 Nathan P. Gossai
Nathan P. Gossai Peter M. Gordon
Peter M. Gordon- 1Division of Pediatric Hematology and Oncology, University of Minnesota, Minneapolis, MN, USA
- 2University of Minnesota Masonic Cancer Center, Minneapolis, MN, USA
Acute lymphoblastic leukemia (ALL) is the most common cancer in children. While survival rates for ALL have improved, central nervous system (CNS) relapse remains a significant cause of treatment failure and treatment-related morbidity. Accordingly, there is a need to identify more efficacious and less toxic CNS-directed leukemia therapies. Extensive research has demonstrated a critical role of the bone marrow (BM) microenvironment in leukemia development, maintenance, and chemoresistance. Moreover, therapies to disrupt mechanisms of BM microenvironment-mediated leukemia survival and chemoresistance represent new, promising approaches to cancer therapy. However, in direct contrast to the extensive knowledge of the BM microenvironment, the unique attributes of the CNS microenvironment that serve to make it a leukemia reservoir are not yet elucidated. Recent work has begun to define both the mechanisms by which leukemia cells migrate into the CNS and how components of the CNS influence leukemia biology to enhance survival, chemoresistance, and ultimately relapse. In addition to providing new insight into CNS relapse and leukemia biology, this area of investigation will potentially identify targetable mechanisms of leukemia chemoresistance and self-renewal unique to the CNS environment that will enhance both the durability and quality of the cure for ALL patients.
Introduction
Acute lymphoblastic leukemia (ALL) represents ~25% of all pediatric cancer diagnoses and, despite significant advances in therapy, it is still a common cause of death in children with cancer (1). While leukemia arises in the bone marrow (BM), it is a systemic disease with a predilection for certain organs such as the central nervous system (CNS). Prior to the advent of CNS-directed leukemia therapies, CNS leukemia developed in over half of pediatric leukemia patients (2–4). Moreover, despite current CNS-directed therapies that often include high-dose systemic chemotherapy, intrathecal chemotherapy, and cranial irradiation in a subset of children, CNS relapse accounts for ~30% of initial relapses in some clinical trials and occurs in ~2–8% of children with leukemia (5–7). Neither the factors that determine the site of leukemia relapse (CNS, BM, other extramedullary, or combined sites) nor why BM relapse confers a significantly worse prognosis than CNS relapse is well understood (8).
Furthermore, sparing cranial irradiation from all but the most high-risk patients, or even all patients in some recent clinical trials (9, 10), requires intensification of the other CNS-directed therapies. Although the elimination of radiation negates the risk of secondary brain tumors and neuroendocrine failure, CNS-directed chemotherapy carries the risk of seizures, encephalopathy, and neurocognitive toxicities that can include significant and persistent impairments in intelligence, processing speed, memory, academics, executive function, and attention (11–18). An extensive literature comprehensively reviews the clinical aspects of CNS leukemia, thus it will not be addressed in this mini-review (5–7, 19).
Rigorous basic science and clinical data demonstrate that leukemia cell-autonomous factors play a critical role in leukemia biology (20–23). The composition and organization of the BM is highly complex with multiple different cell types and soluble factors interacting with and influencing leukemia cells. These elements create distinct niches, which exert unique and functionally important effects on leukemia development, quiescence, maintenance, and chemoresistance (24–33). Accordingly, strategies to disrupt mechanisms of BM microenvironment-mediated chemoresistance or quiescence represent new, promising approaches to cancer therapy (34).
The role of the CNS in regulating leukemia survival and chemoresistance is much less well understood than the BM microenvironment, but is also likely to be important for both understanding leukemia biology as well as developing more effective and less toxic therapies. As the BM and CNS are distinct environments, research investigating the role of the BM in leukemia is unlikely to uniformly translate to the CNS. In this mini-review, we will describe emerging evidence supporting a role for the CNS in regulating critical aspects of leukemia biology and highlight how this area of investigation may translate into more effective and less toxic therapies for patients.
Approaches for Studying CNS Leukemia
Defining the role of the CNS microenvironment in leukemia requires experimental approaches for studying and characterizing leukemia cells in the CNS. Fortunately, many previously developed approaches for studying the BM microenvironment in leukemia and normal hematopoiesis have been, or can be, adapted to the CNS microenvironment (Figure 1A). In vitro co-culture of leukemia cells with CNS-derived cells can be used to examine the effect of both direct cell–cell interactions as well as soluble factors on leukemia biology (35–37). Similarly, in vitro transwell assays that assess the migration of leukemia cells across endothelial or choroid plexus (CP) cells can be used to model the process of leukemia migration across the blood–brain or blood–cerebral spinal fluid (CSF) barriers, respectively (38–41). Cerebral organoids, three-dimensional in vitro cultures that model brain organogenesis, represent a new and powerful model system that could also potentially be exploited for expanding our understanding of CNS leukemia (42). Genetically engineered mouse leukemia models and human leukemia xenografts develop CNS leukemia with a frequency and in anatomic patterns that recapitulate human CNS leukemia (43, 44). Accordingly, these in vivo systems are powerful tools for biologic discovery as well as therapeutic drug testing. Finally, leukemia cells isolated from the CSF of leukemia patients can be used to confirm and extend knowledge gained in the laboratory. A significant, but not insurmountable, challenge is that only about ~15% of children have morphologic evidence of CNS leukemia at time of diagnosis (45, 46). Moreover, the number of leukemia cells that can be isolated from the typical volume of CSF obtained during a lumbar puncture may be limited. However, with cutting-edge technologies that require only a small number of cells, or single cells, one can envision increasingly sophisticated questions being addressed using primary leukemia cells obtained from CSF samples of newly diagnosed or relapsed patients.
