 Stavros Giaglis
Stavros Giaglis Sinuhe Hahn
Sinuhe Hahn Paul Hasler
Paul Hasler- 1Laboratory of Prenantal Medicine, Department of Biomedicine, University Clinics Basel, Basel, Switzerland
- 2Department of Rheumatology, Kantonsspital Aarau, Aarau, Switzerland
- 3University Women’s Hospital, University Clinics Basel, Basel, Switzerland
Neutrophil extracellular trap (NET) formation represents a form of cell death distinct from apoptosis or necrosis, by which invading pathogens are simultaneously entangled and potentially eliminated. Increased NET formation is observed in systemic lupus erythematosus (SLE), rheumatoid arthritis, antineutrophil cytoplasmic antibody-associated small vessel vasculitis, antiphospholipid antibody syndrome (APS), and psoriasis. NETs contribute to the pathogenesis of autoimmunity by exposing cryptic autoepitopes, which may facilitate the generation of autoantibodies, induce the production of interferons, and activate the complement cascade. In SLE, augmented disease activity and renal disease are associated with increased NET formation, so that NETs could serve as a marker for the monitoring of disease activity. NETs can additionally cause endothelial cell damage and death and stimulate inflammation in atheromatous plaques, adding to the accelerated atherosclerosis witnessed in autoimmune disease. Since NETs induce production of interferons, assessing the extent of NET formation might facilitate the prediction of IFN-alpha levels and identification of SLE patients with presumably better responses to anti-IFN-alpha therapies or other novel therapeutic concepts, such as N-acetyl-cysteine and inhibitors of DNase 1 and peptidylarginine deiminase 4 (PAD4), which also target NETs. In summary, the study of NETs provides a novel approach to the understanding of autoimmune disease pathogenesis in childhood and opens new vistas in the development of sensitive disease markers and targeted therapies.
Introduction
Neutrophils play a key role in innate immunity with multiple strategies for defending the host against pathogens. Neutrophils initially migrate to the site of infection and exert their critical roles through phagocytosis, degranulation and secretion of antimicrobial factors, production of reactive oxygen species (ROS), and release of neutrophil extracellular traps (NETs) (1–3). Although the primary function of neutrophils is to protect the body from hazardous microbes, the influx of neutrophils to an inflamed site can contribute directly to tissue damage and the progression to chronic inflammation. Neutrophils mediate a pro-inflammatory response to tissue injury or infection by releasing cytokines that recruit and stimulate other immune cells and promote the activation of adaptive immunity. Neutrophil granulocytes have been traditionally viewed as short-lived cells that die at sites of inflammation; increasing evidence, however, suggests that they can expand their life span upon specific stimuli and transmigrate away from inflammatory loci (2–4).
