 Elizabeth Forsythe
Elizabeth Forsythe Joanna Kenny
Joanna Kenny Chiara Bacchelli
Chiara Bacchelli- Genetics and Genomic Medicine Programme, UCL Great Ormond Street Institute of Child Health, University College London, London, United Kingdom
Bardet–Biedl syndrome is a rare autosomal recessive multisystem disorder caused by defects in genes encoding for proteins that localize to the primary cilium/basal body complex. Twenty-one disease-causing genes have been identified to date. It is one of the most well-studied conditions in the family of diseases caused by defective cilia collectively known as ciliopathies. In this review, we provide an update on diagnostic developments, clinical features, and progress in the management of Bardet–Biedl syndrome. Advances in diagnostic technologies including exome and whole genome sequencing are expanding the spectrum of patients who are diagnosed with Bardet–Biedl syndrome and increasing the number of cases with diagnostic uncertainty. As a result of the diagnostic developments, a small number of patients with only one or two clinical features of Bardet–Biedl syndrome are being diagnosed. Our understanding of the syndrome-associated renal disease has evolved and is reviewed here. Novel interventions are developing at a rapid pace and are explored in this review including genetic therapeutics such as gene therapy, exon skipping therapy, nonsense suppression therapy, and gene editing. Other non-genetic therapies such as gene repurposing, targeted therapies, and non-pharmacological interventions are also discussed.
Introduction
Bardet–Biedl syndrome (BBS), sometimes known as Laurence–Moon–Bardet-Biedl syndrome, is a rare autosomal recessive ciliopathy characterized by rod-cone dystrophy, learning difficulties, polydactyly, obesity, genital malformations, and renal abnormalities.
In the 1880s, a family with retinitis pigmentosa, obesity, and intellectual impairment was described by doctors Laurence and Moon. The affected family members later went on to develop a spastic paraparesis. In 1920 and 1922, respectively, doctors Bardet and Biedl independently described two families with obesity, retinitis pigmentosa, and polydactyly. From 1925, the syndrome was known as Laurence–Moon–Bardet–Biedl syndrome, but there was disagreement as to whether they were the same entity. Later, it was considered as two entities, Laurence–Moon and Bardet–Biedl syndromes, but mutations in known BBS genes have been seen in families with both syndromes (1, 2). Today, it is most usually known as BBS.
It is a pleiotropic disorder and has a prevalence of around 1:100,000 in North America and Europe, but it is significantly more common in certain isolated communities including Newfoundland (1:18,000) (2) and Kuwaiti Bedouins (1: 13,500) (3, 4). In the last 2 decades, 21 BBS genes (BBS1–BBS21) (5–7) have been identified, mutations in which account for 80% of cases with a clinical diagnosis of BBS (1). Table 1 outlines the 21 BBS genes.
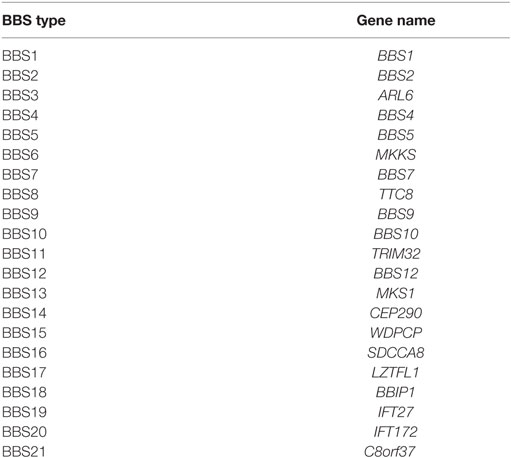
Table 1. Known genes causing BBS.
Mutations in BBS1 and BBS10 account for the majority of genotypes (~51 and ~20%, respectively) in Northern Europe and North America (4).
Bardet–Biedl syndrome proteins localize to the primary cilium/basal body complex, a ubiquitously expressed highly evolutionarily conserved organelle functioning primarily for cell-to-cell signaling. The genes that cause BBS can also cause other ciliopathies, with the classic example being CEP290, which can cause Joubert syndrome, Leber congenital amaurosis, Meckel syndrome, and Senior-Loken syndrome in addition to BBS (8).
Diagnosing BBS
Bardet–Biedl syndrome is a pleiotropic disorder and diagnosis is based on the presence of at least four major features or three major features and at least two minor features in accordance with the diagnostic criteria published by Beales et al. (9). Figure 1 demonstrates the clinical features associated with BBS and highlights the relative frequencies at which these features are observed.
