 Elizabeth A. LaMarca
Elizabeth A. LaMarca Samuel K. Powell1,2,3,5
Samuel K. Powell1,2,3,5 Schahram Akbarian
Schahram Akbarian Kristen J. Brennand
Kristen J. Brennand- 1Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 4Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Medical Scientist Training Program, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 6Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Human-induced pluripotent stem cells (hiPSCs) have revolutionized our ability to model neuropsychiatric and neurodegenerative diseases, and recent progress in the field is paving the way for improved therapeutics. In this review, we discuss major advances in generating hiPSC-derived neural cells and cutting-edge techniques that are transforming hiPSC technology, such as three-dimensional “mini-brains” and clustered, regularly interspersed short palindromic repeats (CRISPR)-Cas systems. We examine specific examples of how hiPSC-derived neural cells are being used to uncover the pathophysiology of schizophrenia and Parkinson’s disease, and consider the future of this groundbreaking research.
Introduction
Neuropsychiatric and neurodegenerative diseases are major contributors to the global burden of disease (1). Improved techniques to model these diseases and inform patient-specific treatment regimens are essential, as many current strategies have considerable limitations. Animal models cannot fully address the unique and complex pathophysiology of human neuropsychiatric disease, so findings are not always transferrable to the human condition. Human postmortem studies typically represent an advanced stage of disease and can be hindered by confounding factors, such as prescription and nonprescription drug use. These approaches also lack the capacity to predict patient-specific clinical outcomes to therapeutic drugs. Human-induced pluripotent stem cells (hiPSCs) are uniquely qualified to address these issues, as limitless patient-derived cells can be used to model human disease, screen drugs, and even generate tissues for transplantation. In this review, we will discuss how hiPSC technology has developed into a powerful platform for studying human neuropsychiatric diseases, focusing on schizophrenia and Parkinson’s disease (PD) as representative examples.
A Brief History of hiPSC Research
In 2006, Shinya Yamanaka and his student Kazutoshi Takahashi demonstrated that iPSCs could be generated from mouse fibroblast cells via retroviral delivery of just four transcription factors—c-Myc, Klf4, Oct3/4, and Sox2 (2). One year later, the first human iPSCs were generated independently by a number of groups (3, 4). The discovery of iPSCs was pivotal for the broad scientific community for several reasons: this was the first evidence that intact, adult cells could be reprogrammed to a pluripotent state; a new avenue of research was opened that was free of the moral and legal controversy surrounding embryonic stem cells (ESCs), and it was now feasible to generate large quantities of isogenic, patient-derived cells that can be differentiated into almost any cell type. Today, highly efficient, non-integrating reprogramming strategies (5–7) are widely used for studies of disease modeling and cell replacement therapy.
Methods and Advances in Generating hiPSC-Derived Neural Progenitor Cells (NPCs) and Neurons
To understand how neural stem cells (NSCs), NPCs, and neurons can be generated from hiPSCs, it is helpful to first briefly review human neural development in vivo, outlined in Box 1.
Box 1
Approximately three weeks after fertilization, gastrulation occurs, in which the early embryo divides into the three germ layers; these will ultimately give rise to every organ in the body: the innermost layer, the endoderm, will form the epithelial lining of the gut and respiratory tract; the middle layer, the mesoderm, will form muscle and connective tissue; and the outermost layer, the ectoderm, will form the nervous system and the outer parts of the body, such as skin. Neurulation occurs next, which begins with the secretion of growth factors from a cylinder of mesodermal cells called the notochord. The notochord secretes Noggin, which inhibits a member of the transforming growth factor-β family, bone morphogenetic protein (BMP)-4, which would otherwise direct cells to form skin. Noggin thus allows cells to continue along their default neuroectodermal lineage and fold inward to form the neural tube (Figure 1). The inside of the neural tube goes on to become the ventricles of the brain, while the lining of the ventricles (i.e., the ventricular zone) gives rise to proliferative cells that migrate and differentiate in the intermediate layer. Various secreted morphogens along the dorsoventral and rostrocaudal axes of the neural tube allow cells to differentiate into functionally distinct and anatomically segregated classes of neurons, giving rise posteriorly to the spinal cord and anteriorly to the three major divisions of the brain: the forebrain (prosencephalon), which will later develop the cerebrum (telencephalon) and thalamic structures (diencephalon), midbrain (mesencephalon), and hindbrain (rhombencephalon), which will later develop the pons, cerebellum (metencephalon), and medulla (myelencephalon). Inductive signaling of sonic hedgehog and retinoic acid is involved in patterning the ventral axis of the neural tube, which give rise to motor neurons, while BMPs and Wnts are involved in patterning the dorsal axis of the neural tube, which primarily give rise to sensory neurons.
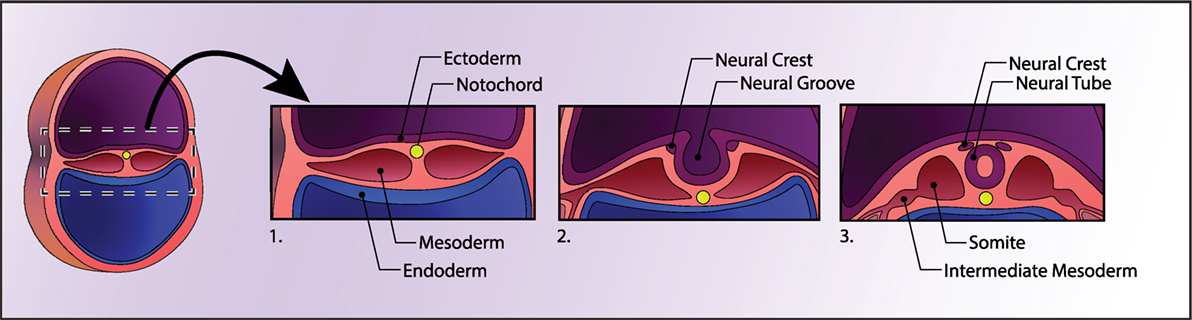
Figure 1. Human neural development. A cross section of the early embryo is depicted on the left. In the first panel, the three germ layers and the notochord are shown. The notochord signals the ectoderm above it to form the neural plate, which then folds into itself to form the neural tube, depicted in panel 3. See Box 1 for details.
Directed Differentiation
Aggregates of human pluripotent stem cells known as embryoid bodies will spontaneously differentiate into cells closely resembling those of all three embryonic germ lineages (8–10) and will further differentiate into neural-like cells at a low efficiency. Neural differentiation can be promoted by either inclusion of growth factors that promote neural differentiation or withdrawal of those factors that prevent it (i.e., mimicking in vivo cell differentiation) in a process known as “directed differentiation.” As mentioned in Box 1, the transforming growth factor-β protein, bone morphogenetic protein (BMP), directs cells away from a neuroectodermal lineage; this occurs via intracellular signaling to the nucleus by SMAD proteins (11). In 2004, it was demonstrated that treatment with the BMP antagonist Noggin (12) dramatically improved neural differentiation. Noggin-mediated SMAD inhibition was later improved by the addition of a second SMAD inhibitor, SB431542, resulting in a technique known as dual SMAD inhibition to promote neural differentiation (13). This technique is highly efficient, leading to 80% conversion to NPCs (13).
By default, NPCs generated by dual SMAD inhibition are characteristic of a rostral, or more primitive, identity (14). Caudalization can be achieved by treatment with an inhibitor of glycogen synthase kinase 3 (GSK3) to activate Wnt signaling; the addition of sonic hedgehog and/or fibroblast growth factor 8 can further direct cells toward a ventral mesencephalic lineage, yielding midbrain dopaminergic (DA) neurons (15) at yields of up to 80% [well reviewed in Ref. (16)]. These DA neurons express transcriptional profiles best aligned with midbrain DA identity, display appropriate electrophysiological activity, and restore lost functionality in animal models of PD [e.g., Ref. (15, 17–20)].
Directed differentiation protocols have several potential limitations, including a protracted differentiation period, the need for costly growth factors that can vary from lot to lot, and the generation of a heterogeneous population of cells that most closely resemble fetal forebrain neurons (21, 22). These issues can be partially circumvented via direct conversion of somatic cells into neurons in a strategy known as “neuronal induction.”
Neural Induction
Neuronal induction was first achieved by overexpressing a set of three transcription factors known as the BAM factors, Brn2 (also known as Pou3f2), Ascl1, and Myt11, in mouse fibroblasts (23, 24). The same group later showed that this strategy is also effective for human fibroblasts when combined with the transcription factor neurogenic differentiation factor 1 (NEUROD1) (25), although these induced neurons were unable to form synapses without a mouse cortical neuron substrate unless expressed with specific microRNAs (26, 27). Specific neuronal subtypes (28–31) or NPCs (32–35) can be induced through the addition of cell-type-specific transcription factors. More recently, human astrocytes have been induced in vitro, and mouse astrocytes in vivo, into mDAergic neurons through overexpression of NeuroD1, Ascl1, Lmx1a, and the microRNA miR218 (36).
In 2013, Zhang et al. showed that it was possible to induce nearly 100% pure populations of neurons from ESCs or hiPSCs via forced overexpression of just a single transcription factor, either Neurogenin2 (NGN2) or NEUROD1 (37), yielding functional synapses within just two weeks. Subsequently, Ho et al. applied this technique to induced neurons from hiPSC-derived NPCs, which are easier to culture and more amenable to high-throughput drug screens (38).
Three-Dimensional (3D) “Mini-Brains” in a Dish
Two-dimensional neural culture systems have vastly expanded our understanding of neurological development and disease, but lack the ability to accurately model the cytoarchitecture and network connectivity of the 3D human brain. Within the last decade, 3D neural culture systems have begun to address this issue and are leading to improved disease modeling approaches.
