 Jens Christian König
Jens Christian König Andrea Titieni
Andrea Titieni Martin Konrad
Martin Konrad The NEOCYST Consortium
The NEOCYST Consortium
- Department of General Pediatrics, University Children’s Hospital Münster, Münster, Germany
Hereditary cystic kidney diseases comprise a complex group of genetic disorders representing one of the most common causes of end-stage renal failure in childhood. The main representatives are autosomal recessive polycystic kidney disease, nephronophthisis, Bardet–Biedl syndrome, and hepatocyte nuclear factor-1beta nephropathy. Within the last years, genetic efforts have brought tremendous progress for the molecular understanding of hereditary cystic kidney diseases identifying more than 70 genes. Yet, genetic heterogeneity, phenotypic variability, a lack of reliable genotype–phenotype correlations and the absence of disease-specific biomarkers remain major challenges for physicians treating children with cystic kidney diseases. To tackle these challenges comprehensive scientific approaches are urgently needed that match the ongoing “revolution” in genetics and molecular biology with an improved efficacy of clinical data collection. Network for early onset cystic kidney diseases (NEOCYST) is a multidisciplinary, multicenter collaborative combining a detailed collection of clinical data with translational scientific approaches addressing the genetic, molecular, and functional background of hereditary cystic kidney diseases. Consisting of seven work packages, including an international registry as well as a biobank, NEOCYST is not only dedicated to current scientific questions, but also provides a platform for longitudinal clinical surveillance and provides precious sources for high-quality research projects and future clinical trials. Funded by the German Federal Government, the NEOCYST collaborative started in February 2016. Here, we would like to introduce the rationale, design, and objectives of the network followed by a short overview on the current state of progress.
Background
Hereditary cystic kidney diseases comprise a group of slowly progressive, chronically debilitating disorders with a high level of complexity. Despite rare individually, as a group they represent one of the most common causes of end-stage renal failure in childhood and, therefore, have an enormous socioeconomic impact. The main representatives of this group of diseases are autosomal recessive polycystic kidney disease (ARPKD), nephronophthisis and nephronophthisis-related ciliopathies (NPH/NPH-RC), Bardet–Biedl syndrome (BBS), and hepatocyte nuclear factor-1beta (HNF1B) nephropathy (1). The incidence ranges from 1:5,000 to 1:100,000 and the estimated overall prevalence is about 300–450 children in Germany (2–7).
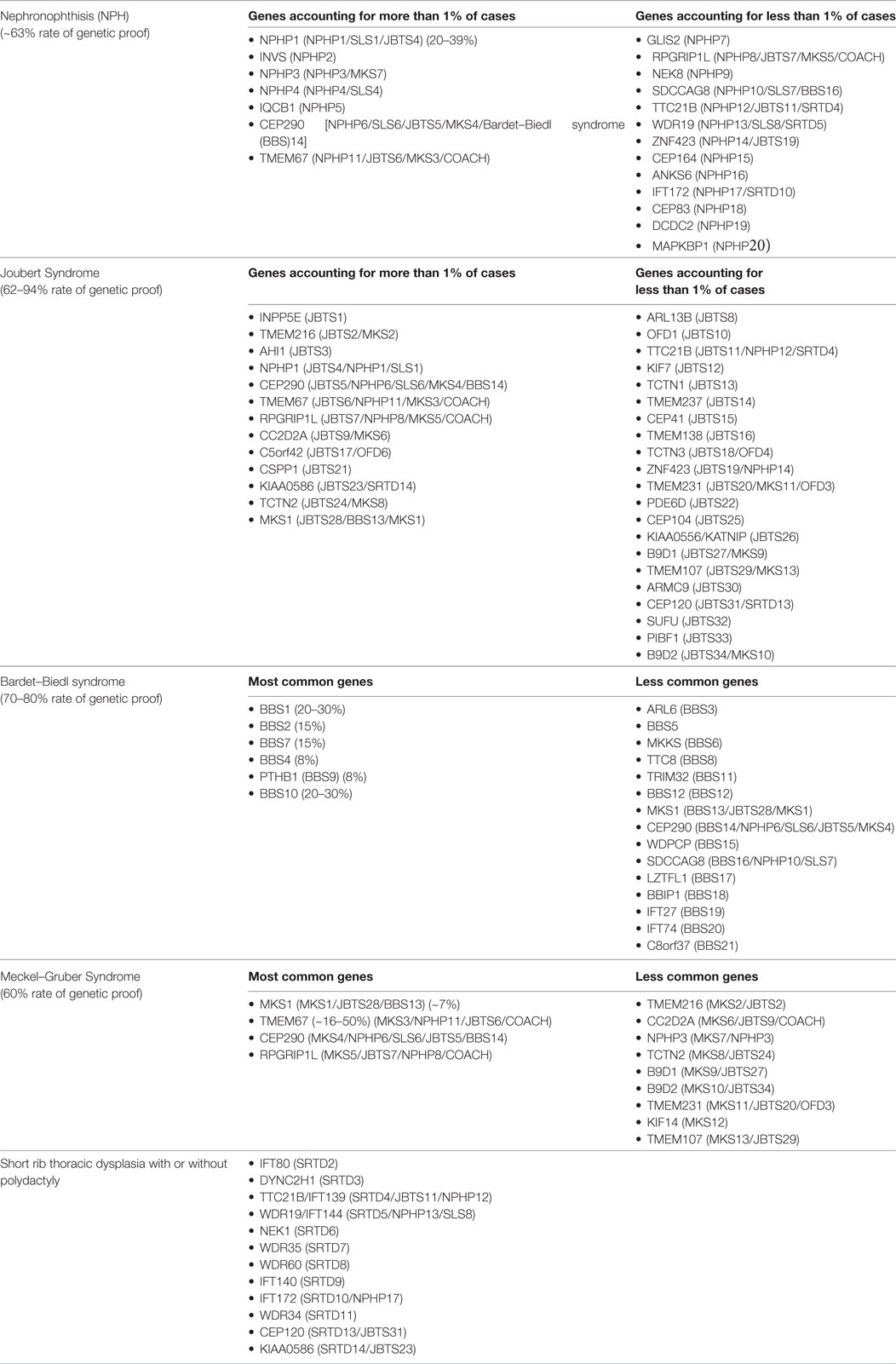
Most early onset cystic kidney diseases are inherited as an autosomal recessive trait. One important exception is the HNF1B nephropathy, which follows autosomal dominant inheritance (6). Within the past 20 years scientific efforts have brought tremendous progress for the molecular understanding of hereditary cystic kidney diseases. To date, mutations in more than 70 individual genes have been identified (Table 1) (8). Yet, genetic heterogeneity is a major problem since, there is a significant amount of genetic as well as phenotypic