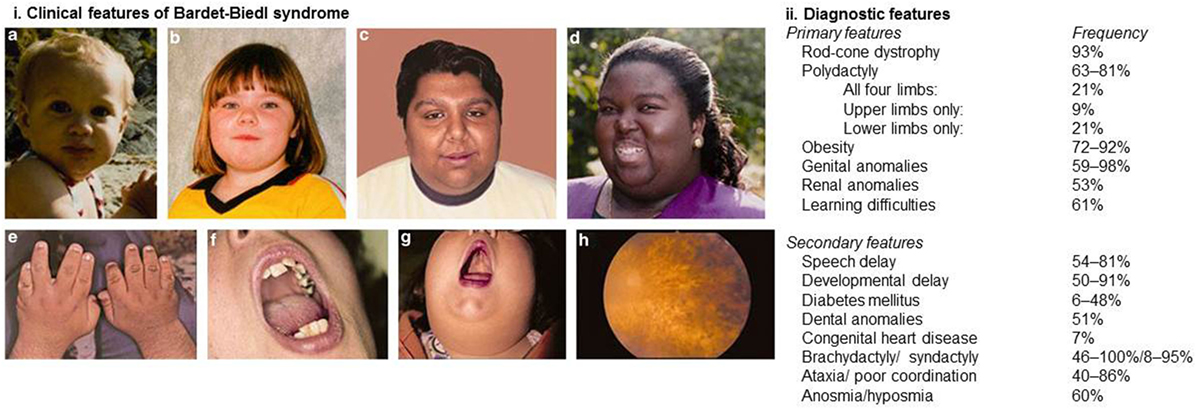
Figure 1. Clinical and diagnostic features of Bardet–Biedl syndrome. (i) Clinical features associated with Bardet–Biedl syndrome. (A–D) Typical facial features are often subtle and not always present. Typical facial features include malar hypoplasia, a depressed nasal bridge, deep set eyes, and retrognathia. (E) Brachydactyly. (F) Dental crowding. (G) High palate. (H) Rod-cone dystrophy. (ii) Diagnostic features of Bardet–Biedl syndrome. At least four major features or three major and two minor features are required to make a clinical diagnosis. Informed consent was obtained and republished with permission (4).
Molecular confirmation of BBS has evolved over the last decade from targeted sequencing of common genetic variants, including the common BBS1 p.M390R, BBS2 p.Y24X, BBS2 p.R275X, and BBS10 c.91fsX5 mutations to next-generation sequencing gene panels containing all known BBS genes. The frequency at which molecular confirmation is achieved has increased accordingly from approximately 40–80% (1).
The age at which patients are diagnosed is extremely variable and is driven by the age of onset of symptomatic rod-cone dystrophy. While this may manifest in infancy, it is more usually seen between the ages of 5 and 10 years of age and typically presents with night blindness (9). Isolated polydactyly at birth or obesity, generally seen from infancy, do not usually prompt referral. Siblings of affected children are generally diagnosed earlier. Antenatal diagnosis is extremely rare in the absence of a family history, but BBS may be suspected from the identification of echogenic kidneys and polydactyly on ultrasound scanning. Children presenting with renal anomalies or renal failure may be diagnosed earlier than those without, but there are insufficient data to confirm this. A subset of individuals present with isolated rod-cone dystrophy with notable absence of other BBS-related features and are often diagnosed in adulthood. These individuals are now being picked up because of the introduction of panel-based genetic testing and major diagnostic studies such as the UK 100,000 genomes project (10) and the Deciphering Developmental Disorders (exome) study (11). They were previously overlooked as there are many causes of rod-cone dystrophy, and it was not understood that BBS genes could cause this feature in isolation.
Currently, diagnostic gene panels are the diagnostic tool of choice. The use of whole exome sequencing (WES) and whole genome sequencing (WGS) may increase coverage, aid in the discovery of novel genes, and allow for the identification of non-coding variants. However, along with increased expense, disadvantage of the more advanced diagnostic sequencing techniques is the identification of pathogenic variants in non-BBS genes and of variants of unknown significance (VUS) in BBS genes (12). This can result in a diagnostic conundrum in particular where patients manifest only one or two non-specific major diagnostic criteria such as obesity and/or learning difficulties. The use of WES and WGS requires careful consenting of patients and having a plan in place to deal with VUS and unlooked for results.
Variable expressivity is the hallmark of BBS (8); patients with the same genotype and even siblings frequently manifest symptoms differently. As a result, although genotype–phenotype correlations exist on a population basis, it is not possible to make individual predictions about symptomatic manifestations (13). As a group, patients with mutations in BBS1 are usually less severely affected than patients with mutations in other BBS genes. On average, they develop visual deterioration later in life (14), are less likely to develop renal disease (13), and more likely to have a better endocrine biochemical profile (15) with a lower prevalence of metabolic syndrome (16, 17). It is not possible to delineate if this is a consequence of the common missense mutation BBS1 p.M390R identified in 80% of Northern European patients (4) or if the milder phenotype is representative of an overall less severe phenotype associated with BBS1 mutations (17).
Suggestion that BBS would be a candidate for triallelic inheritance, whereby a third mutation is required to either manifest the condition or adding mutational load, has gathered limited evidence (18–20). In practice, the phenotypic variability observed in patients with the same genotype and within families is likely to reflect a complex interplay between multiple genetic factors and environmental influences.
In the future, it may be possible to identify phenotypic modifiers and further elucidate the cause for variability through analysis of the “Omics” (genomics, epigenomics, transcriptomics, proteomics, and metabolomics) whereby the complex interplay of genes, transcription, protein expression, and metabolism is considered as part of the phenotypic analysis (21, 22).
Renal Disease in BBS
Bardet–Biedl syndrome has classically been associated with polycystic kidney disease, a typical feature of ciliopathies with renal manifestations (13, 16, 23–29). The prevalence of renal disease in BBS has been estimated at 53–82% (13, 16, 25). A recent study of 350 patients from the United Kingdom estimated that 50% of patients will develop functional renal disease and demonstrated that cystic or dysplastic disease only accounts for 30% of patients with renal disease, where the remainder have hydronephrosis, scarred or atrophic kidneys, loss of corticomedullary function, or developmental abnormalities (13). Around 8% of patients go on to develop end-stage renal disease requiring dialysis or transplantation (13). The majority of patients who develop end-stage renal disease do so in early childhood (before the age of 5), and in most cases, deterioration is rapid with frequent requirement for dialysis within the first year of life (13). Some patients develop sudden renal failure in adulthood for unknown reasons, and a further group of patients develop end-stage kidney disease as a result of comorbidities including type 2 diabetes and hypertension (13). The prevalence of these comorbidities is thought to be higher in BBS patients than the normal population and in 1 study of 69 patients were found in 22 and 35%, respectively (17). The risk of type 2 diabetes relates to obesity and is treated using standard protocols. Many patients with structural renal abnormalities do not go on to develop functional renal disease (17).
