 Veronica Bertini1†
Veronica Bertini1† Angelo Valetto1*†
Angelo Valetto1*† Alessia Azzarà1
Alessia Azzarà1 Annalisa Legitimo2
Annalisa Legitimo2 Giuseppe Saggese2,3Rita Consolini2Alessandro Orsini3Alice Bonuccelli3
Giuseppe Saggese2,3Rita Consolini2Alessandro Orsini3Alice Bonuccelli3
- 1Cytogenetics and Molecular Genetics Unit, Azienda Ospedaliero-Universitaria Pisana, Pisa, Italy
- 2Laboratory of Immunology, University of Pisa, Pisa, Italy
- 3Section of Pediatric Neurology, Azienda Ospedaliero-Universitaria Pisana, Pisa, Italy
“22q11 deletion syndrome” (22q11DS) is a rare genetic syndrome, in which most patients share the same deletion, but their clinical features may vary a great deal. The genetic mechanisms underlying the variable expressivity and reduced penetrance of 22q11DS still have to be fully elucidated. Epilepsy has been reported in about 15.2% of the patients; however, few studies have focused on this topic, and in most cases, a detailed epileptic profile is missing. Since only a minority of patients experience epileptic seizures, 22q11deletion can be considered a predisposing factor, which is not sufficient “per se” to cause epilepsy; to date, no candidate gene for epilepsy has been identified in the deleted region. We report on a 6-year-old girl with 22q11DS presenting a form of epilepsy that can be classified as “Panayiotopoulos syndrome.” Array CGH revealed an additional microduplication of 172 kb in 2q37, harboring three genes. One of these, DGKD (diacylglycerol kinase delta), is interrupted by the distal breakpoint of the duplication. DGKD encodes a cytoplasmic enzyme that phosphorylates diacylglycerol to produce phosphatidic acid. This is an important second messenger in a pathway of lipid signaling that has been implicated in epilepsy and other neurological diseases. Disruption of DGKD by a t(X;2) has been previously reported in a patient with epilepsy. The 2q37 microduplication was inherited from her mother, who never experienced epileptic seizures, thus this imbalance is not “per se” sufficient to cause epilepsy. It can be hypothesized that the epileptic phenotype is provoked by the simultaneous presence of 22q11.2 deletion and 2q37 duplication. It has been shown that rare additional copy-number variations (CNVs) outside the 22q11.2 region may modulate the risk of congenital heart defects. It is possible that also for the epileptic phenotype, the additional CNVs may represent an important modifying factor underlying the variable expressivity and incomplete penetrance in the 22q11DS.
Background
“22q11 deletion syndrome” (22q11DS) is a rare genetic syndrome with a reported prevalence of about 1:4,000 live births. Although most of the patients share exactly the same deletion of nearly 3 Mb in 22q11.2, their clinical features may be highly variable ranging from a severe phenotype to a very mild one (1). The genetic mechanisms underlying the variable expressivity and reduced penetrance of 22q11DS still have to be fully elucidated.
Typical symptoms include multiple developmental anomalies such as congenital heart defects (CHD), palatal, renal, gastrointestinal, ophthalmological, dental, and skeletal abnormalities as well as hypocalcemia, immunodeficiency, and endocrine dysfunction (1). Structural central nervous system (CNS) abnormalities, like polymicrogyria, cerebral atrophy, cerebellar hypoplasia, focal cortical dysplasia, and subependymal heterotopia, are also reported (2). Neuropsychiatric (NP) manifestations such as neurodevelopment disorders (developmental delay, cognitive impairment, and speech disturbances) and psychiatric disorders (attention deficit hyperactivity disorder, schizophrenia, and mood disorders) are often present (2–4).
Among the NP manifestations, seizures are conspicuous events, but in the majority of cases, they can be a secondary manifestation of other syndrome-associated features, such as hypocalcemia, hypoxia/ischemia secondary to CHD, and fever with recurrent infections. In the literature, the epileptic seizure incidence is estimated from about 10% (5) to 21% (6). In a recent article, the incidence of epilepsy in a cohort of 145 22q11DS children was reported to be 15.2%; excluding patients with “structural epilepsy” (CNS abnormalities), the prevalence of “genetic epilepsy” is 8.3% (2).
