 Alexander Weigert
Alexander Weigert Bernd Hoppe
Bernd Hoppe- Division of Pediatric Nephrology, Department of Pediatrics, University Children’s Hospital, Bonn, Germany
Nephrolithiasis, urolithiasis, and nephrocalcinosis (NC) have become common causes of hospitalization and referral to pediatric outpatient clinics. It is of utmost importance to start with diagnostic evaluation directly after the first passage of a kidney stone, or if NC is diagnosed, in each pediatric patient. This is necessary, as in about 80% of children a metabolic reason for stone disease is detected. Current treatment options are scarce and mainly include general measures like an increased fluid intake or elevating the solubility of a lithogenic substance. According to the given lithogenic risk factor(s), specific treatment options are available and are being summarized in this review. Furthermore, an outlook on potential future treatment options, including innovative strategies such as mRNA-based or recombinant enzyme substitution therapy, is given.
Introduction
Nephrolithiasis (NL) and urolithiasis (UL) describe solid stones appearing in the kidney (NL) or in the lower urinary tract (UL). The term nephrocalcinosis (NC) expresses deposits of calcium salts within the renal tubules, the tubular epithelium, and/or the interstitium (1). NC is also classified ultrasonographically due to the anatomic area involved: cortical and diffuse NC or medullary NC, respectively, the latter subdivided according to the degree of echogenicity as medullary NC grade I-III (2). All three entities have become common causes of hospitalization and presentation in pediatric outpatient clinics (3). Although exact numbers for prevalence and incidence rates are still not known, it is said, that numbers increase in pediatrics, as it was shown in the adult population. For example, in the US, the incidence of NL is estimated to be 36–57 per 100,000 population (4). Since in up to 40% of children the diagnosis is made incidentally (for example, after a first or recurrent urinary tract infection) due to the high proportion of unspecific symptoms, the accurate incidence might be underestimated.
Nephrolithiasis affects children of all ages. During the first decade of life, boys are more frequently prone to develop NL, while girls are more frequently observed to develop kidney stones in the second decade of life (5). Clinical presentation is highly variable and depends on the age of affected children, e.g., failure to thrive in infants, or typical flank pain in the older child/adolescent patient (6). If present, newly diagnosed microhematuria or macrohematuria can be the first clinical sign for NL.
Decision-making on treatment regimens should be based on thorough evaluation of the underlying risk factors, as metabolic disorders are found in ~80% of children with kidney stones/NC (7). Diagnostic evaluation thus includes examination of metabolic disorders leading to elevated urinary excretion of a lithogenic factor, or to decreased excretion of an anti-lithogenic substance, but also anatomical abnormalities, urinary tract infections and prematurity, insufficient fluid uptake (8), and, with increasing importance, obesity (9). The 24-h urine is the most important tool in diagnostic workup of NL/NC, since blood examination usually remains unremarkable.
Overall, treatment options are scarce and include general measures such as increased fluid intake and a balanced diet with avoidance of excessive sodium intake. According to the given lithogenic risk factor(s), specific treatment options are already available. However, the current armament to treat the (pediatric) patient with a metabolic disease, thereby resulting in severe risk of kidney stone development or progressive NC, is minor.
General Risk Factors
Dietary excesses, e.g., for oxalate containing food, or simply as hypercaloric diet (“metabolic syndrome”), may also influence the development of NL. While increased sodium intake can lead to an increased urinary calcium excretion, low calcium diet might promote increased intestinal oxalate absorption and thus resulting in secondary hyperoxaluria (10, 11).
