 Hiroto Inaba
Hiroto Inaba Elizabeth M. Azzato2
Elizabeth M. Azzato2 Charles G. Mullighan
Charles G. Mullighan- 1Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 2Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, United States
Acute lymphoblastic leukemia (ALL) is the most common type of cancer in children. In recent Total Therapy studies conducted at St. Jude Children’s Research Hospital, children with ALL had a 5-year overall survival of around 94%. This is the result of a combination of risk stratification based on the biological features of the leukemic cells and the response to treatment (as assessed by the detection of minimal residual disease), treatment modification based on pharmacodynamic and pharmacogenomic data, and improved supportive care. However, innovative approaches are required to further improve survival to as close to 100% as possible and to reduce the adverse effects of treatment. Next-generation sequencing of leukemic cell DNA and RNA, as well as of germline DNA, can identify submicroscopic genetic structural changes and sequence alterations that contribute to leukemogenesis. Next-generation sequencing data can be used to define new ALL subtypes, to help improve treatment response and reduce adverse effects, and to identify novel prognostic markers and therapeutic targets to facilitate personalized precision medicine. In this article, we describe our approach to detecting targetable lesions in patients with ALL by next-generation sequencing and explain how we integrate the sequencing data into the treatment of these patients.
Acute Lymphoblastic Leukemia (ALL) Treatment at St. Jude Children’s Research Hospital
The survival rates of children and adolescents with ALL treated on Total Therapy study protocols at St. Jude Children’s Research Hospital (St. Jude) have improved considerably over time (1). In the St. Jude Total XV study (NCT00137111), which enrolled patients with B-cell ALL (B-ALL) [including Philadelphia chromosome (Ph)- or BCR-ABL1-positive ALL] or T-cell ALL (T-ALL) who were aged 1–18 years at diagnosis, 5-year event-free survival was increased to 87% and overall survival to 94%. In this protocol, prophylactic cranial irradiation was completely replaced by risk-adapted intrathecal chemotherapy and the incidence of isolated CNS relapse was reduced to 2.7% (2), which is comparable to that in previous St. Jude studies and other collaborative studies that incorporated irradiation for up to 33% of patients (3, 4). However, more effective treatment strategies are needed to further improve survival.
Although the outcomes for patients with St. Jude low-risk ALL [equivalent to National Cancer Institute (NCI) standard-risk ALL] are now excellent, there is still a need for substantial improvement in the cure rates for patients with St. Jude standard-risk and high-risk ALL (equivalent to NCI high-risk and very high-risk ALL, respectively). Our latest frontline ALL treatment protocol, Total XVII (NCT03117751), incorporates novel precision-medicine strategies based on inherited and leukemia/lymphoma-specific genomic features and targeted treatment approaches.
Philadelphia Chromosome–Like (Ph-Like) ALL
Ph-like ALL (also known as BCR-ABL1–like ALL), a recently described subtype of B-ALL, is characterized by a gene-expression profile similar to that of Ph-positive ALL; a variety of genetic alterations that activate tyrosine kinase signaling; the mutation of lymphoid transcription factor genes such as IKZF1 (in 70%–80% of cases); and a poor outcome (5–8). Because the observed kinase-activating alterations are potentially targetable with clinically available tyrosine kinase inhibitors (TKIs), many groups studying leukemia are implementing the diagnosis of Ph-like ALL in prospective clinical trials.
In Ph-like ALL, there are ABL-class rearrangements involving ABL1, ABL2, CSF1R, PDGFRA, or PDGFRB, resulting in the expression of fusion genes (Table 1) (7). Multiple fusion partner genes have been reported, but in each case the fusion involves the kinase as the downstream partner and, therefore, preserves the kinase domain.
