 Amy N. Sexauer
Amy N. Sexauer Sarah K. Tasian
Sarah K. Tasian- 1Dana-Farber Cancer Institute, Boston, MA, United States
- 2Boston Children’s Hospital, Department of Pediatrics, Division of Pediatric Hematology/Oncology/Stem Cell Transplant, Boston, MA, United States
- 3Children’s Hospital of Philadelphia, Division of Oncology, Center for Childhood Cancer Research, Philadelphia, PA, United States
- 4Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Acute myeloid leukemia (AML) is the second most common leukemia of childhood and is associated with high rates of chemotherapy resistance and relapse. Clinical outcomes for children with AML treated with maximally intensive multi-agent chemotherapy lag far behind those of children with the more common acute lymphoblastic leukemia, demonstrating continued need for new therapeutic approaches to decrease relapse risk and improve long-term survival. Mutations in the FMS-like tyrosine kinase-3 receptor gene (FLT3) occur in approximately 25% of children and adults with AML and are associated with particularly poor prognoses. Identification and development of targeted FLT3 inhibitors represents a major precision medicine paradigm shift in the treatment of patients with AML. While further development of many first-generation FLT3 inhibitors was hampered by limited potency and significant toxicity due to effects upon other kinases, the more selective second- and third-generation FLT3 inhibitors have demonstrated excellent tolerability and remarkable efficacy in the relapsed/refractory and now de novo FLT3-mutated AML settings. While these newest and most promising inhibitors have largely been studied in the adult population, pediatric investigation of FLT3 inhibitors with chemotherapy is relatively recently ongoing or planned. Successful development of FLT3 inhibitor-based therapies will be essential to improve outcomes in children with this high-risk subtype of AML.
Introduction
Acute myeloid leukemia (AML) is a group of biologically heterogeneous diseases that comprise 20% of pediatric and 80% of adult acute leukemias (1, 2). It is estimated that 21,380 people in the United States will be diagnosed with AML in 2017, and 10,590 of these patients will die from leukemia (3). While outcomes for children with de novo AML have improved over the past several decades, event-free survival (EFS) and overall survival (OS) remain suboptimal at approximately 60 and 70%, respectively (4). Relapsed disease and poor response to salvage therapy remain significant hurdles in achieving cure.
FMS-like tyrosine kinase-3 (FLT3; CD135) is a 993 amino acid single transmembrane type III receptor tyrosine kinase in the same family as the structurally similar stem cell growth factor receptor c-KIT (CD117), colony-stimulating factor-1 receptor (CSF1R; CD115), and platelet-derived growth factor receptor (PDGFR) (5–8). FLT3 has a single extracellular ligand-binding domain with five immunoglobulin-like folds, a juxtamembrane domain, and a single cytoplasmic tyrosine kinase domain separated by a kinase insert region (Figure 1A). FLT3 signaling plays a critical role in hematopoiesis and is expressed on CD34+ hematopoietic stem/progenitor cells, but its surface expression is lost during cellular differentiation (7, 9, 10). Normally, the FLT3 receptor is stimulated by FLT3 ligand, leading to receptor dimerization with subsequent activation of its tyrosine kinase domain, autophosphorylation, and binding of SH2 domain-containing proteins. Activated FLT3 then phosphorylates downstream targets, including STAT5, SHIP, and SHP-2, and signals through critical oncogenic pathways such as Ras/Raf/MAPK and PI3K/Akt/mTOR (5, 6, 11) (Figure 1B).
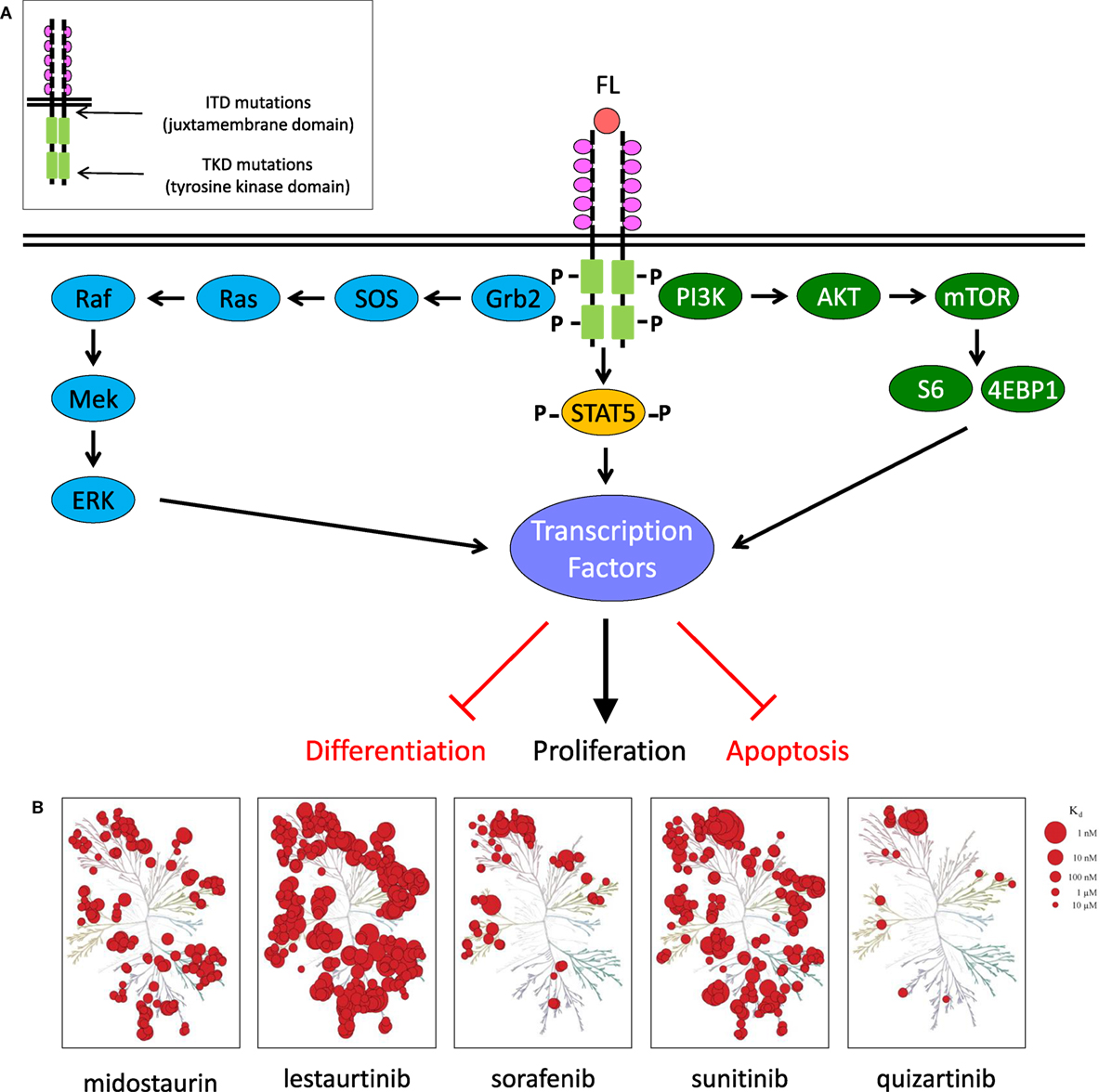
Figure 1. FLT3 signaling in acute myeloid leukemia and clinically relevant FLT3 tyrosine kinase inhibitors. (A) FLT3 receptor and downstream signaling targets schema. (B) KinomeScan dendrograms (http://lincs.hms.harvard.edu/kinomescan/) demonstrating relative potency and selectivity of FLT3 inhibitors [adapted from Zarrinkar et al. (12); used with permission]. Interactions with Kd <3 µM are shown. Red circles designate kinases bound. Circle size specifies relative binding affinity. Dendrogram data are not available for ponatinib, crenolanib, or gilteritinib.