The molecular mechanistic pathways leading to renal disease in BBS remain unelucidated (29). It has been suggested that aberrant mTOR signaling may contribute to the development of cystic kidney disease (28). Another theory proposes that ciliary dysfunction leads to aberrant non-canonical Wnt signaling and planar cell polarity, which may contribute to the development of cysts (30). Molecular evidence supporting these theories remains limited. Furthermore, they do not explain why only some patients develop renal dysplasia or indeed the heterogeneous types of renal disease developed by patients.
Current Management of BBS
Bardet–Biedl syndrome is currently treated symptomatically focusing in particular on aggressive management of diabetes, hypertension, and metabolic syndrome to minimize the secondary impact that these conditions have on vulnerable organ systems already affected by BBS, in particular the eyes and kidneys (17). Weight management is a continual struggle for the majority of patients (31). Some elect to have bariatric surgery (32) while others take antiobesity medication, but for the majority of patients, dietetic input provides the safest and most effective weight loss strategy (33).
In the UK, patients who attend the national BBS clinics are invited to attend a multidisciplinary clinic for annual review by a geneticist, ophthalmologist, nephrologist, endocrinologist, psychologist, dietitian, speech and language therapist, nurse, and a patient support group representative. This provides a platform for regular review and individualized risk assessment in particular with reference to renal and endocrine deterioration. All patients are genotyped using a service-developed diagnostic gene panel. The clinic also provides an opportunity for research into the natural progression of BBS, and it is expected that patients will eventually be stratified according to genotype and their need for clinical follow-up.
Future Therapies for BBS
The last decade has seen significant advances in the development of therapeutic modalities, which could potentially be applicable to patients with BBS and related ciliopathies. However, the large number of disease-causing genes and private mutations, which are those seen in only a single family, present a unique challenge in developing genetic therapies for BBS (34). Figure 2 outlines potential future therapies, which are likely to benefit patients with BBS in the future.
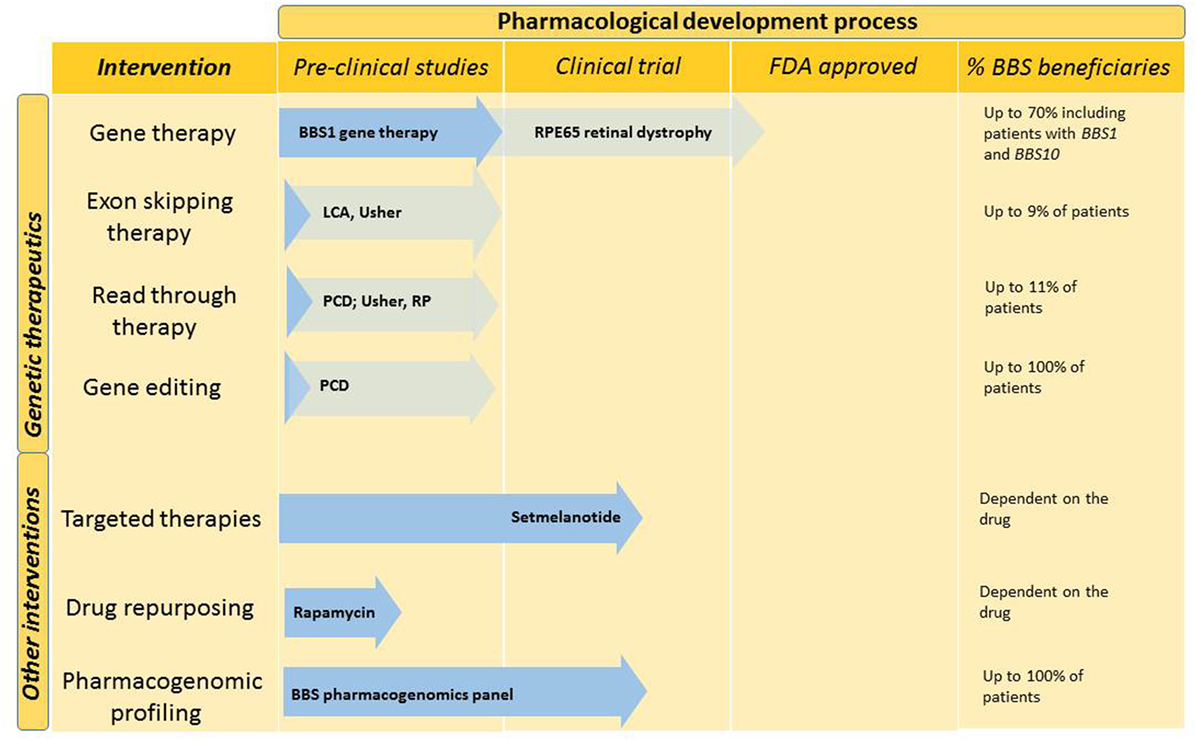
Figure 2. Future interventions and stage in the pharmacological development process. Genetic therapies and other pharmacological interventions are under development for BBS. Dark blue arrows demonstrate the stage to which BBS-specific interventions have been developed. Other ciliopathy relevant developments are indicated in light blue. The last column indicates the percentage of BBS patients who could benefit from this type of intervention. BBS, Bardet–Biedl syndrome; LCA, leber congenital amaurosis; PCD, primary ciliary dyskinesia; RP, retinitis pigmentosa; US, Usher syndrome.