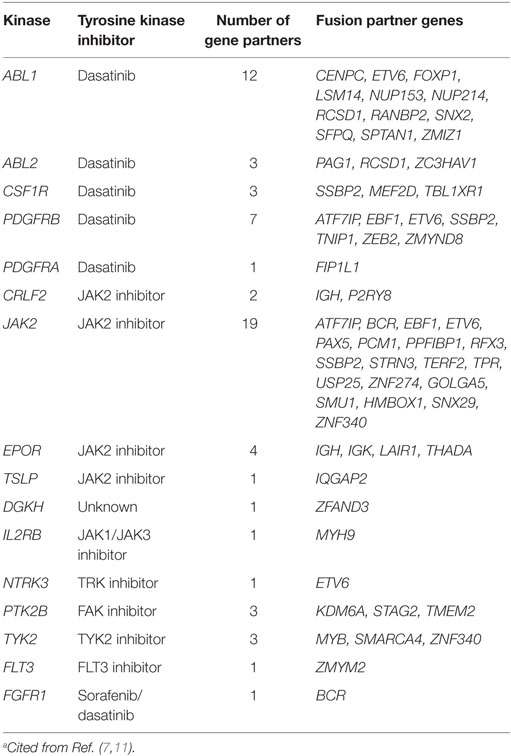
Table 1. Representative kinase rearrangements and therapeutic targets in Ph-like ALL.a
The JAK–STAT signaling pathway is also an important mediator of signals from hematopoietic cytokine receptors, and its activation is frequently detected in Ph-like ALL (7, 9). JAK2/EPOR rearrangements include more than 10 different fusions of JAK2 that preserve the JAK2 kinase domain, along with rearrangements of EPOR with the immunoglobulin heavy (IGH) or kappa (IGK) loci that result in their overexpression and the activation of receptor signaling (Table 1). Approximately 50% of patients with Ph-like ALL have a CRLF2 rearrangement in the Xp/Yp pseudoautosomal region 1 (10). CRLF2 encodes the receptor for thymic stromal lymphopoietin. Rearrangement of CRLF2 into the IGH locus at 14q32 or a focal deletion immediately upstream of the gene results in the fusions IGH-CRLF2 and P2RY8-CRLF2, respectively, leading to CRLF2 overexpression. Approximately 50% of CRLF2 rearrangements are accompanied by activating JAK1 or JAK2 mutations. In addition to these rearrangements, up to 15% of pediatric patients with Ph-like ALL have mutations that activate the JAK–STAT signaling pathway. These mutations include those in genes encoding cytokine receptors (IL7R or IL2RB); activating mutations in the Janus kinase genes themselves (JAK1 or JAK3); and mutations that impair the function of negative regulators of JAK–STAT signaling (SH2B3) (7, 11).
Diagnostic Evaluation of Newly Diagnosed ALL
To capture these relevant alterations in the Total XVII protocol, diagnostic testing involves not only established morphologic, immunophenotyping, and molecular genetic approaches but also next-generation sequencing diagnostics that are performed as the clinical standard of care for all consented patients.
Initially, the DNA index of each patient sample is measured by flow cytometry, and the samples are screened for selected ALL fusions (i.e., TCF3-PBX1, ETV6-RUNX1, MLL rearrangement, and BCR-ABL1) by RT-PCR and fluorescence in situ hybridization (FISH) (Figure 1). Concurrently, the samples undergo RNA sequencing (RNA-Seq) for fusion detection, using a validated in-house de novo assembly and fusion–detection algorithm. This broad approach allows the detection of additional known and novel fusion transcripts, including ABL-class fusions, which are reported by day 15 to enable therapy stratification (Table 1).
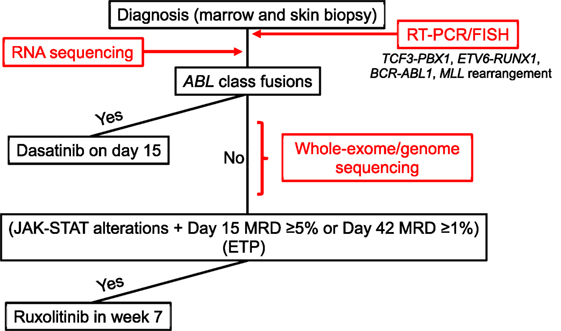
Figure 1. Diagnostic procedure for and treatment of B- and T-acute lymphoblastic leukemia (ALL) with targetable lesions and early T-cell precursor ALL in Total XVII. FISH, fluorescence in situ hybridization; MRD, minimal residual disease.
At study initiation, patients are also offered integrative whole-genome, whole-exome, and whole-transcriptome sequencing, performed in a College of American Pathologists (CAP)/Clinical Laboratory Improvement Amendments-accredited laboratory (Figure 1). The analysis pipeline interrogates tumor and germline sequence information to detect copy-number variations, structural variations, fusion transcripts, single-nucleotide variants, and insertions and deletions, integrating data from all three platforms. As this testing requires the use of paired germline genomic information derived from skin biopsy or remission blood samples, clinical consent must be obtained. This clinical comprehensive genomic-sequencing approach is used to identify all somatic genetic alterations relevant to ALL. These include sequence variants, which are not evaluated in the initial RNA-Seq analysis because of time constraints and the complexity of the analysis, especially those variants that result in kinase-activating lesions amenable to targeting with TKIs. Comprehensive sequencing can also provide information on structural and copy-number variants not captured by earlier analyses (e.g., deletions involving SH2B3 or IKZF1), which may be especially relevant to Ph-like ALL and actionable with TKIs. Comprehensive sequencing data are reported by days 28–42 of remission induction. After diagnostic testing is complete, patients are reapproached for germline testing. If consent is obtained for constitutional testing, data from the previously sequenced germline sample are reanalyzed to detect alterations in cancer predisposition genes.