Wild-type FLT3 is overexpressed in most cases of B-lymphoblastic leukemia and AML and in a smaller percentage of T-lineage ALL and chronic myeloid leukemia (CML) in blast crisis (13). Mutations in FLT3 are one of the most common genetic alterations in AML and are associated with high rates of relapse in adults and children (14–16). Activating FLT3 mutations are classified into two types: (1) internal tandem duplication (FLT3–ITD) mutations, which are 3–400 bp in-frame duplications located in the juxtamembrane domain and (2) activating point mutations, which are found in the tyrosine kinase domain (FLT3–TKD) and most often involve residue D835 (17–19). ITD and TKD mutations occur in approximately 25 and 10% of adult AML cases, respectively (20, 21). Recent studies have reported similar incidence of ITD and activating TKD mutations in childhood AML (15, 22–25). Numerous clinical trials have demonstrated inferior clinical outcomes in patients with FLT3–ITD AML (14–16, 26, 27).
Adults with newly diagnosed AML are generally treated with cytarabine- and anthracycline-based induction chemotherapy followed by consolidation therapy. Allocation to subsequent hematopoietic stem cell transplant (HSCT) is usually based on cytogenetic risk stratification and transplant eligibility status (2). With this approach, 5-year OS for adults with AML is approximately 40%. However, several studies have demonstrated significantly decreased duration of first remission (CR1) and 5-year OS of approximately 15% in adults with FLT3–ITD AML versus those without FLT3 mutations (28–32). Children with FLT3–ITD AML treated on Children’s Cancer Group and Pediatric Oncology Group trials fared similarly poorly with 30% 4-year EFS when treated with conventional multi-agent chemotherapy (15). Higher mutant-to-wild-type FLT3 allelic ratios have also been associated with increasingly inferior outcomes in children treated on Dutch Children’s Oncology Group and Children’s Oncology Group (COG) studies (15, 33). In a recent subgroup analysis, the COG phase 3 trial AAML0531 reported decreased relapse rates in children with FLT3–ITD AML with addition of the CD33-targeting antibody-drug conjugate gemtuzumab ozogamicin to standard chemotherapy (16, 34), demonstrating potential for improved clinical outcomes in this high-risk patient population with inclusion of targeted therapies.
Given the significant negative prognostic effects of FLT3–ITD mutations in AML and the relative frequency of these alterations, therapeutic targeting of aberrant FLT3 signaling has been a major research focus with goals of decreasing relapse and improving survival. Tyrosine kinase inhibitors (TKIs) are small molecules that inhibit the enzymatic activity of tyrosine kinases and block downstream signaling activation. Treatment of adults with CML with the SRC/ABL inhibitor imatinib is one of the major early successes of modern precision medicine (35, 36). Imatinib targets the oncogenic BCR–ABL fusion protein resulting from t(9;22) (Philadelphia chromosome; Ph+) by inhibiting the active site of the ABL1 kinase. Treatment of patients with CML and Ph+ ALL with imatinib or related TKIs markedly improved remission rates and long-term survival versus prior interferon and chemotherapy and is now considered standard-of-care therapy (37–40). Similarly, FLT3 inhibitor treatment of patients with FLT3-mutated AML has been investigated for the past decade with promising results of several studies recently reported. This review discusses the current landscape of and future potential for clinical testing of FLT3 inhibitors in adults and children with FLT3-mutated AML (Table 1).