Genetic Therapies
A particular focus for therapeutic intervention in the ciliopathies has been the development of therapy for rod-cone dystrophy (34–36). The eye offers an attractive organ for therapeutic intervention in BBS due to the ease of access, the presence of a control (other) eye, the small amount of tissue that needs to be infiltrated, and a window of opportunity to develop treatment as patients typically do not develop symptoms before mid to late childhood (37).
Novel disease models are being generated with the potential to develop in vitro organ systems for the assessment of new therapies. A promising development is the generation of induced pluripotent stem cells (38–43). These cells are generated when adult cells are reprogrammed and subsequently differentiated into another cell type through the addition of growth factors (41). A number of cell types have been used for reprogramming including dermal fibroblasts (44), renal epithelial cells (45), keratinocytes (46), and peripheral blood cells (47).
Urine-derived renal epithelial cells have been used to model ciliopathies, such as Joubert syndrome to assess the effect of potential therapeutics (48, 49) and BBS to derive mechanistic insights into disease pathogenesis (unpublished data). This is a particularly attractive model for many ciliopathies including BBS as it is non-invasive and offers an organ-specific relevant disease model.
To our knowledge, currently, there are no efforts in progress to develop genetic therapies targeted to the renal manifestations of BBS. This is likely to be a function of a number of issues including that the onset of renal manifestations is often antenatal precluding a therapeutic window of opportunity. Furthermore, the natural history of renal disease in BBS is not well understood, and the cause of renal disease can be both primary (e.g., cystic renal disease) and secondary to metabolic syndrome, hypertension, or diabetes all of which occur more frequently in BBS than the general population (13). In addition, the kidney is more difficult to target with genetic therapies but may prove to be a more amenable target for pharmacological therapies.
Gene Therapy
Traditional gene replacement therapy has achieved significant success in the treatment of ciliopathy-related eye diseases including Usher syndrome and Leber congenital amaurosis in recent years (50–52). The premise involves the generation of viral or non-viral vectors carrying a wild-type gene of choice with the aim of integrating the gene into the host genome. The highest chance of success is achieved in diseases where only a small amount of healthy gene expression is required to generate a phenotypic effect (53).
A major challenge in developing genetic therapies for BBS is the generation of a long lasting therapy. A successful example of this is the retinal gene therapy (Luxturna), which has been successfully developed for RPE65-associated Leber congenital amaurosis. It is likely to obtain approval from the Food and Drug Administration (FDA) in 2018 as the first gene therapy for ocular disease. Extensive optimization was required before RPE65 gene therapy could be launched for FDA approval due to concerns about the sustainability and long-term maintenance of visual function (36, 37, 54).
Work on retinal gene replacement therapy for BBS is ongoing in animal models, and recent efforts were published demonstrating encouraging results in knock-in mouse models with the most common genotype in humans BBS1 p.M390R (55). Viral AAV vectors were generated containing the wild-type Bbs1 construct and injected subretinally rescuing BBSome formation and rhodopsin localization and showing trends toward improved electroretinogram function in mice (55).
Challenges of developing safe and effective gene therapy include ensuring that the gene expression is proportionate to the requirements of the host organism avoiding overexpression and thus cell toxicity (36, 51, 55), identifying an appropriate time to administer therapy (36) (in the case of rod-cone dystrophy ideally before photoreceptor death), avoiding generation of an immune response, and developing safe and effective vectors (53).
Readthrough Therapy
Readthrough therapy exploits the natural inconsistency of the genetic proofreading mechanism in the process of RNA translation into protein (56). Nonsense mutations account for an estimated 11% of the total mutational load in BBS (own unpublished data) and lead to premature termination of protein synthesis and subsequent degradation via nonsense mediated decay (57). Readthrough therapy acts by destabilizing the translational ribosome’s response to a nonsense mutation, hence allowing the insertion of a near cognate amino-acyl-tRNA. This allows translation to continue and a full-length protein to be produced (58). The effect mimics that of a missense mutation in the same locus, which may result in a less severe clinical phenotype. Readthrough therapy has been applied to a number of different conditions and been tested in clinical trials for both cystic fibrosis (59–61) and Duchenne muscular dystrophy (62, 63). The effect of readthrough therapy has been assessed on some ciliopathies at the preclinical stage including primary ciliary dyskinesia (64), Usher syndrome (65), and retinitis pigmentosa (RP2) (66).
Exon Skipping Therapy
Exon skipping therapy operates at the level of RNA transcription allowing the transcriptional machinery to “skip” exons containing undesirable genetic sequences (67, 68). Antisense oligonucleotides are designed to target an exon/intron of interest. This allows for a novel splicing product that may retain much of its wild-type function depending on the quantity and importance of functional motifs coded for by the exon. This form of genetic therapy is often used to target mutations, which disrupt the genetic reading frame and may otherwise result in a complete lack of functional protein. Successful application of exon skipping therapy has been developed to the level of clinical trials for Duchenne muscular dystrophy and spinal muscular atrophy (69, 70). Exon skipping therapy for ciliopathies has been developed to the stage of preclinical trials for Leber Congenital Amaurosis and Usher syndrome (67, 71). In BBS, this form of therapy could benefit up to 9% of patients. The primary challenge is that many mutations suitable for exon skipping therapy in BBS are private and thus require truly individualized therapy. The common frameshift mutation in BBS (BBS10 c.91fsX5) is not a suitable candidate as the gene contains only two exons.