General Measures
Fluid and Diet
A MUST for all stone patients is a high fluid intake: more than 1.5–2 L × 1.73 m2 body surface area per day, distributed over the entire day, is recommended. This prevents peaks of high concentration levels of a given soluble. However, dietary recommendations should be handled carefully. Excessive dietary sodium uptake has to be avoided, as it promotes calcium excretion (12). Calcium restriction can lead to an increased intestinal oxalate absorption and hence urinary oxalate excretion. Because of that and since calcium restriction might lead to low bone mineral density, this is an obsolete procedure in patients with hypercalciuria (13). Protein restriction, as well as excessive protein uptake, should of course be avoided, as it leads to hypercalciuria and hypocitraturia, due to an increased acid load. Keep in mind (in the pediatric patient) that restriction of protein includes the risk of growth retardation! A diet including the risk of metabolic syndrome (high fat and fructose intake) showed an increased risk of NL in epidemiologic studies (14). Vegetables and fruits provide a good source of citrate and potassium (both stone inhibitors) and by that decrease the risk of NL (15). Summing up, children with the risk of NL should stick to a normal and balanced diet.
Crystallization Inhibitors
Citrate and magnesium effectively increase urinary solubility especially of cystine, calcium oxalate and uric acid at given urinary pH levels (uric acid and calcium oxalate > 6.2 < 7.4, cysteine >8). Citrate is metabolized in the liver into bicarbonate, which elevates urinary pH, resulting in a reduced tubular citrate reabsorption. Urinary citrate reduces calcium excretion by 30% and it binds to urinary calcium forming a soluble complex, which reduces the precipitation of calcium with other lithogenic substances (16). Citrate is best delivered as potassium citrate or as sodium potassium citrate. The adequate increase of citrate excretion can be achieved by a dosage of 0.1–0.2 g/kg body weight (0.3–0.6 mmol/kg). Patients with distal RTA usually require higher dosages (0.2–0.3 g/kg body weight), and the dosage has to be adapted to urine pH (17). Since very high urinary pH levels promote calcium phosphate precipitation, urine alkalization therapy has to be monitored!
Hypercalciuria
The most common risk factor for NL in childhood is hypercalciuria (18), which can arise from idiopathic hypercalciuria (19), a multifactorial disease, as well as from genetic disorders (7) or other underlying diseases, such as (primary) hyperparathyroidism.
An example for a genetic reason (for more information, see Table S1 in Supplementary Material) is autosomal dominant hypocalcemic hypercalciuria (ADHH), which is caused by mutations in the calcium-sensing receptor (CaSR) gene, and genes connected to that receptor pathway (Gα11). Electrolytes in these patients show elevated serum phosphate along with low serum calcium and magnesium (20). ADHH is associated with calcification of brain and kidney and with cataract. Other genetic diseases resulting in hypercalciuria is familial hypomagnesemia hypercalciuria and nephrocalcinosis syndrome or are the different types of Dent Disease (Dent 1 and 2). In the latter, a diagnostic hint can be male gender (x-chromosomal recessive) accompanied by low molecular weight proteinuria and severe hypercalciuria (21).
Secondary hypercalciuria can result from medication (e.g., furosemide, vitamin D) or parenteral nutrition [higher daily intake of protein, sodium, phosphorus, and ascorbic acids, leading to significantly increased urinary calcium and oxalate, but lowish citrate excretion and hence urinary calcium oxalate supersaturation (22)].
Current Treatment
When severe hypercalciuria is present, thiazides reduce the urinary calcium excretion, since they increase calcium reabsorption in the distal and proximal tubule (23). Thiazides also are capable of encountering a reduced bone density in patients with hypercalciuria (24). Daily dosage is 0.5–1 mg/kg body weight, twice a day. Side effects include hypotension and hypokalemia. In case of hypokalemia, amiloride (a potassium-sparing and calcium-lowering diuretic) should be added (25).
Future Treatment Options
Experimental (Animal) Studies
In knock-in mutant mice of the calcium-sensing receptor (CaSR), which mimics ADHH, calcilytic therapy has been shown to reduce urinary calcium excretion, which was able to prevent renal calcification. Calcilytics are CaSR antagonists capable of improving serum calcium and phosphate, due to stimulating PTH secretion (26).
In Dent disease, first experience with bone marrow transplantation in Clcn5 knockout mice, a model for Dent disease, showed an improvement of protein-, calci-, and glucosuria as well as reduced polyuria. This most likely results from bone marrow-derived cells engaged in kidney interstitium (27).