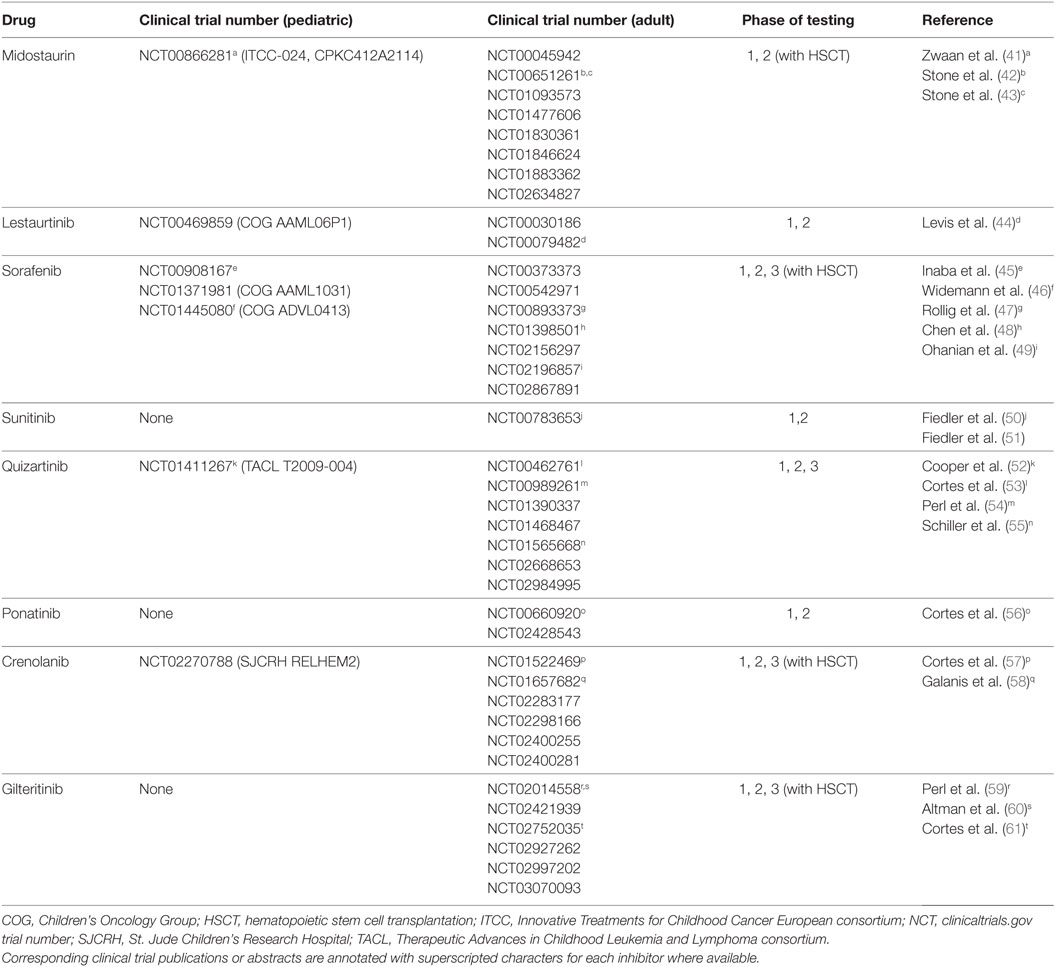
Table 1. Current clinical trials of FLT3 inhibitors in children and adults with AML.
Current FLT3 TKIs IN Clinical Use
Midostaurin
Midostaurin (PKC412) is a first-generation oral FLT3 inhibitor initially named due to its inhibitory effects upon protein kinase C. Midostaurin was subsequently recognized as a promiscuous kinase inhibitor with strong inhibitory effects also against the vascular endothelial growth factor receptor (VEGFR), PDGFRα and β, spleen tyrosine kinase (SYK), c-KIT, and FLT3 (62). Midostaurin has been studied extensively in adults with relapsed/refractory AML. Initially, midostaurin monotherapy was observed to induce a “blast response” (≥50% reduction in blast counts in peripheral blood and/or bone marrow) in 70% of adults with FLT3–ITD AML and in 30–40% with FLT3-wild-type AML (62, 63). However, achievement of long-term remission was rare (63). Subsequent trials, thus, combined midostaurin with induction chemotherapy and reported improved CR rates in patients with FLT3–ITD AML (42). These studies were followed by the Cancer and Leukemia Group B 10603 RATIFY trial, an international double-blind randomized controlled study comparing standard chemotherapy without or with midostaurin in adults (18–59 years) with de novo FLT3–ITD or FLT3–TKD AML. Addition of midostaurin to chemotherapy significantly improved median EFS (8.2 versus 3.0 months) and OS (74.7 versus 25.6 months) compared to patients treated with chemotherapy and placebo (43). The precise impact of HSCT and potential differential responses of midostaurin treatment between FLT3–ITD and TKD patients is not fully known (43, 64). Based on results of this trial, midostaurin was recently approved by the United States Food and Drug Administration (FDA) for use in adults with de novo FLT3-mutated AML (64).
To date, one trial of midostaurin has been conducted in children with leukemia (41). An Innovative Therapies for Children with Cancer European consortium-led phase 1/2 dose escalation study aimed to establish the safety, tolerability, and efficacy of midostaurin in children and adolescents 3 months–18 years of age with either relapsed/refractory FLT3-mutated AML or KMT2A-rearranged ALL (which overexpresses wild-type FLT3). The trial closed early due to inadequate accrual. While the number of subjects studied was too small for more rigorous analysis, 5 of 15 patients with AML and 3 of 13 patients with KMT2A-rearranged ALL had partial or complete responses with midostaurin monotherapy with OS of 3.7 (AML) and 1.4 months (ALL), respectively (41).
Lestaurtinib
Lestaurtinib (CEP-701) is another first-generation multi-kinase inhibitor with activity against FLT3, Janus kinase 2, and tropomyosin receptor kinase A. One randomized trial tested lestaurtinib administration after induction chemotherapy in adult patients with AML in first relapse and demonstrated no survival benefit with lestaurtinib addition versus chemotherapy only (44). However, pharmacodynamic assessment of in vivo signaling inhibition by plasma inhibitory activity (PIA) assays demonstrated that few patients achieved sustained FLT3 inhibition (65), limiting conclusions about the potential efficacy of lestaurtinib in this population (44, 66). The UK AML15 and AML17 trials also studied lestaurtinib addition versus standard chemotherapy in 500 adults with AML harboring FLT3-activating mutations and showed no significant improvement in OS (67). However, PIA assays conducted in this trial correlated with significantly decreased relapse rates in lestaurtinib-treated patients who consistently achieved >85% FLT3 inhibition, further corroborating the importance of pharmacodynamic correlation (67).