More recently, neutrophils were found to form extracellular meshes called NETs, which are composed mainly of DNA, histones, and granular proteases (Figure 1A). NETs were first observed as a unique form of cell death, distinct from apoptosis or necrosis (5), a process further described as NETosis (6, 7). NETs trap and restrain invading pathogens and utilize their highly localized focus of antimicrobial granular peptides to degrade virulent factors and even kill microorganisms (6–8).
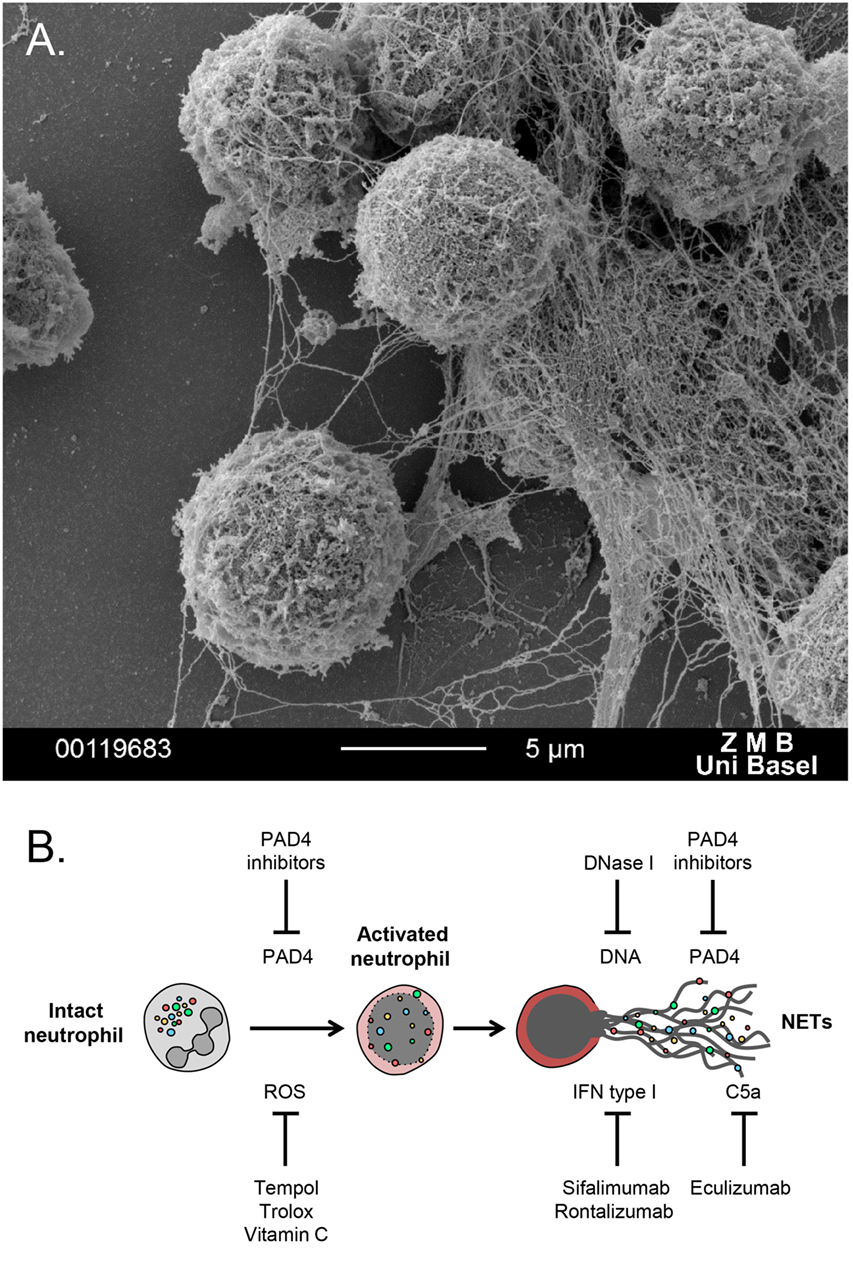
Figure 1. (A) Scanning electron microscopy image of polymorphonuclear neutrophils undergoing NETosis, after in vitro treatment with PMA for 3 h. (B) Schematic overview of potential NETs targeting therapies. A series of antioxidants can reduce generation of ROS, which triggers NET formation. DNase 1 digests extracellular DNA and, thus, degrades NETs. PAD4 inhibitors block histone citrullination, an important event for autoantibody generation in autoimmune disease. Sifalimumab and rontalizumab are monoclonal antibodies to IFN-a, which is a byproduct of NETosis that can further stimulate neutrophil activation and NET release. Eculizumab, a monoclonal antibody to C5, can block complement activation that stimulates formation of NET.
Neutrophil extracellular traps were originally defined being sparked by stimulation with Gram-positive and -negative bacteria, fungi, and parasites, together with agents as IL-8, lipopolysaccharide (LPS), and PMA (6, 7). The most prominent pathway mediating NET formation appears to be NADPH oxidase-generated ROS, with the granular enzymes myeloperoxidase (MPO) and neutrophil elastase (NE) promoting chromatin decondensation and peptidyl arginine deiminase (PAD4) catalyzing histone citrullination (9–12). Moreover, recent data show that NET formation is directly linked to autophagy (13), while regulation of autophagy is closely tied to ROS (14). Most importantly, ROS levels determine whether the autophagy response ends in NETosis (13). The precise ways, however, in which ROS interfere with the signaling network behind NETs and autophagy are only partly understood.
The identification of NETs has been accomplished via microscopic techniques, such as immunofluorescence microscopy, transmission electron microscopy (TEM), and scanning electron microscopy (SEM). Recently, a series of novel immunocyto- and histochemical procedures allowed improved imaging and quantification of NETs. On the one side, in vitro live-cell imaging led to profound new concepts over the mechanisms of NET generation. These include fluorescence microscopy and spectroscopy (6, 15), electron microscopy (15, 16), and lately flow cytometry with simultaneous imaging (17, 18). On the other side, diverse intravital, in vivo, and in situ microscopic techniques led to deeper insights into the role of NET formation during health and disease (12, 19).
Moreover, the deposition of NETs observed in various inflammatory pathologies was associated with the circulating cell-free DNA (cfDNA) levels in biological fluids of the respective patients, such as plasma and serum (15, 20, 21). Therefore, circulatory cfDNA could eventually be utilized as a surrogate marker of NETs in these pathologies, while determination of the DNA levels might facilitate monitoring the disease activity and assessment of the effectiveness of a selected therapeutic strategy.