Recently, Rendu et al. were able to restore normal mRNA and protein levels in fibroblasts of a patient with a deep intronic mutation in the OCRL gene (Lowe syndrome, Dent II), showing RNA-based [mRNA or RNA interference (RNAi)] therapy might be a future approach in these patients (28).
Case Series
For patients with CYP24A1 mutations, leading to idiopathic infantile hypercalcemia (IIH, Table S1 in Supplementary Material), single reports of treatment with rifampicin have been published. Rifampicin acts as a potent inductor of CYP3A4, an inactivator of many xenobiotics, and it was shown that it is capable of reducing urinary calcium excretion, serum calcium levels, and 1,25 (OH)2 D3 (29).
Hyperoxaluria
There are three types of genetic hyperoxaluria: type I (PH I), secondary to mutation in the alanine/glyoxylate aminotransferase (AGT), is characterized through increased urinary excretion of oxalate and glycolate (30), type II (PH II), secondary to mutation in the glyoxylate/hydroxypyruvate reductase (GRHPR), is characterized by elevated oxalate and l-glyceric acid urinary excretion (31) and type III (PH III), secondary to mutation in the 4-hydroxy-2-oxoglutarate aldolase (HOGA 1), characterized by raised urinary excretion of oxalate and hydroxy-oxo-glutarate and/or hydroxy-oxo-glutamate (32). Secondary hyperoxaluria results from an increased intestinal oxalate uptake, due to malabsorptive states such as short bowel disease or chronic inflammatory bowel disease, by increased dietary oxalate intake, or lack of intestinal oxalate degrading bacteria (33).
Current Treatment
There are no approved drugs for the treatment of primary hyperoxaluria (PH). Since most of the oxalate is produced endogenously by the liver, an oxalate-restricted diet is often of limited use to these patients. Treatment in supra-physiological doses (5–20 mg/kg body weight per day) of pyridoxal-phosphate (vitamin B6), as the cofactor for the defective enzyme AGT in type I PH, reduces the endogenous oxalate production and the urinary excretion in about one-third of all PH I patients, especially those with missense mutations (34). Known side effects are polyneuropathy, acne and bullous skin eruptions. For all other PH I and those patients with PH types II and III the current measures, high fluid intake and citrate medication are the only further treatment possibilities.
Patients with PH (I) and end-stage renal failure (ESRF) should be transplanted as soon as possible since no renal replacement therapy is able to remove oxalate adequately. In PH I, combined liver and kidney, or two-timed transplantation, e.g., kidney after liver, are recommended based on the systemic oxalate burden of the patient. Patients with PH II shall only receive kidney transplantation since the defect enzyme is ubiquitous (7). However, just recently a combined liver–kidney transplantation was reported in a PH II patient (35). In PH III only, one patient with ESRF was so far reported (PH III seems to be the mildest and therefore easiest to handle type of PH); hence, transplant strategies were not yet established (36).
Future Treatment Options
Experimental (Animal) Studies
In hyperoxaluric mice, the TNF receptor inhibitor R-7050 was able to delay the progression of NC. The TNFR signal pathway seems to be essential in the adhesion of calcium oxalate crystals (see Figure 1) to the luminal membrane of renal tubules (37).
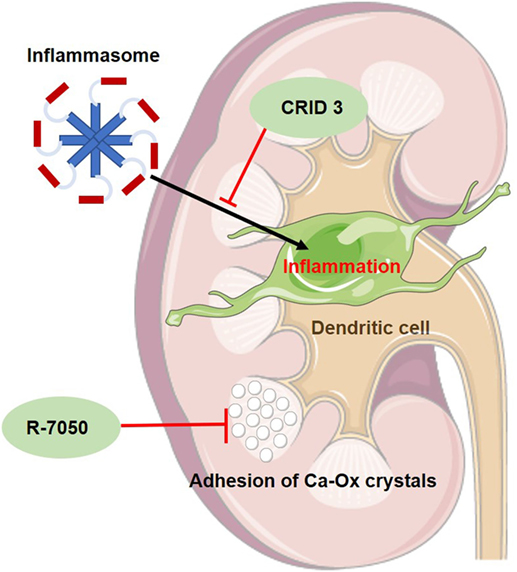
Figure 1. Schematic figure of the mode of action of two new experimental drugs for primary hyperoxaluria I. CRID 3 inhibits inflammasome-mediated inflammation in dendritic cells of the kidney and thus reducing kidney fibrosis. R-7050 is a TNF-receptor inhibitor, delaying the progression of nephrocalcinosis since it seems to prevent adhesion of calcium oxalate (Ca-Ox) crystals to renal tubules.