These findings suggest a link between “genetic epilepsy” and 22q11DS, and this implies that the hemizygous deletion of one or more genes in this specific locus may be associated with epileptogenesis. However, 22q11.2 hemizygous deletion is not sufficient “per se” to cause epilepsy but constitutes a predisposing genetic factor, since only a minority of patients experience epileptic seizures. It may be that a “second hit” is necessary to manifest the epileptic phenotype, but to date, the genetic mechanisms that could increase the risk of epilepsy are not known.
Recently, array CGH has highlighted that humans are much more genetically variable than previously appreciated. To date, nearly 10–15% of the human genome has been annotated as harboring copy-number variations (CNVs), namely microdeletions and microduplications, ranging from 1 kb to several megabases (7). Their presence may contribute significantly toward human phenotypic variability, complex behavioral traits, and disease susceptibility.
Little is known about the contribution of additional CNVs on the phenotypic variability of 22q11DS. A recent article (8) focused on the role of rare CNVs as genetic modifiers predisposing to CHD in a very large cohort of patients with 22q11DS. When these CNVs were carefully examined for their gene content, specific cardiac networks appeared to be overrepresented in 22q11DS CHD cases but not in 22q11DS controls with no cardiac phenotype, suggesting that CNVs outside the 22q11.2 region can modify the risk for CHD in 22q11DS.
We report on a 6-year-old girl with 22q11DS presenting a form of epilepsy that can be classified as “Panayiotopoulos syndrome” (PS). To the best of our knowledge, PS has never been associated with 22q11DS. Array CGH revealed a de novo typical 22q11.2 microdeletion and a rare maternal microduplication of 172 kb in 2q37, which could represent an important factor predisposing to epilepsy. As in the case of CHD, additional CNVs outside the 22q11.2 region may modulate the risk of epileptic seizures in patients with 22q11DS.
Case Report
The girl, the third child of healthy, unrelated parents, was born at full term after a normal pregnancy by cesarean section because of podalic presentation. Her mother had a previous miscarriage at 16 weeks of gestation. The neonatal course was regular. The patient came to our observation at 6 years of age because of recurrent episodes of vomiting. Her weight was 22.3 kg (−0.29 SD), height 118 cm (−0.61 SD), and BMI 16.6 (0.14 SD); she presented dysmorphic features such as a wide mouth with a “Cupid’s bow”-shaped upper lip, epicanthic folds, prognathism and narrow biparietal diameter, and a II–III toe syndactyly.
Parents reported several autonomic partial seizures in sleep (ictal vomiting, retching, pallor, and nausea) sometimes associated with unilateral eye deviation and sometimes with secondary generalization (loss of consciousness and urine incontinence). The episodes started at the age of 4; on that first occasion, she seemed completely unresponsive and flaccid for about 30 min. After that, she began to have episodes of paroxysmal vomiting during the day and then during the night, the frequency of which increased, starting from one episode every 3 months to one episode per month.
The child had also recurrent otitis; the audiological evaluation showed mild bilateral transmission hypoacusis.
She has a mild developmental delay and an attention deficit hyperactivity disorder. At 6 years, a psychometric scale WISC-III (I.Q. tot: 75, V.I.Q.: 70, and P.I.Q.: 85) was administered, which highlighted lower limits of intellectual quality, with a significant discrepancy between verbal and performance skills, with difficulty in accepting, detaining, and recalling verbal information.
During hospital admission, blood chemical and amino acid examinations were performed, as well as brain MRI, cardiac, and abdominal Doppler sonography; all of these test results were normal. In particular, the blood proline value (192 μmol/L) was in the normal range (n.v. 117–332 μmol/L).
Awake electroencephalographic (EEG) showed occipital spikes during eye closure with generalized spike wave complex during sleep. Considering the clinical features and the EEG pattern, we diagnosed an occipital early onset epilepsy syndrome, PS.
