 Rita Consolini
Rita Consolini Giorgio Costagliola
Giorgio Costagliola Davide Spatafora
Davide Spatafora- 1Laboratory of Immunology, Department of Clinical and Experimental Medicine, Division of Pediatrics, University of Pisa, Pisa, Italy
- 2Clinical Immunology and Allergy Unit, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
Primary immune thrombocytopenia (ITP) is the most common cause of thrombocytopenia in children and adolescents and can be considered as a paradigmatic model of autoimmune disease. This second part of our review describes the clinical presentation of ITP, the diagnostic approach and overviews the current therapeutic strategies. Interestingly, it suggests an algorithm useful for differential diagnosis, a crucial process to exclude secondary forms of immune thrombocytopenia (IT) and non-immune thrombocytopenia (non-IT), which require a different therapeutic management. Advances in understanding the pathogenesis led to new therapeutic targets, as thrombopoietin receptor agonists, whose role in treatment of ITP will be discussed in this work.
Introduction
The platelet threshold necessary to make diagnosis of thrombocytopenia has changed during years (1), and for immune thrombocytopenia (ITP) working group thrombocytopenia is defined as a condition characterized by a platelet count lower than 100,000 platelets per microliter (2).
Primary ITP is the most common cause of thrombocytopenia, having an estimated incidence of 4–9 cases out of 100,000 people per year (3–6), with about half of the pediatric cases occurring in previously healthy children.
The first part of this review described the pathogenesis of the disease, with special attention to the role of innate immune system and impairment in megakaryopoiesis (7). In this second part, we focus on the clinical aspects of ITP, particularly on the differential diagnosis and new target therapies. The process of differential diagnosis has the aim to distinguish ITP from secondary IT (caused by infections, immune defects, and other pathologies) and non-IT, particularly inherited diseases, because all these conditions require different treatments.
Therapy of ITP was historically based on the progressive use of immunoglobulins, corticosteroids, and immunosuppressive agents: following the recent advances in the study of the pathogenesis of the disease, new therapeutic targets have been identified, potentially leading to innovative therapeutic strategies.
Classification
Immune thrombocytopenia can be classified according to etiology, disease evolution and age of onset. Etiologic classification will be discussed separately, to introduce the more common forms of secondary IT.
According to disease evolution, it is possible to identify three categories of ITP: newly diagnosed ITP; persistent ITP, still present after 3 months from diagnosis; and chronic ITP, lasting 12 or more months after diagnosis (2), which represents about 20% of the total cases of ITP in childhood (8).
IT affecting young children is typically acute and self-remitting, and primary forms are the most common in this age. Adolescents IT has an intermediate phenotype between childhood-onset and adult-onset forms (9), showing a higher rate of chronicity and a greater percentage of secondary IT.
Etiology of ITP
Etiologic classification divides two categories of ITP: primary ITP and secondary IT.
The primary form of IT, classically defined “idiopathic,” is often seen in childhood and triggered by non-specific viral infections (upper respiratory or gastrointestinal infections): in some cases acute infections by Epstein–Barr virus, cytomegalovirus, parvovirus, rubella, mumps, and varicella have been identified as triggers of ITP (10–12).
Secondary IT has a complex etiology, as specific infections, drugs or vaccinations and immunologic abnormalities, including immunodeficiencies, can be involved in its pathogenesis.
Infections
Infectious diseases caused by HIV, HCV, Helicobacter pylori, and dengue virus can be responsible of secondary IT, usually with chronic course (13–17), trough different mechanisms, like molecular mimicry, modulation of the immune system’s activity or suppression of bone marrow production (18). Association between pulmonary and extra-pulmonary TBC and IT is documented in only a few case reports (19, 20).
Drugs
Secondary IT can be caused by the assumption of drugs and vaccines: drug-induced IT, ascribable in most of cases to the assumption of certain antibiotics, non-steroidal anti-inflammatory drugs, and antivirals, it is often not recognized, resulting in recurrent non-explained episodes of thrombocytopenia (21), that usually show a complete recovery after the withdrawal of the drug. Less commonly than in adulthood, it is possible to observe in childhood the development of heparin-induced thrombocytopenia (22).
Patients who received multiple transfusions are at risk for the development of posttransfusion purpura (PTP), a rare form of secondary IT with a high rate of bleeding: this is more frequent in multiparous female (18), but rare reports of PTP with pediatric onset are described (23).
Immunodeficiency
Immune thrombocytopenia is a possible manifestation of immunodeficiency, particularly common variable immunodeficiency (CVID), selective IgA deficiency, and DiGeorge’s syndrome. Is interesting to underline that, in humoral immune defects, the reduction of platelet count may appear years before the hypogammaglobulinemia (24–26).
Autoimmune Diseases
Systemic autoimmune diseases, such as systemic lupus erythematosus (SLE), Sjogren’s syndrome, and antiphospholipid syndrome, are associated with the development of IT (27): an isolated thrombocytopenia may represent the initial manifestation of SLE, preceding the diagnosis by several years (28, 29).
Current literature reports an association between IT and clinical and subclinical thyroid autoimmune diseases (Hashimoto’s and Basedow–Graves’ diseases), suggesting the presence of an overlap in the pathogenesis of these conditions (30, 31).
Moreover, many lymphoproliferative disorders may cause secondary ITP: in this category, the most frequent disease is autoimmune lymphoproliferative syndrome (32), mostly found in children aged under 3 years.
Neoplasia
Lymphatic malignancies, particularly non-Hodgkin’s lymphoma, represent a cause of IT (33), which rarely can be a paraneoplastic manifestation of a solid neoplasia (34, 35), mostly in adults.
Age-Related Considerations
In neonatal age, the most frequent form of IT is the alloimmune (36), caused by the production of maternal antibodies directed against platelet alloantigens.
In adolescent females, the possibility of an IT secondary to pregnancy should be considered (37).
Other Causes of Thrombocytopenia in Pediatric Age
Other forms of thrombocytopenia occurring during childhood and adolescence could mime ITP and secondary IT, particularly when platelet reduction is the only laboratory finding. Inherited thrombocytopenias, often misdiagnosed as ITP (38), are characterized by impairment in magakaryopoiesis and include a large variety of X-linked and autosomal diseases, commonly presenting with altered platelet size (39). Among them, Wiskott–Aldrich syndrome, caused by the mutation of WAS gene on chromosome X, usually comprehends the association of thrombocytopenia with small platelets, eczema, and immunodeficiency (40).
Thrombocytopenia occurs also in acute leukemia and primary bone marrow failure syndromes as Fanconi anemia, but in these cases, the association with other cytopenias helps making differential diagnosis (41).
Also lisosomial storage disorders, as Gaucher’s and Niemann–Pick’s disease, may present thrombocytopenia at clinical onset, usually accompanied by a considerable splenomegaly (42, 43).
Other less common conditions are indicated in Table 1, which summarizes the most relevant causes of secondary IT and non-IT in childhood and adolescence.
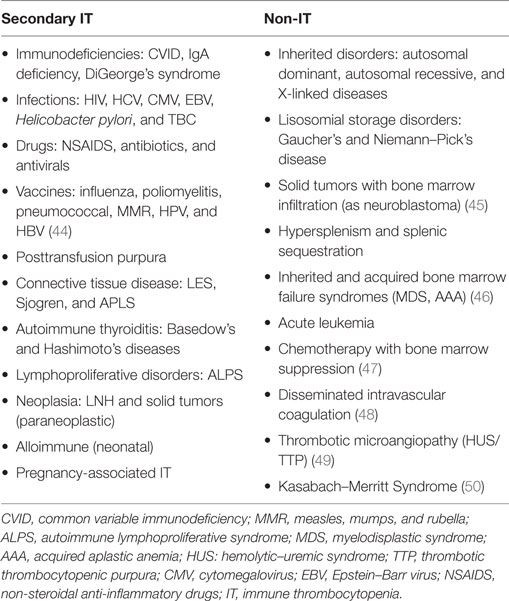
Table 1. Etiology of secondary IT and non-IT in childhood and adolescence.
Clinical Presentation
Immune thrombocytopenia is not rarely asymptomatic, being observed during routinely laboratory evaluations. In symptomatic cases, the most common presenting feature is epistaxis (51), followed by cutaneous and mucosal minor bleeding.