A currently identified and obviously major player in oxalate-induced kidney inflammation is the activation of the inflammasome pathway (38). The inflammasome is a protein complex, which, when activated, activates IL-1β and IL-18 production and thus promoting local inflammation (see Figure 1). In mice with crystal-induced kidney fibrosis, which was induced by an oxalate or adenine-rich diet, a specific inhibitor, CP-456,773 (or CRID3), of the NLRP3 inflammasome pathway was able to delay the progress of kidney fibrosis (39).
In a certain PH I mutation (P11LG170R allele), the functional AGT is mistargeted into mitochondria instead into peroxisomes. Dequalinium chloride (DECA) inhibits mitochondrial protein import into the mitochondrium and restores transportation of AGT into the peroxisome, where it regains its regular function (see Figure 2). This reduces oxalate accumulation, similar to pyridoxal phosphate and even has additive effects with that therapy (40). If other AGT variations lead to a mislocation of the enzyme and could, therefore, be applicable for DECA therapy, is not known yet.
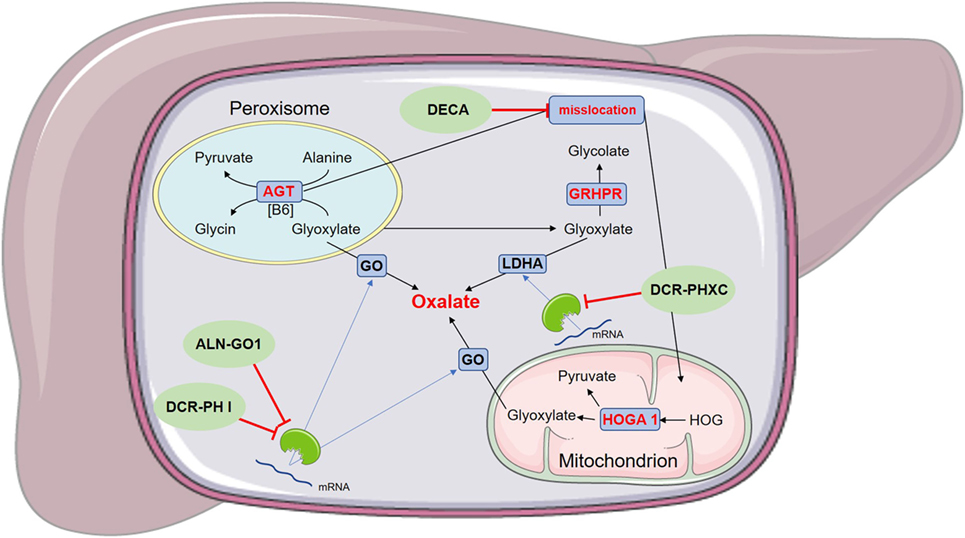
Figure 2. Schematic figure of the mode of action of experimental drugs for primary hyperoxaluria (PH) I. ALN-GO1 and DCR-PH I are RNA interference (RNAi)-based drugs, preventing the translation of glycolate oxidase (GO) and thus reducing endogenous oxalate production. DCR-PHXC as well is an RNAi-based drug targeting the liver-specific lactate dehydrogenase A (LDHA), also reducing endogenous oxalate production. Dequalinium chloride (DECA) prevents the misslocation of alanine:glyoxylate aminotransferase (AGT). This is only applicable for a certain PH I mutation (P11LG170R allele), since other mutations do not cause a misslocation of AGT.