In accordance to the literature data, we decided not to use any antiepileptic drug.
The child underwent an immunologic consultation. A general lymphocyte overview showed normal values comparing to age-matched healthy controls; an extended evaluation of both T cell subpopulations by cytoflorimetric analysis showed that CD4+ and CD8+ effector memory (CD45RA−CD62L−) subset was abnormally high in the first examination.
An auxological and endocrinological consultation showed vitamin D deficiency and normal blood calcium levels (about 10 mg/dL, n.v. 8.6–10.2). The child did not assume supplements or other medications.
After written informed consent, a 180K oligo-array (Agilent, Santa Clara, CA, USA) was performed, according to standard procedures; the analysis was made with the Agilent dedicated software (Cytogenomics, Agilent). The Database of Genomic Variants (DGV) was used to define the variants found. Array CGH tests were also performed on her parents.
Array CGH detected in the proband a de novo ~2.6 Mb deletion in 22q11.2, from position 18,894,835 to position 21,464,119 (hg19 map) and an additional 172 kb duplication in 2q37.1, from position 234,191,549 to position 234,363,450 (Figures 1A,B). FISH with a commercial probe (Cytocell, Tarrytown, NY, USA) was used to confirm the 22q11.2 deletion in the proband.
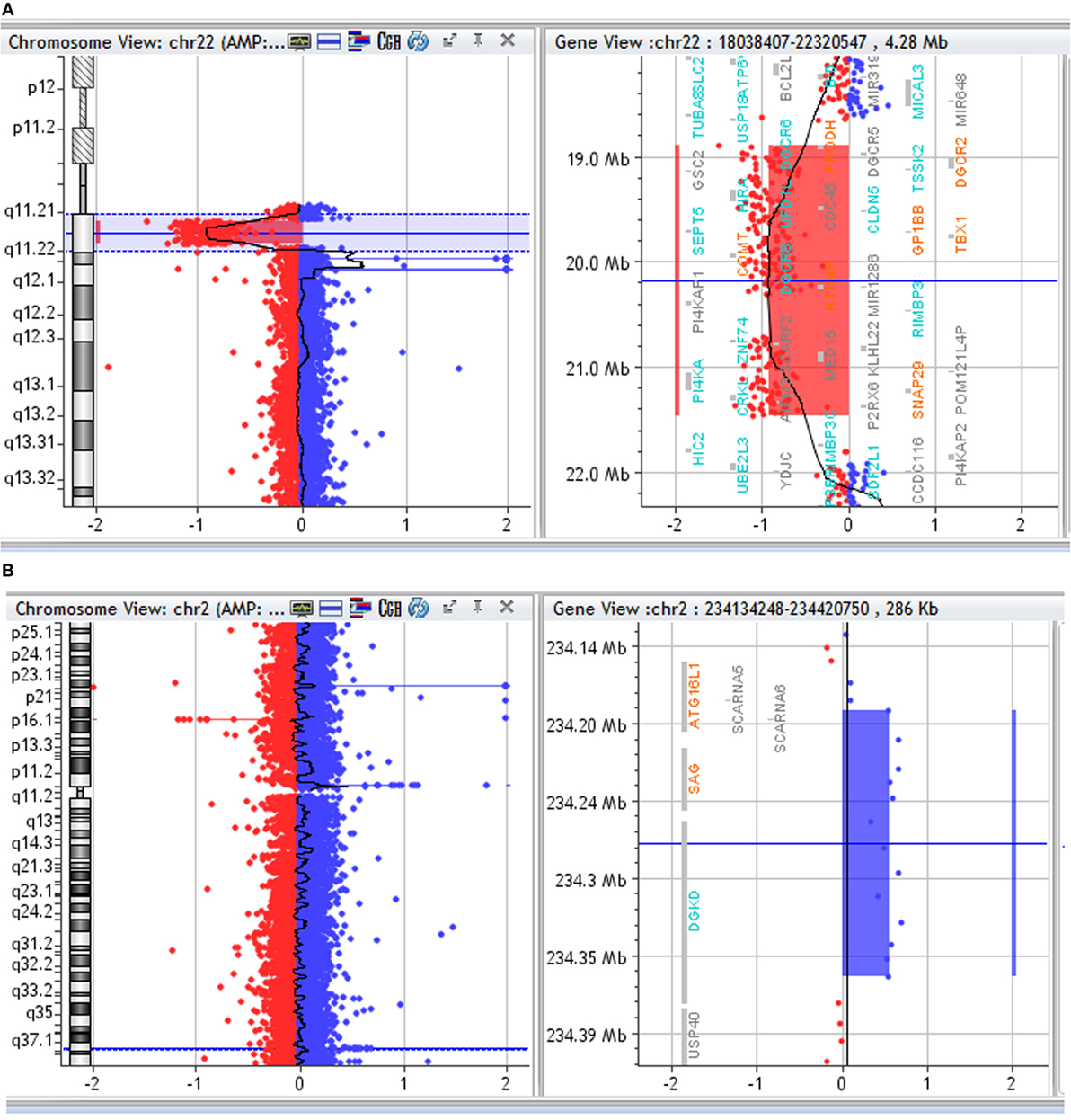