The COG conducted an analogous pilot trial AAML06P1 (NCT00469859) in children and adolescents and young adults (AYAs) <30 years of age with relapsed/refractory AML. These patients were induced with cytarabine and idarubicin, then treated with lestaurtinib. The trial closed after the safety phase demonstrated tolerable combination dosing, but without completion of efficacy phase testing (66). In the pediatric population, lestaurtinib has been better studied in infants with wild-type FLT3-overexpressing KMT2A-rearranged ALL. Despite very promising preclinical data, the randomized COG phase 3 trial AALL0631 (NCT00557193) failed to demonstrate benefit of lestaurtinib addition to chemotherapy in infants with KMT2A-rearranged ALL, although achievement of sustained FLT3 inhibition as measured by PIA assays was variable among patients and across therapy phases (68).
Sorafenib
Sorafenib (BAY 43-9006) is another first-generation pan-kinase inhibitor with activity against Raf, c-KIT, PDGFR, VEGFR, and FLT3 (69). Sorafenib is FDA-approved for the treatment of adults with renal cell, hepatocellular, and thyroid carcinomas (70–72). Initial studies of sorafenib monotherapy in adults with FLT3-mutated AML demonstrated safety and tolerability with minimal toxicity (69, 73). Subsequent trials investigated the safety and efficacy of combining sorafenib with chemotherapy in adults with de novo AML. One phase 2 study at the MD Anderson Cancer Center tested sorafenib with cytarabine and idarubicin in 62 newly diagnosed patients, 19 of whom had FLT3–ITD AML. While response rates were higher in patients with FLT3 mutations, no differences in EFS or OS were observed (74). The successor international phase 2 SORAML trial randomized 267 adults (ages 18–60 years) to induction chemotherapy with cytarabine and daunomycin, followed by high-dose cytarabine consolidation therapy with sorafenib or placebo. Intermediate-risk patients with sibling donors and high-risk patients with any matched donor in first remission were allocated to subsequent allogeneic HSCT. The SORAML trial demonstrated clear benefit in the sorafenib-treated cohort with respect to relapse-free survival (21 months median EFS versus 9 months for placebo-treated patient), although did not improve OS rates (47).
A phase 1 trial conducted at St. Jude Children’s Research Hospital (SJCRH) first studied sorafenib monotherapy in children with relapsed/refractory AML, then in combination with clofarabine and cytarabine. Five of 12 enrolled patients had FLT3–ITD AML. Responses were observed in most patients, including all five FLT3–ITD patients, with complete remission (CR) in four patients, CR with incomplete count recovery (CRi) in two patients, and a partial response in one patient (45). A concomitant COG phase 1 study of sorafenib in children with relapsed/refractory solid tumors or leukemias also identified tolerable dosing in children and reported complete responses in two of eight patients with FLT3–ITD AML, enabling subsequent HSCT (46). The COG phase 3 trial AAML1031 (NCT01371981) is currently assessing the efficacy of non-randomized sorafenib addition to chemotherapy and best available donor HSCT for children and AYAs with de novo FLT3–ITD AML. The study was amended to include a 1-year sorafenib maintenance phase post-HSCT based upon smaller studies reporting potential efficacy of this post-transplant strategy to minimize relapse risk (75). Results from the AAML1031 sorafenib arm are not yet available, although some dosing modifications have been required due to incidence of hand–foot syndrome, hypertension, and cardiac dysfunction (Children’s Oncology Group Myeloid Diseases Committee, unpublished).
Sunitinib
Sunitinib (SU11248), another multi-kinase inhibitor with activity against FLT3, has been studied in adult patients with FLT3-mutated AML with reported similar efficacy as sorafenib (50). Tolerability and preliminary efficacy of sunitinib was assessed in children with relapsed/refractory solid tumors via the COG trials ADVL0413 and ACNS1021 (76, 77). Sunitinib has been studied in a small number of children with FLT3–ITD AML who failed prior sorafenib treatment (78).
Quizartinib
Given the potential for increased toxicities of the first-generation FLT3 inhibitors secondary to effects upon other kinases and/or poor pharmacodynamic properties, second-generation inhibitors with greater anti-FLT3 potency were developed. Quizartinib (AC220) is the first drug specifically designed as a FLT3 inhibitor and has 10–50 times greater in vivo potency than first-generation inhibitors (12, 79, 80). Quizartinib also has moderate activity against c-KIT (81). Initial phase 1 studies of quizartinib monotherapy in adults with relapsed/refractory AML demonstrated tolerability and preliminary efficacy (53), and subsequent phase 2 trials have reported high response rates in both younger and older adults with relapsed/refractory AML (54, 82). Other phase 2 and phase 3 studies are currently investigating the efficacy of quizartinib in combination with chemotherapy. Preclinical and clinical studies have now demonstrated resistance mutations in patients with FLT3–ITD AML treated with quizartinib, particularly the F691L gatekeeper and D835/I836 activation loop mutations (83, 84).
The Therapeutic Advances in Childhood Leukemia/Lymphoma (TACL) consortium conducted a phase 1 trial of quizartinib with cytarabine and etoposide in 17 children (ages 1 month to 21 years) with relapsed/refractory AML or KMT2A-R ALL. Quizartinib was well-tolerated without dose-limiting toxicity, and correlative PIA assays demonstrated near-complete pharmacodynamic inhibition of FLT3 at all tested doses (52). All seven patients with FLT3–ITD AML had marked reduction in medullary leukemia burden with three patients achieving CR or CRi and proceeding to allogeneic HSCT. Similar responses were not observed in children with wild-type FLT3 AML or KMT2A-rearranged ALL (52). These data further support potential improved anti-leukemic activity of more selective FLT3 inhibitors.
Ponatinib
Ponatinib (AP24534) is a third-generation multi-kinase inhibitor with activity against BCR–ABL and FLT3. Ponatinib is currently FDA-approved for treatment of adults with TKI-resistant CML or Ph+ ALL (85). In preclinical studies, ponatinib had significant anti-leukemia activity against AML specimens with FLT3–ITD or TKD mutations, including the F691I gatekeeper (86–88). A current phase 1/2 trial is studying the safety and efficacy of ponatinib in combination with cytarabine in adults with FLT3–ITD AML (NCT02428543). Ponatinib currently has an FDA black box warning regarding serious risk of arterial thrombosis and hepatotoxicity (89). No formal studies of ponatinib in children have been conducted, although anecdotal cases of compassionate usage in pediatrics have been reported (90).