overlap and reliable data on genotype–phenotype correlations are still lacking (9). Moreover, the phenotypic spectrum covered by cystic kidney diseases is highly complex and extremely variable. Although all disease entities are characterized by the development of renal cysts as a common characteristic, clinical courses as well as the onset of chronic renal failure differ significantly. Furthermore, most phenotypes are not limited to the kidneys, but comprise extrarenal organ manifestations (10, 11).
• ARPKD: the clinical presentation of ARPKD is determined by grossly enlarged polycystic kidneys usually diagnosed before birth. Depending on prenatal kidney function, an early oligohydramnion may lead to pulmonary hypoplasia resulting in a perinatal mortality of about 20% in ARPKD. Children surviving the first months of life usually present a preserved renal function for several years. There is obligate hepatic involvement and the clinical presentation of some patients is dominated by a hepatic phenotype rather than by chronic kidney disease (2, 3, 12–14).
• NPH: reduced urinary concentrating capacity, polyuria/polydipsia, and a slowly progressive loss of renal function are the clinical hallmarks of NPH. Urine analysis is typically unremarkable (4). In contrast to ARPKD, kidney volumes are small or normal, and renal cysts have no obligatory feature (15). NPH can be limited to the kidneys, but extrarenal manifestations are found in 20–40% of patients depending on the underlying genetic defect (10). Moreover, many complex syndromes encompass the renal phenotype with NPH, including Senior–Løken syndrome, Joubert-like syndromes, and other NPH-related ciliopathies (16, 17).
• BBS: Bardet–Biedl syndrome is a complex genetic disorder clinically characterized by progressive visual impairment, obesity, developmental delay, postaxial hexadactyly, hypogonadism, and slowly progressive CKD. The clinical presentation is highly variable and can be accompanied by further organ involvement (5). The renal phenotype usually resembles NPH, including polyuria and small- to normal-sized kidneys showing cystic lesions at the corticomedullary junction. However, renal involvement is not mandatory and reported in only 31–42% of BBS (18).
• HNF1B nephropathy: HNF1B is a transcription factor playing a central role in the tissue-specific regulation of gene expression in various organs, such as kidney, liver, biliary duct, pancreas, and genital organs. Initially, mutations in the HNF1B gene were described in association with maturity onset diabetes of the young (MODY type 5). Subsequently, it was shown that in patients with congenital cystic renal dysplasia HNF1B mutations could frequently be identified (renal cysts and diabetes syndrome, RCAD). In contrast to the NPH spectrum, renal cysts may already be detected prenatally, but renal ultrasound presentation is variable including unilateral and bilateral renal dysplasia with different numbers and sizes of cysts. Depending on the extent of renal dysplasia some patients develop renal failure in early childhood while others preserve normal renal function for their whole life (6). Beyond the renal involvement, the clinical spectrum comprises elevated liver enzymes, hyperuricemia, genital tract malformations, and electrolyte disturbances, such as hypomagnesemia and hypokalemia (11).