Genome Editing
Genome engineering offers an attractive future prospect in the management of genetic diseases, allowing DNA to be deleted, replaced, or corrected (72). Targeted endonucleases create double-stranded breaks at specific points in the genome allowing for DNA repair, which can restore the wild-type genotype (43). Promising results have been achieved in cells from patients with the motile ciliopathy primary ciliary dyskinesia where ciliation was restored on replacement of the wild-type DNAH11 sequence (73). Preclinical work and clinical trials for other diseases using genome editing techniques are progressing with significant advances in animal models of epidermolysis bullosa (74). Initial reports of successful gene editing in humans include gene editing on embryos at risk for hypertrophic cardiomyopathy caused by MYBPC3 mutation (75) and treatment-resistant leukemia (75, 76). The potential applications are promising, but this technique requires significant refinement in specificity, efficacy, and safety before this can be applied in a clinical setting. Off target effects, whereby genome engineering may erroneously occur on an unintended gene causing DNA damage are a major challenge in bringing this technique forward (72).
While genome editing can be used to target specific organ systems, it can also be applied ex vivo to correct or insert mutations to directly comparable cell types which apart from the mutation of interest are genetically identical, thus eliminating genetic background noise and providing an ideal model system (77).
Non-Genetic Personalized Therapies
Targeted Therapies
A novel development in the therapeutic landscape for BBS is the application of targeted drug therapies. An example of this is the ongoing work on the melanocortin receptor agonists as potential therapeutic intervention against obesity in BBS (78). Our understanding of how aberrant BBS proteins cause signal disruption is still evolving; however, there is emerging evidence suggesting that BBS results in defects in the hypothalamic leptin–melanocortin axis (79). This in turn causes leptin resistance culminating in obesity (78).
Seo et al. demonstrated that intravenous administration of a melanocortin receptor agonist decreased both body weight and food intake in wild-type and Bbs knockout mice (78). A phase 2/3 clinical trial is in process assessing the effect of novel melanocortin receptor agonist setmelanotide on obesity in BBS and other forms of syndromic obesity (80).
Drug Repurposing
Future therapies may include repurposing of drugs, which are already FDA approved. Early work on Bbs zebrafish indicated that rapamycin may be a candidate for rectifying the renal cystic phenotype (81). Further work on higher animals has not been published, but drug repurposing offers an attractive and economical option for management in a clinical setting since the cost and failure rate of developing novel therapeutics remain high.
Non-Pharmacological Future Interventions
A major challenge in any clinical service is harnessing the advances in technology and managing increasing clinical pressures. To this end, the UK National BBS clinic is implementing several new strategies to meet the increasing need for transparency and access to clinical services.
There is an increasing move to offer patients easy access to their medical notes and to data sharing within the UK National Health Service to optimize patient management (82). These issues are being addressed in the UK National BBS clinic through the development of a cloud-based medical notes system, which is accessible to all clinicians in the service. In the future, patients will be able to access their personal medical records through smart phones or other devices, so that medical information can be accessed wherever they are.
Telemedicine and virtual clinics, whereby patient consultations can take place via a screen in the patient’s own home, are an attractive alternative for BBS patients who are stable and in particular for those with visual impairment where travel can present a considerable obstacle to attending hospital appointments. It also offers an advantage to clinicians where hospital resources and clinic space are at a premium.
Pharmacogenomics
Pharmacogenomic profiling, whereby the effect of the genome on drug response is determined, could offer significant advantages to those patients with complex medical needs who are subject to polypharmacy (83) including BBS patients. Although several private companies offer pharmacogenomics gene panels, this is not currently available in the UK National Health Service, and the evidence base is unclear for many of these private initiatives. However, there is a growing evidence base supporting pharmacogenomic profiling, and this is likely to become available in the National Health Service in the coming years.
Conclusion
Bardet–Biedl syndrome provides a robust model disease for future opportunities in genetic and non-genetic therapeutic management of rare diseases. A significant advantage in the United Kingdom is the presence of the national BBS clinics, which offer a centralized hub for clinical and scientific expertise. The multiorgan effects and wide range of genes and mutation types mean that a number of different genetic therapeutic modalities must be considered, as well as non-genetic pharmacological interventions and non-pharmacological approaches to optimize management of this rare disease in the future.
Author Contributions
EF, JK, CB, and PB made substantial contributions to the writing, drafting, and revision of this manuscript. All authors approved the final published version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
EF is funded by the Medical Research Council. JK is funded in part by Innovate UK. PB is an NIHR Senior Investigator. This work was supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre (GOSH BRC). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
References
1. Forsythe E, Beales PL. Bardet-Biedl syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(R). Seattle, WA (2003).
2. Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A (2005) 132a(4):352–60. doi:10.1002/ajmg.a.30406
3. Farag TI, Teebi AS. High incidence of Bardet Biedl syndrome among the Bedouin. Clin Genet (1989) 36(6):463–4. doi:10.1111/j.1399-0004.1989.tb03378.x
4. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet (2013) 21(1):8–13. doi:10.1038/ejhg.2012.115
5. Heon E, Kim G, Qin S, Garrison JE, Tavares E, Vincent A, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum Mol Genet (2016) 25(11):2283–94. doi:10.1093/hmg/ddw096
6. Khan AO, Decker E, Bachmann N, Bolz HJ, Bergmann C. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet (2016) 37(3):290–3. doi:10.3109/13816810.2015.1066830
7. Schaefer E, Stoetzel C, Scheidecker S, Geoffroy V, Prasad MK, Redin C, et al. Identification of a novel mutation confirms the implication of IFT172 (BBS20) in Bardet-Biedl syndrome. J Hum Genet (2016) 61(5):447–50. doi:10.1038/jhg.2015.162
8. Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol (2017) 241(2):294–309. doi:10.1002/path.4843
9. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet (1999) 36(6):437–46.