The first neural organoid-like structures derived from PSCs were developed using a serum-free, floating culture of embryoid bodies (SFEB) (39), which suffered from low efficiency and inconsistent aggregate sizes. Seeding dissociated mouse ESCs into U-bottomed, low-adhesion plates led to quick reaggregation (SFEBq) of uniformly sized masses and consistent differentiation into cortical neuron-like cells (Figure 2A) (40). By seeding cells in a V-bottom plate in the presence of a rho kinase (ROCK) inhibitor to reduce apoptosis, the SFEBq technique is also suitable for use with human PSCs (41). Importantly, the authors showed that 3D structures were capable of self-organizing into apicobasally polarized neuroepithelial structures. With the addition of rostral-neutralizing factors, organoids can produce polarized radial glia, intermediate progenitors, and both early- and late-born cortical neurons that display evidence of synapse formation (including vesicular glutamate transporter vGLUT1-positive excitatory neurons and dorsal telencephalic-like GABAergic interneurons) (21). Mimicking the laminin-rich basement membrane of epithelial structures with Matrigel improves organoid self-organization (42).
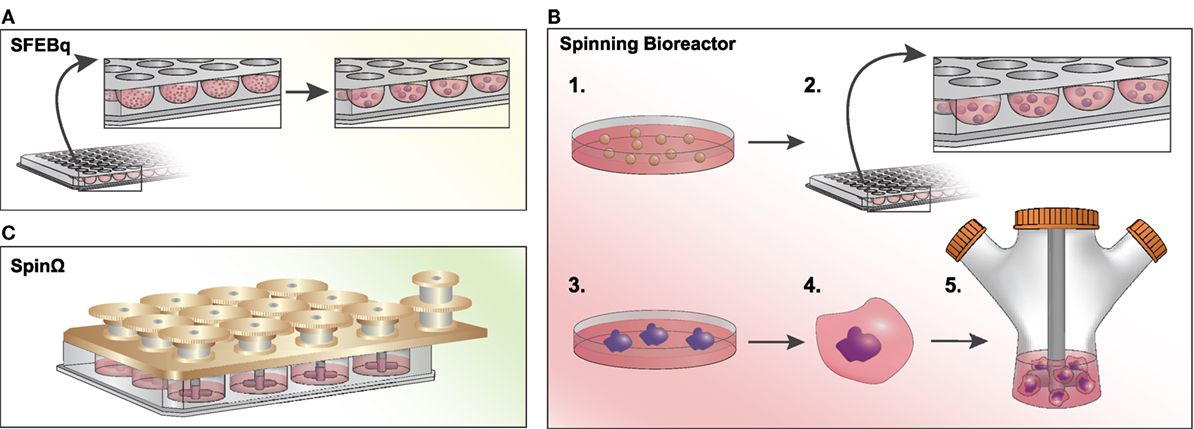
Figure 2. Methods to generate brain organoids in vitro. (A) Serum-free, floating culture of embryoid bodies with quick reaggregation (SFEBq): pluripotent stem cells (PSCs) are dissociated and resuspended in a low-adhesion, U-bottomed 96-well plate in serum-free culture. Cells will quickly reaggregate (within an hour) into uniformly sized masses and will differentiate into neural progenitor cells within ~10–12 days after plating in N2-supplemented media. (B) Spinning Bioreactor: 1. PSCs are dissociated and 2. cultured in a low-adhesion plate in hES media with low bFGF and a ROCK inhibitor. 3. Embryoid bodies are cultured in neural induction medium and begin to form neuroepithelial tissues. 4. Tissues are transferred into Matrigel droplets and four days later are 5. transferred to a spinning bioreactor with differentiation media. (C) SpinΩ: A spinning device is fitted over a standard 12-well culture plate, with 13 interconnecting gears driven by a motor agitating the culture media. Similar to the spinning bioreactor approach, cells are embedded in Matrigel droplets prior to spin culture. This technique significantly reduces the cost and space constraints associated with spinning bioreactors.
In a seminal study by Lancaster et al., large and complex cerebral organoids were generated by seeding neuroectodermal aggregates embedded in Matrigel droplets in a spinning bioreactor to enhance nutrient absorption (Figure 2B) (43). After less than a month, the organoids developed a regional specification analogous to the early fetal brain in vivo. In an attempt to reduce the large volumes and space constraints associated with the spinning bioreactor approach, Qian et al. developed SpinΩ, a mini-bioreactor to generate organoids from hiPSCs (Figure 2C) (44). This study yielded organoids with a human-specific radial glial cell outer layer and all six layers of the cortex clearly defined, as well as several major interneuron subtypes expressing parvalbumin, nNOS, or somatostatin. Furthermore, the authors were able to generate more homogeneous forebrain structures with the addition of a pre-patterning step to initially instruct cell fate direction (44). Cortical spheroids can also be generated without spinning bioreactors or Matrigel through dual SMAD inhibition followed by culture with a neuron differentiation-promoting medium (45). Such spheroids are more homogeneous, containing excitatory pyramidal cell-like deep and superficial cortical neurons, as well as fully integrated astrocytes.
To model the impact of vasculature, hESC-derived NPCs were seeded on peptide-functionalized polyethylene glycol hydrogels, followed by addition of hESC-derived microglial and vascular cells in a time frame that corresponded to the integration of these cells during fetal brain development (46). These organoids formed a vascular network and incorporated both ramified and amoeboid microglial cells, leading to a highly similar transcriptional profile to that of the early fetal brain. Recently, Lancaster et al. combined traditional organoid generation and bioengineering techniques to physically guide 3D differentiation with microfilaments, leading to an increased fidelity of forebrain-like cells and capacity for radial glial migration (47).
Organoids have remarkably similar gene expression patterns to their in vivo fetal brain counterparts (48, 49) and are capable of spatial patterning and cell fate commitment in a similar time frame to that seen in vivo. It is possible to optically clear 3D neural cultures in order to visualize their internal network connectivity while reducing the time and labor associated with standard tissue sectioning and histology, leading to greater scalability (50). Several limitations do exist, however, primarily concerned with the lack of true patterning axes in vitro, precluding the development of more complex self-organizing structures. In addition, although successive techniques have reduced inter-sample variability, this still remains a major limiting factor for assays that require homogeneity among samples such as drug screens. There is a need for future studies to address these issues, potentially by integrating novel bioengineering strategies. For a more detailed review of 3D neural culture techniques and considerations, readers are referred to an excellent review by Kelava and Lancaster (51). Overall, brain organoids seem to faithfully represent human corticogenesis and as such are excellent platforms upon which to study complex neurodevelopmental diseases.
Clustered, Regularly Interspersed Short Palindromic Repeats (CRISPR)
The (CRISPR)/Cas (CRISPR-associated protein) system enables targeted manipulation of genetic material. Initially identified as a key immune defense mechanism in some bacterial species and Archaea (52–54), the CRISPR/Cas9 system involves the production of CRISPR RNAs that bind to Cas endonucleases and cleave invading genetic material (55). Of particular interest to current applications is the type II CRISPR/Cas9 system in which a noncoding transactivating (tracrRNA) facilitates binding of complexes consisting of CRISPR RNAs and Cas9 to target loci (56, 57) (Figure 3). In the presence of a protospacer adjacent motif (58) that is compatible with Cas endonuclease binding, this complex cleaves the genetic material to which it is bound (59, 60). In modern applications, the tracrRNA and cRNA are typically synthesized as a single “guide RNA” (gRNA) (55). A commonly used endonuclease is the Streptococcus pyogenes Cas9 (Sp Cas9) (61) but several others exist [reviewed in Ref. (62)]. In addition to those enabling targeted cutting of genetic material, novel Cas9 endonucleases have been generated for further applications. “Nickase Cas9” (nCas9) is an endonuclease in which one of the two nuclease domains has been mutated such that only one of the target DNA strands is cut (63), which enhances target specificity by nature of the fact that only adjacent nCas9-mediated cuts generate the desired double-stranded break (63–65). Furthermore, the mutation of both nuclease domains creates a null or “dead” Cas9 (66) to which several transcriptional effectors can be fused for the modulation of gene expression at loci of interest (67–69).
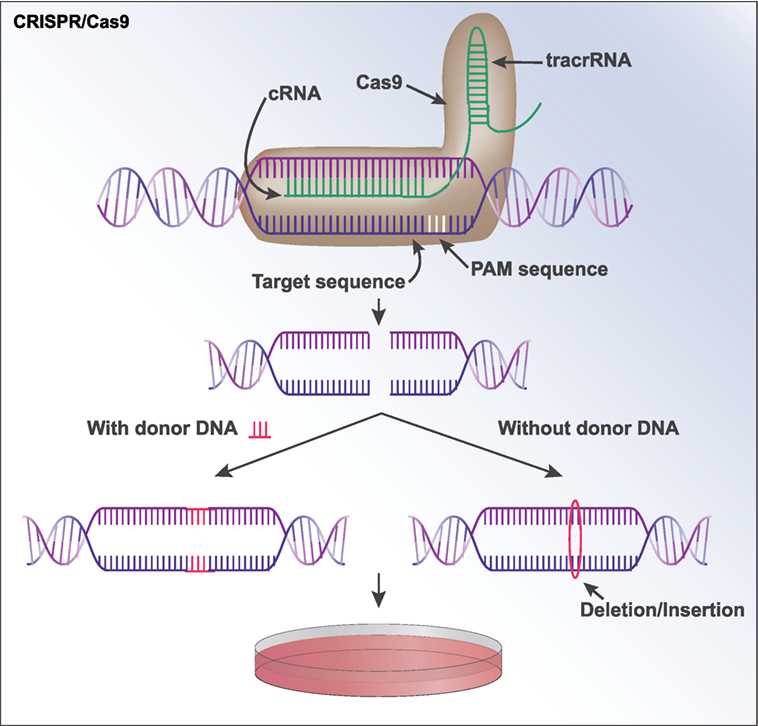
Figure 3. CRISPR/Cas9. A sequence of DNA can be modified by CRISPR/Cas9 through the formation of a guide RNA (gRNA) that contains an RNA complementary to the target DNA sequence. The gRNA additionally contains a noncoding transactivating RNA (tracrRNA) that functions as a “scaffold” to bind to Cas9. The gRNA and Cas9 form a complex and is brought to the target sequence, where Cas9 binds to a specific protospacer adjacent motif (58). Cas9 then cuts both strands of the DNA. If the user supplies a “donor sequence,” it will serve as a repair template and be incorporated at the break site in a process known as homology-directed repair. If no donor sequence is provided, indels are often created as the cell repairs the double-strand break via non-homologous end joining. This can knock out the gene of interest, as the indels are often capable of creating frameshift mutations. The modified sequence can now be expanded in cells in culture.