Although they play a key role in the defense against pathogens, NETs may cause undesirable effects to the host. Most importantly, there has been increased interest in the role of neutrophils and NETs in autoimmunity. Augmented NET formation was first described in preeclampsia and ANCA-associated vasculitis, and successively in a series of autoimmune conditions, including psoriasis, SLE, antiphospholipid antibody syndrome (APS), and rheumatoid arthritis (RA) (15, 22–26) (Table 1).
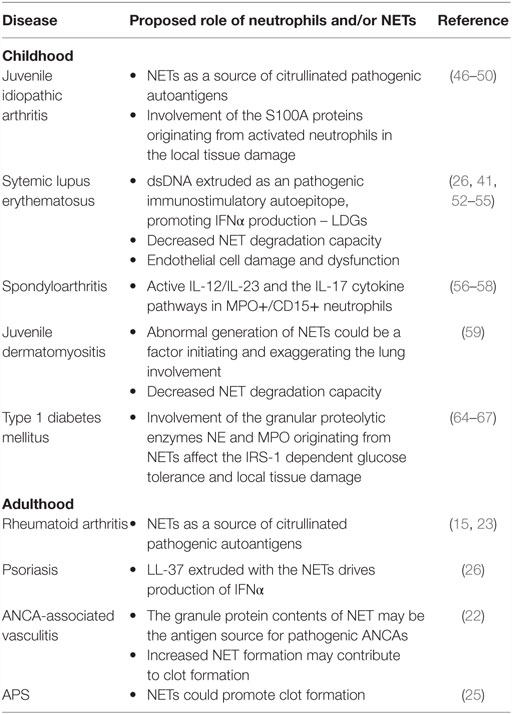
Table 1. Involvement of neutrophils and NETs in autoimmune diseases during childhood and adulthood.
Neutrophil extracellular trap are an obvious source of nuclear material. Among these are a range of cytoplasmic and extracellular citrullinated antigens, well-established targets of anticitrullinated protein antibodies (ACPAs) found in RA (15, 23). Not only do the protein contents of NETs serve as the targets for autoantibody and immune complex formation but they also induce further NETosis, resulting in a harmful positive-feedback loop. These factors form an inflammatory microenvironment that may trigger a strong autoimmune response in individuals with the corresponding susceptibility (27, 28).
Pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα) and interleukin-17 (IL-17), as well as autoantibodies stimulate the formation of NETs and also affect their protein composition (23). Additionally, NETs have been shown to stimulate autoimmunity via the production of interferons and activation of the complement cascade. Antimicrobial granular proteolytic enzymes and the DNA contents of NETs induce plasmacytoid dendritic cell (pDC) interferon α (IFNα) production in SLE and psoriasis, as well as in the case of invading viruses (24). Interferons activate both innate and adaptive immune systems, inducing a Th1 immune response and stimulating B cells toward the generation of autoantibodies.
In addition to the elevated NET formation in patients with autoimmune diseases, increased NET stability and defects in the clearance of NETs have also been observed, particularly in SLE and ANCA-associated vasculitis. In a subset of SLE and ANCA-associated vasculitis patients, a decreased capacity for NET clearance due low DNase 1 activity as well as the presence of DNase 1 inhibitors has been described. Moreover, antibodies may prevent DNase 1 access to NETs, thus protecting them from degradation (25, 29–31). Finally, complement appears to directly interact with NETs and inhibit DNase 1-mediated NET degradation, while non-degraded NETs activate complement in vitro (32). Taken together, a detrimental positive-feedback loop seems to activate complement by the formation of NETs, which, in turn, increases NETs even further by preventing their degradation.
Neutrophil extracellular trap represent a relatively novel therapeutic target in autoimmune disease. There are several key events throughout neutrophil activation toward NETosis, which could be aimed to inhibit NET formation (Figure 1B). Given the involvement of ROS and oxidative stress in NETosis and autoimmunity, the utilization of antioxidants, such as Trolox, Tempol (33), and vitamin C (34), which prevent NET release, could be introduced for treating autoimmune inflammatory diseases. Another target for blocking NETosis is PAD4, inhibitors of which might ameliorate disease manifestations but also improve endothelial dysfunction (35). Recombinant DNase 1 could potentially degrade NETs, leading to efficient clearance of harmful debris. Finally, a series of recombinant human monoclonal antibodies are shown to restrain NET formation, acting indirectly on molecules released from the activated neutrophils together with NETs. These recombinant mAbs could be administered for neutralization of the cytokine byproducts of the NETotic process, such as IFN and members of the complement system (36, 37).