Genetic and Phenotypic Heterogeneity
Although the clinical characteristics of the different hereditary cystic kidney diseases appear quite discriminative, there is significantly genetic as well as phenotypic overlap that hampers an early diagnosis and an individual clinical management (Figure 1) (1). The tremendous progress that has been achieved within the genetic field had major impact on the classification of cystic kidney diseases. However, the newly generated insights seem to further complicate the clinical situation for physicians dealing with affected individuals: it has become increasingly evident that so far well-defined clinical entities can be caused by mutations in multiple genes. Even in ARPKD, which for a long time has been assumed to be a single gene disease, modern NGS-based sequencing techniques just recently were able to identify a new genetic cause encoding a ciliary transition zone protein (26). Also, mutations in the same gene can cause very different phenotypes that range from lethal early embryonic multivisceral manifestations to single organ involvement starting in adolescence (Figure 1) (1, 27). Thus, recent advances in genomics challenged the classical Mendelian conditions and highlighted the genetic complexity of hereditary cystic kidney diseases and related ciliopathies (28). This complexity has been attributed to allelic heterogeneity, locus heterogeneity, reduced penetrance, variable expressivity, modifier genes, and/or environmental factors (29).
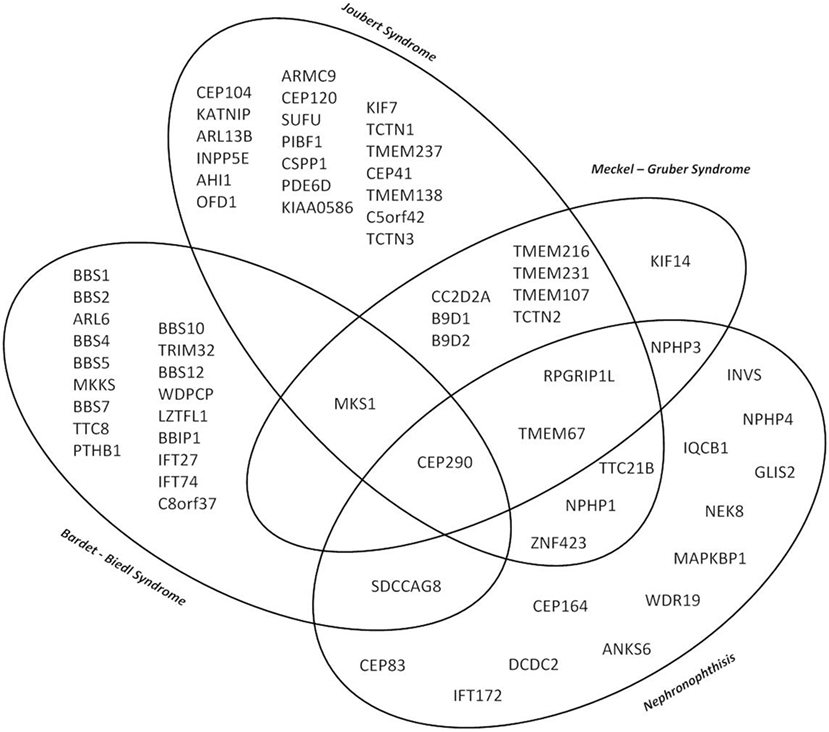
Figure 1. Venn diagram illustrating the major genetic and phenotypic overlap in hereditary ciliopathies featuring cystic kidney disease. So far well-defined clinical entities can be caused by mutations in multiple genes, whereas at the same time mutations in the same gene can cause very different phenotypes depending on the type of mutation ranging from isolated nephronophthisis (NPH) to lethal early embryonic multivisceral manifestations like Meckel–Gruber syndrome.
Based on these newly generated insights it has become clear that the “diagnostic odyssey” experienced by patients does not end with the identification of a disease-causing genotype (30). Neither does the traditional approach using textbook signs nor symptoms to guide diagnosis and management seem to be sufficient any longer (31). Rather a systematic and deep phenotyping approach supplementary to careful genotyping is critical in order to discover nonobvious phenotypes and to determine a precise diagnosis. Thus, physicians addressing hereditary cystic kidney diseases should combine both—detailed, multisystemic phenotyping and careful assessment of genotype pathogenicity—in order to capture all facets of the underlying disease (30). Against this background, it will be important to overcome the financial restraints associated with modern NGS-based sequencing currently hampering a comprehensive genetic characterization.
Molecular Understanding
The genetic discoveries revolutionized our molecular understanding of cystic kidney diseases. The observation that most genes that have been identified so far encode proteins that co-localize in primary cilia and even interact as functional ciliary clusters suggested the existence of a common pathophysiological pathway and lead to the so-called ciliary hypothesis (Figure 2) (32, 33). However, different molecular mechanisms have been described to be altered in renal cyst formation, including cell-proliferation, apoptosis, DNA repair, fluid secretion into the cyst lumen, altered apico-basal cell polarity, directional cell migration, cell–cell adhesion, interaction with the extracellular matrix and ciliary function. Additionally, various intracellular signaling pathways were identified to be activated in epithelia developing cystic lesion, such as intracellular calcium signaling, cAMP triggered fluid secretion, the wnt-, hedgehog, mTOR-, notch-, YAP-hippo-, and other pathways. Whether or not these observations are related to each other and how the mentioned mechanisms could be connected with an altered ciliary function remains poorly understood and is the topic of ongoing research. Details on these topics are beyond the scope of this perspective. However, this has been excellently reviewed by Ong et al. recently (34–36).