The CRISPR/Cas9 system can be used in a variety of ways to study neurological and psychiatric diseases in stem cell-based models (70, 71). One particularly promising application is the manipulation of genetic material to explore the role of putative allelic variations in neurological and psychiatric conditions. Introduction of a PD-associated single nucleotide polymorphism (SNP) in an enhancer region of alpha-synuclein (SNCA) altered histone posttranslational modifications at this site and increased the expression of SNCA (72). In the case of Fragile X Syndrome, deletion of the disease-causing trineucleotide repeat expansion of CGG in the FMR1 gene in hiPSC neurons derived from patients with FXS resulted in normalized expression of FMR1 (73). The generation of NPCs and neurons (74) and cerebral organoids (75) with heterozygous deletions in chromodomain helicase DNA-binding protein 8 (CHD8), a gene implicated in ASD-associated de novo mutations (76–78), provided support for a role of Wnt signaling in ASD pathophysiology. Beyond genomic-editing approaches, a nuclease null or dead Cas9 fused to transcriptional regulators can be employed to alter the expression of several genes implicated in neuropsychiatric disease (79, 80). Overall, these reports are robust demonstrations of the feasibility of CRISPR/Cas9 approaches in disease modeling with hiPSC-derived neurons.
Advances in the Field of Neuropsychiatric hiPSC-Based Disease Modeling
Schizophrenia
Schizophrenia is a severe, complex genetic psychiatric disorder that affects approximately 1% of the population worldwide, primarily characterized by positive, negative, and cognitive symptom domains (81). Large-scale genetic studies have identified 108 common variant loci (82) and eight copy number variations (83) that are significantly associated with schizophrenia. Here, we will discuss how hiPSCs have been used to dissect the pathophysiology associated with genetic risk for schizophrenia, focusing on well-documented phenotypic consequences.
22q11.2
The 22q11.2 region of the genome is flanked by two low-copy repeat segments that are susceptible to non-allelic homologous recombination. Deletions in this region are often at these “breakpoints” and typically occur de novo, as a result of aberrant recombination during meiosis (84). 22q11.2 deletion as a risk allele for schizophrenia has been confirmed at genome-wide significance in a cohort of ~40,000 cases and controls, and interestingly, duplications of 22q11.2 were suspected to have a protective effect in controls (83, 85). Deletions of 22q11.2 have also been implicated in autism spectrum disorders, attention-deficit/hyperactivity disorder, and anxiety and affective disorders (86).
Abnormal gene (87) and microRNA expression (88) occurs in hiPSC-derived 22q11.2 del neurons, with an enrichment for genes and microRNA targets involved in neuropsychiatric disorders. hiPSC-derived 22q11.2 neurons further show impaired ability to differentiate into neurons, abnormal neurite outgrowth, migration, neuron-to-glia ratios, and have smaller neurosphere sizes (89). The downstream targets of DGCR8 (contained in the 22q11.2 region and essential for microRNA biosynthesis), miR-106a, and the miR-17/92 cluster were reduced in patient-derived neurospheres; this finding implicated their target p38a, which drives glial differentiation, and could potentially explain the abnormal neuron-to-glia ratios observed (89). In addition to dysregulated gene expression, 22q11.2 del hiPSC neurons have higher levels of the LINE-1 retrotransposon in synaptic genes, consistent with observations in 22q11.2 del patient postmortem brains (90). To date, the lack of genetically engineered rescue of 22q11.2 deletions in hiPSC neurons has limited efforts to identify the causal gene(s) within the region responsible for these molecular and cellular phenotypes.
Disrupted in Schizophrenia 1 (DISC1)
The discovery of the DISC1 gene dates back to 2000, following a nearly 30-year longitudinal study of a Scottish family with a history of psychiatric illness (91, 92). Researchers identified a balanced translocation between chromosome 1 and chromosome 11 in 37 of 69 biologically related, karyotyped family members, 29 of whom were diagnosed with a psychiatric illness (93). This translocation was found to disrupt two genes on chromosome 1, DISC1 and DISC2 (91). DISC1 is an important regulator of embryonic and adult neurogenesis, neurite outgrowth, and synapse formation (94–96), serving as a cytoskeletal scaffolding protein and a modulator of GSK3β and Wnt signaling (97–99).
Human-induced pluripotent stem cell-derived neurons from two related patients with a frameshift mutation in DISC1 had impaired synaptic vesicle release relative to two clinically unaffected family members without the mutation (100). Using transcription activator-like effector nucleases (TALENs), the authors were able to reverse the respective synaptic phenotypes by editing in the mutation into control lines and correcting it in patient lines, thereby establishing a direct link between DISC1 mutation and synaptic dysfunction. DISC1 regulation at the synapse has been further elucidated by Kassan et al., who found that both mouse and hiPSC-derived neurons have an increased expression of DISC1 and other synaptic proteins when caveolin-1 (Cav-1) is overexpressed under the control of a neuron-specific synapsin promoter and decreased when Cav-1 is knocked out (KO) (101). The authors restored the expression of DISC1 and the other synaptic proteins by re-introducing the synapsin-driven Cav-1 vector into Cav-1-KO neurons.
In addition to synapse function, DISC1 disruption is also deleterious for normal processes involved in mitosis and neural cell fate commitment. DISC1 regulates the attachment of Ndel1 to the kinetochore during mitosis, and the t(1; 11) translocation disrupts this binding complex (102). By introducing a peptide that specifically interferes with the binding of Ndel1 to DISC1 (but does not affect the binding of either DISC1 or Ndel1 to other proteins) in hiPSC-derived forebrain organoids, Ye et al. showed that this complex is important for the proliferation of NSCs, and its disruption leads to delayed cell-cycle progression (103). In line with this finding, hiPSC-derived forebrain organoids from a schizophrenia patient containing a frameshift mutation in the C-terminal region of DISC1 showed prolonged cell-cycle progression during mitosis. The role of DISC1 in neural cell fate commitment was demonstrated by Srikanth et al., who used TALEN- and CRISPR–Cas9 to mutate DISC1 in hiPSCs (104), reporting aberrant neural differentiation and increased Wnt signaling. Given its many protein-binding interactions, understanding the mechanism by which this very rare mutation impacts neuronal function may ultimately inform a larger understanding of schizophrenia risk.
Antipsychotic Drug Modeling
Human-induced pluripotent stem cell-derived neural cells can be used with relative ease to probe the efficacy and mode of action of pharmaceuticals both for large populations and for individual patients. In the first study demonstrating the feasibility of using hiPSC-derived neural cells to model schizophrenia and to study antipsychotic drug effects in vitro, deficits in neuronal connectivity were partially reversed by culture with the first-generation antipsychotic loxapine, which additionally raised levels of several glutamate receptors in the patient cell lines (105). Similarly, Asada et al. found that antipsychotic drugs promote neural differentiation in hiPSC-derived cells (106). In two complementary studies, Paulsen et al. showed that hiPSC-derived neural cells had significantly increased levels of extramitochondrial oxygen consumption and levels of harmful reactive oxygen species (ROS) compared to controls (107), together with elevated levels of potassium and the putative antioxidant zinc (108). Treatment with the mood stabilizer Valproate increased extramitochondrial oxygen flow and normalized both ROS and zinc levels in the patient NPCs.
It may also prove possible to probe the underlying mechanism of discordant antipsychotic drug response with hiPSC-derived neural cells. hiPSC-derived neurons generated from a pair of monozygotic twins with treatment-resistant schizophrenia (only one of whom responded to clozapine) showed unique gene expression patterns, particularly with respect to homophilic cell adhesion, in the responder relative to the non-responder both basally and during treatment with clozapine (109). Consistent with this, Maschietto et al. found many differentially expressed genes in hiPSC-derived NPCs from a patient with treatment-resistant schizophrenia, many of which are also involved in cell adhesion (110).
Overall, these studies (outlined in Table 1) demonstrate the clinical utility of hiPSC-derived neural cells in the study of both schizophrenia and its treatment. More personalized preclinical models could mitigate costly clinical trial failures, as individual patient-derived hiPSC neurons could be treated with arrays of different antipsychotics to test drug efficacy before prescribing to patients.