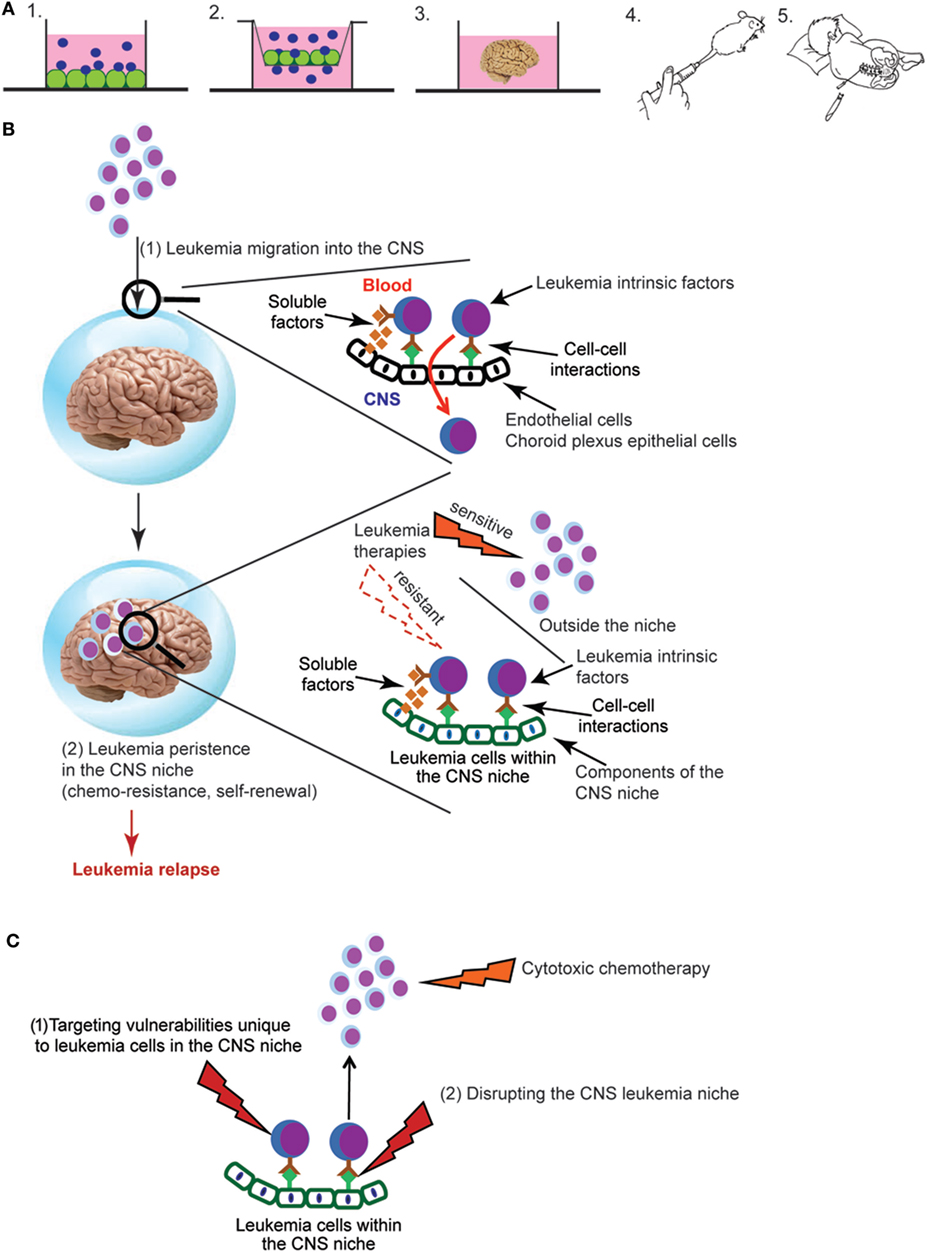
Figure 1. (A) Experimental approaches for studying central nervous system (CNS) leukemia. Complementary experimental approaches for studying CNS leukemia include (1) in vitro co-culture of leukemia and CNS-derived cells, such as meningeal, glial, or choroid plexus (CP) cells, (2) trans-well migration assays of leukemia cells across either endothelial cells or CP epithelial cells, (3) cerebral organoids that are grown in vitro from human induced pluripotent stem cells, (4) in vivo murine models that include either genetically engineered mouse models or xenotransplantation of human leukemia cells into immunodeficient mice, or (5) patient-derived leukemia samples isolated from either the bone marrow or cerebral spinal fluid (CSF). Leukemia cells are shown in blue and CNS-derived cells in green. (B) Overview of key aspects of the pathophysiology of CNS leukemia and relapse. Leukemia cells first breach the blood–brain and/or blood–CSF barriers, illustrated by the blue sphere (1). Leukemia cells must then persist in the CNS and escape the effects of chemotherapy and immune surveillance in order to lead to relapse (2). Identifying the leukemia cell extrinsic factors (soluble factors, cell–cell interactions) that mediate both of these processes as well as their effects on critical aspects of leukemia biology are active areas of investigation. (C) Approaches for targeting leukemia cells in the CNS niche. New therapeutic opportunities will be identified as studies to (i) define the influence of the CNS niche on critical aspects of leukemia biology, such as chemoresistance, self-renewal, and quiescence and (ii) identify CNS-derived factors that protect and maintain leukemia cells in the CNS niche. Directly targeting vulnerabilities unique to leukemia cells in the CNS niche (1) or disrupting the interactions between leukemia cells and the niche (2) represent novel approaches to leukemia therapy.