Severe bleeding rates are more common in childhood compared to adult patients: a review by Neunert et al. showed that severe bleeding occurs in 20.2% of children and 9.6% of adults (52).
The most severe bleeding event is ITP-associated intracranial hemorrhage (ICH): it represents a rare cause of pediatric stroke (53), affecting only 0.4% of children with ITP (52), but complicated by elevated mortality rates, reported between 12 and 25% in recent studies (54–58). ICH, as other severe bleedings, occurs mostly in children with other features of bleeding and in patients with a platelet count lower than 10,000/μl (59) and is often preceded by a precipitant factor, like head trauma (55, 60, 61).
An anamnesis positive for previous minor bleeding is a risk factor for severe haemorrhagic events (52), while the clinical relevance of an occult hemorrhage, often identified in urinary tract, is not completely defined, as studies reported different conclusions about the association with future overt bleeding and ICH (55, 62).
Diagnosis
Diagnosis of ITP remains one of exclusion, and differential diagnosis with secondary forms is crucial, because in these cases thrombocytopenia may be less responsive to conventional therapy, but only to the treatment of primary cause.
Interview should focus on the potential triggers of ITP (assumption of drugs, vaccines, and transfusions) and risk factors for secondary forms, as the presence of weight loss, chronic infections (HIV and HCV) and other immune-mediated disease. It is also important to investigate elements suggestive of inherited thrombocytopenia, as previous bleedings and positive familiar history.
During physical assessment, the clinician must search potential sites of bleeding (cutaneous and mucosal) and identify signs suggestive for secondary IT or other pathologies, by examining the presence of hepatosplenomegaly, abdominal masses, lymphoadenopathies, and bone pain.
Furthermore, we analyze the diagnostic the most relevant laboratory investigations in ITP, to introduce our diagnostic algorithm.
Laboratory Investigations
Full Blood Count with Citrate and Reticulocyte Determination
First laboratory step, has the role of excluding a pseudothrombocytopenia (EDTA related) (63) and the presence of other cytopenias. Patients with severe bleeding could show anemia, related to blood loss. In case of multiple cytopenias, diagnosis of ITP is unlikely and becomes mandatory to investigate for acute leukemia, lymphomas, bone marrow failure (aplastic anemia), and neoplastic infiltration of bone marrow.
Mean Platelet Volume (MPV)
Mean platelet volume is useful in the first laboratory assessment and is normal or slightly high in patients with ITP, while it shows alterations (marco- or micro-thrombocythemia) in almost all patients with inherited thrombocytopenia. Due to the lack of validated thresholds, this exam is significant in presence of a great difference in MPV, as it happens in inherited thrombocytopenia, in which platelet volume can be 50–100% higher than normal values (64). In some particular cases, as mono- and biallelic Bernard–Soulier syndrome (65) and MYH-9-related disease, giant platelets may not be recognized by automatic counters, underestimating MPV and platelet count (66, 67).
Peripheral Blood Smear
It may demonstrate alterations on red blood cells (for example, schistocytes in HUS/TTP) and white cells (blasts in leukemia), which exclude diagnosis of ITP (68). Moreover, analysis of blood smear could be useful to identify alterations of platelet size and correct measurement of their diameters. Many of congenital thrombocytopenias have also changes in platelet morphology, recognizable with blood smear (69). The interpretation of blood smear requires experience, and access may be limited in resource poor regions, making it not always applicable as a first-line tool.
Reticulated Platelet Count (RPLT)
First described in 1969 (70), reticulated platelets are immature platelets circulating in the blood, containing a residual RNA. They can be analyzed with flow cytometry and give an indirect determination of thrombopoietic rate (71). Lack of standardization of methods and definition of threshold values make difficult the interpretation of RPLT value (72): even if the applicability of RPLT determination is not completely defined, recent works conclude that this is a promising tool to distinguish thrombocytopenia caused by bone marrow hypoproduction to that one due to platelet destruction (73). Moreover, a study by Thomas-Kaskel et al. demonstrated the correlation between reticulated platelet count and response to treatment (74).
Rh (D) Typing
This exam should be performed in those patients candidate for receiving therapy with anti-D immunoglobulins (75–77). Currently, this treatment is no longer licensed in Europe (51), where Rh typing is not yet recommended.
Autoantibodies
Antiplatelet antibodies showed absence of specificity for ITP, and therefore the determination is not routinely recommended (78). Other autoantibodies, particularly antinuclear (ANA) and antithyroid antibodies, may have a diagnostic role in identifying secondary IT or, respectively, patients at risk of developing chronic thrombocytopenia and thyroid diseases. Testing for these antibodies is particularly useful in patients with persistent or chronic ITP, as discussed in the Section “Prognosis and Sequelae.”
Bone Marrow Examination
The analysis of bone marrow of a patient with ITP would show an increase in number of megakaryocytes and absence of alterations in other cellular lines. In patients with isolated thrombocytopenia, diagnosis of acute leukemia or lymphoma is unlikely, and bone marrow biopsy and aspirate are rarely useful. Several authors agree affirming that bone marrow biopsy and aspirate must be performed in children and adolescents with atypical findings for ITP (51, 79–81).
Moreover, bone marrow examination should be performed in patients with absence of response to standard treatments, before the beginning of second-line therapies and the execution of splenectomy (82).
Algorithm for Differential Diagnosis
The algorithm we suggest for differential diagnosis of new onset thrombocytopenia, shown in Figure 1, is composed by one clinical step and three laboratory and radiological steps and is primarily directed to patients without acute and severe bleeding. We focused particularly on the exclusion of short-term life-threatening conditions, such as acute leukemia, lymphomas, and other neoplasia, while chronic infections and systemic autoimmune disease with partial expression are investigated in later steps. The algorithm is progressive, and therefore the investigations included in steps 3 and 4 are usually indicated only in patients with persistent and chronic ITP.
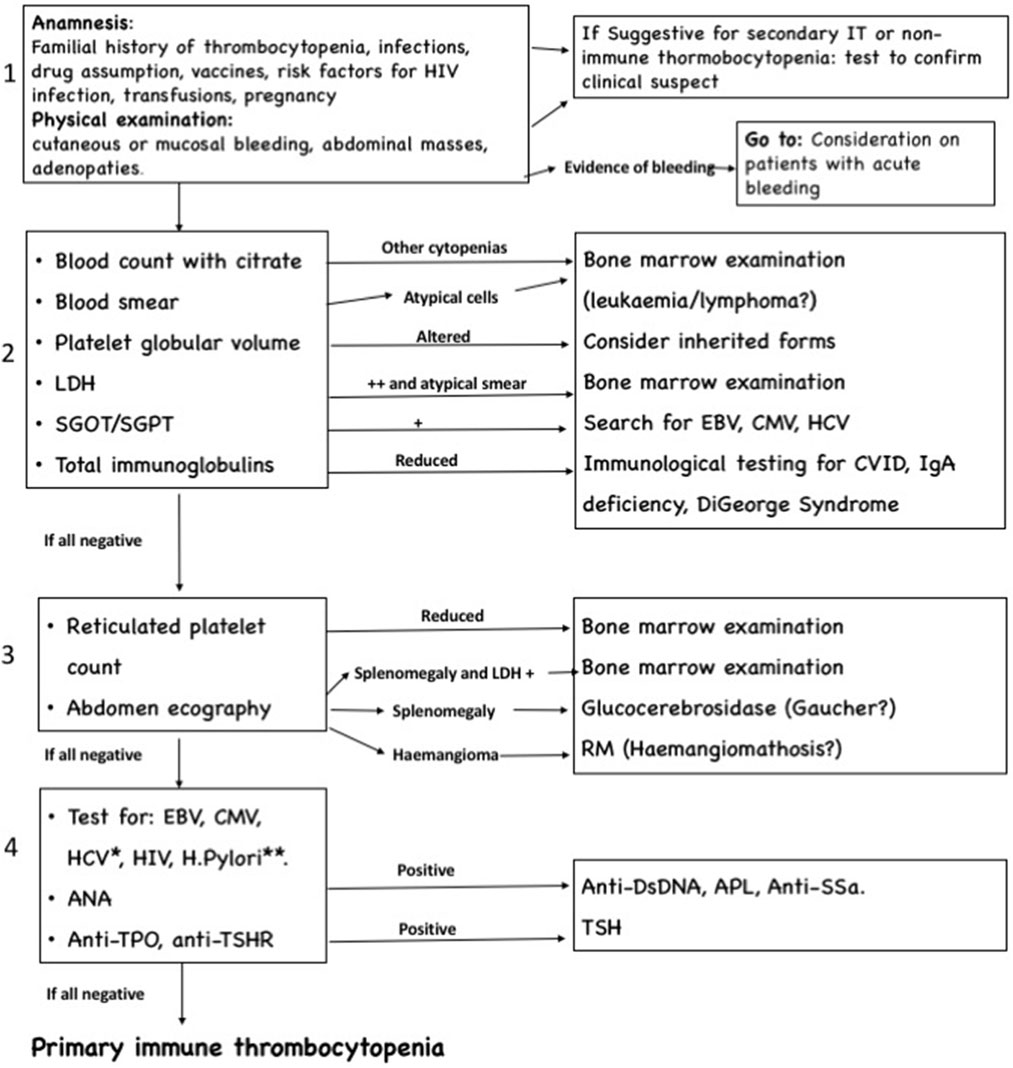
Figure 1. Algorithm for differential diagnosis of immune thrombocytopenia, composed by four progressive steps. *Testing for Epstein–Barr virus (EBV), cytomegalovirus (CMV), and HCV is recommended if not done in previous step. **Helycobacter pylori testing should be performed in high prevalence area or clinical suspect.