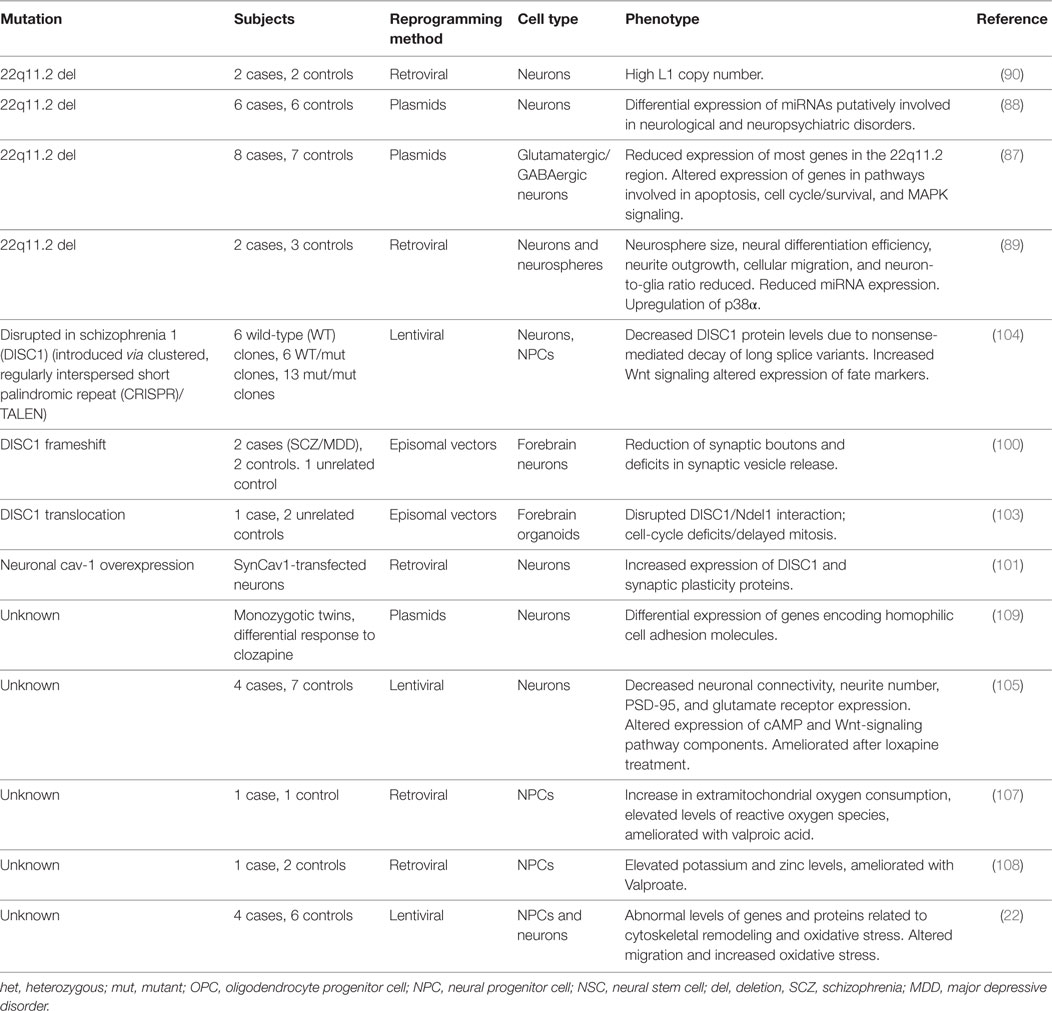
Table 1. Studies of human-induced pluripotent stem cell models of schizophrenia.
Parkinson’s Disease
Parkinson’s disease is a neurological disorder characterized by hallmark motor symptoms of rigidity, tremor, bradykinesia, and postural instability (111). In addition to these, numerous nonmotor symptoms can be observed, including cognitive impairment, mood disorders, psychosis, fatigue, autonomic dysfunction, olfactory abnormalities, and gastrointestinal dysfunction (112). During the many decades of active research on this disorder, a persistent challenge has been the lack of model systems that allow the examination of cellular and molecular deficits in living neurons from patients with PD and an inability to perform meaningful drug screens on human tissue. In this section, we review many studies that have addressed these problems by generating hiPSC-derived neurons from patients with defined genetic mutations associated with PD. These findings have demonstrated a variety of pathological disturbances in PD and have paved the way toward therapeutic innovation.
Leucine-Rich Repeat Kinase 2 (LRRK2)
Mutations in LRRK2 are associated with both idiopathic and familial PD (113, 114). In the first report describing the generation of hiPSCs from a patient with a LRRK2 G2019S mutation, differentiated DA neurons displayed an abnormal accumulation of alpha-synuclein, altered expression patterns of genes involved in oxidative stress, and an increased susceptibility to chemical stressors (115). Neurons generated from patients with the same LRRK2 mutation or an R1441C mutation exhibited dysfunction of several mitochondrial pathways, some of which could be rescued with compounds, including an LRRK2 inhibitor, identified from pharmacologic screens (116). A similar report showed mitochondrial DNA damage in DA neurons with either LRRK2 mutation, which could be alleviated with gene correction (117). Furthermore, the targeted inhibition of mitochondrial fission prevented elevated autophagy and mitochondrial pathology in LRRK2 neurons (118).
Deficits in LRRK2-mediated arrest of dysfunctional mitochondria in G2019S-mutant neurons may be mediated by the outer mitochondrial membrane protein Miro, as G2019S-mutant neurons exhibited abnormal retention of Miro and persistence of dysfunctional mitochondria, leading to neurodegenerative phenotypes that could be improved through reduction in Miro levels (119). A later report also found abnormal mitochondrial dynamics that were associated with a decreased Sirtuin2 activity (120). In others, midbrain DA neurons derived from patients with idiopathic and LRRK2 G2019S PD demonstrated disrupted autophagy and neurite growth patterns (121, 122). NPCs generated from LRRK2 G2019S patients degenerated more compared to wild-type controls in a passage-dependent manner, which was associated with progressive nuclear damage and the acquisition of epigenetic markers associated with aging (31). In addition, neurons generated from patients with LRRK2 mutation and sporadic PD had DA neuron-specific hypermethylation and alteration in gene expression profiles, including aberrant expression of transcription factors with potential relevance to PD (123).
Genetic correction experiments ameliorated the neurodegenerative phenotypes in LRRK2 neurons and provided evidence for the involvement of the ERK-signaling pathway in LRRK2 mutation-mediated neuropathology; furthermore, disease-related neuronal dysfunction could be improved by the inhibition of the ERK pathway (124). In an alternative approach, the generation of sensory neurons from LRRK2 G2019S hiPSCs implicated abnormal neurite outgrowth and altered calcium responses to depolarization as pathologies driving some of the nonmotor symptoms of PD (125), and these abnormalities could be alleviated with pharmacologic inhibition of LRRK2. Finally, both R1441G- and G2019S LRRK2-mutated neurons exhibited altered NF-kB transcriptional responses (126), and gene expression profiles have also been explored in LRRK2 G2019S neural organoids (127). Beyond the LRRK2 mutations already mentioned, hiPSC lines have also been generated from patients with different LRRK2 mutations, including R1398H, R1628P, S1647T, and N551K (128–131). Overall, these studies provide robust demonstration of the ability of hiPSC approaches to reveal disease-associated pathologies in a mutation-specific manner.
PINK1 and Parkin
Mutations in both PTEN-induced putative kinase 1 (PINK1) (132) and Parkin (133) are implicated in autosomal recessive forms of familial PD. These two proteins interact in a mitochondrial-autophagy pathway in which PINK1 recruits Parkin to damaged mitochondria (134, 135) and Parkin subsequently ubiquitinates mitochondrial proteins, targeting them for autophagy (136, 137).
Generation of hiPSC-derived midbrain DA neurons from patients with Parkin mutations revealed an increased oxidative stress and a spontaneous dopamine release mediated by increased transcription of monoamine oxidases (138). Later reports found accumulation of alpha-synuclein and abnormalities in mitochondrial dynamics in PINK1- and PARK2-mutant hiPSC-derived neurons (139, 140). PINK1-mutant neurons exhibited a deficient recruitment of Parkin (141) and an altered Parkin-mediated ubiquitination in response to mitochondrial depolarization (142). Mitochondrial abnormalities were also seen in DA neurons from PARK2 patient-specific mutant and isogenic PARK2-knockout hiPSC lines (143). Different investigators have also shown a heightened sensitivity to metal toxins including manganese (144) and copper (145) in PARK2-mutant hiPSC-derived NPCs. Both TH+ and non-TH+ neurons with PARK2 mutation have several alterations in neurite morphology (146), and a later report documented disrupted microtubule stability (147). Parkin-mutant neurons also exhibit abnormalities in endosomal processes and trafficking (148), as well as disrupted calcium shuttling between mitochondrial and endoplasmic reticulum (ER) (149).
Alpha-Synuclein
Alpha-synuclein, encoded by the SNCA gene is the key component of Lewy body inclusions, which are a pathological hallmark of PD (150); numerous mutations in SNCA have been associated with the disease (151–153). Human ESC lines with point mutations in SNCA, isogenic controls, and SNCA-mutation-associated PD patient hiPSC lines have been generated to provide a platform to study perturbations in alpha-synuclein in a targeted fashion (154). Cortical neurons generated from hiPSC lines of patients with alpha-synuclein mutations exhibited increased nitrosative and ER stress and accumulation of ER degradation substrates; following this observation, a small molecule identified in a yeast screen reversed some of these abnormal phenotypes (155). On the other hand, DA neurons generated from alpha-synuclein-mutant hiPSC patient cells also showed nitrosative stress, which led to Mef2c nitrosylation and a subsequent decreased activity of the neuroprotective peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC1alpha) pathway; such dysregulation led to mitochondrial damage and subsequent apoptosis (156). Neurons generated from hiPSC lines with a variety of SNCA mutations contained altered ratios of alpha-synuclein tetramers to monomers, providing further evidence for the abnormal accumulation of alpha-synuclein as a pathologic hallmark of PD (157). Strikingly, the introduction of a PD-associated SNP in an enhancer region of SNCA enhanced H3K27ac at this region and led to an increased alpha-synuclein expression and altered transcription factor binding in hiPSC-derived neurons (72).
Triplication of SNCA results in a completely penetrant and pernicious form of PD with dementia (158), and midbrain DA neurons generated from an SNCA triplication patient line produced approximately double the normal levels of alpha-synuclein (159). These types of cells also exhibit an increased susceptibility to oxidative stress (160) and lysosomal dysfunction (161). Similar SCNA triplication cell lines also had diminished capabilities to differentiate into both GABAergic and DA neurons, with a corresponding decrease in differentiation-promoting genes (162). Furthermore, the same study documented abnormal cation currents, decreased connectivity, and dramatically altered action potential profiles in neurons with SNCA triplication (162).
Overall, these studies of patient-specific PD hiPSC neurons (outlined in Table 2) have revealed an array of similarities and differences between DA neurons carrying different PD genes, suggesting that a targeted genotype-first therapeutic approach might also advance treatments for PD.