Other collaborative groups have also developed diagnostic strategies for Ph-like ALL. For example, the Children’s Oncology Group (COG) uses tiered screening algorithms, employing TaqMan low-density array cards for NCI high-risk B-ALL, followed by sequential genomic profiling (e.g., evaluation of CRLF2 expression, FISH for CRLF2 rearrangement and Sanger sequencing for JAK1/JAK2 mutations for cases with high CRLF2 expression, and RT-PCR and/or transcriptome sequencing for cases with low CRLF2 expression) (12). However, our comprehensive analysis can identify genetic alterations other than ABL-class fusions and those associated with JAK-STAT activation, and it provides the flexibility to interrogate emerging new prognostic and predictive markers in real time and to accommodate new risk classifications or the incorporation of new agents.
Treatment of ALL with Targetable Lesions
Of the patients with B-ALL treated on the St. Jude Total XV protocol, 12% had Ph-like ALL, which was associated with high levels of minimal residual disease (MRD) during induction (13). Possibly because of our using MRD-based risk-directed therapy, there were no significant differences in event-free survival or overall survival between patients with and without Ph-like ALL. However, a high proportion (15%) of the patients with Ph-like ALL underwent a transplant because of their high MRD levels (≥1%) at the end of remission induction, whereas only 4% of the other patients with B-ALL underwent a transplant. Incorporating TKIs into the conventional chemotherapy for patients with targetable lesions could help reduce the intensity of the chemotherapy.
The identification of targetable lesions and the use of TKIs have proved effective in the treatment of Ph-positive ALL in children and adults (14, 15), and several case reports have described the effectiveness of imatinib/dasatinib in the treatment of refractory Ph-like ALL with ABL-class fusions (7, 16, 17). In COG studies of NCI high-risk B-ALL, dasatinib has been used to treat patients with ABL-class fusions (AALL1131, NCT02883049), and the JAK inhibitor ruxolitinib is administered to those with CRLF2 rearrangements and/or other JAK–STAT pathway alterations (AALL1521, NCT02723994) (12).
Dasatinib was used to treat Ph-positive ALL in our Total XVI protocol and was well-tolerated by patients (18). In the Total XVII study, dasatinib will be given to patients with ABL-class chimeric fusions (i.e., those involving ABL1, ABL2, CSF1R, PDGFRA, or PDGFRB) that are identified by RNA-Seq. Because of our previous experience with dasatinib and its safety profile (18), administration of this drug will start on day 15 of remission induction, regardless of the patient’s MRD level (Figure 1).
Clinical experience of administering ruxolitinib in combination with conventional chemotherapy in children is limited, as is data on its safety profile, and a comprehensive identification of the genetic alterations associated with activation of the JAK–STAT pathway will take 28–42 days. Thus, ruxolitinib will be administered after remission-induction therapy (i.e., 6 weeks after diagnosis) to patients with a rearrangement, mutation, or genomic deletion (e.g., of EPOR, IL7R, JAK1, JAK2, JAK3, or SH2B3) that results in the activation of JAK–STAT signaling and who also have MRD of 5% or more on day 15 of induction or 1% or more at the end of induction. Ruxolitinib will be administered with discontinuous dosing (2 weeks on and 2 weeks off), and a dose-finding phase is included in the study. In patients with MRD (≥0.01%) at the end of induction, subsequent MRD will be monitored closely, and immunotherapy (e.g., with chimeric antigen receptor T cells) or a hematopoietic stem cell transplant will be considered for those with persistently MRD-positive disease.
Treatment of T-ALL with Targetable Lesions and Early T-Cell Precursor ALL
The Total XVII protocol enrolls patients with T-ALL, and targetable genetic alterations and activated signaling pathways also represent important opportunities for precision-medicine approaches to treating T-ALL. ABL-class fusions, with NUP214, SLC9A3R1, ETV6, or MBNL1 as partner genes, are present in patients with T-ALL (19, 20), and dasatinib will be given to those patients. Mutations that lead to JAK–STAT signaling alterations (e.g., those in JAK3, IL7R, or PTPN2) have been seen in approximately 25% of patients with T-ALL and are prevalent in the groups with LX1/TLX3 or HOXA mutations (20). Early T-cell precursor ALL (ETP-ALL) accounts for 10–15% of T-ALL cases. ETP-ALL is characterized by a distinct immunophenotype, with expression of myeloid/stem-cell markers, a high frequency of JAK–STAT-activating mutations (20), biochemical evidence of activated JAK–STAT signaling (21), and exquisite sensitivity to the JAK inhibitor ruxolitinib in preclinical models (22). Thus, ruxolitinib will be given to patients with the ETP immunophenotype, as well as to other patients with T-ALL who exhibit high MRD levels (≥5% on day 15 or ≥1% at the end of remission induction) and have JAK–STAT alterations. Hematopoietic stem cell transplant will be considered for patients with persistently MRD-positive disease.