Ongoing Human Clinical Trials
Oral therapy with Oxalobacter formigenes (Oxabact, Oxthera AB, Sweden), an anaerobic bacterium that degrades oxalate for its sole carbon source (41), promotes the removal of endogenously produced oxalate via the intestinal tract (see Figure 3). Activation of the intestinal oxalate transporter and a high concentration gradient from blood to intestinal lumen induces an oxalate shift with the possibility that oxalate can be metabolized by intestinal Oxalobacter. First, data showed conflicting results. A Phase I study led to a significant reduction in urinary oxalate excretion (42), whereas other studies over a longer period of time showed no significant results (43, 44). Since the interpersonal effect of O. formigenes seems to be variable, and especially depending on patient’s compliance, further studies according to the efficacy of O. formigenes are currently being conducted. Nevertheless, ad hoc interpretation of study results showed a positive effect on kidney function over time. In addition, all recent Oxalobacter trials made obvious that urinary oxalate excretion might not be the perfect endpoint for a treatment study in patients with PH. Therefore, a further study will evaluate a variety of parameters, mostly focusing on plasma oxalate follow-up and amelioration or prevention of systemic oxalate deposition. A study with PH I patients on maintenance hemodialysis is ongoing and preliminary results show improvement of plasma oxalate levels, as well of systemic oxalate burden of those patients being compliant.
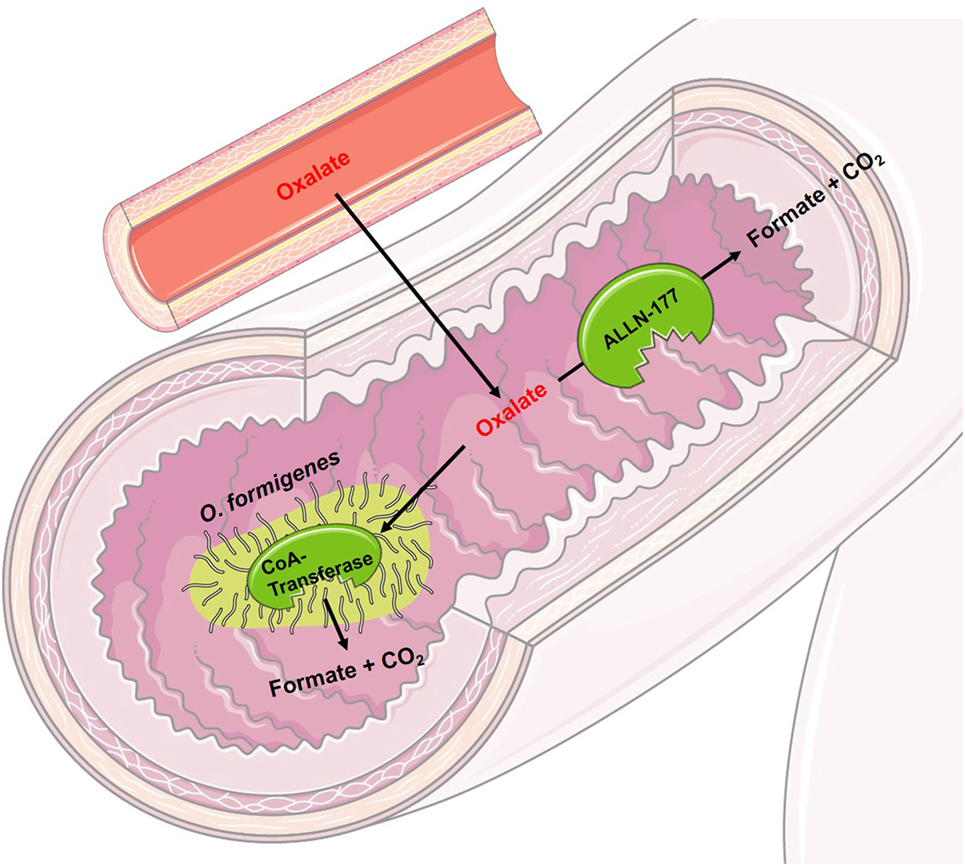
Figure 3. Schematic figure of the mode of operation of experimental drugs for primary hyperoxaluria I. Oxalobacter formigenes uses oxalate as its sole carbon source. Orally administered, it degrades intraluminal oxalate in the intestine. By a concentration gradient and through activation of the intestinal oxalate transporter, oxalate is transported from the blood into the intestinal lumen. ALLN-177 is a recombined, microbial oxalate decarboxylase leading to the same intraluminal effect as O. Formigenes, however, maybe unable to increase the shift of blood oxalate into the intestinal lumen, as it cannot directly activate the intestinal oxalate transporter.