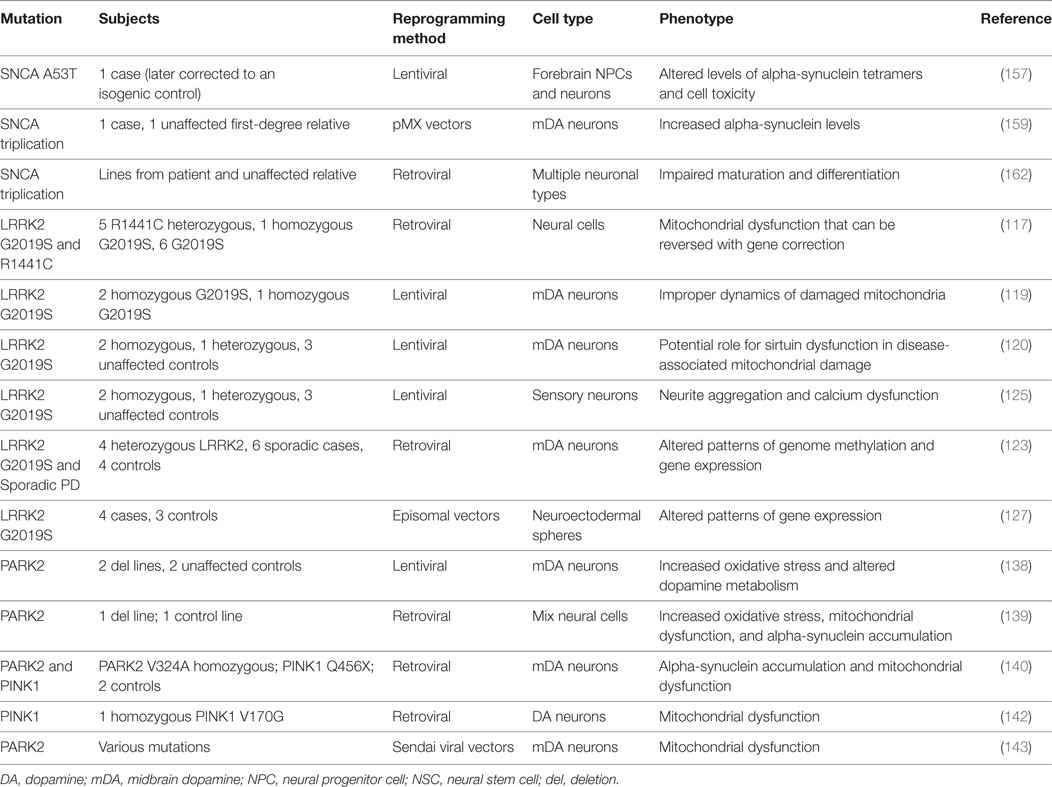
Table 2. Studies of human-induced pluripotent stem cell (hiPSC) models of Parkinson’s disease (PD).
Challenges of hiPSC Disease Modeling and Future Perspectives
Despite recent advances in hiPSC differentiation protocols, several obstacles still remain. hiPSC-derived neurons are currently unable to mature past a fetal stage, which can be limiting for studies of diseases that primarily affect the adult brain, such as PD. In order to address this issue, a consensus must first be reached concerning the measurable hallmarks that define cellular aging. Lopez-Otin et al. proposed nine such hallmarks that can be used as screens for cellular aging, including altered intracellular communication, cellular senescence, deregulated nutrient-sensing, epigenetic alterations, genome instability, loss of proteostasis, mitochondrial dysfunction, stem cell exhaustion, and telomere attrition (163).
The overexpression of progerin, a protein involved in the premature aging disorder Hutchinson–Gilford progeria, in hiPSC-derived mDA neurons leads to several age-related deficits, such as mitochondrial abnormality and AKT deregulation (164). Although progerin overexpression is unable to recapitulate all aspects of normal aging, it may prove especially useful for modeling late-onset disorders in vitro. Neural induction strategies can be used to accelerate differentiation, and coculture of neurons with astrocytes (165) or astrocyte-conditioned medium (166, 167) has been shown to promote electrophysiological maturation. More studies are needed to assess whether these techniques are capable of accelerating other characteristics of cell maturation, such as late-stage neuronal markers and morphology (168).
Sample heterogeneity between differentiation strategies (169, 169) and laboratories (170) remains a large issue for hiPSC research. Optimizing and standardizing differentiation protocols across laboratories are thus necessary to permit the generation of accurate, reproducible, and scalable data. Several large-scale hiPSC initiatives, such as the Innovative Medicines Initiative EU Centralized iPSC Repository, the New York Stem Cell Foundation, the California Institute for Regenerative Medicine, Kyoto University Center for iPS Cell Research & Application, Progenicyte, HipSci, StemBANCC, and the National Institute of Mental Health (171) have been founded to create biorepositories of well-characterized hiPSC lines from control and patient groups for widespread distribution to researchers and clinicians around the world. Such large-scale initiatives will help to increase the statistical power of hiPSC-based studies, which have historically suffered from low sample sizes and variability among samples and replicates. Automated culture systems can also help to reduce sample variability (172). Furthermore, two recent studies demonstrated the need for increased donor lines in hiPSC-based studies in order to minimize sample variability (168, 169), even at the expense of reducing the number of replicated hiPSC lines per individual.
Since the inception of hiPSC technology, researchers have made tremendous progress in engineering these in vitro systems to more closely resemble the molecular and electrophysiological properties of human cells in vivo. Recently, several studies have reported improved culture techniques that permit enhanced network-wide electrophysiological properties in hiPSC-derived neural cells (173, 174). In addition, Kuijlaars et al. showed that coculture with human primary astrocytes leads to increased neuronal network activities, such as bursting frequency and calcium oscillation synchronization over time (175). Other advances in the area of biomaterials (176, 177) are improving the efficiency, scalability, and fidelity of hiPSC-derived neuronal differentiation strategies (177). As these innovations continue to improve hiPSC technology, researchers will be better equipped to model the complex phenotypes associated with neuropsychiatric and neurodegenerative illnesses.
The CRISPR/Cas system is undoubtedly revolutionizing hiPSC research. Off-target effects present a major drawback, but can be mitigated by optimizing gRNA design or by using high-fidelity alternatives to the classic Cas9 endonuclease (178). More importantly, the commonly used lentiviral delivery strategy is highly efficient but prone to random genomic integration, limiting its clinical applicability. Alternative, non-integrating approaches have been proposed, such as adeno-associated viruses and nonviral methods (178–180), which may provide safer alternatives that can be used in a clinical setting. CRISPR-based clinical trials aimed at treating human immunodeficiency virus, Epstein–Barr-associated malignancies, gastrointestinal infection, and multiple forms of cancer are already underway in China, as well as for several forms of cancer in the USA (www.clinicaltrials.gov). In these studies, cells will be removed from the patient and modified with CRISPR, then will be reintroduced into the patient. In addition to the enormous benefit provided by generating hiPSC-based models of disease, in the future it may be possible to introduce patient-derived hiPSCs modified by CRISPR (to introduce corrective or protective mutations) into patients to treat neurodegenerative illnesses. Stem cell transplants have already been used with success in rodent models of neural disease (15, 181–185), which gives hope that human iPSC transplants will soon become a practicable therapy.
Conclusion
As we have described here, hiPSCs are an enormously powerful tool for neuropsychiatric disease modeling. The ability to generate nearly limitless quantities of patient-derived neurons, combined with CRISPR/Cas9 gene editing, allows researchers to introduce or correct disease-relevant mutations in order to study the pathophysiology of disease risk. 3D culture systems will ultimately yield disease models that more faithfully reflect the complex interconnectivity of the human brain. By studying hiPSC-derived neurons from patients with defined genetic mutations associated with particular illnesses, disease mechanisms can be explored in living tissue on a genetic background specific to the patient. As pathological phenotypes are identified, patient-derived neurons can be used for pharmacological drug screening. For these reasons, hiPSCs provide an invaluable research tool for investigators and a source of hope for patients struggling with neurological and psychiatric diseases.
Definitions
Positive, Negative, and Cognitive Symptom Domains of Schizophrenia
Positive symptoms of schizophrenia include functions or behaviors that are not present in healthy individuals, but are present in patients. These include hallucinations, delusions, abnormal movement, and thought disorders. Negative symptoms include functions or behaviors that are not present in patients, which typically are in healthy individuals. Such examples are flat affect, alogia, anhedonia, avolition, and asociality. The cognitive symptom domain includes disruptions in executive function, working memory, attention, and learning.
Low-Copy Repeat Segments
Segments of highly repetitive DNA can give rise to chromosomal rearrangements.
Non-Allelic Homologous Recombination
Exchange of genetic material is between similar or identical regions of DNA within different alleles.
Primary Neural Cultures
Neural cells are derived from the brain in vivo (postmortem or biopsy) and cultured in vitro.
First-Generation Antipsychotic
An older class of antipsychotics that primarily act on the DA system, particularly blocking the action of dopamine 2 receptors (e.g., chlorpromazine and haloperidol), and are associated with movement-related extrapyramidal side effects and tardive dyskinesia.
(Clozapine) Second-Generation Antipsychotic
A newer class of antipsychotics with actions at a variety of neurotransmitter receptors, most notably DA and serotonergic receptors, along with significant antihistaminic and anticholinergic activity (e.g., clozapine, olanzapine, quetiapine, and risperidone). Although second-generation antipsychotics generally cause lower rates of extrapyramidal side effects than do the first generation of antipsychotics, their use is associated with high rates of metabolic adverse effects. In the case of clozapine, the risk of a rare but potentially life-threatening blood condition (agranulocytosis) has limited its use to treatment-refractory cases.
Author Contributions
EL, SP, and KB conceived the concept and structure of the paper; EL and SP wrote the manuscript; KB critically revised the manuscript; KB and SA supervised the preparation and submission of the manuscript; EL prepared the figures. All the authors reviewed and approved the final manuscript for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
KB is a New York Stem Cell Foundation—Robertson Investigator. The Brennand Laboratory is supported by a Brain and Behavior Young Investigator Grant, National Institute of Health (NIH) grants R01MH101454 and R01MH106056, and the New York Stem Cell Foundation. The Akbarian laboratory is supported by R01MH106056.