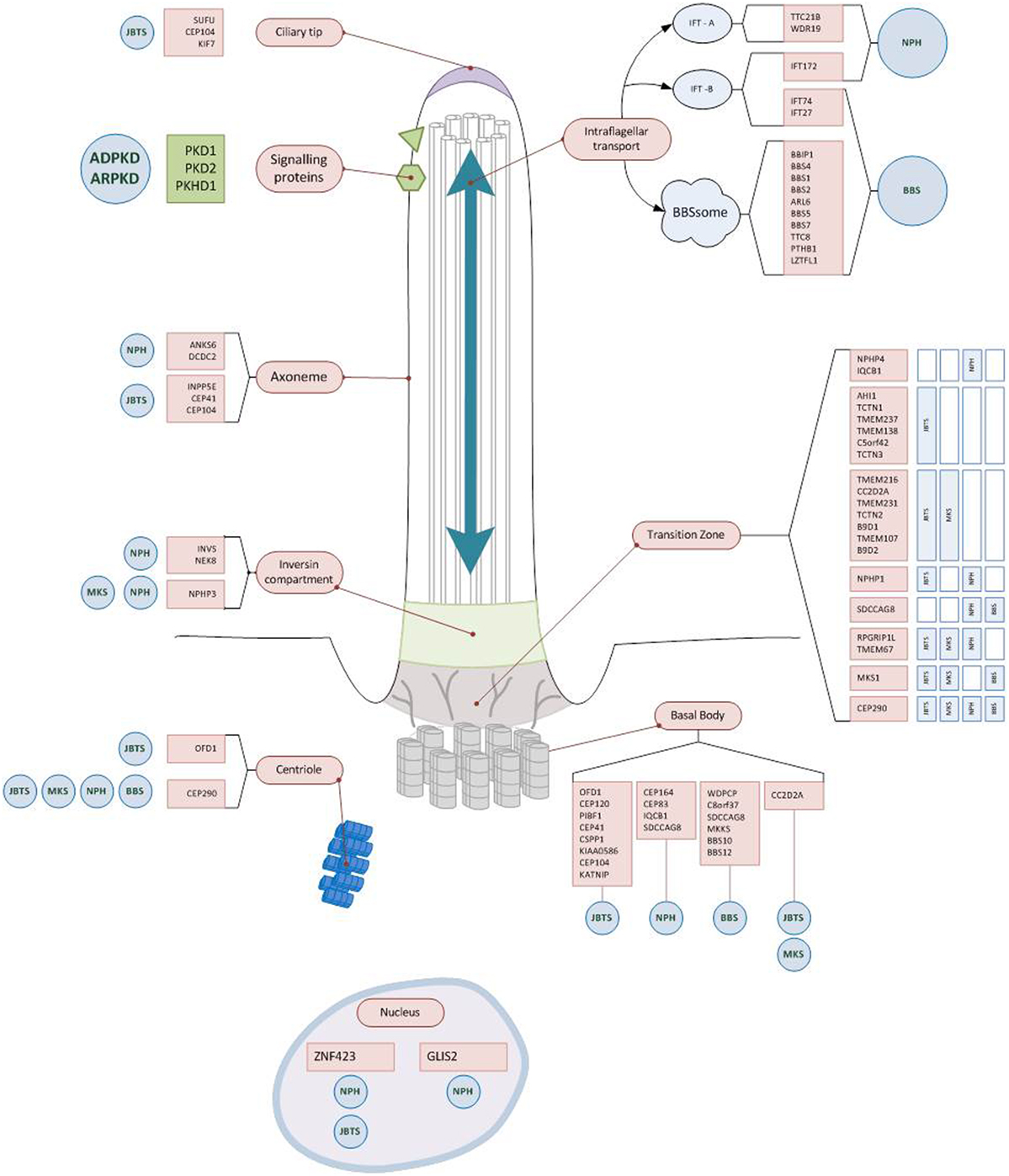
Figure 2. Ciliary localization of the proteins encoded by genes causing early onset cystic kidney diseases. Almost all gene products mutated in hereditary cystic kidney diseases are located at the primary cilium, the ciliary base, or the basal body. Some of the proteins form functional units (Polycystin1/2; BBSom; Nephrocystin-4,5) and are found at characteristic parts of the cilium (e.g., ciliary base). While for some of the gene mutations there is a clear impact on regular ciliary function (e.g., disrupting intraflagellar transport), for most others the exact pathophysiological mechanisms still remain to be unraveled. Abbreviations: NPH, nephronophthisis; JBTS, Joubert syndrome; BBS, Bardet–Biedl syndrome; MKS, Meckel–Gruber syndrome; ARPKD, autosomal recessive polycystic kidney disease; ADPKD, autosomal dominant polycystic kidney disease.