ALLN-177 (Allena Pharmaceuticals, USA) is a recombinant, microbial enzymatic oxalate decarboxylase, which degrades oxalate in the gastrointestinal tract (see Figure 3). It has been shown that ALLN-177 is able to reduce the urinary oxalate excretion in healthy persons (45). If ALLN-177 can help patients with primary or secondary hyperoxaluria is currently under investigation. Degrading of dietary oxalate will obviously be possible; however, removal of endogenously produced oxalate via the intestinal tract might be tricky to achieve with such a medication. On overt, concentration gradient of oxalate (blood vs. intestinal tract) might lead to secretion of oxalate into the intestinal lumen; however, an activation of the intestinal oxalate transporter, as it is seen with Oxalobacter treatment, is still under debate.
Another therapeutic approach is the administration of ALN-GO1 (Alnylam Pharmaceuticals, USA), an investigational RNAi medication. RNAi function is based on small RNA molecules (small interfering RNA, siRNAi), which bind to cytoplasmatic enzymes and form a highly specific working complex, that decomposes mRNA and thus prevents the translation of that mRNA into the subsequent protein (46). ALN-GO1 targets the glycolate oxidase (GO) mRNA (see Figure 2), preventing the translation from mRNA into the working protein and thus reducing the development of glyoxylate and hence the production of oxalate. A study on animals showed a reduction of urinary oxalate excretion in mice and non-human primates by up to 98%, after multiple subcutaneous administrations (47). Initial results of a phase I Study of ALN-GO1, as presented at the 17th Congress of the International Pediatric Nephrology Association (IPNA), ALN-GO1 was able to silence up to 80% of the GO mRNA, without serious adverse events in healthy subjects (48). Preliminary results of the ongoing phase I/II study of ALN-GO1, presented at the American Society of Nephrology (ASN) annual meeting in 2017, showed a reduction of urinary oxalate excretion, up to 50%, in PH I patients without treatment-related serious adverse events (48).
Another RNAi, which was initially investigated in a phase I study, is DCR-PH1 (Dicerna Pharmaceuticals, USA). DCR-PH1 also prevents the translation of GO (see Figure 2). After having shown its capability of reducing urinary oxalate in animal models, both healthy volunteers and human patients with PH I are now being enrolled in a Phase II study (49), which was later interrupted. Dicerna Pharmaceuticals just recently applied for a phase I study of DCR-PHXC, another RNAi-based therapy, targeting the lactate dehydrogenase A (LDHA) (see Figure 2). In animal models, DCR-PHXC was able to silence the liver LDHA and thus preventing an excessive oxalate production (50). This medication would hence be able for treatment of patients with all types of PH.
Cystinuria
Cystinuria, one of the most frequent autosomal-recessive inherited genetic disorders (prevalence: 1:7,000), is responsible for about 5–10% of all pediatric kidney stones. A defective tubular reabsorption leads to an increased urinary excretion of the dibasic amino acids cystine, ornithine, lysine, and arginine, but only cystine is able to promote stone formation since the other acids are highly soluble in urine (51).
Current Treatment
The main goal of therapy for patients with cystinuria is urine alkalization. Cystine has a higher solubility at a pH above 8. Chelating agents such as d-penicillamine and alphamercaptopropionyl-glycine (MPG) destroy the bond between two cysteine molecules, which separately have a higher solubility than combined with cystine. Both are equally effective and should be administered with 20–40 mg/kg body weight (52). Side effects include rash, exanthema, arthralgia, thrombocytopenia, polymyositis, and nephritic syndrome. MPG seems to have fewer side effects than d-penicillamine (53), which furthermore reduces the level of pyridoxine and therefore has to be replaced during therapy. The ACE inhibitor captopril has a similar effect like MPG, but fewer side effects (54). A urinary cystine excretion above 720 mg/day justifies a treatment with 75–150 mg of captopril per day, although the effect is variable (55). Ascorbic acid in high doses (3–5 g/day) not only can decrease the urinary cystine excretion but can increase endogenous oxalate production and therefore urinary oxalate excretion, so no clear dosage recommendation can be given for that therapy (56). A careful protein reduced diet is recommended since the contained methionine is metabolized to cystine.