Crenolanib
Crenolanib (CP-868596) was originally designed as a PDGFR inhibitor, although later studies also demonstrated its potency as a FLT3 inhibitor. Due to its short half-life, crenolanib requires thrice-daily dosing. Early data suggest that this third-generation TKI has robust activity against both FLT3–ITD and FLT3–TKD mutations, including those that confer resistance to quizartinib (58, 91, 92). Crenolanib monotherapy has been studied in several early-phase trials in adults with relapsed/refractory FLT3-mutated AML with encouraging activity (93), and combination trials in patients with newly diagnosed FLT3–ITD AML have demonstrated promising results (94).
Tolerable pediatric dosing of crenolanib monotherapy was established via a phase 1 trial conducted at SJCRH in children with central nervous system gliomas, which have activated PDGFR signaling (95). The current SJCRH RELHEM2 phase 1 trial (NCT02270788) is assessing the safety of combined crenolanib and sorafenib in children with relapsed or refractory hematologic malignancies.
Gilteritinib
Gilteritinib (ASP2215), the newest third-generation oral FLT3 inhibitor, is the most potent and selective FLT3 inhibitor developed to date with moderate additional activity against the AXL kinase. In preclinical studies, gilteritinib has in vitro anti-FLT3–ITD activity that equals or surpasses that of other FLT3 inhibitors previously discussed. Gilteritinib is also active against FLT3–TKD resistance mutations and does not appreciably inhibit c-KIT (96), thereby potentially avoiding the myelosuppressive effects of quizartinib that have been observed in clinical trials (97). A first-in-human phase 1/2 trial evaluated gilteritinib monotherapy in adults with relapsed/refractory AML. This study reported excellent tolerability of gilteritinib and a 30% CR/CRi rate in heavily pretreated patients with many patients achieving deep molecular remission (59, 60). Gilteritinib is now under investigation in adults with relapsed and refractory FLT3-mutated AML via a randomized double-blinded phase 3 registration trial (NCT02997202) (Table 1). The FDA also recently granted orphan drug designation for gilteritinib for patients with FLT3-mutated AML. A pediatric development program for gilteritinib is planned.
Future Directions
Despite maximal therapeutic intensification and significant improvements in supportive care, more than one-third of children with AML still die from leukemia or associated complications. FLT3-mutated AML is a particularly high-risk leukemia subtype in both adults and children, and the potential for selective FLT3 inhibitors to decrease relapse risk and improve cure rates is alluring. Initial trials of first-generation FLT3 inhibitors have validated FLT3 as a viable therapeutic target in AML and expedited FDA approval of midostaurin for adults with FLT3-mutated AML is a major recent achievement (64). However, poor pharmacokinetic properties or unfavorable toxicities of many multi-kinase inhibitors have limited usage in some patients. Trials of more selective second- and third-generation FLT3 inhibitors in adults with relapsed/refractory AML have established safety and tolerability of TKI monotherapy and in combination with chemotherapy, as well as exciting potential efficacy (14).
It is probable, perhaps certain, that children with FLT3 signaling-driven AML will similarly benefit from addition of FLT3 inhibition to chemotherapy. The newest and more selective inhibitors, quizartinib, crenolanib, and gilteritinib, have demonstrated very promising activity in adults with relapsed/refractory and newly diagnosed AML, but have been minimally or not studied to date in children. These agents merit broader clinical investigation in pediatrics. Emerging data from sorafenib maintenance studies also demonstrate the potential importance of such therapeutic strategies in the post-HSCT setting (75, 98). In addition, preclinical studies of FLT3-targeting chimeric antigen receptor T cell immunotherapy have demonstrated potent anti-leukemia activity in cell line (99, 100) and patient-derived xenograft models (Tasian, unpublished), further validating FLT3 as a robust therapeutic target in childhood AML.
Acquisition of resistance mutations following FLT3 inhibitor therapy remains a major source of treatment failure, although the incidence of such mutations in children with AML is not fully known. It is plausible that combining FLT3 inhibitors with chemotherapy may decrease the incidence of resistance mutations that occur with inhibitor monotherapy, analogous to lower mutation rates often observed in children with Ph+ ALL treated with TKI and chemotherapy (101, 102). However, major challenges exist in the study of new drugs in the pediatric population, including the relative rarity and genetic heterogeneity of childhood AML, rapid disease progression which may hamper trial enrollment, and the ability to partner with pharmaceutical companies to access novel agents for study in young children. Nonetheless, prospective clinical evaluation of exciting next-generation FLT3 inhibitors specifically in children with FLT3-mutated AML is ongoing or on the imminent horizon. Such important clinical investigation is critical to improve remission and decrease relapse in this highest-risk population of children with AML, potentially also reducing the significant toxicities associated with salvage therapy.
Author Contributions
ANS and SKT wrote and edited the manuscript. Both authors approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
ANS is supported by 5T32HL007574 from the National Institutes of Health/National Heart, Lung, and Blood Institute. SKT is supported by K08CA184418 from the National Institutes of Health/National Cancer Institute.
References
1. Rubnitz JE. How I treat pediatric acute myeloid leukemia. Blood (2012) 119(25):5980–8. doi:10.1182/blood-2012-02-392506
2. Rowe JM, Tallman MS. How I treat acute myeloid leukemia. Blood (2010) 116(17):3147–56. doi:10.1182/blood-2010-05-260117
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin (2017) 67(1):7–30. doi:10.3322/caac.21387
4. Alexander TB, Wang L, Inaba H, Triplett BM, Pounds S, Ribeiro RC, et al. Decreased relapsed rate and treatment-related mortality contribute to improved outcomes for pediatric acute myeloid leukemia in successive clinical trials. Cancer (2017) 123(19):3791–8. doi:10.1002/cncr.30791
5. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood (2002) 100(5):1532–42. doi:10.1182/blood-2002-02-0492
6. Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia (2003) 17(9):1738–52. doi:10.1038/sj.leu.2403099
7. Small D, Levenstein M, Kim E, Carow C, Amin S, Rockwell P, et al. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci U S A (1994) 91(2):459–63. doi:10.1073/pnas.91.2.459
8. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer (2003) 3(9):650–65. doi:10.1038/nrc1169
9. Matthews W, Jordan CT, Wiegand GW, Pardoll D, Lemischka IR. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell (1991) 65(7):1143–52. doi:10.1016/0092-8674(91)90010-V
10. Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC. FLT3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood (1996) 88(9):3383–90.