Autoimmune Conditions in Childhood
The role of neutrophils and NETs in pediatric rheumatology is currently gaining increasing attention. Childhood rheumatic disorders are categorized into autoimmune and autoinflammatory conditions (38). Neutrophil infiltration into the tissues is present in both autoinflammatory diseases characterized by activation of the innate immune system, and classic autoimmune diseases are characterized by the presence of autoantibodies and autoreactive T cells. This makes the common basic effector pathways evident. In the pediatric setting, special attention must be given to the possibility of congenital immune deficiencies that present with features of autoimmune disease. For instance, children may develop manifestations of inflammatory bowel disease (IBD) with neutrophilic inflammation due to primary genetic defects in the innate immune system, including chronic granulomatous disease (CGD) with mutations in NADPH oxidase (39).
Neutrophils and NETs in JIA
Juvenile idiopathic arthritis (JIA) is the most common chronic autoimmune disease of childhood (40). JIA represents a group of entities that start prior to puberty with incident arthritis lasting at least 6 weeks without any additionally identified cause (41). The 2001 ILAR classification criteria divide JIA into several disease categories based on the number and type of affected joints and the presence of systemic presentations (42). Although different JIA categories share common pathogenic mechanisms and therapeutic strategies, they are distinct entities, and the role of neutrophils in pathogenesis likely differs by JIA subtype as suggested by both heterogeneous clinical characteristics and underlying genetics.
A series of factors indicate that oligoarticular and rheumatoid factor (RF)-negative polyarticular JIA are typical autoimmune diseases, including the linkage with specific HLA haplotypes, as well as the presence in the synovial fluid of specific memory Th1 lymphocytes (43, 44). Genome wide association studies have detected associations with a series of loci that overlap with classic autoimmune diseases, including adult RA, multiple sclerosis (MS), and type I diabetes mellitus (T1D) (43, 45).
Gene expression analyses in neutrophils from patients suffering from RF-negative polyarticular JIA revealed modulation of IL-8- and IFNγ-regulated genes (46). Alleles related to calcium flux and ROS generation have been reported in affected neutrophils (47). These abnormalities distinguished JIA from healthy controls, but not active from inactive disease, suggesting the existence of additional intrinsic cellular defects. Furthermore, there is evidence for chronic activation of peripheral neutrophils in pediatric patients with polyarticular disease like in adult RA (46).
S100A proteins are zinc and calcium-binding proteins that represent up to half of the total cytosolic protein in phagocytes. Neutrophils and monocytes produce the S100A8/S100A9 heterodimer, also known as calprotectin, while S100A12 is primarily produced by activated neutrophils. Upon binding the receptors for advanced glycation end products (RAGE), S100A proteins activate endothelial cells, inducing expression of adhesion molecules and chemoattractants, thus increasing leukocyte recruitment (48). Calprotectin also triggers toll-like receptor 4 (TLR4), promoting secretion of pro-inflammatory cytokines, such as IL-6, IL-1β, and TNFα (49). Levels of calprotectin and S10012A rise to very high levels within inflamed joints, but increased serum levels have been also reported, reflecting a state of intense neutrophil activation (39).
Interestingly, calprotectin was found to be the major antifungal component in NETs. Absence of calprotectin resulted in the complete loss of antifungal activity in vitro and in vivo (50). Moreover, reconstituted NET formation after gene therapy for human CGD was associated with rapid resolution of pulmonary aspergillosis in vivo, underlining the role of functional NADPH oxidase in NET formation and calprotectin release for antifungal activity (51). The circulating levels of S100A seem to have an even higher impact in systemic JIA, in which neutrophils are the primary effector cells. Clinical symptoms, such as fever, rashes, hepatosplenomegaly and serositis, and the underlying genetic background of the affected individuals, place this condition firmly into the “autoinflammatory disease” category, with IL-1 and IL-6 playing major roles in the manifestations of this disorder (44).
Although clearly implicated in JIA, the influence of neutrophils and NETs on pathogenesis and the utility of biomarkers for neutrophil activity require further studies.
Neutrophils and NETs in SLE
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease with multi-organ involvement characterized by autoantibody generation and deposition of immune complexes in the tissues. The main emphasis in SLE pathogenesis has been laid on the adaptive immune system. Nevertheless, recent studies have brought attention to the role of the innate immune arm of defense, and neutrophils in particular (41).