Trafficking of Leukemia Cells to the CNS
The CNS is an immunologically privileged site that is isolated from the blood system by blood–brain and blood–CSF barriers. Although the process by which normal leukocytes traffic into the CNS has been well studied and reviewed extensively (47–51), this process is less well understood in the context of leukemia. Additionally, tight junctions between epithelial, rather than endothelial, cells provide the blood–CSF barrier within the CP (52). It is not clear that principles governing trans-endothelial migration will apply to the CP. Based on patient autopsy reports and murine leukemia xenotransplantation studies, a rudimentary anatomical and temporal course of leukemia invasion of the CNS can be posited (2, 43). After transiting CP epithelial cells and/or meningeal postcapillary venules, leukemia cells are initially localized to the leptomeninges on the surface of the brain and within the CSF. Leukemia cells then migrate into the deeper meningeal tissues surrounding vessels in the cortex and white matter (Virchow–Robin or perivascular spaces). Only late in the disease is the pial-glial membrane destroyed and leukemia cells identified within the brain parenchyma.
Leukemia Migration
The contributions of leukemia cell intrinsic and extrinsic factors to CNS trafficking remain incompletely understood (Figure 1B). Yet, an increasing number of leukemia genes and pathways, as well as leukemia cell autonomous factors, have been identified as playing a role in CNS trafficking (Table 1). Not surprisingly, factors implicated in trans-endothelial migration of leukemia cells and leukocytes outside of the CNS also appear to play a role in crossing the blood–brain or blood–CSF barriers. For example, myosin II, a cytoskeleton class II non-muscle myosin motor protein, has been shown to be important for T-cell extravasation (53, 54). Using a pre-B ALL murine leukemia model, Wigton et al. showed that myosin-IIA depletion or inhibition with either shRNA or blebbistatin, respectively, significantly decreased leukemia infiltration into the CNS as a result of impaired trans-endothelial extravasation (55). Similarly, known inhibitors of T-cell leukemia migration diminished the ability of leukemia cells to cross CP epithelial cells in a transwell assay designed to mimic the blood–CSF barrier (38). Using similar in vitro co-culture and transwell assays, with brain-derived endothelial cells rather than CP cells, Akers et al. showed VE-cadherin expression by leukemia cells enhanced adhesion to endothelial cells while PECAM-1 expression enhanced adhesion to, and migration through, endothelial cells (39). Together, these studies suggest that many of the cellular mechanisms governing the trans-endothelial migration of leukocytes and leukemia cells outside the CNS are likely to also apply to the transit of leukemia cells across the blood–brain and blood–CSF barriers.

Table 1. Selected genes and pathways that have been associated with central nervous system (CNS) leukemia.
Leukemia Genes and Pathways Implicated in CNS Migration
Focusing more specifically on the trafficking of leukemia cells to the CNS, Buonamici et al. used murine T-cell leukemia models involving expression of the oncogenic, intracellular Notch1 fragment in hematopoietic progenitors combined with gene expression profiling to identify the chemokine receptor CCR7 as an essential adhesion molecule required for the infiltration of leukemic T-cells into the CNS (44). Silencing of CCR7 in leukemia cells, or one of its ligands CCL19 in mice, specifically diminished infiltration of the CNS, but not other tissues, in both murine and xenotransplantation leukemia models. The other ligand for CCR7, CCL21, was undetectable in mouse brain sections and presumed to be less important for leukemia migration into the CNS. Interestingly, deletion of CCR7 in two models of B-cell leukemia failed to inhibit CNS infiltration, suggesting that CCR7 function may be specific for Notch1-induced T-cell leukemia. Complementing this genomic approach, Holland et al. used semiquantitative proteomics to compare the plasma membrane protein composition of an invasive pre-B ALL cell line that resulted in CNS leukemia when transplanted into NOD-SCID mice versus two pre-B ALL cell lines with less invasive behaviors (56). Proteins upregulated on the membrane of the more invasive leukemia cell line classified into a number of biological classes likely functionally relevant for leukemia trafficking to the CNS, including cytoskeletal organization, adhesion, migration, invasion, signaling, and endocytosis. Finally, further functional characterization of three of the differentially expressed proteins, asparaginyl endopeptidase, intercellular adhesion molecule 1, and ras-related C3 botulinum toxin substrate 2, suggested a complex role for multiple proteins and cellular processes in the pathogenesis of CNS disease in pre-B-cell ALL.
Soluble Factors Implicated in CNS Migration
Cytokines and chemokines also likely play a role in the trafficking of leukemia cells to the CNS. Interleukin-15 (IL-15) single-nucleotide polymorphisms and/or mRNA levels have been implicated in leukemia chemoresistance as well as likelihood of CNS disease in leukemia (57–59). While the role for IL-15 in leukemia proliferation and survival is discussed in the next section, IL-15 also upregulates p-selectin glycoprotein ligand-1 (PSGL-1) and CXCR3 levels in leukemia cells (60). Both PSGL-1 and CXCR3 have been implicated in the migration of leukocytes across the blood–CSF barrier and may play a similar role in leukemia (61, 62). However, it is worth noting that other xenotransplantation studies of primary pre-B ALL samples failed to identify a chemokine receptor signature that correlated with CNS invasiveness (43).