Future Treatment Options
Experimental (Animal) Studies
In cystinuria, several substances have been examined on their ability to inhibit cystine crystal growth. One of the first was l-cystine dimethyl ester (CDME), which was able to reduce cystine stone size, but not urinary cystine excretion in a knockout mouse model (57). The l-cystine diamides l-cystine bismorpholide and l-cystine bis (N′-methylpiperazide) were in vitro more powerful in increasing cystine solubility, than CDME. l-cystine bis (N′-methylpiperazide) has shown its ability to reduce stone formation in cystinuria knockout mice and thus providing a possible new treatment option in cystinuria (58).
Another substance that was able to increase cystine solubility in a knockout mouse model is α-lipoic acid. Zee et al. report a reduction of stone formation due to α-lipoic acid as nutritional supplementation (59). The effect of α-lipoic acid on kidney stone recurrence is now being evaluated in a human clinical trial (NCT02910531) (60).
Ongoing Human Clinical Trials
A currently recruiting phase II trial is investigating the safety and effectiveness of bucillamine (NCT02942420). Bucillamine is a drug developed from tioprinin, currently used as an antirheumatic agent and, acting as a Thiol donor, which might be capable of binding cysteine from urine and thus reducing the risk of stone formation (61).
A pilot study (NCT02538016) on the effect and safety of tolvaptan, a vasopressin antagonist, is also currently being conducted (62).
Purine Stones
Purine stones can result from hyperuricosuria secondary to tumor lysis syndrome, or rarer, from genetic defects (e.g., Lesch–Nyhan syndrome), or enzymatic defects such as 2,8-dihydroxadeninuria (63) or xanthinuria, which is based on xanthine oxidase deficiency (64).
Current Treatment (Uric Acid, 2,8-Dihydroxyadenine, Xanthine)
Uric acid stones can also be treated best with urine alkalinization. Urine pH should be kept above 6.5 and excessive protein intake should be avoided. In severe cases, Allopurinol, an inhibitor of xanthine oxidase, can be applied, since it reduces serum uric acid. Dosage must be carefully triggered since it can lead to relevant xanthinuria. Fluid intake, leading to a urinary output of at least 2–3 l per day is recommended (65). Allopurinol also can be used in 2,8-Dihydroxyadeninuria, as well as hyperhydration and dietary restriction of adenine and purine. If allopurinol is not applicable, e.g., due to allergic reactions, single cases with successive treatment with febuxostat have been reported (66) and clinical experience supports these reports.
Patients suffering from xanthinuria do not benefit from urine alkalization. Increased fluid uptake still remains the only effective therapeutic measure (64).
Infectious Stones
Urinary tract infections with urease-producing bacteria (e.g., proteus species) lead to the formation of struvite stones. The bacteria are capable of hydrolyzing ammonia into ammonium ions, resulting in an elevated urinary pH (67). This promotes the formation of carbonate ions and the production of trivalent phosphate ions, both major components of struvite stones.
Current Treatment
Children suffering from infectious stones must be treated with adequate antibiotics. Formatted stones require an extraction procedure. If the stone stays in situ, it provides an optimal nidus for bacterial growth and therefore increases the risk of re-infection.
Hypocitraturia
Hypocitraturia is known to be a common risk factor in preterm infants (68). In older children, it is most prevalent in some parts of the world (e.g., Turkey). It is also a characteristic finding in complete distal renal tubular acidosis (d-RTA) and can be found in patients with metabolic acidosis, hypokalemia, urinary tract infections, and malabsorption syndromes (69).