The clinical step remains fundamental: in case of severe bleeding signs, it is mandatory to treat the patient, and the required investigations are different (see Consideration on Patients with Acute Bleeding). Moreover, if anamnesis or physical assessment shows elements indicative for secondary IT (abdominal masses and adenopathies), the algorithm becomes not necessary, and the laboratory and radiological approach must start with investigations directed to confirm the etiologic hypothesis suggested by the clinical findings.
The second step comprehends laboratory exams directed to identify the conditions that more frequently cause secondary IT and non-ITP, including inherited thrombocytopenia, infections, immunodeficiency, and lymphoid malignancies.
The third step includes an abdominal echography, useful to recognize alterations in liver, spleen, and abdominal lymph nodes, not always appreciable during clinical examination. This step also considers the determination of reticulated platelet count: despite the lack of standardization of values and difficulties in interpretations, this investigation, when available, can give important information about thrombopoietic rate, and thus remains an option to consider.
The last step comprehends investigations for autoimmune diseases and chronic infections. Determination of ANA is also important to predict the evolution to a chronic form (see Prognosis and Sequelae).
Consideration on Patients with Acute Bleeding
In this case, primary diagnostic approach should exclude conditions, such as HUS/TTP, DIC, antiphospholipid syndrome, coagulation abnormalities, and neoplasia (promyelocytic leukemia). First-step analyses include determination of full blood count, blood smear (if available), coagulation tests, APL, LDH, and D-dimer, accompanied by the evaluation of the bleeding site (echography, endoscopy, and neuroimaging). In case of negative results or resolution of the bleeding episode, it is possible to apply the diagnostic algorithm discussed above.
Therapy
About two out of three pediatric patients with ITP show a spontaneous improvement in platelet count in 6 months without necessity of medical treatment, and those remissions are usually sustained. Most of patients with newly diagnosed ITP do not show signs of bleeding, and can be managed with a “watch and see” strategy (83–86).
There is no absolute consensus about the platelet threshold necessary to start treatment in ITP: 1996 guidelines of the American Society of Hematology recommended to treat patients with a platelet count lower than 10,000/μl and minor purpura or those one with a count lower 20,000/μl and significant bleeding (87). An update published in 2011 suggested that children without bleeding or with mild bleeding should be managed only with observations, regardless of platelet count (88). Despite these recommendations, most patients with low risk of bleeding are currently treated (89).
First-line Treatment
Prednisone–Prednisolone
All guidelines support the use of corticosteroids in the first-line treatment of ITP. Oral prednisone is often effective in inducing response in pediatric patients when administered at doses of 1–2 mg/kg for 7–14 days and maintains efficacy also at higher doses (4 mg/kg/day) for 3 or 4 days, raising platelet count over 50,000/μl in the first 72 h in 72–88% of patients (78, 90, 91).
However, due to the adverse effects of a prolonged treatment with corticosteroids in children, those drugs must be used only for short periods, to maintain a hemostatic platelet count (78).
Intravenous Immunoglobulins (IVIg)
Immunoglobulins have been used for ITP since 1981 (92, 93), for the effect of modulation on immune system. The treatment induces a raise in platelet count in 80% of pediatric patients, obtaining an effect in the first 48 h more frequent than corticosteroids (94). IVIg are usually administered in a single dose of 0.8–1 g/kg, with the chance of using a second dose in case of incomplete response, even if also lower doses (0.6 g/kg) are reported to be effective (95). Adverse effects include headache and fever and are more common when used doses are greater than 1 g/kg for consecutive days (91).
Intravenous Anti-D Immunoglobulin
Rh-positive children could receive short infusions of anti-D immunoglobulin, with a recommended dose of 50–75 µg/kg (78). This therapeutic strategy has a response rate greater than 50% and acts more rapidly than IVIG (76, 77, 96, 97).
However, in patients with comorbidity, the treatment has been associated with severe hemolysis, acute renal failure, and disseminated intravascular coagulation, and therefore anti-D immunoglobulin administration should require a careful selection of patients and post-therapy monitoring, as concluded by Despotovic et al. (98).
Second-line Therapies
High-Dose Corticosteroids
High-dose methylprednisolone has been used as an alternative to IVIg, showing comparable response rates (99, 100).
Dexamethasone (28–40 mg/m2/day) has been used in pediatric patients with chronic refractory ITP, obtaining response rates greater than 80%, with and a mean duration of the response of 26 months (101): moreover, psychiatric adverse effects, such as insomnia and aggressive behavior, are extremely frequent (102), and this makes dexamethasone only a second-line therapeutic alternative.
Rituximab
This anti-CD20 antibody, used in other autoimmune diseases and B-cell lymphoma, has been used in chronic refractory ITP often showing response rates greater than 60% (103–106), even though in a study by Bennett et al. only 31% of patients responded (107). However, follow-up studies have shown that sustained response is uncommon (108, 109), and safety profile is unclear.
Danazol
This attenuated androgen is successfully used in second-line treatment of adult patients with ITP, particularly in elderly patients (110). There are only a few studies about its use in pediatric patients, showing a good effectiveness without significant adverse reactions. Unfortunately, danazol can accelerate bone growth, and this limits its applicability in prepuberal patients (111, 112).
Thrombopoietin Receptor Agonists (TPO-RAs)
Since the discovery of the role of thrombopoietin (TPO) in ITP several thrombopoietic drugs was tested (113), and in 2008 FDA approved two TPO receptor agonists for non-responsive ITP in adults: romiplostim and eltrombopag (114, 115). Romiplostim acts on TPO-binding subunit of the receptor and is administered subcutaneously weekly (116). It is not yet approved for childhood-onset ITP, although in several studies it showed a 50–80% response rate, without severe adverse effects (117–125).
Eltrombopag acts binding the transmembrane domain of TPO receptor and is administered orally daily (116). It showed response rates greater than 60% in two randomized trials, associated with a good tolerability (126, 127), so in 2015 FDA has approved it for the use in childhood-onset disease. Reported adverse effects consist in an increased risk of hepatic damage and cataract.
Recently, avatrombopag, a new drug with an eltrombopag-like mechanism of action, was included in clinical trials for adults, showing response rate similar to other TPO-RAs in absence of severe adverse effects (128). In summary, TPO-RAs seem to be safe end effective also in childhood-onset refractory ITP.
Use of Immunosuppressors
There are only a few studies investigating the role of immunosuppressive agents, single or in combination, in patients with refractory ITP, and experience in childhood is not enough strong to give specific recommendations (78). Azathioprine, used in several autoimmune pediatric diseases, is still an option for the treatment of adult patients with ITP, particularly in chronic ITP and when splenectomy is contraindicated or has been ineffective (129). Response is detectable after about 4 months, and adverse effects, such as posttreatment leukemia, are extremely rare (130).