In contrast to arthritis, where neutrophils are considered to be generally primed, even in the circulation (15, 23), there is evidence that neutrophils from SLE patients show compromised activation and reduced capacities for oxidative burst, chemotaxis, adhesion, and phagocytosis (26, 41). Lupus neutrophils, however, might present an activated phenotype locally in the vasculature, where they contribute to the development of vasculitis by damaging the endothelium (26, 41, 52). A possible reason is most likely the presence of a special neutrophil subset in the circulation, determined by a distinctive gene expression profile, termed low-density neutrophils (LDGs). Although deficient in some typical neutrophil effector functions (degranulation, phagocytosis), LDGs exert a pro-inflammatory phenotype as measured by cytokine release (including type I IFNs) and enhanced capacity for endothelial damage (53). Compared with normal density neutrophils, LDGs have an unusual tendency toward NET formation in the absence of any secondary stimuli and externalize various immunostimulatory proteins (53). LDGs show an enhanced capacity to stimulate pDCs to secrete type I interferon, a major cytokine implicated in lupus (24, 54). Moreover, NETs are a possible source of autoepitopes in lupus (41). Otherwise, neutrophils from pediatric SLE patients exert increased apoptosis that could also act as a source of autoantigens. In particular, serum from pediatric lupus patients induces neutrophil apoptosis, which is abrogated by granulocyte-macrophage colony-stimulating factor (GM-CSF) treatment, suggesting a potential therapeutic benefit of GM-CSF in SLE patients (55).
In conclusion, most of the current evidence supports a role of neutrophils in the pathogenesis of this disease, particulary by LDGs.
Neutrophils and NETs in Spondyloarthritis
Calprotectin levels are found increased in enthesitis-related arthritis (ERA). These elevations, though, are less than in other types of juvenile arthritis, suggesting a reduced role for neutrophils in spondyloarthritis (56). Recent studies underline the significance of the IL-12/IL-23 and the IL-17 cytokine pathways in the pathogenesis of the disease (57). Remarkably, MPO-positive CD15-positive neutrophils were foremost in producing IL-17, suggesting an unexpected role for the innate immune system (58). In this context, cytokine production may be the main driver of neutrophil mediated inflammation rather than NET formation.
Neutrophils and NETs in Juvenile Dermatomyositis
The pathogenesis of the systemic autoimmune disorders dermatomyositis (DM) and polymyositis (PM) remains unclear. Patients suffering from DM and PM were shown to be exposed to large amounts of NETs (59). Compared to control subjects, these patients exhibited a significantly enhanced capacity for inducing NETs, which was supported by elevated levels of plasma LL-37 and cfDNA in the circulation (59). More importantly, DM and PM patients did not show an adequate ability to clear NETs sufficiently due to the low activity of DNase 1.
Moreover, patients suffering from DM and PM presenting with interstitial lung disease exhibited the lowest NETs degradation capacity in vitro. DNase 1 activity in patients with disease-specific autoantibodies was significantly lower than in patients without, while treatment with glucocorticoids seemed to improve this condition (59). This finding suggests that abnormal regulation of NETs may be involved in the disease pathogenesis and could be one of the factors that initiate and exaggerate lung involvement.
Neutrophils and NETs in Type 1 Diabetes Mellitus
Metabolic diseases represent a leading health challenge (60). Microvascular complications, including diabetic nephropathy, retinopathy, and neuropathy, are major causes of morbidity and mortality in individuals suffering from diabetes worldwide. Current therapies are still inadequate, as high glucose levels promote activation, dysfunction, and apoptosis of both intravascular and extravascular immune cells (61).
Several recent studies revealed unexpected roles for neutrophils in metabolic conditions. It has long been acknowledged that obesity, a major cause of type 2 diabetes mellitus (T2D) and metabolic syndrome broadly, exhibits a distinct pro-inflammatory character that contributes to insulin resistance (62). Neutrophils are present in increased numbers in obese patients and migrate into the adipose tissue (63). Moreover, a strong increase of neutrophil numbers in the adipose tissue, accompanied by elevated release of NE and high NE activity is confirmed (64). Most importantly, inhibition of NE lead to a remarkable improvement of glucose tolerance and insulin sensitivity in vivo. NE degrades insulin receptor substrate 1 (IRS-1) in hepatocytes, inhibiting the insulin-driven signal transduction, akin to a mechanism proposed previously in lung cancer cells (65).
Furthermore, a role for neutrophils in the pathogenesis of autoimmune type 1 diabetes (T1D) was implied, which is usually diagnosed in children and young adults, previously known as juvenile diabetes. Neutrophils are involved by the triggering of pDC cytokine production and subsequent activation of the adaptive immune system (66). This study, among others, placed neutrophils high in the rank of possible players involved in the pathogenesis of autoimmunity, adding a crucial influence in shaping the adaptive immune response (15, 27, 54). In this context, a recent report confirmed a clear elevation of NE and proteinase 3 (PR3) in T1D patients, which was associated with elevated formation of NETs (21). These findings support a role of neutrophil activation and augmented protease activities in the early pathogenesis of β-cell autoimmunity and also suggest that circulating NE and PR3 could serve as sensitive diagnostic markers for T1D (21).
