086/302417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Han J, Yuan P, Yang H, Zhang J, Soh BS, Li P, et al. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature (2010) 463(7284):1096–100. doi:10.1038/nature08735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Tumpel S, Sanz-Ezquerro JJ, Isaac A, Eblaghie MC, Dobson J, Tickle C. Regulation of Tbx3 expression by anteroposterior signalling in vertebrate limb development. Dev Biol (2002) 250(2):251–62. doi:10.1006/dbio.2002.0762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet (1996) 14(3):357–60. doi:10.1038/ng1196-357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of smoothened. Nature (2002) 418(6900):892–7. doi:10.1038/nature00989
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell (2001) 105(6):781–92. doi:10.1016/S0092-8674(01)00385-3
75. Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature (2002) 418(6901):979–83. doi:10.1038/nature01033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol (2011) 27:513–37. doi:10.1146/annurev-cellbio-092910-154048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Biesecker LG. What you can learn from one gene: GLI3. J Med Genet (2006) 43(6):465–9. doi:10.1136/jmg.2004.029181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Radhakrishna U, Bornholdt D, Scott HS, Patel UC, Rossier C, Engel H, et al. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; no phenotype prediction from the position of GLI3 mutations. Am J Hum Genet (1999) 65(3):645–55. doi:10.1086/302557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Liem KF Jr, Ashe A, He M, Satir P, Moran J, Beier D, et al. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol (2012) 197(6):789–800. doi:10.1083/jcb.201110049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate smoothened functions at the primary cilium. Nature (2005) 437(7061):1018–21. doi:10.1038/nature04117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet (2005) 1(4):e53. doi:10.1371/journal.pgen.0010053.eor
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Ocbina PJ, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn (2008) 237(8):2030–8. doi:10.1002/dvdy.21551
83. Kos S, Roth K, Korinth D, Zeilinger G, Eich G. Hydrometrocolpos, postaxial polydactyly, and hypothalamic hamartoma in a patient with confirmed Pallister-Hall syndrome: a clinical overlap with McKusick-Kaufman syndrome. Pediatr Radiol (2008) 38(8):902–6. doi:10.1007/s00247-008-0870-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Biesecker LG. The Greig cephalopolysyndactyly syndrome. Orphanet J Rare Dis (2008) 3:10. doi:10.1186/1750-1172-3-10
85. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol (2011) 26(7):1039–56. doi:10.1007/s00467-010-1731-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Stearns FW. One hundred years of pleiotropy: a retrospective. Genetics (2010) 186(3):767–73. doi:10.1534/genetics.110.122549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Haddad V, Aboura A, Tosca L, Guediche N, Mas AE, L’Hermine AC, et al. Tetrasomy 13q31.1qter due to an inverted duplicated neocentric marker chromosome in a fetus with multiple malformations. Am J Med Genet A (2012) 158A(4):894–900. doi:10.1002/ajmg.a.35258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Hahm GK, Barth RF, Schauer GM, Reiss R, Opitz JM. Trisomy 2p syndrome: a fetus with anencephaly and postaxial polydactyly. Am J Med Genet (1999) 87(1):45–8. doi:10.1002/(SICI)1096-8628(19991105)87:1<45::AID-AJMG9>3.0.CO;2-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Hayashi S, Okamoto N, Makita Y, Hata A, Imoto I, Inazawa J. Heterozygous deletion at 14q22.1-q22.3 including the BMP4 gene in a patient with psychomotor retardation, congenital corneal opacity and feet polysyndactyly. Am J Med Genet A (2008) 146A(22):2905–10. doi:10.1002/ajmg.a.32519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Higginbottom MC, Mascarello JT, Hassin H, McCord WK. A second patient with partial deletion of the short arm of chromosome 3: karyotype 46,XY,del(3)(p25). J Med Genet (1982) 19(1):71–3. doi:10.1136/jmg.19.1.71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Lurie IW, Schwartz MF, Schwartz S, Cohen MM. Trisomy 7p resulting from isochromosome formation and whole-arm translocation. Am J Med Genet (1995) 55(1):62–6. doi:10.1002/ajmg.1320550117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Pfeiffer RA, Santelmann R. Limb anomalies in chromosomal aberrations. Birth Defects Orig Artic Ser (1977) 13(1):319–37.
93. Ounap K, Ilus T, Bartsch O. A girl with inverted triplication of chromosome 3q25.3 – > q29 and multiple congenital anomalies consistent with 3q duplication syndrome. Am J Med Genet A (2005) 134(4):434–8. doi:10.1002/ajmg.a.30134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Paskulin GA, Riegel M, Machado Rosa RF, Graziadio C, Gazzola Zen PR. Interstitial deletion of 7q31.32 – > q33 secondary to a paracentric inversion of a maternal chromosome 7. Eur J Med Genet (2011) 54(2):181–5. doi:10.1016/j.ejmg.2010.10.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Makrythanasis P, Gimelli S, Bena F, Dahoun S, Morris MA, Antonarakis SE, et al. Homozygous deletion of a gene-free region of 4p15 in a child with multiple anomalies: could biallelic loss of conserved, non-coding elements lead to a phenotype? Eur J Med Genet (2012) 55(1):63–6. doi:10.1016/j.ejmg.2011.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Lumaka A, Van Hole C, Casteels I, Ortibus E, De Wolf V, Vermeesch JR, et al. Variability in expression of a familial 2.79 Mb microdeletion in chromosome 14q22.1-22.2. Am J Med Genet A (2012) 158A(6):1381–7. doi:10.1002/ajmg.a.35353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Kessel E, Pfeiffer RA, Baisch C. [Postaxial polydactyly: a symptom of partial trisomy of the long arm of chromosome 13. Two new observations with 46, XX, t (22;13) (q13;q31) and 46, XY, Dup (13) (pter-q34:q22-qter) (author’s transl)]. Klin Padiatr (1980) 192(1):85–90. doi:10.1055/s-2008-1033863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: postaxial polydactyly, molecular approach, hedgehog protein, sonic hedgehog, signal pathways, gene, cilia, investigation, approach
Citation: Verma PK and El-Harouni AA (2015) Review of literature: genes related to postaxial polydactyly. Front. Pediatr. 3:8. doi: 10.3389/fped.2015.00008
Received: 20 October 2014; Accepted: 26 January 2015;
Published online: 11 February 2015.
Edited by:
Meow-Keong Thong, University of Malaya, MalaysiaReviewed by:
Corrado Romano, IRCCS Associazione Oasi Maria Santissima, ItalyFeodora Stipoljev, Clinical Hospital “Sveti Duh”, Croatia
Thomy J. L. De Ravel, University Hospitals Leuven, Belgium
Copyright: © 2015 Verma and El-Harouni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashraf A. El-Harouni, Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia e-mail:YWhhcm91bmkxMkBnbWFpbC5jb20=