The Role of the CNS Microenvironment in Leukemia Biology
While defining the mechanisms by which leukemia cells traffic from the blood to the CNS is important for understanding the biology and pathophysiology of leukemia, a number of observations may diminish its importance for understanding CNS relapse. First, ~15% of patients show evidence of CNS leukemia by morphological examinations of CSF (45, 46). However, more sensitive examinations of CSF using PCR or flow cytometry detect leukemia cells in up to ~40% of patients at diagnosis (63–66). Second, high rates (~50–75%) of CNS leukemia developed in patients prior to the development of adequate CNS-directed therapies (2–4). Third, ~80% of mice transplanted with human, primary B-cell precursor leukemia cells developed CNS leukemia despite the majority of leukemia samples coming from patients without morphological evidence of CNS leukemia (43). Fourth, clonal tracking of xenotransplanted leukemia cells demonstrated that the composition of leukemia cells in the CNS was polyclonal and similar in composition to the spleen or femur (43). Finally, CNS leukemia relapses occur despite cranial irradiation and/or high-dose systemic and intrathecal chemotherapy that either overcome or bypass the blood–brain barrier, respectively (10). Together, these observations support a model in which the ability of leukemia cells to persist in the CNS and escape the effects of chemotherapy and immune surveillance may contribute more to relapse and therapy resistance than the ability of leukemia cells to migrate from the BM and blood to the CNS (Figure 1B). Further supporting this hypothesis, Akers et al. showed that co-culture of leukemia cells with astrocytes, CP epithelial cells, or meningeal cells enhanced leukemia cell resistance to dexamethasone, cytarabine, and methotrexate-induced cell death (35). Notably, these drugs play an important role in CNS leukemia therapy and prophylaxis. Their work also suggested that both soluble factors secreted by the CNS-derived cells and adhesion-mediated signaling contributed to chemoresistance.
CNS Relapses in Pre-B ALL with t(1;19) Translocation
Mechanisms of leukemia chemoresistance in the CNS have also been studied in the context of leukemia bearing the t(1;19) translocation. This translocation occurs in ~5% of pre-B ALL, is associated with an increased risk for CNS relapse, and has been associated with increased expression of the Mer receptor kinase (37, 67, 68). Mer kinase is involved in multiple physiological processes including cell survival, migration, and differentiation (69). Its overexpression or ectopic expression has also been implicated in a wide array of cancers. High Mer-expressing t(1;19) leukemia cells co-cultured with CNS-derived cells exhibit G0/G1 cell cycle arrest, suggestive of dormancy or quiescence, as well as methotrexate chemoresistance (37). Moreover, high Mer expression in t(1;19) leukemia cells increase CNS involvement in murine xenografts and correlated with CNS leukemia at diagnosis in leukemia patients. Finally, Mer kinase inhibitors have been developed and could represent a novel therapy in leukemia patients with the t(1;19) translocation and high Mer expression (69).
Influence of IL-15 on CNS Leukemia
As described in the prior section, the cytokine IL-15 has been implicated in CNS leukemia. In addition to upregulating genes in leukemia cells implicated in CNS trafficking, IL-15 also enhances leukemia proliferation through an effect on the Raf/Ras/ERK signaling pathway (60). This stimulation of leukemia growth was maximal under conditions of low or no serum supplementation, which the authors speculate may mimic the low-protein composition of CSF (60). Further supporting this possibility, high levels of IL-15 have been detected in the CSF and serum of patients with neuro-inflammatory disorders (70). The ability of IL-15 to regulate NK cell development, survival, and activation may provide another, indirect mechanism by which IL-15 influences CNS leukemia. NK cells are a component of the innate immune system with an important role in cancer immune surveillance (71). Frishman-Levy et al. used murine leukemia models and xenografts to show that expression of IL-15 by leukemia cells is associated with the activation of NK cells (72). However, while activated NK cells attenuated the growth of leukemia cells in the periphery via a NKG2D receptor-mediated mechanism, NK cells fail to effectively enter the CNS and, as a result, poorly control CNS leukemia. While this mechanism has yet to be demonstrated in patients, analysis of BM samples from pediatric leukemia patients showed high levels of the NKG2D receptor in infiltrating NK cells in patients with CNS leukemia (72).
The CNS Niche Influences Leukemia Gene Expression Profiles
Further supporting an important role for the CNS microenvironment in leukemia biology, it has recently been shown that the CNS niche imparts unique and functionally important gene expression changes in both leukemia cells lines and primary xenografts (36, 73). In these experiments, human pre-B leukemia cells isolated from the BM and CNS microenvironments of the same mice were subjected to gene expression profiling analyses. Gene set enrichment analyses and functional annotation of the differentially expressed genes revealed that the genes were involved in multiple pathways important for cancer biology, including MAPK, RAS, apoptosis, as well as adaptation to hypoxia with enhanced quiescence and downregulation of oxidative phosphorylation. Additionally, genes dysregulated in leukemia cells isolated from the CNS were shown to be functionally important to leukemia biology. One study demonstrated that upregulation of the gene PBX1 in leukemia cells in the CNS microenvironment conferred enhanced leukemia chemoresistance and self-renewal properties (36). The other study showed that targeting VEGFA, one of the most upregulated genes in CNS-derived leukemia cells, with the VEGF neutralizing antibody bevacizumab reduced the extent of leukemia involvement in the CNS of mice (73). Furthermore, elevated CSF levels of VEGFA have been identified in patients with CNS leukemia (74). The authors speculate that VEGFA may further enhance migration of leukemia cells into the CNS through its effects on increasing endothelial, and potentially blood–brain, permeability. Together, these data support the proposition that the CNS provides a unique leukemia niche that influences leukemia biology.