Network for Early Onset Cystic Kidney Diseases (NEOCYST)—A Comprehensive Approach on Hereditary Cystic Kidney Diseases
Despite all the achievements mentioned above, enormous challenges remain to be solved: the complex phenotypic spectrum, genetic heterogeneity, a lack of reliable genotype–phenotype correlations, a limited molecular understanding and the absence of disease-specific biomarkers still hamper an early diagnosis and individual counseling. Data from international registry studies like the NEPHREG or the ARegPKD registry will certainly help to define robust genotype–phenotype associations by increasing sample sizes and following individual disease courses in a longitudinal fashion (10, 37). However, in order to achieve a deeper clinical as well as molecular understanding, international collaborative efforts comprising various subspecialists will be necessary to move knowledge forward especially in the context of rare diseases. A detailed molecular understanding will be fundamental in order to identify new therapeutic targets and develop disease specific treatment approaches. Therefore, future research activities should go one step further and try to verify the hypotheses gained from cell culture experiments in specimen harvested from actual patients.
To address all these questions, the NEOCYST was initiated in 2016 in order to match the ongoing “revolution” in genetics and molecular biology with an improved efficacy of clinical data collection. Here, we would like to introduce the structure, the goals, and the individual work-packages of the NEOCYST collaborative accompanied by a short overview on the current state of progress.
Design
Network for early onset cystic kidney diseases is a multidisciplinary, multicenter observational study of hereditary cystic kidney diseases in childhood. It combines a detailed and comprehensive clinical data collection with translational scientific approaches covering the genetic, molecular, and functional background.
Aims
The primary goal of the NEOCYST consortium is to improve the clinical situation and the management of children with hereditary cystic kidney diseases. In order to reach this goal a multimodal approach was chosen including:
• A detailed and comprehensive longitudinal characterization of different hereditary cystic kidney diseases in childhood.
• The establishment of reliable genotype–phenotype correlations.
• The identification of new cystic disease genes.
• An improved molecular understanding.
• The identification of disease-specific marker proteins.
• A standardized clinical management based on standard-of-care-guidelines.
• The implementation of a national biobank generating a platform for future research projects.
Study Cohort
Eligible are patients with the diagnosis of ARPKD, isolated NPH or NPH-related ciliopathies, BBS, and HNF1B-nephropathy. While for the first four study, inclusion is justified either by clinical or genetic criteria, HNF1B-nephropathy has to be confirmed genetically in order to avoid phenotypic overlap with other urinary tract malformations. For the clinical diagnosis of NPH at least two of the following criteria have to be met: (i) characteristic clinical course with polyuria/polydipsia; (ii) chronic kidney disease; (iii) kidney ultrasound or biopsy suggestive of NPH (24); (iv) pedigree compatible with autosomal recessive inheritance. The clinical diagnosis of BBS is based on the diagnostic criteria according to Beales et al. (38). Phenotypes reassembling a NPH-related ciliopathy are classified by the clinical criteria published by Bergmann (1).
Exclusion criteria encompass the definite genetic or clinical diagnosis of other cystic kidney diseases, in particular autosomal dominant polycystic kidney disease (ADPKD) which is addressed by other clinical registry studies.
Patient inclusion is preceded by a written informed consent and an approval by the local ethics committee of each contributing center, guaranteeing accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines.
Work Packages
To address the goals mentioned above the NEOCYST consortium comprises seven work packages (Figure 3). Subsequently, we will outline the main topics covered by these work-packages:
• Clinical registry study: the web-based NEOCYST registry is a newly created retro- and prospective clinical registry providing a detailed genetic and phenotypic characterization of all hereditary cystic kidney diseases in one common database collecting cross-sectional as well as longitudinal data. Three pre-existing registries on NPH-RC,1 BBS, and HNF1B were merged to build the fundaments of this new database. Additionally, technical bridgehead components have been implemented that allow a direct comparative data analysis with the international ARPKD registry2 (37). Thereby, NEOCYST is the first registry study providing a comprehensive approach to early onset hereditary cystic kidney diseases and allowing back-to-back analyses on similarities and differences of the individual disease entities. At the same time, the database supplies technical features that enable external international cooperation and provide a platform for further research projects and future clinical trials throughout Europe.
• Genetic characterization: as the majority of NPH/NPH-RC patients as well as some ARPKD and BBS patients are still genetically unsolved, the NEOCYST network has set out to genetically characterize as many patients as possible, including potential genetic modifiers. The genetic approach applied consists of NGS-based panel diagnostics covering more than 100 ciliary genes followed by whole exome sequencing for those patients in whom no convincing mutations can be identified.
• Molecular biology: the molecular focus of the NEOCYST collaborative is on the characterization of an altered ciliary structure and function, intracellular signaling pathways as well as cell programming and cell adhesion processes. These topics get addressed by internationally acknowledged experts in the field of hereditary cystic kidney diseases (for details see NEOCYST consortium section below). Using conventional cell cultures as well as urine-derived renal epithelial cells (UREC) from patients carrying different mutations, modern techniques, such as proteomics, 3D spheroid models, cell (re)programming, and genomic engineering are applied to analyze different signaling pathways and their impact on cystogenesis in vivo and in vitro. Additionally, in order to characterize altered ciliary processes in hereditary cystic kidney diseases, nasal brushes from affected patients are generated and techniques like high-frequency video microscopy, freeze-fracture analysis, and immunohistological staining are applied to motile as well as immotile cilia. Finally, it is a goal to identify disease-specific proteome marker profiles from patients’ spot urine using a blinded prospective approach based on capillary electrophoresis coupled with mass spectrometry.