Figure 1. (A) Array CGH profile of chromosome 22. Array CGH of the proband showing chromosome 22 on the left and the 22q11.2 deletion highlighted on the right. (B) Array CGH profile of chromosome 2. Array CGH of the proband showing chromosome 2 on the left and the 2q37 duplication highlighted on the right.
The 2q37.1 duplication was inherited from her healthy mother who did not present any neurological disease except for recurrent headaches.
Putative interaction networks among genes harbored in the 22q11.2 and in the 2q37.1 region were built by the GeneMANIA1 with default parameters. Physical, co-expression, and pathway gene–gene interactions were evaluated (9). As shown in Figure 2, the network revealed that DGKD is co-expressed with PRODH, HIRA, GNB1L, C22orf29, ARVCF, MED15, and LZTR1 and that it interacts with GNB1L. Details about the functional role of these genes are reported in Table 1.
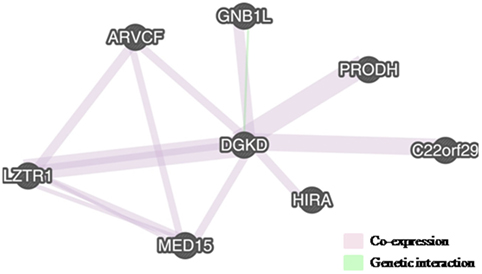
Figure 2. Interactions between DGKD and the genes located in 22q11.2, as shown by GENEMania. DGKD shows a co-expression with seven genes in 22q11.2 (PRODH, HIRA, GNB1L, C22orf29, ARVCF, MED15, and LZTR1) (pink lines) and functionally interacts with GNB1L (green line).
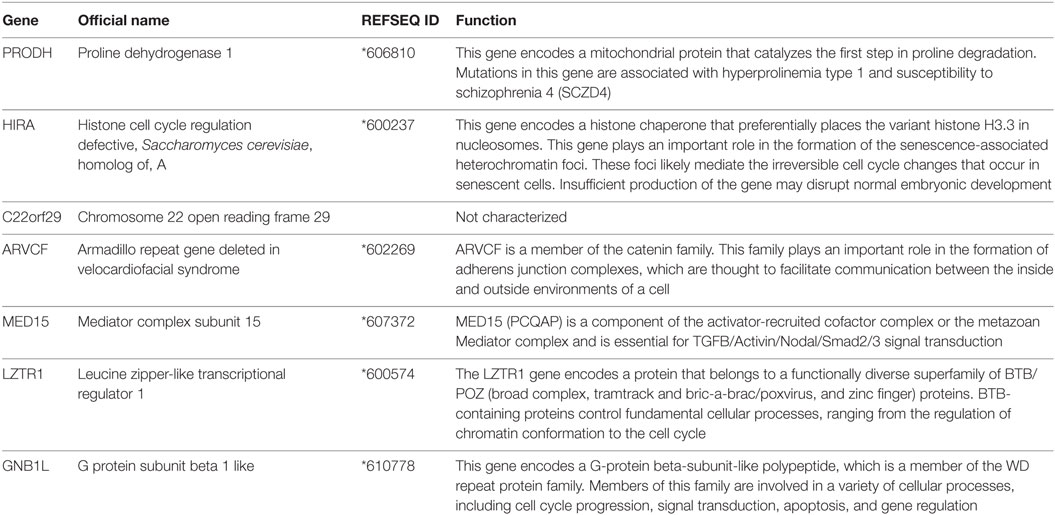
Table 1. Description of the functional role of the genes co-expressed with DGKD.