References
1. Global Burden of Disease Study Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2015) 386:743–800. doi:10.1016/S0140-6736(15)60692-4
2. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126:663–76. doi:10.1016/j.cell.2006.07.024
3. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131:861–72. doi:10.1016/j.cell.2007.11.019
4. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (2007) 318:1917–20. doi:10.1126/science.1151526
5. Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell (2010) 7:618–30. doi:10.1016/j.stem.2010.08.012
6. Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci (2009) 85:348–62. doi:10.2183/pjab.85.348
7. Lin SL, Chang DC, Chang-Lin S, Lin CH, Wu DT, Chen DT, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA (2008) 14:2115–24. doi:10.1261/rna.1162708
8. Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med (2000) 6:88–95.
9. Martin GR, Evans MJ. Differentiation of clonal lines of teratocarcinoma cells: formation of embryoid bodies in vitro. Proc Natl Acad Sci U S A (1975) 72:1441–5. doi:10.1073/pnas.72.4.1441
10. Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol (1985) 87:27–45.
11. Wrana JL, Attisano L. The Smad pathway. Cytokine Growth Factor Rev (2000) 11:5–13. doi:10.1016/S1359-6101(99)00024-6
12. Pera MF, Andrade J, Houssami S, Reubinoff B, Trounson A, Stanley EG, et al. Regulation of human embryonic stem cell differentiation by BMP-2 and its antagonist Noggin. J Cell Sci (2004) 117:1269–80. doi:10.1242/jcs.00970
13. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol (2009) 27:275–80. doi:10.1038/nbt.1529
14. Suzuki IK, Vanderhaeghen P. Is this a brain which I see before me? Modeling human neural development with pluripotent stem cells. Development (2015) 142:3138–50. doi:10.1242/dev.120568
15. Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature (2011) 480:547–51. doi:10.1038/nature10648
16. Arenas E, Denham M, Villaescusa JC. How to make a midbrain dopaminergic neuron. Development (2015) 142:1918–36. doi:10.1242/dev.097394
17. Kirkeby A, Grealish S, Wolf DA, Nelander J, Wood J, Lundblad M, et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep (2012) 1:703–14. doi:10.1016/j.celrep.2012.04.009
18. Doi D, Samata B, Katsukawa M, Kikuchi T, Morizane A, Ono Y, et al. Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Reports (2014) 2:337–50. doi:10.1016/j.stemcr.2014.01.013
19. Grealish S, Diguet E, Kirkeby A, Mattsson B, Heuer A, Bramoulle Y, et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell (2014) 15:653–65. doi:10.1016/j.stem.2014.09.017
20. Kikuchi T, Morizane A, Doi D, Magotani H, Onoe H, Hayashi T, et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinsons disease model. Nature (2017) 548:592–6. doi:10.1038/nature23664
21. Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A (2012) 109:12770–5. doi:10.1073/pnas.1202944109
22. Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry (2015) 20:361–8. doi:10.1038/mp.2014.22
23. Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature (2010) 463:1035–41. doi:10.1038/nature08797
24. Pfisterer U, Wood J, Nihlberg K, Hallgren O, Bjermer L, Westergren-Thorsson G, et al. Efficient induction of functional neurons from adult human fibroblasts. Cell Cycle (2011) 10:3311–6. doi:10.4161/cc.10.19.17584
25. Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, et al. Induction of human neuronal cells by defined transcription factors. Nature (2011) 476:220–3. doi:10.1038/nature10202
26. Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA, et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell (2011) 9:113–8. doi:10.1016/j.stem.2011.07.002
27. Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature (2011) 476:228–31. doi:10.1038/nature10323
28. Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature (2011) 476:224–7. doi:10.1038/nature10284
29. Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A (2011) 108:10343–8. doi:10.1073/pnas.1105135108
30. Son EY, Ichida JK, Wainger BJ, Toma JS, Rafuse VF, Woolf CJ, et al. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell (2011) 9:205–18. doi:10.1016/j.stem.2011.07.014
31. Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature (2012) 491:603–7. doi:10.1038/nature11557
32. Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, et al. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci U S A (2011) 108:7838–43. doi:10.1073/pnas.1103113108
33. Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T, et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell (2012) 10:473–9. doi:10.1016/j.stem.2012.03.003
34. Ring KL, Tong LM, Balestra ME, Javier R, Andrews-Zwilling Y, Li G, et al. Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell (2012) 11:100–9. doi:10.1016/j.stem.2012.05.018
35. Tian C, Ambroz RJ, Sun L, Wang Y, Ma K, Chen Q, et al. Direct conversion of dermal fibroblasts into neural progenitor cells by a novel cocktail of defined factors. Curr Mol Med (2012) 12:126–37. doi:10.2174/156652412798889018
36. Rivetti di Val Cervo P, Romanov RA, Spigolon G, Masini D, Martin-Montanez E, Toledo EM, et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat Biotechnol (2017) 35:444–52. doi:10.1038/nbt.3835
37. Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron (2013) 78:785–98. doi:10.1016/j.neuron.2013.05.029
38. Ho SM, Hartley BJ, Tcw J, Beaumont M, Stafford K, Slesinger PA, et al. Rapid NGN2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods (2016) 101:113–24. doi:10.1016/j.ymeth.2015.11.019
39. Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, et al. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci (2005) 8:288–96. doi:10.1038/nn1402
40. Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell (2008) 3:519–32. doi:10.1016/j.stem.2008.09.002
41. Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A (2013) 110:20284–9. doi:10.1073/pnas.1315710110
42. Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature (2011) 472:51–6. doi:10.1038/nature09941
43. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (2013) 501:373–9. doi:10.1038/nature12517
44. Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell (2016) 165:1238–54. doi:10.1016/j.cell.2016.04.032
45. Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods (2015) 12:671–8. doi:10.1038/nmeth.3415
46. Schwartz MP, Hou Z, Propson NE, Zhang J, Engstrom CJ, Santos Costa V, et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc Natl Acad Sci U S A (2015) 112:12516–21. doi:10.1073/pnas.1516645112
47. Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, et al. Guided self-organization and cortical plate formation in human brain organoids. Nat Biotechnol (2017) 35:659–66. doi:10.1038/nbt.3906
48. Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature (2017) 545:48–53. doi:10.1038/nature22047
49. Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A (2015) 112:15672–7. doi:10.1073/pnas.1520760112
50. Boutin ME, Hoffman-Kim D. Application and assessment of optical clearing methods for imaging of tissue-engineered neural stem cell spheres. Tissue Eng Part C Methods (2015) 21:292–302. doi:10.1089/ten.tec.2014.0296
51. Kelava I, Lancaster MA. Stem cell models of human brain development. Cell Stem Cell (2016) 18:736–48. doi:10.1016/j.stem.2016.05.022
52. Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science (2008) 321:960–4. doi:10.1126/science.1159689
53. Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature (2010) 468:67–71. doi:10.1038/nature09523
54. Koonin EV, Krupovic M. Evolution of adaptive immunity from transposable elements combined with innate immune systems. Nat Rev Genet (2015) 16:184–92. doi:10.1038/nrg3859
55. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337:816–21. doi:10.1126/science.1225829
56. Zetsche B, Gootenberg JS, Abudayyeh OO I, Slaymaker M, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell (2015) 163:759–71. doi:10.1016/j.cell.2015.09.038
57. Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, et al. Discovery and functional characterization of diverse class 2 CRISPR–Cas systems. Mol Cell (2015) 60:385–97. doi:10.1016/j.molcel.2015.10.008
58. Hoffman GE, Hartley BJ, Flaherty E, Ladran I, Gochman P, Ruderfer D, et al. Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with signatures from post mortem adult brains. Nat Commun (2017) 8:2225. doi:10.1038/s41467-017-02330-5
59. Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology (2009) 155:733–40. doi:10.1099/mic.0.023960-0
60. Shah SA, Erdmann S, Mojica FJ, Garrett RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol (2013) 10:891–9. doi:10.4161/rna.23764
61. Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lecrivain AL, Bzdrenga J, et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR–Cas systems. Nucleic Acids Res (2014) 42:2577–90. doi:10.1093/nar/gkt1074
62. Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem (2016) 85:227–64. doi:10.1146/annurev-biochem-060815-014607
63. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol (2013) 31:833–8. doi:10.1038/nbt.2675
64. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell (2013) 154:1380–9. doi:10.1016/j.cell.2013.08.021
65. Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, et al. Efficient genome modification by CRISPR–Cas9 nickase with minimal off-target effects. Nat Methods (2014) 11:399–402. doi:10.1038/nmeth.2857
66. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell (2013) 152:1173–83. doi:10.1016/j.cell.2013.02.022
67. Kiani S, Chavez A, Tuttle M, Hall RN, Chari R, Ter-Ovanesyan D, et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat Methods (2015) 12:1051–4. doi:10.1038/nmeth.3580
68. Vojta A, Dobrinic P, Tadic V, Bockor L, Korac P, Julg B, et al. Repurposing the CRISPR–Cas9 system for targeted DNA methylation. Nucleic Acids Res (2016) 44:5615–28. doi:10.1093/nar/gkw159
69. Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell (2016) 167(233–47):e17. doi:10.1016/j.cell.2016.08.056