In pediatric age, cyclosporine is used in several conditions (organ transplants, autoimmune hepatitis, acquired aplastic anemia, juvenile dermatomyositis, and nephrotic syndrome) while its applicability in ITP is not completely defined. In adult patients, this drug often shows positive response rates both in single therapy and in association with steroids, with possibility of sustained remission after discontinuation of treatment (131, 132). Despite the lack of evidence in childhood-onset ITP and the necessity of further studies, these data support the potential utility of immunosuppressive agents as a second-line treatment in refractory ITP.
Splenectomy
Several studies reported a response in almost 85% of patients after splenectomy, with a 20–25% of relapses during the following years (133–135). Many works investigated the role of potential predictors of response to splenectomy in children and adults and suggested that female sex, younger age, response to steroids, and higher platelet count could be positive prognostic determinants, although the role of response to steroids is not univocally accepted (135–141).
Patients who received splenectomy are at risk of developing relevant adverse effects, particularly infections and sepsis by capsulated bacteria, with reported mortality rates of 3% (133), and thus the procedure is rarely recommended in children (78), being usually performed only in selected cases.
New Therapeutic Targets
There are ongoing trials about other classes of drugs for ITP, currently limited to application in adulthood. New potential targets are represented by interaction between T-cells and antigen-presenting cells (anti-CD40L antibodies) (142), platelet phagocytosis [SYK inhibitors and interference with FcR binding on macrophages (143)], activation of B-cells (anti-CD52 or alemtuzumab) (144) and T-cells [anti-IL-2R or daclizumab (145)], and TH1 expansion (anti-CD16) (146, 147). Figure 2 summarizes the new therapeutic targets for ITP and the corresponding drug classes.
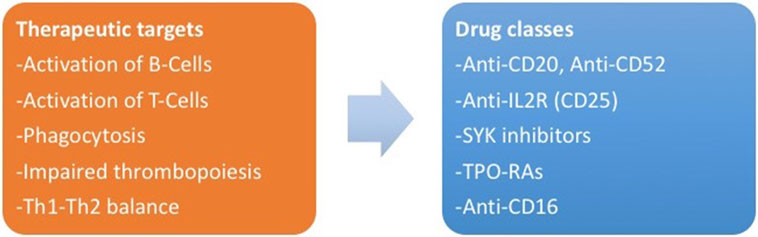
Figure 2. (Left) The new therapeutic targets identified by the study of pathogenesis of immune thrombocytopenia. (Right) Corresponding drug classes.
Emergency Treatment in Childhood
In both adult and childhood-onset disease, in cases of life-threatening hemorrhage or organ damage, it is necessary to obtain a rapid raise in platelet count, to reduce bleeding risk. Consensus document of 2010 recommends to administer a high dose of platelet (two to three times larger than usual), accompanied with intravenous corticosteroids and IVIg. In particular cases, such as unstoppable bleeding, it is possible to consider the execution of a splenectomy in emergency (78).
Prognosis and Sequelae
Immune thrombocytopenia in childhood is usually self-remitting, while there is the possibility of reactivation of the disease following viral infections or other triggers. The major sequelae of acute episodes are represented by permanent neurologic damages, defined as epilepsy, cognitive and learning disorder, and paresis, that can be detected in patients surviving the acute event of ICH (55, 148).
Adolescents are more likely to develop a chronic disease: a recent work by Heitink-Pollè et al. identified some predictors of chronic ITP, including female sex, age >11, insidious onset, absence of a trigger (infection or vaccination), and ANA positivity (149).
Patients with chronic ITP can undercover secondary misdiagnosed forms, as chronic infections, CVID, or other autoimmune pathologies, suggesting that this subset of patients need continuous reevaluation and discussion of primary diagnosis. Moreover, rarely, these patients may show an evolution to SLE: since there are no specific predictors of this progression (150), a study by Panzer et al. suggested that combined assessment of ANA and anti-DsDNA may have a role in identifying subjects at higher risk (151), who need to be periodically monitored.
Conclusion
The centenary of ITP traced history characterized of new progressive knowledge, making it a paradigmatic model of autoimmune disease. In this issue, a second part of a revision of ITP story, we focused on ITP diagnostic approach. By combining physical examinations and laboratory findings, we designed a diagnostic algorithm, to dissect the complex diagnosis, substantially based on the exclusion of the multiple possible concurrent causes of thrombocytopenia. We described conventional therapy of ITP and focused on the new therapeutic targets.
Author Contributions
All the authors contributed to the work presented in this paper, wrote and reviewed the paper, and provided approval of the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ruggeri M, Fortuna S, Rodeghiero F. Heterogeneity of terminology and clinical definitions in adult idiopathic thrombocytopenic purpura: a critical appraisal from a systematic review of the literature. Haematologica (2008) 93(1):98–103. doi:10.3324/haematol.11582
2. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood (2009) 113(11):2386–93. doi:10.1182/blood-2008-07-162503
3. Schoonen WM, Kucera G, Coalson J, Li L, Rutstein M, Mowat F, et al. Epidemiology of immune thrombocytopenic purpura in the general practice research database. Br J Haematol (2009) 145(2):235–44. doi:10.1111/j.1365-2141.2009.07615.x
4. Segal JB, Powe NR. Prevalence of immune thrombocytopenia: analyses of administrative data. J Thromb Haemost (2006) 4(11):2377–83. doi:10.1111/j.1538-7836.2006.02147.x
5. Deane S, Teuber SS, Gershwin ME. The geoepidemiology of immune thrombocytopenic purpura. Autoimmun Rev (2010) 9(5):A342–9. doi:10.1016/j.autrev.2009.11.020
6. Higashigawa M, Maeyama T, Yoshino A, Matsuda K, Ito M, Maji T, et al. Incidence of childhood primary immune thrombocytopenic purpura. Pediatr Int (2015) 57(5):1041–3. doi:10.1111/ped.12788
7. Consolini R, Legitimo A, Caparello MC. The centenary of immune thrombocytopenia – part 1: revising nomenclature and pathogenesis. Front Pediatr (2016) 4:102. doi:10.3389/fped.2016.00102
8. British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol (2003) 120(4):574–96. doi:10.1046/j.1365-2141.2003.04131.x
9. Lowe EJ, Buchanan GR. Idiopathic thrombocytopenic purpura diagnosed during the second decade of life. J Pediatr (2002) 141(2):253–8. doi:10.1067/mpd.2002.125909
10. Yenicesu I, Yetgin S, Ozyurek E, Aslan D. Virus-associated immune thrombocytopenic purpura in childhood. Pediatr Hematol Oncol (2002) 19(6):433–7. doi:10.1080/08880010290097233
11. Smalisz-Skrzypczyk K, Romiszewski M, Matysiak M, Demkow U, Pawelec K. The influence of primary cytomegalovirus or Epstein-Barr virus infection on the course of idiopathic thrombocytopenic purpura. Adv Exp Med Biol (2016) 878:83–8. doi:10.1007/5584_2015_162
12. Kossiva L, Kyriakou D, Mitsioni A, Garoufi A. Acute renal failure in a child with thrombocytopenic purpura caused by acute Epstein-Barr virus infection after treatment with anti-D immunoglobulin. Pediatr Emerg Care (2013) 29(6):748–50. doi:10.1097/PEC.0b013e318294f3a5
13. Shah I. Immune thrombocytopenic purpura: a presentation of HIV infection. J Int Assoc Provid AIDS Care (2013) 12(2):95–7. doi:10.1177/1545109712462068
14. Pacifico L, Osborn JF, Tromba V, Romaggioli S, Bascetta S, Chiesa C. Helicobacter pylori infection and extragastric disorders in children: a critical update. World J Gastroenterol (2014) 20(6):1379–401. doi:10.3748/wjg.v20.i6.1379
15. Abdollahi A, Shoar S, Ghasemi S, Zohreh OY. Is Helicobacter pylori infection a risk factor for idiopathic thrombocytopenic purpura in children? Ann Afr Med (2015) 14(4):177–81. doi:10.4103/1596-3519.153357
16. Sakuraya M, Murakami H, Uchiumi H, Hatsumi N, Akiba T, Yokohama A, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol (2002) 68(1):49–53. doi:10.1034/j.1600-0609.2002.00509.x
17. Ramirez-Fonseca T, Segarra-Torres A, Jaume-Anselmi F, Ramirez-Rivera J. Dengue fever: a rare cause of immune thrombocytopenia. Bol Asoc Med P R (2015) 107(2):51–3.