Discussion
We report on a 6-year-old girl with 22q11DS presenting prolonged autonomic seizures (nausea, retching, ictal vomiting, pallor, and urine incontinence) that fits the criteria of PS.
Epilepsy is reported in 22q11DS, but few studies have focused on this topic, and most of them do not address epilepsy phenotype in more detail. In a recent article (5), a careful revision of the literature on 22q11DS patients with epilepsy and the description of six new cases have been made; among the 53 individuals reported in PubMed, 47% were diagnosed with focal epilepsy (including cases of Rolandic and atypical absence epilepsy), 41% had the diagnosis of genetic generalized epilepsy (including cases with myoclonic features and juvenile myoclonic epilepsy), 6% had myoclonic jerks and unspecified epilepsy, and 6% had unspecified epilepsy without myoclonic features. To the best of our knowledge, PS has never been associated with 22q11DS.
Epilepsy of the proband can be defined as “genetic” since neither structural CNS abnormalities nor metabolic conditions predisposing to epilepsy were found. In 22q11DS, “genetic epilepsy” has a prevalence of about 8.3%, suggesting that one or more genes in this specific locus may be associated to epileptogenesis (2).
This case shows the typical deletion between the low-copy repeats A and D in 22q11.2, where about 50 RefSeq genes are harbored. Neither ion channel genes nor genes whose hemizygous deletion causes epilepsy have been identified. The only gene that can be associated with epilepsy is PRODH (Proline Dehydrogenase 1), whose homozygous deletion causes type I hyperprolinemia, characterized by a high proline level in blood and various NP symptoms, including epileptic seizures (10). In this proband, PRODH has a functional allele since her proline level was normal.
However, it is worth noting that some other genes in the 22q11 deletion are involved in brain function and development, like SNAP29 (synaptosomal-associated protein 29) and RTN4R (reticulon four receptor, NOGO RECEPTOR) (11, 12). A third gene, DGCR8 (microprocessor complex subunit 8), encodes a subunit of the miRNA microprocessor complex and has a genome-wide effect on the biogenesis of micro-RNAs, which are particularly expressed in the mammalian brain (13). Therefore, it is possible that the hemizygous expression of multiple genes involved in the development of neuronal circuits can explain the broad spectrum of structural abnormalities and the coexistence of several NP manifestations, including epilepsy.
Since only a minority of patients experience epileptic seizures, the hemizygous deletion of 22q11 is not sufficient “per se” to cause epilepsy, and a “second hit” is necessary. In the present case, in addition to the 22q11 deletion, the array CGH data showed a 172-kb duplication in 2q37.1, where three genes are harbored: ATG16L1 (Autophagy 16-like 1), SAG (S-antigen; retina and pineal gland), and DGKD (diacylglycerol kinase delta).
Several evidences support a role of DGKD in epileptogenesis.
DGKD encodes a cytoplasmic enzyme that phosphorylates diacylglycerol (DAG) to produce phosphatidic acid (PA). Both DAG and PA are important second messengers in a pathway of lipid signaling that has been implicated in epilepsy and other neurological diseases. Of special interest, in this respect, is DAG-gated transient receptor potential cation channels (e.g., TRPC3, -6, and -7), in which DAG binding directly opens the channels (14).