Moreover, NETs were recently shown to impair wound healing, particularly in diabetes, where neutrophils are more susceptible to form NETs. Inhibition of NET formation or cleaving NETs with DNase 1 was shown to improve wound healing and reduce NET-driven chronic diabetic inflammation (67). Finally, high glucose and hyperglycemia increase NET release and circulating markers of NETosis, respectively, providing a link between neutrophils, inflammation, and tissue damage in T2D (68), while MPO inhibition is the suggested potential strategy for prevention and treatment of insulin resistance (69).
Conclusion
The study of neutrophils and NETs offer insight into the pathogenesis of multiple autoimmune diseases, since they are of high relevance in a broad series of such disorders. There are yet only few studies reporting on the role of neutrophils in pediatric autoimmunity; however, there is currently growing interest in relating the findings in adult disease to pediatrics.
Although neutrophils are the most common cells in the synovial fluid during arthritis, the understanding of their role in pathogenesis is only emerging. Studies of neutrophils in oligo- and polyarticular JIA reveal intrinsic activity-independent abnormalities in gene expression. Evidence of activation of circulating neutrophils, such as S100A proteins and surrogate markers for NET formation, appear as promising biomarkers for overt and subclinical disease activity. Nevertheless, prospective studies regarding the practical application of these neutrophil-related biomarkers are required.
Distinct activated neutrophil subsets and altered apoptosis have been described in lupus. NETs may be the source of key autoantigens and potent inducers of IFNα. The importance of the IL-12/IL-23 and IL-17 cytokine pathways in the neutrophil-mediated pathogenesis of spondyloarthritis were recently implied.
Dermatomyositis is currently considered as a NET-related pathology due to the detection of overt NET formation and the reduced NETs clearance capacity.
Moreover, in type 1 diabetes mellitus, activated neutrophils contribute to the pathogenetic process, especially via the granular proteolytic enzymes NE and MPO, which may originate from enhanced NET formation.
In summary, neutrophil NETs are shown to be of high relevance in the pathobiology of a series of childhood autoimmune disorders and provide the potential for developing successful disease markers and ultimately novel targeted effective therapeutics, such as DNase 1, PAD inhibitors, and anti-IFN therapies. Clarifying the remaining issues will pose novel implications for a broad range of pediatric rheumatologic conditions’ diagnosis and treatment.
Author Contributions
SG devised and wrote the manuscript. SH and PH critically reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Microscopy Core Facility of the Biocenter, University of Basel and Chanchal Sur Chowdhury for their assistance with the scanning electron microscopy imaging.
Funding
SG is supported by funds from the Cantonal Hospital of Aarau and the University Hospital of Basel.
References
1. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6(3):173–82. doi:10.1038/nri1785
2. Giaglis S, Stoikou M, Grimolizzi F, Subramanian BY, van Breda SV, Hoesli I, et al. Neutrophil migration into the placenta: good, bad or deadly? Cell Adh Migr (2016) 10(1–2):208–25. doi:10.1080/19336918.2016.1148866
3. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11(8):519–31. doi:10.1038/nri3024
4. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol (2014) 9:181–218. doi:10.1146/annurev-pathol-020712-164023
5. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol (1996) 59(2):229–40.
6. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi:10.1126/science.1092385
7. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol (2007) 5(8):577–82. doi:10.1038/nrmicro1710
8. Hahn S, Giaglis S, Chowdhury CS, Hosli I, Hasler P. Modulation of neutrophil NETosis: interplay between infectious agents and underlying host physiology. Semin Immunopathol (2013) 35(4):439–53. doi:10.1007/s00281-013-0380-x
9. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood (2011) 117(3):953–9. doi:10.1182/blood-2010-06-290171
10. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep (2014) 8(3):883–96. doi:10.1016/j.celrep.2014.06.044
11. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol (2008) 180(3):1895–902. doi:10.4049/jimmunol.180.3.1895
12. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191(3):677–91. doi:10.1083/jcb.201006052
13. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21(2):290–304. doi:10.1038/cr.2010.150
14. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci (2011) 36(1):30–8. doi:10.1016/j.tibs.2010.07.007
15. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther (2014) 16(3):R122. doi:10.1186/ar4579
16. Krautgartner WD, Vitkov L. Visualization of neutrophil extracellular traps in TEM. Micron (2008) 39(4):367–72. doi:10.1016/j.micron.2007.03.007
17. Gavillet M, Martinod K, Renella R, Harris C, Shapiro NI, Wagner DD, et al. Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. Am J Hematol (2015) 90(12):1155–8. doi:10.1002/ajh.24185
18. Zhao W, Fogg DK, Kaplan MJ. A novel image-based quantitative method for the characterization of NETosis. J Immunol Methods (2015) 423:104–10. doi:10.1016/j.jim.2015.04.027
19. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun (2015) 6:6673. doi:10.1038/ncomms7673
20. Sur Chowdhury C, Hahn S, Hasler P, Hoesli I, Lapaire O, Giaglis S. Elevated levels of total cell-free DNA in maternal serum samples arise from the generation of neutrophil extracellular traps. Fetal Diagn Ther (2016). doi:10.1159/000444853
21. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes (2014) 63(12):4239–48. doi:10.2337/db14-0480
22. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi:10.1038/nm.1959
23. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5(178):178ra140. doi:10.1126/scitranslmed.3005580
24. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi:10.1126/scitranslmed.3001180
25. Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol (2014) 32(1):66–70.
26. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187(1):538–52. doi:10.4049/jimmunol.1100450
27. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol (2012) 189(6):2689–95. doi:10.4049/jimmunol.1201719
28. Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol (2012) 198(5):773–83. doi:10.1083/jcb.201203170
29. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107(21):9813–8. doi:10.1073/pnas.0909927107
30. Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol (2014) 25(5):990–7. doi:10.1681/ASN.2013060606
31. Leffler J, Gullstrand B, Jönsen A, Nilsson JÅ, Martin M, Blom AM, et al. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res Ther (2013) 15(4):R84. doi:10.1186/ar4264
32. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188(7):3522–31. doi:10.4049/jimmunol.1102404
33. Vorobjeva NV, Pinegin BV. Effects of the antioxidants trolox, tiron and tempol on neutrophil extracellular trap formation. Immunobiology (2016) 221(2):208–19. doi:10.1016/j.imbio.2015.09.005
34. Mohammed BM, Fisher BJ, Kraskauskas D, Farkas D, Brophy DF, Fowler AA III, et al. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients (2013) 5(8):3131–51. doi:10.3390/nu5083131
35. Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis (2015) 74(12):2199–206. doi:10.1136/annrheumdis-2014-205365
36. Mathian A, Hie M, Cohen-Aubart F, Amoura Z. Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs (2015) 75(8):835–46. doi:10.1007/s40265-015-0394-x
37. van Bijnen ST, Wouters D, van Mierlo GJ, Muus P, Zeerleder S. Neutrophil activation and nucleosomes as markers of systemic inflammation in paroxysmal nocturnal hemoglobinuria: effects of eculizumab. J Thromb Haemost (2015) 13(11):2004–11. doi:10.1111/jth.13125
38. van Kempen TS, Wenink MH, Leijten EF, Radstake TR, Boes M. Perception of self: distinguishing autoimmunity from autoinflammation. Nat Rev Rheumatol (2015) 11(8):483–92. doi:10.1038/nrrheum.2015.60
39. Carneiro-Sampaio M, Coutinho A. Early-onset autoimmune disease as a manifestation of primary immunodeficiency. Front Immunol (2015) 6:185. doi:10.3389/fimmu.2015.00185
40. Macaubas C, Nguyen K, Milojevic D, Park JL, Mellins ED. Oligoarticular and polyarticular JIA: epidemiology and pathogenesis. Nat Rev Rheumatol (2009) 5(11):616–26. doi:10.1038/nrrheum.2009.209
41. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol (2011) 7(12):691–9. doi:10.1038/nrrheum.2011.132
42. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol (2004) 31(2):390–2.