18. Cines DB, Liebman H, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol (2009) 46(1 Suppl 2):S2–14. doi:10.1053/j.seminhematol.2008.12.005
19. Arora SK, Shah D. Severe thrombocytopenia secondary to tubercular lymphadenitis. Ann Trop Paediatr (2010) 30(2):157–9. doi:10.1179/146532810X12703902516365
20. Akyildiz B, Uzel N, Yanni D, Citak A, Karabocuoglu M, Ucsel R. Immune thrombocytopenic purpura associated with pulmonary tuberculosis. Turk J Pediatr (2009) 51(3):271–4.
21. Reese JA, Nguyen LP, Buchanan GR, Curtis BR, Terrell DR, Vesely SK, et al. Drug-induced thrombocytopenia in children. Pediatr Blood Cancer (2013) 60(12):1975–81. doi:10.1002/pbc.24682
22. Takemoto CM, Streiff MB. Heparin-induced thrombocytopenia screening and management in pediatric patients. Hematology (2011) 2011:162–9. doi:10.1182/asheducation-2011.1.162
23. Kashyap R, Venkatesh S, Choudhry VP. Post transfusion purpura in a thalassemic child. Indian Pediatr (1997) 34(3):246–7.
24. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program (2009) 1:139–43. doi:10.1182/asheducation-2009.1.139
25. Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al. Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun Rev (2016) 15(9):877–82. doi:10.1016/j.autrev.2016.07.011
26. Heeney MM, Zimmerman SA, Ware RE. Childhood autoimmune cytopenia secondary to unsuspected common variable immunodeficiency. J Pediatr (2003) 143(5):662–5. doi:10.1067/S0022-3476(03)00445-1
27. Liu Y, Chen S, Sun Y, Lin Q, Liao X, Zhang J, et al. Clinical characteristics of immune thrombocytopenia associated with autoimmune disease: a retrospective study. Medicine (2016) 95(50):e5565. doi:10.1097/MD.0000000000005565
28. Bashal F. Hematological disorders in patients with systemic lupus erythematosus. Open Rheumatol J (2013) 7:87–95. doi:10.2174/1874312901307010087
29. Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (2010) 49(12):2243–54. doi:10.1093/rheumatology/keq269
30. Cheung E, Liebman HA. Thyroid disease in patients with immune thrombocytopenia. Hematol Oncol Clin North Am (2009) 23(6):1251–60. doi:10.1016/j.hoc.2009.08.003
31. Cordiano I, Betterle C, Spadaccino CA, Soini B, Girolami A, Fabris F. Autoimmune thrombocytopenia (AITP) and thyroid autoimmune disease (TAD): overlapping syndromes? Clin Exp Immunol (1998) 113(3):373–8. doi:10.1046/j.1365-2249.1998.00677.x
32. Li P, Huang P, Yang Y, Hao M, Peng H, Li F. Updated understanding of autoimmune lymphoproliferative syndrome (ALPS). Clin Rev Allergy Immunol (2016) 50(1):55–63. doi:10.1007/s12016-015-8466-y
33. Stern M, Buser AS, Lohri A, Tichelli A, Nissen-Druey C. Autoimmunity and malignancy in hematology – more than an association. Crit Rev Oncol Hematol (2007) 63(2):100–10. doi:10.1016/j.critrevonc.2007.02.002
34. Krauth MT, Puthenparambil J, Lechner K. Paraneoplastic autoimmune thrombocytopenia in solid tumors. Crit Rev Oncol Hematol (2012) 81(1):75–81. doi:10.1016/j.critrevonc.2011.02.004
35. Nenova IS, Valcheva MY, Beleva EA, Tumbeva DY, Yaneva MP, Rancheva EL, et al. Autoimmune phenomena in patients with solid tumors. Folia Med (2016) 58(3):195–9. doi:10.1515/folmed-2016-0026
36. Curtis BR. Recent progress in understanding the pathogenesis of fetal and neonatal alloimmune thrombocytopenia. Br J Haematol (2015) 171(5):671–82. doi:10.1111/bjh.13639
37. Townsley DM. Hematologic complications of pregnancy. Semin Hematol (2013) 50(3):222–31. doi:10.1053/j.seminhematol.2013.06.004
38. Bader-Meunier B, Proulle V, Trichet C, Debray D, Gabolde M, Yvart J, et al. Misdiagnosis of chronic thrombocytopenia in childhood. J Pediatr Hematol Oncol (2003) 25(7):548–52. doi:10.1097/00043426-200307000-00010
39. Balduini CL, Savoia A. Genetics of familial forms of thrombocytopenia. Hum Genet (2012) 131(12):1821–32. doi:10.1007/s00439-012-1215-x
40. Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott-Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet (2014) 7:55–66. doi:10.2147/TACG.S58444
41. Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol (2017) 177(4):526–42. doi:10.1111/bjh.14535
42. Thomas AS, Mehta AB, Hughes DA. Diagnosing Gaucher disease: an on-going need for increased awareness amongst haematologists. Blood Cells Mol Dis (2013) 50(3):212–7. doi:10.1016/j.bcmd.2012.11.004
43. Wasserstein MP, Desnick RJ, Schuchman EH, Hossain S, Wallenstein S, Lamm C, et al. The natural history of type B Niemann-Pick disease: results from a 10-year longitudinal study. Pediatrics (2004) 114(6):e672–7. doi:10.1542/peds.2004-0887
44. Perricone C, Ceccarelli F, Nesher G, Borella E, Odeh Q, Conti F, et al. Immune thrombocytopenic purpura (ITP) associated with vaccinations: a review of reported cases. Immunol Res (2014) 60(2–3):226–35. doi:10.1007/s12026-014-8597-x
45. Rastogi P, Naseem S, Varma N, Das R, Ahluwalia J, Sachdeva MU, et al. Bone marrow involvement in neuroblastoma: a study of hemato-morphological features. Indian J Hematol Blood Transfus (2015) 31(1):57–60. doi:10.1007/s12288-014-0405-1
46. Erlacher M, Strahm B. Missing cells: pathophysiology, diagnosis, and management of (Pan)cytopenia in childhood. Front Pediatr (2015) 3:64. doi:10.3389/fped.2015.00064
47. Kuter DJ. Managing thrombocytopenia associated with cancer chemotherapy. Oncology (Williston Park) (2015) 29(4):282–94.
48. Boral BM, Williams DJ, Boral LI. Disseminated intravascular coagulation. Am J Clin Pathol (2016) 146(6):670–80. doi:10.1093/ajcp/aqw195
49. Shen YM. Clinical evaluation of thrombotic microangiopathy: identification of patients with suspected atypical hemolytic uremic syndrome. Thromb J (2016) 14(Suppl 1):19. doi:10.1186/s12959-016-0114-0
50. Sarihan H, Mocan H, Abeys M, Akyazici R, Cay A, Imamoglu M. Kasabach-Merritt syndrome in infants. Panminerva Med (1998) 40(2):128–31.
51. Labarque V, Van Geet C. Clinical practice: immune thrombocytopenia in paediatrics. Eur J Pediatr (2014) 173(2):163–72. doi:10.1007/s00431-013-2254-6
52. Neunert C, Noroozi N, Norman G, Buchanan GR, Goy J, Nazi I, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost (2015) 13(3):457–64. doi:10.1111/jth.13019
53. Giroud M, Lemesle M, Madinier G, Manceau E, Osseby GV, Dumas R. Stroke in children under 16 years of age. Clinical and etiological difference with adults. Acta Neurol Scand (1997) 96(6):401–6. doi:10.1111/j.1600-0404.1997.tb00306.x
54. Tarantino MD, Danese M, Klaassen RJ, Duryea J, Eisen M, Bussel J. Hospitalizations in pediatric patients with immune thrombocytopenia in the United States. Platelets (2016) 27(5):472–8. doi:10.3109/09537104.2016.1143923
55. Psaila B, Petrovic A, Page LK, Menell J, Schonholz M, Bussel JB. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood (2009) 114(23):4777–83. doi:10.1182/blood-2009-04-215525
56. So MY, Li CH, Lee AC, Kwong NS. Intracranial haemorrhage among Chinese children with immune thrombocytopenia in a Hong Kong regional hospital. Hong Kong Med J (2013) 19(2):129–34.