• Biobank: due to the paucity of patients characterized by an early onset renal failure, biological samples of such patients are highly precious. Thus, to ensure current as well as future research projects, the NEOCYST collaborative includes a biobank for storing urine, blood, and respiratory epithelial cells. All samples get stored at the Hannover Unified Biobank, one of the most modern biobanks in Europe. It is characterized by a high degree of automation and state-of-the-art biobank infrastructure according to standard operating procedures with data safety concepts, pseudonymization tools, sample identification via 2D bar codes and integrated IT systems that enable high quality and safety standards.
• Standard-of-care guidelines: so far, the clinical management of patients with hereditary cystic kidney diseases is mainly based on local physician’s experience and differs quite considerably between different centers. Due to the limited number of patients, the experience of most clinical centers is based on only a few single cases and clinical guidelines on the management of hereditary cystic kidney diseases are scarcely available. Thus, NEOCYST has set itself the task to develop and elaborate recommendations on different topics concerning hereditary cystic kidney diseases based on the given evidence in the literature as well as experts’ opinions.
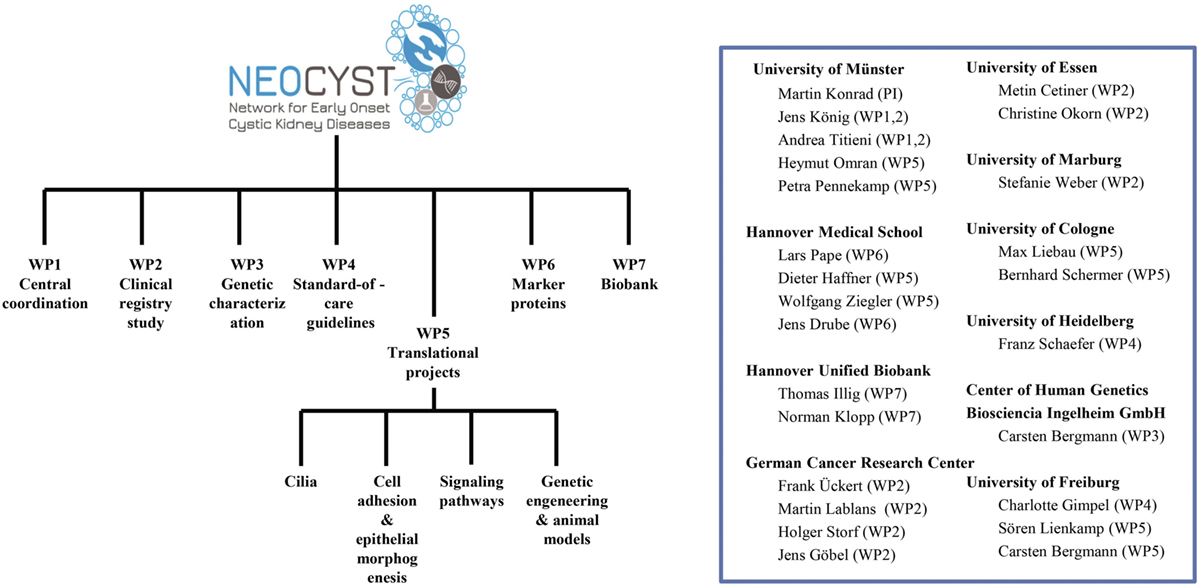
Figure 3. Organizational structure of the network for early onset cystic kidney diseases (NEOCYST) collaborative.
Current Status
The NEOCYST project started in February 2016 after initiation and approval of funding by the Federal Ministry of Education and Research (BMBF). After 24 months crucial milestones have been reached.
First of all, a homepage3 has been installed addressing patients, clinicians, and researchers to the same extent. Valuable information on the NEOCYST collaborative, the study goals as well as the medical background is provided in German and English language. A download domain contains all necessary study documents.
Furthermore, in September 2017 the technical implementation of the new online NEOCYST database has been completed merging three pre-existing clinical registries. Up-to-date 344 patients have been included comprising 194 patients with NPH and related ciliopathies, 88 patients with BBS, and 62 patients with HNF1B nephropathy. Especially, regarding HNF1B a rapid increase of participants has been observed ever since HNF1B became a part of the regular genetic screening in patients with hereditary cystic kidney diseases. However, we still assume a high number of unreported cases anticipating a further expansion of the study cohort within the next few years. The database is accessible via the NEOCYST-homepage (see text footnote 3).
Major success has been achieved by the genetic workgroup in identifying DZIP1L as a new cause for early onset cystic kidney diseases. DZIP1L encodes a protein located at the ciliary-transition-zone leading to a phenotype mimicking ARPKD when mutated (26). Additionally, mutations in MKS1 so far only associated with lethal Meckel–Gruber syndrome were found also to be responsible for a milder phenotype resembling Joubert syndrome accompanied by agenesis of the corpus callosum (39).