11. Scheijen B, Griffin JD. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene (2002) 21(21):3314–33. doi:10.1038/sj.onc.1205317
12. Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood (2009) 114(14):2984–92. doi:10.1182/blood-2009-05-222034
13. Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood (1996) 87(3):1089–96.
14. Pratz KW, Levis M. How I treat FLT3-mutated AML. Blood (2017) 129(5):565–71. doi:10.1182/blood-2016-09-693648
15. Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood (2006) 108(12):3654–61. doi:10.1182/blood-2006-03-009233
16. Tarlock K, Alonzo TA, Gerbing RB, Raimondi SC, Hirsch BA, Sung L, et al. Gemtuzumab ozogamicin reduces relapse risk in FLT3/ITD acute myeloid leukemia: a report from the Children’s Oncology Group. Clin Cancer Res (2016) 22(8):1951–7. doi:10.1158/1078-0432.CCR-15-1349
17. Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia (1996) 10(12):1911–8.
18. Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia (2015) 29(12):2390–2. doi:10.1038/leu.2015.165
19. Tarlock K, Alonzo TA, Loken MR, Gerbing RB, Ries RE, Aplenc R, et al. Disease characteristics and prognostic implications of cell-surface FLT3 receptor (CD135) expression in pediatric acute myeloid leukemia: a report from the Children’s Oncology Group. Clin Cancer Res (2017) 23(14):3649–56. doi:10.1158/1078-0432.CCR-16-2353
20. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood (2001) 97(8):2434–9. doi:10.1182/blood.V97.8.2434
21. Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol (2001) 113(4):983–8. doi:10.1046/j.1365-2141.2001.02850.x
22. Meshinchi S, Stirewalt DL, Alonzo TA, Zhang Q, Sweetser DA, Woods WG, et al. Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood (2003) 102(4):1474–9. doi:10.1182/blood-2003-01-0137
23. Tarlock K, Hansen ME, Hylkema T, Ries R, Farrar JE, Guidry Auvil J, et al. Discovery and functional validation of novel pediatric specific FLT3 activating mutations in acute myeloid leukemia: results from the COG/NCI target initiative. Blood (2015) 126(23):87.
24. Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters – an analysis of 3082 patients. Blood (2008) 111(5):2527–37. doi:10.1182/blood-2007-05-091215
25. Liang DC, Shih LY, Hung IJ, Yang CP, Chen SH, Jaing TH, et al. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia (2003) 17(5):883–6. doi:10.1038/sj.leu.2402928
26. Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood (2002) 99(12):4326–35. doi:10.1182/blood.V99.12.4326
27. Manara E, Basso G, Zampini M, Buldini B, Tregnago C, Rondelli R, et al. Characterization of children with FLT3-ITD acute myeloid leukemia: a report from the AIEOP AML-2002 study group. Leukemia (2017) 31(1):18–25. doi:10.1038/leu.2016.177
28. Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood (1999) 93(9):3074–80.
29. Kottaridis PD, Gale RE, Linch DC. Prognostic implications of the presence of FLT3 mutations in patients with acute myeloid leukemia. Leuk Lymphoma (2003) 44(6):905–13. doi:10.1080/1042819031000067503
30. Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood (2007) 109(2):431–48. doi:10.1182/blood-2006-06-001149
31. Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res (2001) 61(19):7233–9.
32. Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood (2002) 100(13):4372–80. doi:10.1182/blood-2002-05-1440
33. Zwaan CM, Meshinchi S, Radich JP, Veerman AJ, Huismans DR, Munske L, et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood (2003) 102(7):2387–94. doi:10.1182/blood-2002-12-3627
34. Gamis AS, Alonzo TA, Meshinchi S, Sung L, Gerbing RB, Raimondi SC, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol (2014) 32(27):3021–32. doi:10.1200/JCO.2014.55.3628
35. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med (2001) 344(14):1031–7. doi:10.1056/NEJM200104053441401
36. Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med (2006) 355(23):2408–17. doi:10.1056/NEJMoa062867
37. Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a Children’s Oncology Group study. J Clin Oncol (2009) 27(31):5175–81. doi:10.1200/JCO.2008.21.2514
38. Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, Slayton WB, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia (2014) 28(7):1467–71. doi:10.1038/leu.2014.30
39. Slayton WB, Kairalla JA, Schultz KR, Devidas M, Helian S, Pulsipher M, et al. Outcomes of dasatinib plus intensive chemotherapy or stem cell transplant (SCT) for Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) on Children’s Oncology Group AALL0622. J Clin Oncol (2015) 33(Suppl; abstract 10006). doi:10.1200/jco.2015.33.15_suppl.10006
40. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med (2017) 376(10):917–27. doi:10.1056/NEJMoa1609324
41. Zwaan CM, Söderhäll S, Brethon B, Luciani M, Rizzari C, Sternberg D, et al. A phase 1/2, open-label, dose-escalation study of midostaurin in pediatric patients (Pts) with relapsed or refractory (R/R) acute leukemia: final results of study ITCC-024 (CPKC412A2114). Blood (2015) 126(23):2564.
42. Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia (2012) 26(9):2061–8. doi:10.1038/leu.2012.115
43. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med (2017) 377(5):454–64. doi:10.1056/NEJMoa1614359
44. Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood (2011) 117(12):3294–301. doi:10.1182/blood-2010-08-301796
45. Inaba H, Rubnitz JE, Coustan-Smith E, Li L, Furmanski BD, Mascara GP, et al. Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J Clin Oncol (2011) 29(24):3293–300. doi:10.1200/JCO.2011.34.7427
46. Widemann BC, Kim A, Fox E, Baruchel S, Adamson PC, Ingle AM, et al. A phase I trial and pharmacokinetic study of sorafenib in children with refractory solid tumors or leukemias: a Children’s Oncology Group Phase I Consortium report. Clin Cancer Res (2012) 18(21):6011–22. doi:10.1158/1078-0432.CCR-11-3284
47. Rollig C, Serve H, Huttmann A, Noppeney R, Muller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol (2015) 16(16):1691–9. doi:10.1016/S1470-2045(15)00362-9
48. Chen YB, Li S, Lane AA, Connolly C, Del Rio C, Valles B, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for FMS-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant (2014) 20(12):2042–8. doi:10.1016/j.bbmt.2014.09.007
49. Ohanian M, Garcia-Manero G, Levis MJ, Jabbour E, Daver NG, Borthakur G, et al. Sorafenib plus 5-azacytidine (AZA) in older untreated FLT3-ITD mutated AML. J Clin Oncol (2017) 35(15_suppl):7029.
50. Fiedler W, Kayser S, Kebenko M, Janning M, Krauter J, Schittenhelm M, et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol (2015) 169(5):694–700. doi:10.1111/bjh.13353
51. Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O’Farrell AM, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood (2005) 105(3):986–93. doi:10.1182/blood-2004-05-1846
52. Cooper TM, Cassar J, Eckroth E, Malvar J, Sposto R, Gaynon P, et al. A phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: a therapeutic advances in childhood leukemia & lymphoma (TACL) study. Clin Cancer Res (2016) 22(16):4014–22. doi:10.1158/1078-0432.CCR-15-1998
53. Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol (2013) 31(29):3681–7. doi:10.1200/JCO.2013.48.8783
54. Perl AE, Dohner H, Rousselot PH, Marie J-P, Martinelli G, Shah NP, et al. Efficacy and safety of quizartinib (AC220) in patients age ≥ 70 years with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia (AML). J Clin Oncol (2013) 31(15_suppl):7023.
55. Schiller GJ, Tallman MS, Goldberg SL, Perl AE, Marie J-P, Martinelli G, et al. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukemia. J Clin Oncol (2014) 32(15_suppl):7100.
56. Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med (2012) 367(22):2075–88. doi:10.1056/NEJMoa1205127
57. Cortes JE, Kantarjian HM, Kadia TM, Borthakur G, Konopleva M, Garcia-Manero G, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. J Clin Oncol (2016) 34(15_suppl):7008.
58. Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood (2014) 123(1):94–100. doi:10.1182/blood-2013-10-529313
59. Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol (2017) 18(8):1061–75. doi:10.1016/S1470-2045(17)30416-3
60. Altman JK, Perl AE, Cortes JE, Smith CC, Litzow MR, Hill JE, et al. Deep molecular response to gilteritinib to improve survival in FLT3 mutation-positive relapsed/refractory acute myeloid leukemia. J Clin Oncol (2017) 35(15_suppl):7003.
61. Cortes JE, Altman J, Ritchie EK, Larson RA, Claxton D, Minden MD, et al. A phase II/III, multicenter, open-label, 3-arm study of gilteritinib, gilteritinib plus azacitidine, or azacitidine alone in the treatment of newly diagnosed FLT3 mutation-positive acute myeloid leukemia (AML) patients ineligible for intensive induction chemotherapy. J Clin Oncol (2017) 35(15_suppl):TS7068.
62. Gallogly MM, Lazarus HM. Midostaurin: an emerging treatment for acute myeloid leukemia patients. J Blood Med (2016) 7:73–83. doi:10.2147/JBM.S100283
63. Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol (2010) 28(28):4339–45. doi:10.1200/JCO.2010.28.9678
64. Levis M. Midostaurin approved for FLT3-mutated AML. Blood (2017) 129(26):3403–6. doi:10.1182/blood-2017-05-782292
65. Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood (2006) 108(10):3477–83. doi:10.1182/blood-2006-04-015743
66. Bernstein ML. Targeted therapy in pediatric and adolescent oncology. Cancer (2011) 117(10 Suppl):2268–74. doi:10.1002/cncr.26050
67. Knapper S, Russell N, Gilkes A, Hills RK, Gale RE, Cavenagh JD, et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood (2017) 129(9):1143–54. doi:10.1182/blood-2016-07-730648
68. Brown P, Kairalla J, Wang C, Dreyer Z, Salzer W, Sorenson M, et al. Addition of FLT3 inhibitor lestaurtinib to post-induction chemotherapy does not improve outcomes in MLL-rearranged infant acute lymphoblastic leukemia (All): AALL0631, a Children’s Oncology Group Study. Pediatric Blood & Cancer; SIOP Annual Meeting 2016. Dublin (2016).
69. Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst (2008) 100(3):184–98. doi:10.1093/jnci/djm328
70. Auclair D, Miller D, Yatsula V, Pickett W, Carter C, Chang Y, et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia (2007) 21(3):439–45. doi:10.1038/sj.leu.2404508
71. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med (2007) 356(2):125–34. doi:10.1056/NEJMoa060655
72. Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res (2006) 12(24):7271–8. doi:10.1158/1078-0432.CCR-06-1249
73. Pratz KW, Cho E, Levis MJ, Karp JE, Gore SD, McDevitt M, et al. A pharmacodynamic study of sorafenib in patients with relapsed and refractory acute leukemias. Leukemia (2010) 24(8):1437–44. doi:10.1038/leu.2010.132
74. Ravandi F, Arana Yi C, Cortes JE, Levis M, Faderl S, Garcia-Manero G, et al. Final report of phase II study of sorafenib, cytarabine and idarubicin for initial therapy in younger patients with acute myeloid leukemia. Leukemia (2014) 28(7):1543–5. doi:10.1038/leu.2014.54
75. Tarlock K, Chang B, Cooper T, Gross T, Gupta S, Neudorf S, et al. Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer (2015) 62(6):1048–54. doi:10.1002/pbc.25437
76. Dubois SG, Shusterman S, Ingle AM, Ahern CH, Reid JM, Wu B, et al. Phase I and pharmacokinetic study of sunitinib in pediatric patients with refractory solid tumors: a Children’s Oncology Group study. Clin Cancer Res (2011) 17(15):5113–22. doi:10.1158/1078-0432.CCR-11-0237
77. Wetmore C, Daryani VM, Billups CA, Boyett JM, Leary S, Tanos R, et al. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: a Children’s Oncology Group study ACNS1021. Cancer Med (2016) 5(7):1416–24. doi:10.1002/cam4.713
78. Baker SD, Zimmerman EI, Wang YD, Orwick S, Zatechka DS, Buaboonnam J, et al. Emergence of polyclonal FLT3 tyrosine kinase domain mutations during sequential therapy with sorafenib and sunitinib in FLT3-ITD-positive acute myeloid leukemia. Clin Cancer Res (2013) 19(20):5758–68. doi:10.1158/1078-0432.CCR-13-1323
79. Chao Q, Sprankle KG, Grotzfeld RM, Lai AG, Carter TA, Velasco AM, et al. Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. J Med Chem (2009) 52(23):7808–16. doi:10.1021/jm9007533
80. Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood (2010) 115(7):1425–32. doi:10.1182/blood-2009-09-242859
81. Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Dohner H, Dohner K, Schittenhelm MM. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol Cancer (2013) 12:19. doi:10.1186/1476-4598-12-19
82. Abdelall W, Kantarjian HM, Borthakur G, Garcia-Manero G, Patel KP, Jabbour EJ, et al. The combination of quizartinib with azacitidine or low dose cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: interim report of a phase I/II trial. Blood (2016) 128(23):1642.