57. Elalfy M, Elbarbary N, Khaddah N, Abdelwahab M, El Rashidy F, Hassab H, et al. Intracranial hemorrhage in acute and chronic childhood immune thrombocytopenic purpura over a ten-year period: an Egyptian multicenter study. Acta Haematol (2010) 123(1):59–63. doi:10.1159/000262293
58. Choudhary DR, Naithani R, Mahapatra M, Kumar R, Mishra P, Saxena R. Intracranial hemorrhage in childhood immune thrombocytopenic purpura. Pediatr Blood Cancer (2009) 52(4):529–31. doi:10.1002/pbc.21728
59. Arnold DM. Bleeding complications in immune thrombocytopenia. Hematol Am Soc Hematol Educ Program (2015) 2015:237–42. doi:10.1182/asheducation-2015.1.237
60. Butros LJ, Bussel JB. Intracranial hemorrhage in immune thrombocytopenic purpura: a retrospective analysis. J Pediatr Hematol Oncol (2003) 25(8):660–4. doi:10.1097/00043426-200308000-00017
61. Lilleyman JS. Intracranial haemorrhage in idiopathic thrombocytopenic purpura. Paediatric haematology forum of the British Society for Haematology. Arch Dis Child (1994) 71(3):251–3. doi:10.1136/adc.71.3.251
62. Flores A, Buchanan GR. Occult hemorrhage in children with severe ITP. Am J Hematol (2016) 91(3):287–90. doi:10.1002/ajh.24264
63. Dumont P, Goussot V, David A, Lizard S, Riedinger JM. Identification and validation of a factor of commutability between platelet counts performed on EDTA and citrate. Ann Biol Clin (2017) 75(1):61–6. doi:10.1684/abc.2016.1211
64. Balduini CL, Noris P. Mean platelet volume for distinguishing between inherited thrombocytopenias and immune thrombocytopenia – response to Beyan. Br J Haematol (2013) 163(3):413–4. doi:10.1111/bjh.12504
65. Savoia A, Pastore A, De Rocco D, Civaschi E, Di Stazio M, Bottega R, et al. Clinical and genetic aspects of Bernard-Soulier syndrome: searching for genotype/phenotype correlations. Haematologica (2011) 96(3):417–23. doi:10.3324/haematol.2010.032631
66. Noris P, Klersy C, Gresele P, Giona F, Giordano P, Minuz P, et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: a multicentric, real life study. Br J Haematol (2013) 162(1):112–9. doi:10.1111/bjh.12349
67. Balduini CL, Pecci A, Savoia A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br J Haematol (2011) 154(2):161–74. doi:10.1111/j.1365-2141.2011.08716.x
69. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood (2004) 103(2):390–8. doi:10.1182/blood-2003-05-1742
70. Ingram M, Coopersmith A. Reticulated platelets following acute blood loss. Br J Haematol (1969) 17(3):225–9. doi:10.1111/j.1365-2141.1969.tb01366.x
71. Jimenez MM, Guedan MJ, Martin LM, Campos JA, Martinez IR, Vilella CT. Measurement of reticulated platelets by simple flow cytometry: an indirect thrombocytopoietic marker. Eur J Intern Med (2006) 17(8):541–4. doi:10.1016/j.ejim.2006.03.006
72. Hoffmann JJ. Reticulated platelets: analytical aspects and clinical utility. Clin Chem Lab Med (2014) 52(8):1107–17. doi:10.1515/cclm-2014-0165
73. Dusse LM, Freitas LG. Clinical applicability of reticulated platelets. Clin Chim Acta (2015) 439:143–7. doi:10.1016/j.cca.2014.10.024
74. Thomas-Kaskel AK, Mattern D, Kohler G, Finke J, Behringer D. Reticulated platelet counts correlate with treatment response in patients with idiopathic thrombocytopenic purpura and help identify the complex causes of thrombocytopenia in patients after allogeneic hematopoietic stem cell transplantation. Cytometry B Clin Cytom (2007) 72(4):241–8. doi:10.1002/cyto.b.20163
75. Tarantino MD, Madden RM, Fennewald DL, Patel CC, Bertolone SJ. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr (1999) 134(1):21–6. doi:10.1016/S0022-3476(99)70367-7
76. Tarantino MD, Young G, Bertolone SJ, Kalinyak KA, Shafer FE, Kulkarni R, et al. Single dose of anti-D immune globulin at 75 microg/kg is as effective as intravenous immune globulin at rapidly raising the platelet count in newly diagnosed immune thrombocytopenic purpura in children. J Pediatr (2006) 148(4):489–94. doi:10.1016/j.jpeds.2005.11.019
77. Scaradavou A, Woo B, Woloski BM, Cunningham-Rundles S, Ettinger LJ, Aledort LM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood (1997) 89(8):2689–700.
78. Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood (2010) 115(2):168–86. doi:10.1182/blood-2009-06-225565
79. Cooper N. A review of the management of childhood immune thrombocytopenia: how can we provide an evidence-based approach? Br J Haematol (2014) 165(6):756–67. doi:10.1111/bjh.12889
80. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost (2002) 8(1):73–6. doi:10.1177/107602960200800110
81. Ahmad Z, Durrani NU, Hazir T. Bone marrow examination in ITP in children: is it mandatory? J Coll Physicians Surg Pak (2007) 17(6):347–9. doi:06.2007/JCPSP.347349
82. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol (2007) 14(5):520–5. doi:10.1097/MOH.0b013e3282ab98f2
83. Sutor AH, Harms A, Kaufmehl K. Acute immune thrombocytopenia (ITP) in childhood: retrospective and prospective survey in Germany. Semin Thromb Hemost (2001) 27(3):253–67. doi:10.1055/s-2001-15255
84. Neunert CE, Buchanan GR, Imbach P, Bolton-Maggs PH, Bennett CM, Neufeld EJ, et al. Severe hemorrhage in children with newly diagnosed immune thrombocytopenic purpura. Blood (2008) 112(10):4003–8. doi:10.1182/blood-2008-03-138487
85. Buchanan GR, de Alarcon PA, Feig SA, Gilchrist GS, Lukens JN, Moertel CL, et al. Acute idiopathic thrombocytopenic purpura – management in childhood. Blood (1997) 89(4):1464–5.
86. Bolton-Maggs PH, Dickerhoff R, Vora AJ. The nontreatment of childhood ITP (or “the art of medicine consists of amusing the patient until nature cures the disease”). Semin Thromb Hemost (2001) 27(3):269–75. doi:10.1055/s-2001-15256
87. George JN, Woolf SH, Raskob GE, Wasser JS, Aledort LM, Ballem PJ, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood (1996) 88(1):3–40.
88. Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood (2011) 117(16):4190–207. doi:10.1182/blood-2010-08-302984
89. Witmer CM, Lambert MP, O’Brien SH, Neunert C. Multicenter cohort study comparing U.S. management of inpatient pediatric immune thrombocytopenia to current treatment guidelines. Pediatr Blood Cancer (2016) 63(7):1227–31. doi:10.1002/pbc.25961
90. Carcao MD, Zipursky A, Butchart S, Leaker M, Blanchette VS. Short-course oral prednisone therapy in children presenting with acute immune thrombocytopenic purpura (ITP). Acta Paediatr Suppl (1998) 424:71–4.
91. Blanchette VS, Luke B, Andrew M, Sommerville-Nielsen S, Barnard D, de Veber B, et al. A prospective, randomized trial of high-dose intravenous immune globulin G therapy, oral prednisone therapy, and no therapy in childhood acute immune thrombocytopenic purpura. J Pediatr (1993) 123(6):989–95. doi:10.1016/S0022-3476(05)80400-7
92. Imbach P, Barandun S, d’Apuzzo V, Baumgartner C, Hirt A, Morell A, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet (1981) 1(8232):1228–31. doi:10.1016/S0140-6736(81)92400-4
93. Imbach P, Barandun S, Baumgartner C, Hirt A, Hofer F, Wagner HP. High-dose intravenous gammaglobulin therapy of refractory, in particular idiopathic thrombocytopenia in childhood. Helv Paediatr Acta (1981) 36(1):81–6.