The number of biological samples stored in the biobank is continuously growing and substantial progress has been achieved by the projects attended to the molecular biology of cystic kidney diseases. Methods that have been established in another context were successfully transferred to the scientific issues addressed by the NEOCYST project. Detailed imaging and structural analyses of primary and motile cilia have been elaborated and brought to a new scale. First results suggest that structural abnormalities and differences in ciliary protein composition specific for the underlying genetic defect. Furthermore, urine-derived renal epithelial cells, URECs have been established as a valid model on studying cell polarity and epithelial morphogenesis in cystic kidney diseases producing distinguishable patterns of cell clusters. Most results are still preliminary and part of an ongoing research process that will be the subject of future publications.
So far, 19 manuscripts from the NEOCYST cohort have been published or accepted for publication—including a positional paper on the “perinatal management of early onset cystic kidney diseases” (40). Two further positional papers on “imaging of early onset cystic kidney diseases” and “management of early onset ADPKD” have been finalized and are about to be published.
Conclusion
Since initiation in February 2016, the NEOCYST collaborative has made substantial progress in addressing clinical, genetic, and molecular questions related to hereditary cystic kidney diseases. However, the mentioned achievements just represent the first steps of an ongoing process and further scientific initiatives and funding as well as international cooperation will be needed in order to answer these questions. Thus, any participation by international centers is warmly welcome. By setting up the infrastructure of an international clinical registry accompanied by several biological projects and the longtime storage of biomaterial, NEOCYST provides a platform that guarantees a detailed collection of precious longitudinal clinical data going along with further high-quality research approaches and enabling future clinical trials.
Neocyst Consortium
C. Bergmann, Ingelheim, Germany; M. Cetiner, Essen, Germany; J. Drube, Hannover, Germany; C. Gimpel, Freiburg, Germany; J. Göbel, Frankfurt, Germany; D. Haffner, Hannover, Germany; T. Illig, Hannover, Germany; N. Klopp, Hannover, Germany; J. König, Münster, Germany; M. Konrad, Münster, Germany; M. Lablans, Heidelberg, Germany; M. C. Liebau, Cologne, Germany; S. Lienkamp, Freiburg, Germany; C. Okorn, Essen, Germany; H. Omran, Münster, Germany; L. Pape, Hannover, Germany; P. Pennekamp, Münster, Germany; F. Schaefer, Heidelberg, Germany; B. Schermer, Cologne, Germany; H. Storf, Frankfurt, Germany; A. Titieni, Münster, Germany; F. Ückert, Heidelberg, Germany; S. Weber, Marburg, Germany; W. Ziegler, Hannover, Germany.
Author Contributions
JK, AT, and MK drafted the manuscript. MK is the principle investigator and JK the central coordinator of the described consortium.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors thank GPN for their support. The NEOCYST consortium is funded by the German Federal Ministry of Research and Education (BMBF, grant 01GM1515).
Footnotes
References
1. Bergmann C. Educational paper: ciliopathies. Eur J Pediatr (2012) 171(9):1285–300. doi:10.1007/s00431-011-1553-z
2. Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol (2002) 13:2384–98. doi:10.1097/01.ASN.0000028643.17901.42
3. Bergmann C, Zerres K. Early manifestations of polycystic kidney disease. Lancet (2007) 369(9580):2157. doi:10.1016/S0140-6736(07)61005-8
4. Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol (2009) 20(1):23–35. doi:10.1681/ASN.2008050456
5. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet (2013) 21(1):8–13. doi:10.1038/ejhg.2012.115
6. Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol (2006) 17(2):497–503. doi:10.1681/ASN.2005101040
7. Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol (2006) 17(10):2864–70. doi:10.1681/ASN.2006030277
8. Arts HH, Knoers NV. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol (2013) 28(6):863–74. doi:10.1007/s00467-012-2259-9
9. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol (2011) 26(7):1039–56. doi:10.1007/s00467-010-1731-7
10. König J, Kranz B, König S, Schlingmann KP, Titieni A, Tönshoff B, et al. Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol (2017) 12(12):1974–83. doi:10.2215/CJN.01280217
11. Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypers: the kidney and beyond. Pediatr Nephrol (2016) 31(5):707–14. doi:10.1007/s00467-015-3142-2
12. Dell KM. The spectrum of polycystic kidney disease in children. Adv Chronic Kidney Dis (2011) 18(5):339–47. doi:10.1053/j.ackd.2011.05.001
13. Sweeney WE Jr, Avner ED. Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol (2011) 26:675–92. doi:10.1007/s00467-010-1656-1
14. Büscher R, Büscher AK, Weber S, Mohr J, Hegen B, Vester U, et al. Clinical manifestations of autosomal recessive polycystic kidney disease (ARPKD): kidney-related and non-kidney-related phenotypes. Pediatr Nephrol (2014) 29(10):1915–25. doi:10.1007/s00467-013-2634-1
15. Blowey DL, Querfeld U, Geary D, Warady BA, Alon U. Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol (1996) 10:22–4. doi:10.1007/BF00863431
16. Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol (2007) 18(6):1855–71. doi:10.1681/ASN.2006121344
17. Wolf M. Nephronophthisis and related syndromes. Curr Opin Pediatr (2015) 27(2):201–11. doi:10.1097/MOP.0000000000000194
18. Forsythe E, Sparks K, Best S, Borrows S, Hoskins B, Sabir A, et al. Risk factors for severe renal disease in Bardet-Biedl Syndrome. J Am Soc Nephrol (2017) 28(3):963–70. doi:10.1681/ASN.2015091029