Complementing these xenotransplantation approaches, gene expression profiling of leukemia cells isolated from the BM of high risk pediatric pre-B cell leukemia patients identified genes and pathways, including WNT, JAK, NF-κB, and B-cell receptor signaling, which distinguish patients with varying extents of CNS leukemia (CNS1-3) at the time of diagnosis (75). Although this work identifies genes and pathways that may either increase CNS homing or facilitate leukemia survival in the CNS, it does not identify the effects of the CNS niche on the leukemia transcriptome, as it utilized BM samples. In an attempt to define the effects of the CNS niche on leukemia cells, van der Velden et al. recently described a unique gene expression pattern in pre-B cell precursor ALL cells isolated from the CSF of patients with isolated CNS relapse when compared with leukemia cells isolated from the BM of patients at diagnosis (76). Moreover, for 5/8 patients with isolated CNS relapse data, they also had gene expression data from the patients’ leukemia cells isolated from the BM at time of diagnosis. However, while the samples were paired for patients, they were obtained at different times (i.e., diagnosis and CNS relapse). Unsupervised clustering analysis showed that the CSF-derived ALL samples were transcriptionally distinct from the BM ALL samples. Pathway analyses of the differentially expressed genes showed enrichment of genes involved in cellular development, cell death/survival, and several signaling pathways, including JAK/STAT and MAPK. Finally, a subpopulation of pre-B ALL cells with a “CNS leukemia profile” (SCD gene positive and increased SPP1 gene expression) was identified in the BM of patients that later developed an isolated CNS relapse. In contrast, this population was low (<1%) or absent in all other patients. Moreover, the lack of a correlation between this leukemia subpopulation and morphologic CNS involvement at diagnosis raises the possibility that these leukemia genes and pathways may provide a survival advantage to the leukemia cells residing in the CNS niche rather than enhancing leukemia trafficking to the CNS. However, it is possible that more sensitive approaches for detecting CNS leukemia (PCR, flow cytometry) would have detected CNS disease in patients with a “CNS leukemia profile” at diagnosis and that these genes are important for trafficking to the CNS as well.
Conclusion
Developing more effective therapies for CNS leukemia is crucial to long-term survival and quality of life for ALL patients. Given the importance of the microenvironment in many aspects of leukemia biology, more effective and less toxic therapies will likely only be realized through a better understanding of the effects of the CNS on critical aspects of leukemia biology. Since the BM and CNS are unique niches, research from the BM will not uniformly translate to the CNS leukemia niche. Supporting this, it has been shown that the CNS niche, relative to the BM, imparts unique effects on the leukemia proteome and transcriptome that influence important aspects of leukemia biology including chemoresistance (35, 36, 73). Furthermore, the complex role of the BM niche in leukemia and hematopoiesis suggests that much remains to be learned about role of the CNS niche in leukemia. For example, distinct niches within the BM, such as the endosteal and perivascular, uniquely influence normal hematopoiesis as well as leukemia biology. Accordingly, further defining at a cellular and molecular level, the components of the CNS that harbor and support chemoresistant leukemia cells will provide a foundation for experiments aimed at understanding and targeting the mechanisms by which the CNS niche influences leukemia biology. Similarly, more comprehensive analyses of the leukemia transcriptome, genome, and proteome in the CNS microenvironment will provide a more detailed understanding of the role of the CNS niche in leukemia as well as new insight into CNS relapse and leukemia biology. Finally, we anticipate this area of investigation will ultimately enhance both the durability and quality of the cure for ALL patients by identifying leukemia cell vulnerabilities unique to the CNS niche or approaches for disrupting the CNS niche (Figure 1C). For example, based on our current therapeutic arsenal and understanding of the genes, pathways, and molecules implicated in the pathophysiology of CNS leukemia (Table 1), potential approaches for augmenting CNS-directed therapy include (i) neutralizing or blocking antibodies directed against VEGF (bevacizumab), CCR7, or other adhesion molecules, (ii) Mer tyrosine kinase inhibitors in the setting of a t(1;19), or (iii) metabolic/mitochondrial modulation.
Author Contributions
NG and PG both contributed to the conception, writing, and editing of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Amy Ryan for assistance with drawing the images in Figure 1A.
Funding
PMG: Hyundai Hope on Wheels Young Investigator Award.