94. Beck CE, Nathan PC, Parkin PC, Blanchette VS, Macarthur C. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr (2005) 147(4):521–7. doi:10.1016/j.jpeds.2005.04.032
95. Benesch M, Kerbl R, Lackner H, Berghold A, Schwinger W, Triebl-Roth K, et al. Low-dose versus high-dose immunoglobulin for primary treatment of acute immune thrombocytopenic purpura in children: results of a prospective, randomized single-center trial. J Pediatr Hematol Oncol (2003) 25(10):797–800. doi:10.1097/00043426-200310000-00011
96. Moser AM, Shalev H, Kapelushnik J. Anti-D exerts a very early response in childhood acute idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol (2002) 19(6):407–11. doi:10.1080/08880010290097161
97. El Alfy MS, Mokhtar GM, El-Laboudy MA, Khalifa AS. Randomized trial of anti-D immunoglobulin versus low-dose intravenous immunoglobulin in the treatment of childhood chronic idiopathic thrombocytopenic purpura. Acta Haematol (2006) 115(1–2):46–52. doi:10.1159/000089465
98. Despotovic JM, Lambert MP, Herman JH, Gernsheimer TB, McCrae KR, Tarantino MD, et al. RhIG for the treatment of immune thrombocytopenia: consensus and controversy (CME). Transfusion (2012) 52(5):1126–36. doi:10.1111/j.1537-2995.2011.03384.x
99. Ozer EA, Yaprak I, Atabay B, Turker M, Aksit S, Sarioglu B. Oral cyclic megadose methylprednisolone therapy for chronic immune thrombocytopenic purpura in childhood. Eur J Haematol (2000) 64(6):411–5. doi:10.1034/j.1600-0609.2000.90166.x
100. Ozsoylu S, Irken G, Karabent A. High-dose intravenous methylprednisolone for acute childhood idiopathic thrombocytopenic purpura. Eur J Haematol (1989) 42(5):431–5. doi:10.1111/j.1600-0609.1989.tb01466.x
101. Ben-Yehuda D, Gillis S, Eldor A. Clinical and therapeutic experience in 712 Israeli patients with idiopathic thrombocytopenic purpura. Israeli ITP Study Group. Acta Haematol (1994) 91(1):1–6. doi:10.1159/000204251
102. Kuhne T, Freedman J, Semple JW, Doyle J, Butchart S, Blanchette VS. Platelet and immune responses to oral cyclic dexamethasone therapy in childhood chronic immune thrombocytopenic purpura. J Pediatr (1997) 130(1):17–24. doi:10.1016/S0022-3476(97)70305-6
103. Dai WJ, Zhang RR, Yang XC, Yuan YF. Efficacy of standard dose rituximab for refractory idiopathic thrombocytopenic purpura in children. Eur Rev Med Pharmacol Sci (2015) 19(13):2379–83.
104. Parodi E, Nobili B, Perrotta S, Rosaria Matarese SM, Russo G, Licciardello M, et al. Rituximab (anti-CD20 monoclonal antibody) in children with chronic refractory symptomatic immune thrombocytopenic purpura: efficacy and safety of treatment. Int J Hematol (2006) 84(1):48–53. doi:10.1532/IJH97.E0518
105. Weide R, Feiten S, Friesenhahn V, Heymanns J, Kleboth K, Thomalla J, et al. Outpatient management of patients with immune thrombocytopenia (ITP) by hematologists 1995-2014. Oncol Res Treat (2016) 39(1–2):41–4. doi:10.1159/000442769
106. Rao A, Kelly M, Musselman M, Ramadas J, Wilson D, Grossman W, et al. Safety, efficacy, and immune reconstitution after rituximab therapy in pediatric patients with chronic or refractory hematologic autoimmune cytopenias. Pediatr Blood Cancer (2008) 50(4):822–5. doi:10.1002/pbc.21264
107. Bennett CM, Rogers ZR, Kinnamon DD, Bussel JB, Mahoney DH, Abshire TC, et al. Prospective phase 1/2 study of rituximab in childhood and adolescent chronic immune thrombocytopenic purpura. Blood (2006) 107(7):2639–42. doi:10.1182/blood-2005-08-3518
108. Matsubara K, Takahashi Y, Hayakawa A, Tanaka F, Nakadate H, Sakai M, et al. Long-term follow-up of children with refractory immune thrombocytopenia treated with rituximab. Int J Hematol (2014) 99(4):429–36. doi:10.1007/s12185-014-1541-y
109. Patel VL, Mahevas M, Lee SY, Stasi R, Cunningham-Rundles S, Godeau B, et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood (2012) 119(25):5989–95. doi:10.1182/blood-2011-11-393975
110. Audia S, Godeau B, Bonnotte B. Is there still a place for “old therapies” in the management of immune thrombocytopenia? Rev Med Interne (2016) 37(1):43–9. doi:10.1016/j.revmed.2015.08.007
111. Szmydki-Baran A, Adamowicz-Salach A, Golebiowska-Staroszczyk S. [Danazol – effective second line therapy in idiopathic thrombocytopenic purpura in children. Three case reports]. Med Wieku Rozwoj (2008) 12(4 Pt 2):1130–4.
112. Weinblatt ME, Kochen J, Ortega J. Danazol for children with immune thrombocytopenic purpura. Am J Dis Child (1988) 142(12):1317–9.
113. Ikeda Y, Miyakawa Y. Development of thrombopoietin receptor agonists for clinical use. J Thromb Haemost (2009) 7(Suppl 1):239–44. doi:10.1111/j.1538-7836.2009.03440.x
114. Dmytrijuk A, Robie-Suh K, Rieves D, Pazdur R. Eltrombopag for the treatment of chronic immune (idiopathic) thrombocytopenic purpura. Oncology (Williston Park) (2009) 23(13):1171–7.
115. Jamali F, Lemery S, Ayalew K, Robottom S, Robie-Suh K, Rieves D, et al. Romiplostim for the treatment of chronic immune (idiopathic) thrombocytopenic purpura. Oncology (Williston Park) (2009) 23(8):704–9.
116. Garzon AM, Mitchell WB. Use of thrombopoietin receptor agonists in childhood immune thrombocytopenia. Front Pediatr (2015) 3:70. doi:10.3389/fped.2015.00070
117. Tarantino MD, Bussel JB, Blanchette VS, Despotovic J, Bennett C, Raj A, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet (2016) 388(10039):45–54. doi:10.1016/S0140-6736(16)00279-8
118. Bussel JB, Hsieh L, Buchanan GR, Stine K, Kalpatthi R, Gnarra DJ, et al. Long-term use of the thrombopoietin-mimetic romiplostim in children with severe chronic immune thrombocytopenia (ITP). Pediatr Blood Cancer (2015) 62(2):208–13. doi:10.1002/pbc.25136
119. Pasquet M, Aladjidi N, Guiton C, Courcoux MF, Munzer M, Auvrignon A, et al. Romiplostim in children with chronic immune thrombocytopenia (ITP): the French experience. Br J Haematol (2014) 164(2):266–71. doi:10.1111/bjh.12609
120. Escudero Vilaplana V, Aragones JH, Fernandez-Llamazares CM, Bieler CB, Rodriguez SM, Saez MS. Use of romiplostim for primary immune thrombocytopenia in children. Pediatr Hematol Oncol (2012) 29(2):197–205. doi:10.3109/08880018.2011.629401
121. Mokhtar GM, Tantawy AA, El Sherif NH. Romiplostim therapy in children with unresponsive chronic immune thrombocytopenia. Platelets (2012) 23(4):264–73. doi:10.3109/09537104.2011.619601
122. Bussel JB, Buchanan GR, Nugent DJ, Gnarra DJ, Bomgaars LR, Blanchette VS, et al. A randomized, double-blind study of romiplostim to determine its safety and efficacy in children with immune thrombocytopenia. Blood (2011) 118(1):28–36. doi:10.1182/blood-2010-10-313908
123. Elalfy MS, Abdelmaksoud AA, Eltonbary KY. Romiplostim in children with chronic refractory ITP: randomized placebo controlled study. Ann Hematol (2011) 90(11):1341–4. doi:10.1007/s00277-011-1172-9
124. Ramaswamy K, Hsieh L, Leven E, Thompson MV, Nugent D, Bussel JB. Thrombopoietic agents for the treatment of persistent and chronic immune thrombocytopenia in children. J Pediatr (2014) 165(3):600–5.e4. doi:10.1016/j.jpeds.2014.03.060