19. Parisi M, Glass I. Joubert syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2003). p. 1993–2017.
20. Ece Solmaz A, Onay H, Atik T, Aykut A, Cerrah Gunes M, Ozalp Yuregir O, et al. Targeted multi-gene panel testing for the diagnosis of Bardet Biedl syndrome: identification of nine novel mutations across BBS1, BBS2, BBS4, BBS7, BBS9, BBS10 genes. Eur J Med Genet (2015) 58(12):689–94. doi:10.1016/j.ejmg.2015.10.011
21. Hartill V, Szymanska K, Sharif SM, Wheway G, Johnson CA. Meckel-Gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr (2017) 5:244. doi:10.3389/fped.2017.00244
22. Szymanska K, Berry I, Logan CV, Cousins SR, Lindsay H, Jafri H, et al. Foundermutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia (2012) 1(1):18. doi:10.1186/2046-2530-1-18
23. Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol (2017) 9(3):a028191. doi:10.1101/cshperspect.a028191
24. König J, Ermisch-Omran B, Omran H. Nephronophthisis and autosomal dominant interstitial kidney disease (ADIKD). In: Geary DF, Schaefer F, editors. Pediatric Kidney Diseases. Berlin: Springer Verlag (2016):369–88.
25. Stokman M, Lilien M, Knoers N. Nephronophthisis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2016). p. 1993–2017.
26. Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, Kramer C, et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet (2017) 49(7):1025–34. doi:10.1038/ng.3871
27. Ware SM, Gunay-Aygun M, Hildebrandt F. Spectrum of clinical diseases caused by disorders of primary cilia. Proc Am Thorac Soc (2011) 8(5):444–50. doi:10.1513/pats.201103-025SD
28. Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol (2014) 12:228. doi:10.1186/gb-2011-12-9-228
29. Lemaire M, Parekh RS. A perspective on inherited kidney disease: lessons for practicing nephrologists. Clin J Am Soc Nephrol (2017) 12(12):1914–6. doi:10.2215/CJN.11751017
30. Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet (2016) 89:275–84. doi:10.1111/cge.12654
31. Joly D, Beroud C, Grünfeld JP. Rare inherited disorders with renal involvement-approach to the patient. Kidney Int (2015) 87:901–8. doi:10.1038/ki.2014.432
32. Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Müller D, Thumfart J, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol (2006) 17:2424–33. doi:10.1681/ASN.2005121351
33. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med (2011) 364(16):1533–43. doi:10.1056/NEJMra1010172
34. Ong AC, Devuyst O, Knebelmann B, Walz G; ERA-EDTA Working Group for Inherited Kidney Diseases. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet (2015) 385(9981):1993–2002. doi:10.1016/S0140-6736(15)60907-2
35. Loftus H, Ong AC. Cystic kidney diseases: many ways to form a cyst. Pediatr Nephrol (2013) 28(1):33–49. doi:10.1007/s00467-012-2221-x
36. Ong AC, Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int (2015) 88(4):699–710. doi:10.1038/ki.2015.207
37. Ebner K, Feldkoetter M, Ariceta G, Bergmann C, Buettner R, Doyon A, et al. Rationale, design and objectives of ARegPKD, a European ARPKD registry study. BMC Nephrol (2015) 18(16):22. doi:10.1186/s12882-015-0002-z
38. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet (1999) 36(6):437–46.
39. Bader I, Decker E, Mayr JA, Lunzer V, Koch J, Boltshauser E, et al. MKS1 mutations cause Joubert syndrome with agenesis of the corpus callosum. Eur J Med Genet (2016) 59(8):386–91. doi:10.1016/j.ejmg.2016.06.007
Keywords: hereditary cystic kidney diseases, ciliopathy, nephronophthisis, autosomal recessive polycystic kidney disease, Bardet–Biedl syndrome, hepatocyte nuclear factor-1beta nephropathy
Citation: König JC, Titieni A, Konrad M and the NEOCYST Consortium (2018) Network for Early Onset Cystic Kidney Diseases—A Comprehensive Multidisciplinary Approach to Hereditary Cystic Kidney Diseases in Childhood. Front. Pediatr. 6:24. doi: 10.3389/fped.2018.00024
Received: 18 October 2017; Accepted: 25 January 2018;
Published: 13 February 2018
Edited by:
Miriam Schmidts, Radboud University Nijmegen, NetherlandsReviewed by:
Laura Malaga-Dieguez, New York University School of Medicine, United StatesJulia Hoefele, Technische Universität München, Germany
Copyright: © 2018 König, Titieni, Konrad and the NEOCYST Consortium. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens Christian König, amVucy5rb2VuaWdAdWttdWVuc3Rlci5kZQ==