References
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
2. Price RA, Johnson WW. The central nervous system in childhood leukemia. I. The arachnoid. Cancer (1973) 31:520–33.
3. Price RA. Histopathology of CNS leukemia and complications of therapy. Am J Pediatr Hematol Oncol (1979) 1:21–30.
4. Evans AE, Gilbert ES, Zandstra R. The increasing incidence of central nervous system leukemia in children. (Children’s Cancer Study Group A). Cancer (1970) 26:404–9. doi:10.1002/1097-0142(197008)26:2<404::AID-CNCR2820260222>3.0.CO;2-I
5. Pui CH, Howard SC. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol (2008) 9:257–68. doi:10.1016/S1470-2045(08)70070-6
6. Pui CH. Central nervous system disease in acute lymphoblastic leukemia: prophylaxis and treatment. Hematology Am Soc Hematol Educ Program (2006) 2006:142–6. doi:10.1182/asheducation-2006.1.142
7. Pui CH, Thiel E. Central nervous system disease in hematologic malignancies: historical perspective and practical applications. Semin Oncol (2009) 36:S2–16. doi:10.1053/j.seminoncol.2009.05.002
8. Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia (2008) 22:2142–50. doi:10.1038/leu.2008.251
9. Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med (2009) 360:2730–41. doi:10.1056/NEJMoa0900386
10. Vora A, Andreano A, Pui CH, Hunger SP, Schrappe M, Moericke A, et al. Influence of cranial radiotherapy on outcome in children with acute lymphoblastic leukemia treated with contemporary therapy. J Clin Oncol (2016) 34(9):919–26. doi:10.1200/JCO.2015.64.2850
11. Silverman LB. Balancing cure and long-term risks in acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program (2014) 2014:190–7. doi:10.1182/asheducation-2014.1.190
12. Hijiya N, Hudson MM, Lensing S, Zacher M, Onciu M, Behm FG, et al. Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA (2007) 297:1207–15. doi:10.1001/jama.297.11.1207
13. Krull KR, Brinkman TM, Li C, Armstrong GT, Ness KK, Srivastava DK, et al. Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J Clin Oncol (2013) 31:4407–15. doi:10.1200/JCO.2012.48.2315
14. Bhojwani D, Sabin ND, Pei D, Yang JJ, Khan RB, Panetta JC, et al. Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J Clin Oncol (2014) 32:949–59. doi:10.1200/JCO.2013.53.0808
15. Iyer NS, Balsamo LM, Bracken MB, Kadan-Lottick NS. Chemotherapy-only treatment effects on long-term neurocognitive functioning in childhood ALL survivors: a review and meta-analysis. Blood (2015) 126:346–53. doi:10.1182/blood-2015-02-627414
16. Conklin HM, Krull KR, Reddick WE, Pei D, Cheng C, Pui CH. Cognitive outcomes following contemporary treatment without cranial irradiation for childhood acute lymphoblastic leukemia. J Natl Cancer Inst (2012) 104:1386–95. doi:10.1093/jnci/djs344
17. Pui CH, Cheng C, Leung W, Rai SN, Rivera GK, Sandlund JT, et al. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N Engl J Med (2003) 349:640–9. doi:10.1056/NEJMoa035091
18. Hearps S, Seal M, Anderson V, McCarthy M, Connellan M, Downie P, et al. The relationship between cognitive and neuroimaging outcomes in children treated for acute lymphoblastic leukemia with chemotherapy only: a systematic review. Pediatr Blood Cancer (2017) 64:225–33. doi:10.1002/pbc.26188
19. Winick NJ. The evolution of central nervous system prophylaxis for patients with acute lymphoblastic leukemia. Pediatr Blood Cancer (2015) 62:1877–8. doi:10.1002/pbc.25658
20. Krause DS, Scadden DT. A hostel for the hostile: the bone marrow niche in hematologic neoplasms. Haematologica (2015) 100:1376–87. doi:10.3324/haematol.2014.113852
21. Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood (2015) 126:2443–51. doi:10.1182/blood-2015-07-533588
22. Chiarini F, Lonetti A, Evangelisti C, Buontempo F, Orsini E, Evangelisti C, et al. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: from biology to therapeutic targeting. Biochim Biophys Acta (2016) 1863:449–63. doi:10.1016/j.bbamcr.2015.08.015
23. Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol (2014) 164:767–78. doi:10.1111/bjh.12725
24. Ebinger S, Özdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, Grunert M, et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell (2016) 30:849–62. doi:10.1016/j.ccell.2016.11.002
25. Gordon PM, Dias S, Williams DA. Cytokines secreted by bone marrow stromal cells protect c-KIT mutant AML cells from c-KIT inhibitor-induced apoptosis. Leukemia (2014) 28:2257–60. doi:10.1038/leu.2014.212
26. Weisberg E, Liu Q, Zhang X, Nelson E, Sattler M, Liu F, et al. Selective Akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PLoS One (2013) 8:e56473. doi:10.1371/journal.pone.0056473
27. Bjorklund CC, Baladandayuthapani V, Lin HY, Jones RJ, Kuiatse I, Wang H, et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: therapeutic implications. Leukemia (2014) 28:373–83. doi:10.1038/leu.2013.174
28. Nefedova Y, Landowski TH, Dalton WS. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia (2003) 17:1175–82. doi:10.1038/sj.leu.2402924
29. Alonso S, Su M, Jones JW, Ganguly S, Kane MA, Jones RJ, et al. Human bone marrow niche chemoprotection mediated by cytochrome P450 enzymes. Oncotarget (2015) 6:14905–12. doi:10.18632/oncotarget.3614
30. Hall BM, Gibson LF. Regulation of lymphoid and myeloid leukemic cell survival: role of stromal cell adhesion molecules. Leuk Lymphoma (2004) 45:35–48. doi:10.1080/1042819031000139620
31. Mallampati S, Leng X, Ma H, Zeng J, Li J, Wang H, et al. Tyrosine kinase inhibitors induce mesenchymal stem cell-mediated resistance in BCR-ABL+ acute lymphoblastic leukemia. Blood (2015) 125:2968–73. doi:10.1182/blood-2014-05-576421
32. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood (2016) 128:253–64. doi:10.1182/blood-2015-07-655860
33. Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest (2007) 117:1049–57. doi:10.1172/JCI30235
34. Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell (2015) 16:254–67. doi:10.1016/j.stem.2015.02.014
35. Akers SM, Rellick SL, Fortney JE, Gibson LF. Cellular elements of the subarachnoid space promote ALL survival during chemotherapy. Leuk Res (2011) 35:705–11. doi:10.1016/j.leukres.2010.12.031
36. Gaynes JS, Jonart LM, Zamora EA, Naumann JA, Gossai NP, Gordon PM. The central nervous system microenvironment influences the leukemia transcriptome and enhances leukemia chemo-resistance. Haematologica (2016) 102(4):e136–9. doi:10.3324/haematol.2016.152926
37. Krause S, Pfeiffer C, Strube S, Alsadeq A, Fedders H, Vokuhl C, et al. Mer tyrosine kinase promotes the survival of t(1;19)-positive acute lymphoblastic leukemia (ALL) in the central nervous system (CNS). Blood (2015) 125:820–30. doi:10.1182/blood-2014-06-583062
38. Naumann JA, Gordon PM. In vitro model of leukemia cell migration across the blood-cerebrospinal fluid barrier. Leuk Lymphoma (2016) 58(7):1747–9. doi:10.1080/10428194.2016.1254778
39. Akers SM, O’Leary HA, Minnear FL, Craig MD, Vos JA, Coad JE, et al. VE-cadherin and PECAM-1 enhance ALL migration across brain microvascular endothelial cell monolayers. Exp Hematol (2010) 38:733–43. doi:10.1016/j.exphem.2010.05.001
40. Monnot AD, Zheng W. Culture of choroid plexus epithelial cells and in vitro model of blood-CSF barrier. Methods Mol Biol (2013) 945:13–29. doi:10.1007/978-1-62703-125-7_2
41. Lazarevic I, Engelhardt B. Modeling immune functions of the mouse blood-cerebrospinal fluid barrier in vitro: primary rather than immortalized mouse choroid plexus epithelial cells are suited to study immune cell migration across this brain barrier. Fluids Barriers CNS (2016) 13:2. doi:10.1186/s12987-016-0027-0
42. Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (2013) 501:373–9. doi:10.1038/nature12517
43. Williams MTS, Yousafzai YM, Elder A, Rehe K, Bomken S, Frishman-Levy L, et al. The ability to cross the blood-cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukaemia blasts. Blood (2016) 127(16):1998–2006. doi:10.1182/blood-2015-08-665034
44. Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature (2009) 459:1000–4. doi:10.1038/nature08020
45. Bürger B, Zimmermann M, Mann G, Kühl J, Löning L, Riehm H, et al. Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol (2003) 21:184–8. doi:10.1200/JCO.2003.04.096
46. Mahmoud HH, Rivera GK, Hancock ML, Krance RA, Kun LE, Behm FG, et al. Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med (1993) 329:314–9. doi:10.1056/NEJM199307293290504
47. Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, et al. Immunologic privilege in the central nervous system and the blood-brain barrier. J Cereb Blood Flow Metab (2013) 33:13–21. doi:10.1038/jcbfm.2012.153
48. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol (2012) 33:579–89. doi:10.1016/j.it.2012.07.004
49. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12:623–35. doi:10.1038/nri3265
50. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol (2017) 18:123–31. doi:10.1038/ni.3666
51. Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest (2010) 120:1368–79. doi:10.1172/JCI41911
52. Meeker RB, Williams K, Killebrew DA, Hudson LC. Cell trafficking through the choroid plexus. Cell Adh Migr (2012) 6:390–6. doi:10.4161/cam.21054
53. Jacobelli J, Estin Matthews M, Chen S, Krummel MF. Activated T cell trans-endothelial migration relies on myosin-IIA contractility for squeezing the cell nucleus through endothelial cell barriers. PLoS One (2013) 8:e75151. doi:10.1371/journal.pone.0075151
54. Krummel MF, Friedman RS, Jacobelli J. Modes and mechanisms of T cell motility: roles for confinement and myosin-IIA. Curr Opin Cell Biol (2014) 30:9–16. doi:10.1016/j.ceb.2014.05.003
55. Wigton EJ, Thompson SB, Long RA, Jacobelli J. Myosin-IIA regulates leukemia engraftment and brain infiltration in a mouse model of acute lymphoblastic leukemia. J Leukoc Biol (2016) 100(1):143–53. doi:10.1189/jlb.1A0815-342R
56. Holland M, Castro FV, Alexander S, Smith D, Liu J, Walker M, et al. RAC2, AEP, and ICAM1 expression are associated with CNS disease in a mouse model of pre-B childhood acute lymphoblastic leukemia. Blood (2011) 118:638–49. doi:10.1182/blood-2010-09-307330
57. Cario G, Izraeli S, Teichert A, Rhein P, Skokowa J, Möricke A, et al. High interleukin-15 expression characterizes childhood acute lymphoblastic leukemia with involvement of the CNS. J Clin Oncol (2007) 25:4813–20. doi:10.1200/JCO.2007.11.8166
58. Lin D, Liu C, Xue M, Liu R, Jiang L, Yu X, et al. The role of interleukin-15 polymorphisms in adult acute lymphoblastic leukemia. PLoS One (2010) 5:e13626. doi:10.1371/journal.pone.0013626
59. Wu S, Fischer L, Gökbuget N, Schwartz S, Burmeister T, Notter M, et al. Expression of interleukin 15 in primary adult acute lymphoblastic leukemia. Cancer (2010) 116:387–92. doi:10.1002/cncr.24729
60. Williams MT, Yousafzai Y, Cox C, Blair A, Carmody R, Sai S, et al. Interleukin-15 enhances cellular proliferation and upregulates CNS homing molecules in pre-B acute lymphoblastic leukemia. Blood (2014) 123:3116–27. doi:10.1182/blood-2013-05-499970
61. Hühmer AF, Biringer RG, Amato H, Fonteh AN, Harrington MG. Protein analysis in human cerebrospinal fluid: physiological aspects, current progress and future challenges. Dis Markers (2006) 22:3–26. doi:10.1155/2006/158797
62. Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest (1999) 103:807–15. doi:10.1172/JCI5150
63. Scrideli CA, Queiroz RP, Takayanagui OM, Bernardes JE, Tone LG. Polymerase chain reaction on cerebrospinal fluid cells in suspected leptomeningeal involvement in childhood acute lymphoblastic leukemia: comparison to cytomorphological analysis. Diagn Mol Pathol (2003) 12(3):124–7.