125. Mazzucconi MG, Santoro C, Baldacci E, De Angelis F, Chisini M, Ferrara G, et al. TPO-RAs in pITP: description of a case series and analysis of predictive factors for response. Eur J Haematol (2017) 98(3):242–9. doi:10.1111/ejh.12822
126. Grainger JD, Locatelli F, Chotsampancharoen T, Donyush E, Pongtanakul B, Komvilaisak P, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet (2015) 386(10004):1649–58. doi:10.1016/S0140-6736(15)61107-2
127. Bussel JB, de Miguel PG, Despotovic JM, Grainger JD, Sevilla J, Blanchette VS, et al. Eltrombopag for the treatment of children with persistent and chronic immune thrombocytopenia (PETIT): a randomised, multicentre, placebo-controlled study. Lancet Haematol (2015) 2(8):e315–25. doi:10.1016/S2352-3026(15)00114-3
128. Bussel JB, Kuter DJ, Aledort LM, Kessler CM, Cuker A, Pendergrass KB, et al. A randomized trial of avatrombopag, an investigational thrombopoietin-receptor agonist, in persistent and chronic immune thrombocytopenia. Blood (2014) 123(25):3887–94. doi:10.1182/blood-2013-07-514398
129. Quiquandon I, Fenaux P, Caulier MT, Pagniez D, Huart JJ, Bauters F. Re-evaluation of the role of azathioprine in the treatment of adult chronic idiopathic thrombocytopenic purpura: a report on 53 cases. Br J Haematol (1990) 74(2):223–8. doi:10.1111/j.1365-2141.1990.tb02569.x
130. Yenson PR, Forrest D, Schmiegelow K, Dalal BI. Azathioprine-associated acute myeloid leukemia in a patient with Crohn’s disease and thiopurine S-methyltransferase deficiency. Am J Hematol (2008) 83(1):80–3. doi:10.1002/ajh.21014
131. Emilia G, Morselli M, Luppi M, Longo G, Marasca R, Gandini G, et al. Long-term salvage therapy with cyclosporin A in refractory idiopathic thrombocytopenic purpura. Blood (2002) 99(4):1482–5. doi:10.1182/blood.V99.4.1482
132. Kappers-Klunne MC, van’t Veer MB. Cyclosporin A for the treatment of patients with chronic idiopathic thrombocytopenic purpura refractory to corticosteroids or splenectomy. Br J Haematol (2001) 114(1):121–5. doi:10.1046/j.1365-2141.2001.02893.x
133. Aronis S, Platokouki H, Avgeri M, Pergantou H, Keramidas D. Retrospective evaluation of long-term efficacy and safety of splenectomy in chronic idiopathic thrombocytopenic purpura in children. Acta Paediatr (2004) 93(5):638–42. doi:10.1111/j.1651-2227.2004.tb02989.x
134. Kuhne T, Blanchette V, Buchanan GR, Ramenghi U, Donato H, Tamminga RY, et al. Splenectomy in children with idiopathic thrombocytopenic purpura: a prospective study of 134 children from the Intercontinental Childhood ITP Study Group. Pediatr Blood Cancer (2007) 49(6):829–34. doi:10.1002/pbc.21108
135. Ahmed R, Devasia AJ, Viswabandya A, Lakshmi KM, Abraham A, Karl S, et al. Long-term outcome following splenectomy for chronic and persistent immune thrombocytopenia (ITP) in adults and children: splenectomy in ITP. Ann Hematol (2016) 95(9):1429–34. doi:10.1007/s00277-016-2738-3
136. Shojaiefard A, Mousavi SA, Faghihi SH, Abdollahzade S. Prediction of response to splenectomy in patients with idiopathic thrombocytopenic purpura. World J Surg (2008) 32(3):488–93. doi:10.1007/s00268-007-9399-2
137. Fenaux P, Caulier MT, Hirschauer MC, Beuscart R, Goudemand J, Bauters F. Reevaluation of the prognostic factors for splenectomy in chronic idiopathic thrombocytopenic purpura (ITP): a report on 181 cases. Eur J Haematol (1989) 42(3):259–64. doi:10.1111/j.1600-0609.1989.tb00109.x
138. Aleem A. Durability and factors associated with long term response after splenectomy for primary immune thrombocytopenia (ITP) and outcome of relapsed or refractory patients. Platelets (2011) 22(1):1–7. doi:10.3109/09537104.2010.515697
139. Fabris F, Tassan T, Ramon R, Carraro G, Randi ML, Luzzatto G, et al. Age as the major predictive factor of long-term response to splenectomy in immune thrombocytopenic purpura. Br J Haematol (2001) 112(3):637–40. doi:10.1046/j.1365-2141.2001.02615.x
140. Kwon HC, Moon CH, Cho YR, Kim MC, Kim KH, Han JY, et al. Prognostic factors of response to laparoscopic splenectomy in patients with idiopathic thrombocytopenic purpura. J Korean Med Sci (2005) 20(3):417–20. doi:10.3346/jkms.2005.20.3.417
141. Zheng CX, Zheng D, Chen LH, Yu JF, Wu ZM. Laparoscopic splenectomy for immune thrombocytopenic purpura at a teaching institution. Chin Med J (2011) 124(8):1175–80.
142. Patel VL, Schwartz J, Bussel JB. The effect of anti-CD40 ligand in immune thrombocytopenic purpura. Br J Haematol (2008) 141(4):545–8. doi:10.1111/j.1365-2141.2008.07039.x
143. Shih A, Nazi I, Kelton JG, Arnold DM. Novel treatments for immune thrombocytopenia. Presse Med (2014) 43(4 Pt 2):e87–95. doi:10.1016/j.lpm.2014.02.006
144. Andersson PO, Wadenvik H. Chronic idiopathic thrombocytopenic purpura (ITP): molecular mechanisms and implications for therapy. Expert Rev Mol Med (2004) 6(24):1–17. doi:10.1017/S1462399404008415
145. Fogarty PF, Seggewiss R, McCloskey DJ, Boss CA, Dunbar CE, Rick ME. Anti-interleukin-2 receptor antibody (daclizumab) treatment of corticosteroid-refractory autoimmune thrombocytopenic purpura. Haematologica (2006) 91(2):277–8.
146. Soubrane C, Tourani JM, Andrieu JM, Visonneau S, Beldjord K, Israel-Biet D, et al. Biologic response to anti-CD16 monoclonal antibody therapy in a human immunodeficiency virus-related immune thrombocytopenic purpura patient. Blood (1993) 81(1):15–9.
147. Provan D, Newland AC. Current management of primary immune thrombocytopenia. Adv Ther (2015) 32(10):875–87. doi:10.1007/s12325-015-0251-z
148. Bansal D, Bhamare TA, Trehan A, Ahluwalia J, Varma N, Marwaha RK. Outcome of chronic idiopathic thrombocytopenic purpura in children. Pediatr Blood Cancer (2010) 54(3):403–7. doi:10.1002/pbc.22346
149. Heitink-Pollè KM, Nijsten J, Boonacker CW, de Haas M, Bruin MC. Clinical and laboratory predictors of chronic immune thrombocytopenia in children: a systematic review and meta-analysis. Blood (2014) 124(22):3295–307. doi:10.1182/blood-2014-04-570127
150. Altintas A, Ozel A, Okur N, Okur N, Cil T, Pasa S, et al. Prevalence and clinical significance of elevated antinuclear antibody test in children and adult patients with idiopathic thrombocytopenic purpura. J Thromb Thrombolysis (2007) 24(2):163–8. doi:10.1007/s11239-007-0031-y
Keywords: immune thrombocytopenia, differential diagnosis, diagnostic algorithm, chronic thrombocytopenia, conventional therapy, new therapeutic targets
Citation: Consolini R, Costagliola G and Spatafora D (2017) The Centenary of Immune Thrombocytopenia—Part 2: Revising Diagnostic and Therapeutic Approach. Front. Pediatr. 5:179. doi: 10.3389/fped.2017.00179
Received: 05 May 2017; Accepted: 07 August 2017;
Published: 21 August 2017
Edited by:
Claudio Pignata, University of Naples Federico II, ItalyReviewed by:
Jenny McDade Despotovic, Baylor College of Medicine, United StatesMartina Pigazzi, University of Padua, Italy
Copyright: © 2017 Consolini, Costagliola and Spatafora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Costagliola, Z2lvcmdpby5jb3N0YWdsaW9sYUBob3RtYWlsLmNvbQ==