In Drosophila and mice, expression studies support a role for DGKD in development and function of CNS; EEG assessment of Dgkd knockdown mice revealed abnormal epileptic discharges in half of the mutant mice evaluated (15).
In humans, disruption of the DGKD sequence by a translocation between a chromosome X and a chromosome 2 has been previously reported in a patient with epilepsy (14).
Further data about human DGKD haploinsufficiency have been searched for in two publically available databases that collect pathogenetic imbalances in Decipher,2 ClinVar,3 and HGMD,4 a database that collects human genome mutations. This search did not give additional information, since the cases with only one imbalance involving DGKD (9 deletions and 4 duplications in Decipher, 7 deletions and 12 duplications in ClinVar) have a very large extent with an average size of several megabases, and their clinical description is very limited. No point mutations in DGKD are present in HGMD.
As for the 22q11.2 deletion, the 2q37.1 duplication, which leads to DGKD haploinsufficiency, is not “per se” sufficient to cause epilepsy. In the proband, this duplication is inherited from her mother, who never experienced epileptic seizures; a case with a duplication breaking DGKD sequence is reported in the DGV5 that collects CNVs identified in healthy controls (16).
We have searched for epistatic interactions between genes harbored in the two imbalances. DGKD shows a co-expression with six genes located in 22q11.2 (PRODH, HIRA, C22orf29, ARVCF, MED15, and LZTR1), and it functionally interacts with GNB1L, a gene involved in cell cycle progression, signal transduction, apoptosis, and gene regulation (Figure 2; Table 1).
Even if we cannot exclude that other smaller genetic variants or mutations elsewhere in the genome may play a role, it is likely that in this case the 2q37.1 duplication, which leads to DGKD haploinsufficiency, may represent the “second hit” necessary for the outcome of the epileptic phenotype.
As in the case of CHD, additional rare CNVs outside the 22q11.2 region may represent an important modifying factor underlying the variable expressivity and incomplete penetrance of epileptic seizures in 22q11DS.
Concluding Remarks
Association between 22q11DS and epilepsy is evident; thus, a detailed description of the epileptic phenotype, when present, is mandatory. In larger cohorts of patients, an accurate analysis of the additional rare CNV gene content will be useful to elucidate the genetic mechanisms underlying the pathological phenotype.
Ethics Statement
This study was approved by Pediatric Regional Ethic Committee (Comitato Etico Pediatrico Regionale per la sperimentazione clinica) of the AOU Meyer (Florence, Italy). Number of the folder: CGF-DEL 22q11.2 CR-AOUP. The patient signed an informed consent before genetic testing.
Author Contributions
VB, AV, and AA performed array CGH analysis, contributed to the analyses and data interpretation, and wrote and critically revised the paper. AL and RC collected and interpreted the clinical data, performed the immunological studies, and contributed to the revision of the paper. GS contributed to the clinical collection of data and critically revised the paper. AO and AB performed the neurological assessment, interpreted the clinical data, and critically revised the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Wendy Doherty for her critical revision of the manuscript and English editing. We also thank Alessandra Consani for her technical support.
Funding
This work has been supported by a fund (PRA_2015_0104) Progetto di ricerca di ateneo 2015 (PRA), Università di Pisa.