Conclusion
Our next-generation sequencing approach identifies therapeutic targets to facilitate personalized precision medicine while providing the flexibility to interrogate emerging prognostic and predictive markers in real time. This approach could lead to improved cure rates and reduced toxicities, especially in higher risk patients.
Author Contributions
Conception and design: HI. Manuscript writing and final approval: all authors.
Conflict of Interest Statement
HI obtained drug support from Bristol-Myers Squibb and Incyte Corporation. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YP and handling Editor declared their shared affiliation.
Acknowledgments
We thank Keith A. Laycock, PhD, ELS (St. Jude Children’s Research Hospital), for editorial assistance.
Funding
This work was supported by grant no. CA21765 and R35 CA197695 from the National Institutes of Health and by ALSAC.
References
1. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet (2013) 381:1943–55. doi:10.1016/S0140-6736(12)62187-4
2. Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med (2009) 360:2730–41. doi:10.1056/NEJMoa0900386
3. Pui CH, Pei D, Sandlund JT, Ribeiro RC, Rubnitz JE, Raimondi SC, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia (2010) 24:371–82. doi:10.1038/leu.2009.252
4. Vora A, Andreano A, Pui CH, Hunger SP, Schrappe M, Moericke A, et al. Influence of cranial radiotherapy on outcome in children with acute lymphoblastic leukemia treated with contemporary therapy. J Clin Oncol (2016) 34:919–26. doi:10.1200/JCO.2015.64.2850
5. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med (2009) 360:470–80. doi:10.1056/NEJMoa0808253
6. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol (2009) 10:125–34. doi:10.1016/S1470-2045(08)70339-5
7. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med (2014) 371:1005–15. doi:10.1056/NEJMoa1403088
8. Pui C-H, Roberts KG, Yang JJ, Mullighan CG. Philadelphia chromosome-like acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk (2017) 17:464–70. doi:10.1016/j.clml.2017.03.299
9. Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A (2009) 106:9414–8. doi:10.1073/pnas.0811761106
10. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet (2009) 41:1243–6. doi:10.1038/ng.469
11. Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, et al. High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol (2017) 35:394–401. doi:10.1200/JCO.2016.69.0073
12. Tran TH, Loh ML. Ph-like acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program (2016) 2016:561–6. doi:10.1182/asheducation-2016.1.561
13. Roberts KG, Pei D, Campana D, Payne-Turner D, Li Y, Cheng C, et al. Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol (2014) 32:3012–20. doi:10.1200/JCO.2014.55.4105
14. Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a Children’s Oncology Group study. J Clin Oncol (2009) 27:5175–81. doi:10.1200/JCO.2008.21.2514
15. Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med (2013) 369:1783–96. doi:10.1056/NEJMoa1306494
16. Schwab C, Ryan SL, Chilton L, Elliott A, Murray J, Richardson S, et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood (2016) 127:2214–8. doi:10.1182/blood-2015-09-670166
17. Weston BW, Hayden MA, Roberts KG, Bowyer S, Hsu J, Fedoriw G, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB–positive acute lymphoblastic leukemia. J Clin Oncol (2013) 31:e413–6. doi:10.1200/JCO.2012.47.6770
18. Jeha S, Coustan-Smith E, Pei D, Sandlund JT, Rubnitz JE, Howard SC, et al. Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer (2014) 120:1514–9. doi:10.1002/cncr.28598
19. Quintás-Cardama A, Tong W, Manshouri T, Vega F, Lennon PA, Cools J, et al. Activity of tyrosine kinase inhibitors against human NUP214-ABL1–positive T cell malignancies. Leukemia (2008) 22:1117–24. doi:10.1038/leu.2008.80
20. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet (2017) 49:1211–8. doi:10.1038/ng.3909
21. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature (2012) 481:157–63. doi:10.1038/nature10725
Keywords: acute lymphoblastic leukemia, next-generation sequencing, Philadelphia chromosome-like leukemia, molecularly targeted therapy, early T-cell precursor
Citation: Inaba H, Azzato EM and Mullighan CG (2017) Integration of Next-Generation Sequencing to Treat Acute Lymphoblastic Leukemia with Targetable Lesions: The St. Jude Children’s Research Hospital Approach. Front. Pediatr. 5:258. doi: 10.3389/fped.2017.00258
Received: 08 August 2017; Accepted: 20 November 2017;
Published: 04 December 2017
Edited by:
Brenton Garrett Mar, Dana–Farber Cancer Institute, United StatesReviewed by:
Sarah K. Tasian, University of Pennsylvania, United StatesYana Pikman, Dana–Farber Cancer Institute, United States
Jose Andres Yunes, Centro Infantil Boldrini, Brazil
Copyright: © 2017 Inaba, Azzato and Mullighan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroto Inaba, aGlyb3RvLmluYWJhQHN0anVkZS5vcmc=