43. Hinks A, Cobb J, Marion MC, Prahalad S, Sudman M, Bowes J, et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet (2013) 45(6):664–9. doi:10.1038/ng.2614
44. Lin YT, Wang CT, Gershwin ME, Chiang BL. The pathogenesis of oligoarticular/polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun Rev (2011) 10(8):482–9. doi:10.1016/j.autrev.2011.02.001
45. Thompson SD, Sudman M, Ramos PS, Marion MC, Ryan M, Tsoras M, et al. The susceptibility loci juvenile idiopathic arthritis shares with other autoimmune diseases extend to PTPN2, COG6, and ANGPT1. Arthritis Rheum (2010) 62(11):3265–76. doi:10.1002/art.27688
46. Jarvis JN, Petty HR, Tang Y, Frank MB, Tessier PA, Dozmorov I, et al. Evidence for chronic, peripheral activation of neutrophils in polyarticular juvenile rheumatoid arthritis. Arthritis Res Ther (2006) 8(5):R154. doi:10.1186/ar2048
47. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS One (2014) 9(5):e97088. doi:10.1371/journal.pone.0097088
48. Foell D, Roth J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum (2004) 50(12):3762–71. doi:10.1002/art.20631
49. Vaos G, Kostakis ID, Zavras N, Chatzemichael A. The role of calprotectin in pediatric disease. Biomed Res Int (2013) 2013:542363. doi:10.1155/2013/542363
50. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog (2009) 5(10):e1000639. doi:10.1371/journal.ppat.1000639
51. Bianchi M, Niemiec MJ, Siler U, Urban CF, Reichenbach J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J Allergy Clin Immunol (2011) 127(5):1243–1252e1247. doi:10.1016/j.jaci.2011.01.021
52. Hervier B, Hamidou M, Haroche J, Durant C, Mathian A, Amoura Z. Systemic lupus erythematosus associated with ANCA-associated vasculitis: an overlapping syndrome? Rheumatol Int (2012) 32(10):3285–90. doi:10.1007/s00296-011-2055-z
53. Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol (2013) 35(4):455–63. doi:10.1007/s00281-013-0375-7
54. Knight JS, Kaplan MJ. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol (2012) 24(5):441–50. doi:10.1097/BOR.0b013e3283546703
55. Chiewchengchol D, Midgley A, Sodsai P, Deekajorndech T, Hirankarn N, Beresford MW, et al. The protective effect of GM-CSF on serum-induced neutrophil apoptosis in juvenile systemic lupus erythematosus patients. Clin Rheumatol (2015) 34(1):85–91. doi:10.1007/s10067-014-2800-2
56. Rahman MT, Myles A, Gaur P, Misra R, Aggarwal A. TLR4 endogenous ligand MRP8/14 level in enthesitis-related arthritis and its association with disease activity and TLR4 expression. Rheumatology (2014) 53(2):270–4. doi:10.1093/rheumatology/ket375
57. Smith JA, Colbert RA. Review: the interleukin-23/interleukin-17 axis in spondyloarthritis pathogenesis: Th17 and beyond. Arthritis Rheumatol (2014) 66(2):231–41. doi:10.1002/art.38291
58. Appel H, Maier R, Wu P, Scheer R, Hempfing A, Kayser R, et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther (2011) 13(3):R95. doi:10.1186/ar3370
59. Zhang S, Shu X, Tian X, Chen F, Lu X, Wang G. Enhanced formation and impaired degradation of neutrophil extracellular traps in dermatomyositis and polymyositis: a potential contributor to interstitial lung disease complications. Clin Exp Immunol (2014) 177(1):134–41. doi:10.1111/cei.12319
60. Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med (2005) 56:45–62. doi:10.1146/annurev.med.56.082103.104751
61. Harcourt BE, Penfold SA, Forbes JM. Coming full circle in diabetes mellitus: from complications to initiation. Nat Rev Endocrinol (2013) 9(2):113–23. doi:10.1038/nrendo.2012.236
62. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi:10.1146/annurev-immunol-031210-101322
63. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol (2009) 175(4):1473–82. doi:10.2353/ajpath.2009.080999
64. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18(9):1407–12. doi:10.1038/nm.2885
65. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med (2010) 16(2):219–23. doi:10.1038/nm.2084
66. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med (2013) 19(1):65–73. doi:10.1038/nm.3042
67. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med (2015) 21(7):815–9. doi:10.1038/nm.3887
68. Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, Bonora B, et al. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol (2015) 52(3):497–503. doi:10.1007/s00592-014-0676-x
Keywords: neutrophils, NETs, NETosis, biomarkers, juvenile idiopathic arthritis, SLE
Citation: Giaglis S, Hahn S and Hasler P (2016) “The NET Outcome”: Are Neutrophil Extracellular Traps of Any Relevance to the Pathophysiology of Autoimmune Disorders in Childhood? Front. Pediatr. 4:97. doi: 10.3389/fped.2016.00097
Received: 18 April 2016; Accepted: 30 August 2016;
Published: 13 September 2016
Edited by:
Claudio Pignata, University of Naples Federico II, ItalyReviewed by:
Oskar A. Haas, St. Anna Children’s Hospital, AustriaRamesh Natarajan, Virginia Commonwealth University, USA
Copyright: © 2016 Giaglis, Hahn and Hasler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stavros Giaglis, c3RhdnJvcy5naWFnbGlzQHVuaWJhcy5jaA==