64. Ranta S, Nilsson F, Harila-Saari A, Saft L, Tani E, Söderhäll S, et al. Detection of central nervous system involvement in childhood acute lymphoblastic leukemia by cytomorphology and flow cytometry of the cerebrospinal fluid. Pediatr Blood Cancer (2014) 62:951–6. doi:10.1002/pbc.25363
65. Pine SR, Yin C, Matloub YH, Sabaawy HE, Sandoval C, Levendoglu-Tugal O, et al. Detection of central nervous system leukemia in children with acute lymphoblastic leukemia by real-time polymerase chain reaction. J Mol Diagn (2005) 7:127–32. doi:10.1016/S1525-1578(10)60018-9
66. Martínez-Laperche C, Gómez-García AM, Lassaletta Á, Moscardó C, Vivanco JL, Molina J, et al. Detection of occult cerebrospinal fluid involvement during maintenance therapy identifies a group of children with acute lymphoblastic leukemia at high risk for relapse. Am J Hematol (2013) 88:359–64. doi:10.1002/ajh.23407
67. Hunger SP, Galili N, Carroll AJ, Crist WM, Link MP, Cleary ML. The t(1;19)(q23;p13) results in consistent fusion of E2A and PBX1 coding sequences in acute lymphoblastic leukemias. Blood (1991) 77:687–93.
68. Jeha S, Pei D, Raimondi SC, Onciu M, Campana D, Cheng C, et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia (2009) 23:1406–9. doi:10.1038/leu.2009.42
69. Huey MG, Minson KA, Earp HS, DeRyckere D, Graham DK. Targeting the TAM receptors in leukemia. Cancers (Basel) (2016) 8:101. doi:10.3390/cancers8110101
70. Rentzos M, Rombos A. The role of IL-15 in central nervous system disorders. Acta Neurol Scand (2012) 125:77–82. doi:10.1111/j.1600-0404.2011.01524.x
71. Marçais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:450. doi:10.3389/fimmu.2013.00450
72. Frishman-Levy L, Shemesh A, Bar-Sinai A, Ma C, Ni Z, Frenkel S, et al. Central nervous system acute lymphoblastic leukemia: role of natural killer cells. Blood (2015) 125:3420–31. doi:10.1182/blood-2014-08-595108
73. Kato I, Nishinaka-Arai Y, Nakamura M, Akarca AU, Niwa A, Ozawa H, et al. VEGFA – a new therapeutic target in CNS leukemia. Blood (2016) 128:911.
74. Tang YT, Jiang F, Guo L, Si MY, Jiao XY. Expression and significance of vascular endothelial growth factor A and C in leukemia central nervous system metastasis. Leuk Res (2013) 37:359–66. doi:10.1016/j.leukres.2012.10.008
75. Hicks C, Ramani R, Karlson C, Sitthi-Amorn J, Douglas J, Vijayakumar V, et al. Molecular analysis of central nervous system disease spectrum in childhood acute lymphoblastic leukemia. Clin Med Insights Oncol (2016) 10:5–15. doi:10.4137/CMO.S18180
76. van der Velden VHJ, de Launaij D, de Vries JF, de Haas V, Sonneveld E, Voerman JSA, et al. New cellular markers at diagnosis are associated with isolated central nervous system relapse in paediatric B-cell precursor acute lymphoblastic leukaemia. Br J Haematol (2015) 172:769–81. doi:10.1111/bjh.13887
Keywords: acute lymphoblastic leukemia, central nervous system, chemoresistance, migration, microenvironment, niche
Citation: Gossai NP and Gordon PM (2017) The Role of the Central Nervous System Microenvironment in Pediatric Acute Lymphoblastic Leukemia. Front. Pediatr. 5:90. doi: 10.3389/fped.2017.00090
Received: 20 February 2017; Accepted: 10 April 2017;
Published: 26 April 2017
Edited by:
Brenton Garrett Mar, Dana Farber Cancer Institute, USAReviewed by:
Yong-mi Kim, Children’s Hospital of Los Angeles, USARachel Rau, Baylor College of Medicine, USA
Copyright: © 2017 Gossai and Gordon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter M. Gordon, Z29yZDAwNDdAdW1uLmVkdQ==
†Present address: Nathan P. Gossai, Children’s Minnesota Cancer and Blood Disorders, Minneapolis, MN, USA