70. Powell SK, Gregory J, Akbarian S, Brennand KJ. Application of CRISPR/Cas9 to the study of brain development and neuropsychiatric disease. Mol Cell Neurosci (2017) 82:157–66. doi:10.1016/j.mcn.2017.05.007
71. Muffat J, Li Y, Jaenisch R. CNS disease models with human pluripotent stem cells in the CRISPR age. Curr Opin Cell Biol (2016) 43:96–103. doi:10.1016/j.ceb.2016.10.001
72. Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, et al. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature (2016) 533:95–9. doi:10.1038/nature17939
73. Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep (2015) 13:234–41. doi:10.1016/j.celrep.2015.08.084
74. Wang P, Lin M, Pedrosa E, Hrabovsky A, Zhang Z, Guo W, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol Autism (2015) 6:55. doi:10.1186/s13229-015-0048-6
75. Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism (2017) 8:11. doi:10.1186/s13229-017-0124-1
76. Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature (2012) 485:242–5. doi:10.1038/nature11011
77. O’Roak BJ, Vives L, Fu W, Egertson JD I, Stanaway B I, Phelps G, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (2012) 338:1619–22. doi:10.1126/science.1227764
78. O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature (2012) 485:246–50. doi:10.1038/nature10989
79. Ho SM, Hartley BJ, Flaherty E, Rajarajan P, Abdelaal R, Obiorah I, et al. Evaluating synthetic activation and repression of neuropsychiatric-related genes in hiPSC-derived NPCs, neurons, and astrocytes. Stem Cell Reports (2017) 9:615–28. doi:10.1016/j.stemcr.2017.06.012
80. Heman-Ackah SM, Bassett AR, Wood MJ. Precision modulation of neurodegenerative disease-related gene expression in human iPSC-derived neurons. Sci Rep (2016) 6:28420. doi:10.1038/srep28420
82. Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511:421–7. doi:10.1038/nature13595
83. Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet (2017) 49:27–35. doi:10.1038/ng.3725
84. Scambler PJ. The 22q11 deletion syndromes. Hum Mol Genet (2000) 9:2421–6. doi:10.1093/hmg/9.16.2421
85. Rees E, Kirov G, Sanders A, Walters JT, Chambert KD, Shi J, et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry (2014) 19:37–40. doi:10.1038/mp.2013.156
86. Norkett EM, Lincoln SH, Gonzalez-Heydrich J, D’Angelo EJ. Social cognitive impairment in 22q11 deletion syndrome: a review. Psychiatry Res (2017) 253:99–106. doi:10.1016/j.psychres.2017.01.103
87. Lin M, Pedrosa E, Hrabovsky A, Chen J, Puliafito BR, Gilbert SR, et al. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst Biol (2016) 10:105. doi:10.1186/s12918-016-0366-0
88. Zhao D, Lin M, Chen J, Pedrosa E, Hrabovsky A, Fourcade HM, et al. MicroRNA profiling of neurons generated using induced pluripotent stem cells derived from patients with schizophrenia and schizoaffective disorder, and 22q11.2 Del. PLoS One (2015) 10:e0132387. doi:10.1371/journal.pone.0132387
89. Toyoshima M, Akamatsu W, Okada Y, Ohnishi T, Balan S, Hisano Y, et al. Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl Psychiatry (2016) 6:e934. doi:10.1038/tp.2016.206
90. Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron (2014) 81:306–13. doi:10.1016/j.neuron.2013.10.053
91. Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet (2000) 9:1415–23. doi:10.1093/hmg/9.9.1415
92. St. Clair D, Blackwood D, Muir W, Walker M, St. Clair D, Muir W, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet (1990) 336:13–6. doi:10.1016/0140-6736(90)91520-K
93. Blackwood DHR, Fordyce A, Walker MT, St. Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders—cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet (2001) 69:428–33. doi:10.1086/321969
94. Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, et al. Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell (2007) 130:1146–58. doi:10.1016/j.cell.2007.07.010
95. Ozeki Y, Tomoda T, Kleiderlein J, Kamiya A, Bord L, Fujii K, et al. Disrupted-in-schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci U S A (2003) 100:289–94. doi:10.1073/pnas.0136913100
96. Miyoshi K, Honda A, Baba K, Taniguchi M, Oono K, Fujita T, et al. Disrupted-in-schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry (2003) 8:685–94. doi:10.1038/sj.mp.4001352
97. Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, et al. Disrupted in schizophrenia 1 interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry (2007) 12:74–86. doi:10.1038/sj.mp.4001880
98. Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell (2009) 136:1017–31. doi:10.1016/j.cell.2008.12.044
99. De Rienzo G, Bishop JA, Mao Y, Pan L, Ma TP, Moens CB, et al. Disc1 regulates both beta-catenin-mediated and noncanonical Wnt signaling during vertebrate embryogenesis. FASEB J (2011) 25:4184–97. doi:10.1096/fj.11-186239
100. Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature (2014) 515:414–8. doi:10.1038/nature13716
101. Kassan A, Egawa J, Zhang Z, Almenar-Queralt A, Nguyen QM, Lajevardi Y, et al. Caveolin-1 regulation of disrupted-in-schizophrenia-1 as a potential therapeutic target for schizophrenia. J Neurophysiol (2017) 117:436–44. doi:10.1152/jn.00481.2016
102. Brandon NJ, Handford EJ, Schurov I, Rain JC, Pelling M, Duran-Jimeniz B, et al. Disrupted in Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Mol Cell Neurosci (2004) 25:42–55. doi:10.1016/j.mcn.2003.09.009
103. Ye F, Kang E, Yu C, Qian X, Jacob F, Yu C, et al. DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron (2017) 96:1041–54. doi:10.1016/j.neuron.2017.11.034
104. Srikanth P, Han K, Callahan DG, Makovkina E, Muratore CR, Lalli MA, et al. Genomic DISC1 disruption in hiPSCs alters Wnt signaling and Neural Cell Fate. Cell Rep (2015) 12:1414–29. doi:10.1016/j.celrep.2015.07.061
105. Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature (2011) 473:221–5. doi:10.1038/nature09915
106. Asada M, Mizutani S, Takagi M, Suzuki H. Antipsychotics promote neural differentiation of human iPS cell-derived neural stem cells. Biochem Biophys Res Commun (2016) 480:615–21. doi:10.1016/j.bbrc.2016.10.102
107. Paulsen Bda S, de Moraes Maciel R, Galina A, Souza da Silveira M, dos Santos Souza C, Drummond H, et al. Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplant (2012) 21:1547–59. doi:10.3727/096368911X600957
108. Paulsen Bda S, Cardoso SC, Stelling MP, Cadilhe DV, Rehen SK. Valproate reverts zinc and potassium imbalance in schizophrenia-derived reprogrammed cells. Schizophr Res (2014) 154:30–5. doi:10.1016/j.schres.2014.02.007
109. Nakazawa T, Kikuchi M, Ishikawa M, Yamamori H, Nagayasu K, Matsumoto T, et al. Differential gene expression profiles in neurons generated from lymphoblastoid B-cell line-derived iPS cells from monozygotic twin cases with treatment-resistant schizophrenia and discordant responses to clozapine. Schizophr Res (2017) 181:75–82. doi:10.1016/j.schres.2016.10.012
110. Maschietto M, Tahira AC, Puga R, Lima L, Mariani D, Paulsen Bda S, et al. Co-expression network of neural-differentiation genes shows specific pattern in schizophrenia. BMC Med Genomics (2015) 8:23. doi:10.1186/s12920-015-0098-9
111. Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol (1999) 56:33–9. doi:10.1001/archneur.56.1.33
112. Barone P, Antonini A, Colosimo C, Marconi R, Morgante L, Avarello TP, et al. The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord (2009) 24:1641–9. doi:10.1002/mds.22643
113. Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci (2010) 11:791–7. doi:10.1038/nrn2935
114. Cookson MR, Bandmann O. Parkinson’s disease: insights from pathways. Hum Mol Genet (2010) 19:R21–7. doi:10.1093/hmg/ddq167
115. Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell (2011) 8:267–80. doi:10.1016/j.stem.2011.01.013
116. Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med (2012) 4:141ra90. doi:10.1126/scitranslmed.3003985
117. Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis (2014) 62:381–6. doi:10.1016/j.nbd.2013.10.013
118. Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet (2013) 22:4545–61. doi:10.1093/hmg/ddt301
119. Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, et al. Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell (2016) 19:709–24. doi:10.1016/j.stem.2016.08.002
120. Schwab AJ, Sison SL, Meade MR, Broniowska KA, Corbett JA, Ebert AD. Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Reports (2017) 9(6):1839–52. doi:10.1016/j.stemcr.2017.10.010
121. Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med (2012) 4:380–95. doi:10.1002/emmm.201200215
122. Qing X, Walter J, Jarazo J, Arias-Fuenzalida J, Hillje AL, Schwamborn JC. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and alpha-synuclein modulation in dopaminergic neurons. Stem Cell Res (2017) 24:44–50. doi:10.1016/j.scr.2017.08.013
123. Fernandez-Santiago R, Carballo-Carbajal I, Castellano G, Torrent R, Richaud Y, Sanchez-Danes A, et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med (2015) 7:1529–46. doi:10.15252/emmm.201505439
124. Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell (2013) 12:354–67. doi:10.1016/j.stem.2013.01.008
125. Schwab AJ, Ebert AD. Neurite aggregation and calcium dysfunction in iPSC-derived sensory neurons with Parkinson’s disease-related LRRK2 G2019S mutation. Stem Cell Reports (2015) 5:1039–52. doi:10.1016/j.stemcr.2015.11.004
126. Lopez de Maturana R, Lang V, Zubiarrain A, Sousa A, Vazquez N, Gorostidi A, et al. Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J Neuroinflammation (2016) 13:295. doi:10.1186/s12974-016-0761-x
127. Son MY, Sim H, Son YS, Jung KB, Lee MO, Oh JH, et al. Distinctive genomic signature of neural and intestinal organoids from familial Parkinson’s disease patient-derived induced pluripotent stem cells. Neuropathol Appl Neurobiol (2017) 43:584–603. doi:10.1111/nan.12396
128. Ma D, Ng EY, Zeng L, Lim CY, Zhao Y, Tan EK. Development of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient carrying the N551K variant in LRRK2 gene. Stem Cell Res (2017) 18:51–3. doi:10.1016/j.scr.2016.12.013