10. Samuel GN, Farsides B. Public trust and ‘ethics review’ as a commodity: the case of Genomics England Limited and the UK’s 100,000 genomes project. Med Health Care Philos (2017). doi:10.1007/s11019-017-9810-1
11. Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet (2015) 385(9975):1305–14. doi:10.1016/s0140-6736(14)61705-0
12. Vears DF, Senecal K, Borry P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet (2017) 60(10):553–8. doi:10.1016/j.ejmg.2017.07.016
13. Forsythe E, Sparks K, Best S, Borrows S, Hoskins B, Sabir A, et al. Risk factors for severe renal disease in Bardet-Biedl syndrome. J Am Soc Nephrol (2017) 28(3):963–70. doi:10.1681/ASN.2015091029
14. Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Fielding Hejtmancic J, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol (2012) 130(7):901–7. doi:10.1001/archophthalmol.2012.89
15. Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, et al. Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab (2011) 96(3):E528–35. doi:10.1210/jc.2010-2290
16. Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol (2011) 6(1):22–9. doi:10.2215/CJN.03320410
17. Forsythe E, Sparks K, Hoskins BE, Bagkeris E, McGowan BM, Carroll PV, et al. Genetic predictors of cardiovascular morbidity in Bardet-Biedl syndrome. Clin Genet (2015) 87(4):343–9. doi:10.1111/cge.12373
18. Katsanis N, Eichers ER, Ansley SJ, Lewis RA, Kayserili H, Hoskins BE, et al. BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am J Hum Genet (2002) 71(1):22–9. doi:10.1086/341031
19. Hichri H, Stoetzel C, Laurier V, Caron S, Sigaudy S, Sarda P, et al. Testing for triallelism: analysis of six BBS genes in a Bardet-Biedl syndrome family cohort. Eur J Hum Genet (2005) 13(5):607–16. doi:10.1038/sj.ejhg.5201372
20. Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet (2012) 20(4):420–7. doi:10.1038/ejhg.2011.205
21. Topol EJ. Individualized medicine from prewomb to tomb. Cell (2014) 157(1):241–53. doi:10.1016/j.cell.2014.02.012
22. Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol (2017) 18(1):83. doi:10.1186/s13059-017-1215-1
23. Tieder M, Levy M, Gubler MC, Gagnadoux MF, Broyer M. Renal abnormalities in the Bardet-Biedl syndrome. Int J Pediatr Nephrol (1982) 3(3):199–203.
24. Gourdol O, David L, Colon S, Bouvier R, Ayral A, Aguercif M, et al. [Renal involvement in the Laurence-Moon-Bardet-Biedl syndrome. Apropos of 3 cases]. Pediatrie (1984) 39(3):175–81.
25. Harnett JD, Green JS, Cramer BC, Johnson G, Chafe L, McManamon P, et al. The spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med (1988) 319(10):615–8. doi:10.1056/NEJM198809083191005
26. O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis (1996) 27(6):776–83. doi:10.1016/S0272-6386(96)90513-2
27. Putoux A, Mougou-Zerelli S, Thomas S, Elkhartoufi N, Audollent S, Le MM, et al. BBS10 mutations are common in ‘Meckel’-type cystic kidneys. J Med Genet (2010) 47(12):848–52. doi:10.1136/jmg.2010.079392
28. Marion V, Schlicht D, Mockel A, Caillard S, Imhoff O, Stoetzel C, et al. Bardet-Biedl syndrome highlights the major role of the primary cilium in efficient water reabsorption. Kidney Int (2011) 79(9):1013–25. doi:10.1038/ki.2010.538
29. Putoux A, Attie-Bitach T, Martinovic J, Gubler MC. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol (2012) 27(1):7–15. doi:10.1007/s00467-010-1751-3