Footnotes
References
1. Hacıhamdioğlu B, Hacıhamdioğlu D, Delil K. 22q11 deletion syndrome: current perspective. Appl Clin Genet (2015) 8:123–32. doi:10.2147/TACG.S82105
2. Kim EH, Yum MS, Lee BH, Kim HW, Lee HJ, Kim GH, et al. Epilepsy and other neuropsychiatric manifestations in children and adolescents with 22q11.2 deletion syndrome. J Clin Neurol (2016) 12(1):85–92. doi:10.3988/jcn.2016.12.1.85
3. Montojo CA, Ibrahim A, Karlsgodt KH, Chow C, Hilton AE, Jonas RK, et al. Disrupted working memory circuitry and psychotic symptoms in 22q11.2 deletion syndrome. Neuroimage Clin (2014) 4:392–402. doi:10.1016/j.nicl.2014.01.010
4. Schneider M, Debbané M, Bassett AS, Chow EW, Fung WL, van den Bree M, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 deletion syndrome. Am J Psychiatry (2014) 171(6):627–39. doi:10.1176/appi.ajp.2013.13070864
5. Strehlow V, Swinkels ME, Thomas RH, Rapps N, Syrbe S, Dorn T, et al. Generalized epilepsy and myoclonic seizures in 22q11.2 deletion syndrome. Mol Syndromol (2016) 7:239–46. doi:10.1159/000448445
6. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. Med Genet (1997) 34:798–804. doi:10.1136/jmg.34.10.798
7. Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet (2007) 39(7 Suppl):S48–54. doi:10.1038/ng2092
8. Mlynarski EE, Xie M, Taylor D, Sheridan MB, Guo T, Racedo SE, et al. Rare copy number variants and congenital heart defects in the 22q11.2 deletion syndrome. Hum Genet (2016) 135:273–85. doi:10.1007/s00439-015-1623-9
9. Montojo J, Zuberi K, Rodriguez H, Bader GD, Morris Q. GeneMANIA: fast gene network construction and function prediction for Cytoscape. F1000Res (2014) 3:153. doi:10.12688/f1000research.4572.1
10. Di Rosa G, Nicotera AG, Lenzo P, Spanò M, Tortorella G. Long-term neuropsychiatric follow-up in hyperprolinemia type I. Psychiatr Genet (2014) 24(4):172–5. doi:10.1097/YPG.0000000000000037
11. Pan PY, Cai Q, Lin L, Lu PH, Duan S, Sheng ZH. SNAP-29-mediated modulation of synaptic transmission in cultured hippocampal neurons. J Biol Chem (2005) 280(27):25769–79. doi:10.1074/jbc.M502356200
12. Ramasamy S, Yu F, Hong Yu Y, Srivats H, Dawe GS, Ahmed S. NogoR1 and PirB signaling stimulates neural stem cell survival and proliferation. Stem Cells (2014) 32(6):1636–48. doi:10.1002/stem.1645
13. Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, et al. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell (2014) 28(5):547–60. doi:10.1016/j.devcel.2014.01.032
14. Leach NT, Sun Y, Michaud S, Zheng Y, Ligon KL, Ligon AH, et al. Disruption of diacylglycerol kinase delta (DGKD) associated with seizures in humans and mice. Am J Hum Genet (2007) 80(4):792–9. doi:10.1086/513019
15. Jiang LQ, de Castro Barbosa T, Massart J, Deshmukh AS, Löfgren L, Duque-Guimaraes DE, et al. Diacylglycerol kinase-d regulates AMPK signaling, lipid metabolism, and skeletal muscle energetics. Am J Physiol Endocrinol Metab (2016) 310(1):E51–60. doi:10.1152/ajpendo.00209.2015
Keywords: 22q11 deletion syndrome, epilepsy, array CGH, copy-number variations, 2q37, DGKD
Citation: Bertini V, Valetto A, Azzarà A, Legitimo A, Saggese G, Consolini R, Orsini A and Bonuccelli A (2017) A Case of 22q11 Deletion Syndrome (22q11DS) with a Panayiotopoulos Epileptic Pattern: Are Additional Copy-Number Variations a Possible Second Hit in Modulating the 22q11DS Phenotype? Front. Pediatr. 5:48. doi: 10.3389/fped.2017.00048
Received: 18 November 2016; Accepted: 24 February 2017;
Published: 21 March 2017
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Jaime Imitola, The Ohio State University, USAAndrew McQuillin, University College London, UK
Bulent Hacihamdioglu, Suleymaniye Women Maternity and Child Diseases Training and Research Hospital, Turkey
Copyright: © 2017 Bertini, Valetto, Azzarà, Legitimo, Saggese, Consolini, Orsini and Bonuccelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelo Valetto, YS52YWxldHRvQGFvLXBpc2EudG9zY2FuYS5pdA==
†These authors have contributed equally to this work.