83. Smith CC, Paguirigan A, Jeschke GR, Lin KC, Massi E, Tarver T, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood (2017) 130(1):48–58. doi:10.1182/blood-2016-04-711820
84. Alvarado Y, Kantarjian HM, Luthra R, Ravandi F, Borthakur G, Garcia-Manero G, et al. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer (2014) 120(14):2142–9. doi:10.1002/cncr.28705
85. Jain P, Kantarjian H, Jabbour E, Gonzalez GN, Borthakur G, Pemmaraju N, et al. Ponatinib as first-line treatment for patients with chronic myeloid leukaemia in chronic phase: a phase 2 study. Lancet Haematol (2015) 2(9):e376–83. doi:10.1016/S2352-3026(15)00127-1
86. Gozgit JM, Wong MJ, Wardwell S, Tyner JW, Loriaux MM, Mohemmad QK, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther (2011) 10(6):1028–35. doi:10.1158/1535-7163.MCT-10-1044
87. Zirm E, Spies-Weisshart B, Heidel F, Schnetzke U, Bohmer FD, Hochhaus A, et al. Ponatinib may overcome resistance of FLT3-ITD harbouring additional point mutations, notably the previously refractory F691I mutation. Br J Haematol (2012) 157(4):483–92. doi:10.1111/j.1365-2141.2012.09085.x
88. Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood (2013) 121(16):3165–71. doi:10.1182/blood-2012-07-442871
89. Gainor JF, Chabner BA. Ponatinib: accelerated disapproval. Oncologist (2015) 20(8):847–8. doi:10.1634/theoncologist.2015-0253
90. Nickel RS, Daves M, Keller F. Treatment of an adolescent with chronic myeloid leukemia and the T315I mutation with ponatinib. Pediatr Blood Cancer (2015) 62(11):2050–1. doi:10.1002/pbc.25551
91. Smith CC, Lasater EA, Lin KC, Wang Q, McCreery MQ, Stewart WK, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A (2014) 111(14):5319–24. doi:10.1073/pnas.1320661111
92. Fathi AT. Emergence of crenolanib for FLT3-mutant AML. Blood (2013) 122(22):3547–8. doi:10.1182/blood-2013-10-528992
93. Randhawa JK, Kantarjian HM, Borthakur G, Thompson PA, Konopleva M, Daver N, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood (2014) 124(21):389.
94. Wang ES, Stone RM, Tallman MS, Walter RB, Eckhardt JR, Collins R. Crenolanib, a type I FLT3 TKI, can be safely combined with cytarabine and anthracycline induction chemotherapy and results in high response rates in patients with newly diagnosed FLT3 mutant acute myeloid leukemia (AML). Blood (2016) 128(22):1071.
95. Wetmore C, Broniscer A, Turner D, Wright KD, Pai-Panandiker A, Kun LE, et al. First-in-pediatrics phase I study of crenolanib besylate (CP-868,596-26) administered during and after radiation therapy (RT) in newly diagnosed diffuse intrinsic pontine glioma (DIPG) and recurrent high-grade glioma (HGG). J Clin Oncol (2014) 32(15_suppl):10064.
96. Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood (2017) 129(2):257–60. doi:10.1182/blood-2016-10-745133
97. Galanis A, Levis M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica (2015) 100(3):e77–9. doi:10.3324/haematol.2014.117028
98. Brunner AM, Li S, Fathi AT, Wadleigh M, Ho VT, Collier K, et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br J Haematol (2016) 175(3):496–504. doi:10.1111/bjh.14260
99. Chien CD, Sauter CT, Ishii K, Nguyen SM, Shen F, Tasian SK, et al. Preclinical development of FLT3-redirected chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia. Blood (2016) 128(22):1072.
100. Chen L, Mao H, Zhang J, Chu J, Devine S, Caligiuri MA, et al. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia (2017) 31(8):1830–4. doi:10.1038/leu.2017.147
101. Bernt KM, Hunger SP. Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia. Front Oncol (2014) 4:54. doi:10.3389/fonc.2014.00054
Keywords: acute myeloid leukemia, clinical trials, FLT3, kinase inhibitor, pediatrics
Citation: Sexauer AN and Tasian SK (2017) Targeting FLT3 Signaling in Childhood Acute Myeloid Leukemia. Front. Pediatr. 5:248. doi: 10.3389/fped.2017.00248
Received: 07 September 2017; Accepted: 06 November 2017;
Published: 20 November 2017
Edited by:
Peter Michael Gordon, University of Minnesota, United StatesReviewed by:
E. Anders Kolb, Alfred I. duPont Hospital for Children, United StatesJessica Pollard, Seattle Children’s Hospital, United States
Copyright: © 2017 Sexauer and Tasian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah K. Tasian, dGFzaWFuc0BlbWFpbC5jaG9wLmVkdQ==