30. Tobin JL, Beales PL. Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol (2007) 22(7):926–36.
31. Hamlington B, Ivey LE, Brenna E, Biesecker LG, Biesecker BB, Sapp JC. Characterization of courtesy stigma perceived by parents of overweight children with Bardet-Biedl syndrome. PLoS One (2015) 10(10):e0140705. doi:10.1371/journal.pone.0140705
32. Daskalakis M, Till H, Kiess W, Weiner RA. Roux-en-Y gastric bypass in an adolescent patient with Bardet-Biedl syndrome, a monogenic obesity disorder. Obes Surg. (2010) 20(1):121–5. doi:10.1007/s11695-009-9915-6
33. Mujahid S, Huda MS, Beales P, Carroll PV, McGowan BM. Adjustable gastric banding and sleeve gastrectomy in Bardet-Biedl syndrome. Obes Surg (2014) 24(10):1746–8. doi:10.1007/s11695-014-1379-7
34. Sahel JA, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb Perspect Med (2014) 5(2):a017111. doi:10.1101/cshperspect.a017111
35. Estrada-Cuzcano A, Roepman R, Cremers FP, den Hollander AI, Mans DA. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum Mol Genet (2012) 21(R1):R111–24. doi:10.1093/hmg/dds298
36. Dias MF, Joo K, Kemp JA, Fialho SL, da Silva Cunha A Jr, Woo SJ, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: basic research and clinical perspectives. Prog Retin Eye Res (2017). doi:10.1016/j.preteyeres.2017.10.004
37. Lee H, Lotery A. Gene therapy for RPE65-mediated inherited retinal dystrophy completes phase 3. Lancet (2017) 390(10097):823–4. doi:10.1016/s0140-6736(17)31622-7
38. Jin ZB, Okamoto S, Mandai M, Takahashi M. Induced pluripotent stem cells for retinal degenerative diseases: a new perspective on the challenges. J Genet (2009) 88(4):417–24. doi:10.1007/s12041-009-0063-5
39. Al-Shamekh S, Goldberg JL. Retinal repair with induced pluripotent stem cells. Transl Res (2014) 163(4):377–86. doi:10.1016/j.trsl.2013.11.002
40. Tucker BA, Mullins RF, Stone EM. Stem cells for investigation and treatment of inherited retinal disease. Hum Mol Genet (2014) 23(R1):R9–16. doi:10.1093/hmg/ddu124
41. Nguyen HV, Li Y, Tsang SH. Patient-specific iPSC-derived RPE for modeling of retinal diseases. J Clin Med (2015) 4(4):567–78. doi:10.3390/jcm4040567
42. Wiley LA, Burnight ER, Songstad AE, Drack AV, Mullins RF, Stone EM, et al. Patient-specific induced pluripotent stem cells (iPSCs) for the study and treatment of retinal degenerative diseases. Prog Retin Eye Res (2015) 44:15–35. doi:10.1016/j.preteyeres.2014.10.002
43. Zheng A, Li Y, Tsang SH. Personalized therapeutic strategies for patients with retinitis pigmentosa. Expert Opin Biol Ther (2015) 15(3):391–402. doi:10.1517/14712598.2015.1006192
44. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131(5):861–72. doi:10.1016/j.cell.2007.11.019
45. Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, et al. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc (2012) 7(12):2080–9. doi:10.1038/nprot.2012.115
46. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol (2008) 26(11):1276–84. doi:10.1038/nbt.1503
47. Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell (2010) 7(1):20–4. doi:10.1016/j.stem.2010.06.002
48. Ajzenberg H, Slaats GG, Stokman MF, Arts HH, Logister I, Kroes HY, et al. Non-invasive sources of cells with primary cilia from pediatric and adult patients. Cilia (2015) 4:8. doi:10.1186/s13630-015-0017-x
49. Srivastava S, Ramsbottom SA, Molinari E, Alkanderi S, Filby A, White K, et al. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum Mol Genet (2017) 26(23):4657–67. doi:10.1093/hmg/ddx347
50. McIntyre JC, Williams CL, Martens JR. Smelling the roses and seeing the light: gene therapy for ciliopathies. Trends Biotechnol (2013) 31(6):355–63. doi:10.1016/j.tibtech.2013.03.005
51. Williams CL, Uytingco CR, Green WW, McIntyre JC, Ukhanov K, Zimmerman AD, et al. Gene therapeutic reversal of peripheral olfactory impairment in Bardet-Biedl syndrome. Mol Ther (2017) 25(4):904–16. doi:10.1016/j.ymthe.2017.02.006
52. Zhang W, Li L, Su Q, Gao G, Khanna H. Gene therapy using a miniCEP290 fragment delays photoreceptor degeneration in a mouse model of leber congenital amaurosis. Hum Gene Ther (2017) 29(1):42–50. doi:10.1089/hum.2017.049
53. Moore NA, Morral N, Ciulla TA, Bracha P. Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin Biol Ther (2018) 18(1):37–49. doi:10.1080/14712598.2018.1389886
54. Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med (2015) 372(20):1887–97. doi:10.1056/NEJMoa1414221
55. Seo S, Mullins RF, Dumitrescu AV, Bhattarai S, Gratie D, Wang K, et al. Subretinal gene therapy of mice with Bardet-Biedl syndrome type 1. Invest Ophthalmol Vis Sci (2013) 54(9):6118–32. doi:10.1167/iovs.13-11673
56. Oren YS, Pranke IM, Kerem B, Sermet-Gaudelus I. The suppression of premature termination codons and the repair of splicing mutations in CFTR. Curr Opin Pharmacol (2017) 34:125–31. doi:10.1016/j.coph.2017.09.017
57. Lee HL, Dougherty JP. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther (2012) 136(2):227–66. doi:10.1016/j.pharmthera.2012.07.007
58. Richardson R, Smart M, Tracey-White D, Webster AR, Moosajee M. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp Eye Res (2017) 155:24–37. doi:10.1016/j.exer.2017.01.001