129. Ma D, Ng SH, Zeng L, Zhao Y, Tan EK. Generation of a human induced pluripotent stem cell (iPSC) line carrying the Parkinson’s disease linked LRRK2 variant S1647T. Stem Cell Res (2017) 18:54–6. doi:10.1016/j.scr.2016.12.010
130. Ma D, Tio M, Ng SH, Li Z, Lim CY, Zhao Y, et al. Derivation of human induced pluripotent stem cell (iPSC) line with LRRK2 gene R1398H variant in Parkinson’s disease. Stem Cell Res (2017) 18:48–50. doi:10.1016/j.scr.2016.12.014
131. Ma D, Zhou W, Ng EY, Zeng L, Zhao Y, Tan EK. Reprogramming of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient with a R1628P variant in the LRRK2 gene. Stem Cell Res (2017) 18:45–7. doi:10.1016/j.scr.2016.12.011
132. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science (2004) 304:1158–60. doi:10.1126/science.1096284
133. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature (1998) 392:605–8. doi:10.1038/33416
134. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol (2010) 8:e1000298. doi:10.1371/journal.pbio.1000298
135. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A (2010) 107:378–83. doi:10.1073/pnas.0911187107
136. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol (2010) 191:1367–80. doi:10.1083/jcb.201007013
137. Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP, Lohmann K, et al. Mutations in PINK1 and Parkin impair ubiquitination of mitofusins in human fibroblasts. PLoS One (2011) 6:e16746. doi:10.1371/journal.pone.0016746
138. Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, et al. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun (2012) 3:668. doi:10.1038/ncomms1669
139. Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain (2012) 5:35. doi:10.1186/1756-6606-5-35
140. Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Reports (2016) 7:664–77. doi:10.1016/j.stemcr.2016.08.012
141. Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci (2011) 31:5970–6. doi:10.1523/JNEUROSCI.4441-10.2011
142. Rakovic A, Shurkewitsch K, Seibler P, Grunewald A, Zanon A, Hagenah J, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem (2013) 288:2223–37. doi:10.1074/jbc.M112.391680
143. Shaltouki A, Sivapatham R, Pei Y, Gerencser AA, Momcilovic O, Rao MS, et al. Mitochondrial alterations by Parkin in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Reports (2015) 4:847–59. doi:10.1016/j.stemcr.2015.02.019
144. Aboud AA, Tidball AM, Kumar KK, Neely MD, Ess KC, Erikson KM, et al. Genetic risk for Parkinson’s disease correlates with alterations in neuronal manganese sensitivity between two human subjects. Neurotoxicology (2012) 33:1443–9. doi:10.1016/j.neuro.2012.10.009
145. Aboud AA, Tidball AM, Kumar KK, Neely MD, Han B, Ess KC, et al. PARK2 patient neuroprogenitors show increased mitochondrial sensitivity to copper. Neurobiol Dis (2015) 73:204–12. doi:10.1016/j.nbd.2014.10.002
146. Ren Y, Jiang H, Hu Z, Fan K, Wang J, Janoschka S, et al. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells (2015) 33:68–78. doi:10.1002/stem.1854
147. Cartelli D, Amadeo A, Calogero AM, Casagrande FVM, De Gregorio C, Gioria M, et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol Aging (2018) 61:66–74. doi:10.1016/j.neurobiolaging.2017.09.010
148. Song P, Trajkovic K, Tsunemi T, Krainc D. Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J Neurosci (2016) 36:2425–37. doi:10.1523/JNEUROSCI.2569-15.2016
149. Gautier CA, Erpapazoglou Z, Mouton-Liger F, Muriel MP, Cormier F, Bigou S, et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet (2016) 25:2972–84. doi:10.1093/hmg/ddw148
150. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature (1997) 388:839–40. doi:10.1038/42166
151. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science (1997) 276:2045–7. doi:10.1126/science.276.5321.2045
152. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet (2009) 41:1303–7. doi:10.1038/ng.485
153. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet (2009) 41:1308–12. doi:10.1038/ng.487
154. Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell (2011) 146:318–31. doi:10.1016/j.cell.2011.06.019
155. Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science (2013) 342:983–7. doi:10.1126/science.1245296
156. Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell (2013) 155:1351–64. doi:10.1016/j.cell.2013.11.009
157. Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, et al. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun (2015) 6:7314. doi:10.1038/ncomms9008
158. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science (2003) 302:841. doi:10.1126/science.1090278
159. Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun (2011) 2:440. doi:10.1038/ncomms1453
160. Byers B, Cord B, Nguyen HN, Schule B, Fenno L, Lee PC, et al. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One (2011) 6:e26159. doi:10.1371/journal.pone.0026159
161. Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. Alpha-synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A (2016) 113:1931–6. doi:10.1073/pnas.1520335113
162. Oliveira LM, Falomir-Lockhart LJ, Botelho MG, Lin KH, Wales P, Koch JC, et al. Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis (2015) 6:e1994. doi:10.1038/cddis.2015.318
163. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi:10.1016/j.cell.2013.05.039
164. Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell (2013) 13:691–705. doi:10.1016/j.stem.2013.11.006
165. Johnson MA, Weick JP, Pearce RA, Zhang SC. Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci (2007) 27:3069–77. doi:10.1523/JNEUROSCI.4562-06.2007
166. Rushton DJ, Mattis VB, Svendsen CN, Allen ND, Kemp PJ. Stimulation of GABA-induced Ca2+ influx enhances maturation of human induced pluripotent stem cell-derived neurons. PLoS One (2013) 8:e81031. doi:10.1371/journal.pone.0081031
167. Tang X, Zhou L, Wagner AM, Marchetto MC, Muotri AR, Gage FH, et al. Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem Cell Res (2013) 11:743–57. doi:10.1016/j.scr.2013.05.002
168. Studer L, Vera E, Cornacchia D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell (2015) 16:591–600. doi:10.1016/j.stem.2015.05.004
169. Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet (2018) 50:54–61. doi:10.1038/s41588-017-0005-8
170. Stein JL, de la Torre-Ubieta L, Tian Y, Parikshak NN, Hernandez IA, Marchetto MC, et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron (2014) 83:69–86. doi:10.1016/j.neuron.2014.05.035
171. McKernan R, Watt FM. What is the point of large-scale collections of human induced pluripotent stem cells? Nat Biotechnol (2013) 31:875–7. doi:10.1038/nbt1213-1148c
172. Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods (2015) 12:885–92. doi:10.1038/nmeth.3507
173. Amin H, Maccione A, Marinaro F, Zordan S, Nieus T, Berdondini L. Electrical responses and spontaneous activity of human iPS-derived neuronal networks characterized for 3-month culture with 4096-electrode arrays. Front Neurosci (2016) 10:121. doi:10.3389/fnins.2016.00121
174. Nadadhur AG, Emperador Melero J, Meijer M, Schut D, Jacobs G, Li KW, et al. Multi-level characterization of balanced inhibitory-excitatory cortical neuron network derived from human pluripotent stem cells. PLoS One (2017) 12:e0178533. doi:10.1371/journal.pone.0178533
175. Kuijlaars J, Oyelami T, Diels A, Rohrbacher J, Versweyveld S, Meneghello G, et al. Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Sci Rep (2016) 6:36529. doi:10.1038/srep36529
176. Tong Z, Solanki A, Hamilos A, Levy O, Wen K, Yin X, et al. Application of biomaterials to advance induced pluripotent stem cell research and therapy. EMBO J (2015) 34:987–1008. doi:10.15252/embj.201490756
177. Adil MM, Rodrigues GM, Kulkarni RU, Rao AT, Chernavsky NE, Miller EW, et al. Efficient generation of hPSC-derived midbrain dopaminergic neurons in a fully defined, scalable, 3D biomaterial platform. Sci Rep (2017) 7:40573. doi:10.1038/srep40573
178. Zhang Z, Zhang Y, Gao F, Han S, Cheah KS, Tse HF, et al. CRISPR/Cas9 genome-editing system in human stem cells: current status and future prospects. Mol Ther Nucleic Acids (2017) 9:230–41. doi:10.1016/j.omtn.2017.09.009
179. Han X, Liu Z, Jo MC, Zhang K, Li Y, Zeng Z, et al. CRISPR–Cas9 delivery to hard-to-transfect cells via membrane deformation. Sci Adv (2015) 1:e1500454. doi:10.1126/sciadv.1500454
180. Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res (2014) 24:1020–7. doi:10.1101/gr.171264.113
181. Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry (2013) 18:1193–8. doi:10.1038/mp.2013.111
182. Donegan JJ, Tyson JA, Branch SY, Beckstead MJ, Anderson SA, Lodge DJ. Stem cell-derived interneuron transplants as a treatment for schizophrenia: preclinical validation in a rodent model. Mol Psychiatry (2017) 22:1492–501. doi:10.1038/mp.2016.121
183. Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Paskewitz S, et al. Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci U S A (2014) 111:7450–5. doi:10.1073/pnas.1316488111
184. Michelsen KA, Acosta-Verdugo S, Benoit-Marand M, Espuny-Camacho I, Gaspard N, Saha B, et al. Area-specific reestablishment of damaged circuits in the adult cerebral cortex by cortical neurons derived from mouse embryonic stem cells. Neuron (2015) 85:982–97. doi:10.1016/j.neuron.2015.02.001
Keywords: induced pluripotent stem cells, schizophrenia, Parkinson’s disease, neural induction, neural differentiation
Citation: LaMarca EA, Powell SK, Akbarian S and Brennand KJ (2018) Modeling Neuropsychiatric and Neurodegenerative Diseases With Induced Pluripotent Stem Cells. Front. Pediatr. 6:82. doi: 10.3389/fped.2018.00082
Received: 09 January 2018; Accepted: 15 March 2018;
Published: 03 April 2018
Edited by:
Sofia Beatriz Lizarraga, University of South Carolina, United StatesReviewed by:
Maria Lehtinen, Harvard University, United StatesAditi Deshpande, University of California, San Francisco, United States
Copyright: © 2018 LaMarca, Powell, Akbarian and Brennand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristen J. Brennand, a3Jpc3Rlbi5icmVubmFuZEBtc3NtLmVkdQ==