59. Mutyam V, Du M, Xue X, Keeling KM, White EL, Bostwick JR, et al. Discovery of clinically approved agents that promote suppression of cystic fibrosis transmembrane conductance regulator nonsense mutations. Am J Respir Crit Care Med (2016) 194(9):1092–103. doi:10.1164/rccm.201601-0154OC
60. Mutyam V, Libby EF, Peng N, Hadjiliadis D, Bonk M, Solomon GM, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros (2017) 16(1):24–9. doi:10.1016/j.jcf.2016.09.005
61. Xue X, Mutyam V, Thakerar A, Mobley J, Bridges RJ, Rowe SM, et al. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum Mol Genet (2017) 26(16):3116–29. doi:10.1093/hmg/ddx196
62. Finkel RS, Flanigan KM, Wong B, Bonnemann C, Sampson J, Sweeney HL, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One (2013) 8(12):e81302. doi:10.1371/journal.pone.0081302
63. Reinig AM, Mirzaei S, Berlau DJ. Advances in the treatment of Duchenne muscular dystrophy: new and emerging pharmacotherapies. Pharmacotherapy (2017) 37(4):492–9. doi:10.1002/phar.1909
64. Bukowy-Bieryllo Z, Dabrowski M, Witt M, Zietkiewicz E. Aminoglycoside-stimulated readthrough of premature termination codons in selected genes involved in primary ciliary dyskinesia. RNA Biol (2016) 13(10):1041–50. doi:10.1080/15476286.2016.1219832
65. Goldmann T, Overlack N, Wolfrum U, Nagel-Wolfrum K. PTC124-mediated translational readthrough of a nonsense mutation causing Usher syndrome type 1C. Hum Gene Ther (2011) 22(5):537–47. doi:10.1089/hum.2010.067
66. Schwarz N, Carr AJ, Lane A, Moeller F, Chen LL, Aguila M, et al. Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. Hum Mol Genet (2015) 24(4):972–86. doi:10.1093/hmg/ddu509
67. Garanto A, Chung DC, Duijkers L, Corral-Serrano JC, Messchaert M, Xiao R, et al. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet (2016) 25(12):2552–63. doi:10.1093/hmg/ddw118
68. Garanto A, Collin RWJ. Design and in vitro use of antisense oligonucleotides to correct pre-mRNA splicing defects in inherited retinal dystrophies. Methods Mol Biol (2018) 1715:61–78. doi:10.1007/978-1-4939-7522-8_5
69. Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med (2011) 364(16):1513–22. doi:10.1056/NEJMoa1011367
70. Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol (2013) 12(5):435–42. doi:10.1016/s1474-4422(13)70061-9
71. Slijkerman RW, Vache C, Dona M, Garcia-Garcia G, Claustres M, Hetterschijt L, et al. Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol Ther Nucleic Acids (2016) 5(10):e381. doi:10.1038/mtna.2016.89
72. Lux CT, Scharenberg AM. Therapeutic gene editing safety and specificity. Hematol Oncol Clin North Am (2017) 31(5):787–95. doi:10.1016/j.hoc.2017.05.002
73. Lai M, Pifferi M, Bush A, Piras M, Michelucci A, Di Cicco M, et al. Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. J Med Genet (2016) 53(4):242–9. doi:10.1136/jmedgenet-2015-103539
74. Wu W, Lu Z, Li F, Wang W, Qian N, Duan J, et al. Efficient in vivo gene editing using ribonucleoproteins in skin stem cells of recessive dystrophic epidermolysis bullosa mouse model. Proc Natl Acad Sci U S A (2017) 114(7):1660–5. doi:10.1073/pnas.1614775114
75. Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature (2017) 548(7668):413–9. doi:10.1038/nature23305
76. Reardon S. First CRISPR Clinical Trial Gets Green Light from US Panel (2017). Available from: https://www.nature.com/news/first-crispr-clinical-trial-gets-green-light-from-us-panel-1.20137
77. Huai C, Jia C, Sun R, Xu P, Min T, Wang Q, et al. CRISPR/Cas9-mediated somatic and germline gene correction to restore hemostasis in hemophilia B mice. Hum Genet (2017) 136(7):875–83. doi:10.1007/s00439-017-1801-z
78. Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet (2009) 18(7):1323–31. doi:10.1093/hmg/ddp031
79. Mason K, Page L, Balikcioglu PG. Screening for hormonal, monogenic, and syndromic disorders in obese infants and children. Pediatr Ann (2014) 43(9):e218–24. doi:10.3928/00904481-20140825-08
80. Rhythm Pharmaceuticals. Rhythm Expands Phase 2 Clinical Trials of Setmelanotide to the Treatment of Bardet-Biedl Syndrome Obesity. Boston, MA: Rhythm Pharmaceuticals (2017).
81. Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol (2008) 23(11):2095–9. doi:10.1007/s00467-008-0898-7
82. Armstrong S. Data, data everywhere: the challenges of personalised medicine. BMJ (2017) 359:j4546. doi:10.1136/bmj.j4546
Keywords: Bardet–Biedl syndrome, genetic therapies, pharmacogenomics, genome editing, targeted therapies, drug repurposing
Citation: Forsythe E, Kenny J, Bacchelli C and Beales PL (2018) Managing Bardet–Biedl Syndrome—Now and in the Future. Front. Pediatr. 6:23. doi: 10.3389/fped.2018.00023
Received: 14 December 2017; Accepted: 25 January 2018;
Published: 13 February 2018
Edited by:
Miriam Schmidts, Radboud University Nijmegen, NetherlandsReviewed by:
Diana Valverde, University of Vigo, SpainJohn Andrew Sayer, Newcastle University, United Kingdom
Copyright: © 2018 Forsythe, Kenny, Bacchelli and Beales. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Forsythe, ZWxpemFiZXRoLmZvcnN5dGhlQHVjbC5hYy51aw==