Current Treatment
Hypocitraturia can be encountered by giving potassium citrate (1 mEq/kg daily). This treatment successfully reduces stone reoccurrence (70). Since hypocitraturia is often accompanied by other abnormal urine findings, these must be treated according to their entity.
Renal Tubular Acidosis (RTA)
Renal tubular acidosis is characterized by an impaired H+ excretion, hypercalciuria, and hypocitraturia (17). This leads to early onset of NL and is accompanied by loss of hearing. Incomplete distal RTA causes kidney stones without clear acidosis being present.
Nephrolithiasis in children is seldom purely drug related (71), but certain drugs increase the risk of NL. Two pathomechanisms might lead to NL: drugs excreted by the kidney with poor solubility (e.g., indinavir, acyclovir, TMP or sulfadiazine) can either provide a direct nidus for stone formation (72) or increase the excretion of urinary lithogenic substances (e.g., furosemide induces hypercalciuria in preterm infants, carboanhydrase inhibitors result in hypocitraturia and hypercalciuria) (73).
Current Treatment
The continuous supplementation of alkali is the only therapeutic approach in all forms of RTA. This can be achieved by administration of sodium and potassium bicarbonate, or citrate salts. Therapeutic goal is a serum bicarbonate above 20 mEq/L in infants an >22 mEq/L in children, keeping in mind the amount of required alkali decreases with coming of age: 5–8 mEq/kg/day for infants, 3–4 mEq/kg/day for children, and 1–2 mEq/kg/day for adults (74).
Future Treatment Options
Ongoing Human Clinical Trials
Six-month data presented at the ASN annual meeting in 2017 showed promising results of a phase III trial for ADV7103 (bicarbonate and citrate in 2-mm granules), a new agent in the treatment of dRTA. ADV7103 is a slow release medication of alkaline citrate, which provides equilibrate alkali dosing over 12 h. A dosage of ADV7103 twice a day was able to maintain a normal blood bicarbonate level and improved quality of life in patients with dRTA (75). This is in contrast to the current alkaline medication, where both blood bicarbonate, as well as urine citrate excretion may fluctuate over the day and depend much more on the timing of medication (here >2 times a day). Advicenne pharmaceuticals is now seeking market authorization for ADV7103 and was granted orphan drug designation already by the EU in 06/2017. In addition to the potential treatment in dRTA, a phase II/III trial for the use of ADV7103 in patients with cystinuria is currently being initiated (76).
Conclusion and Outlook
The current therapeutic possibilities for patients with NL or NC are minor, promising research is on the rise to evaluate new treatment options. They range from therapeutic manipulation of a substance’ urine solubility (e.g., in cystinuria), mRNA-based approaches (e.g., PH and Lowe syndrome) to soluble degrading enzymes or bacteria (in PH). Many of these therapeutical chances have only been tested in vitro or in animal models. The way to get them into human use still may be long, but patients’ distress is urging scientist to proceed with their work (20, 21, 77–93).
Author Contributions
Both authors contributed equally to this paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fped.2018.00098/full#supplementary-material.
References
1. Geary DF, Schaefer F. Comprehensive Pediatric Nephrology. St. Louis, MO: Mosby (2008). XVII, 1020 S p.
2. Dick PT, Shuckett BM, Tang B, Daneman A, Kooh SW. Observer reliability in grading nephrocalcinosis on ultrasound examinations in children. Pediatr Radiol (1999) 29(1):68–72. doi:10.1007/s002470050539
3. VanDervoort K, Wiesen J, Frank R, Vento S, Crosby V, Chandra M, et al. Urolithiasis in pediatric patients: a single center study of incidence, clinical presentation and outcome. J Urol (2007) 177(6):2300–5. doi:10.1016/j.juro.2007.02.002
4. Routh JC, Graham DA, Nelson CP. Epidemiological trends in pediatric urolithiasis at United States freestanding pediatric hospitals. J Urol (2010) 184(3):1100–4. doi:10.1016/j.juro.2010.05.018
5. Novak TE, Lakshmanan Y, Trock BJ, Gearhart JP, Matlaga BR. Sex prevalence of pediatric kidney stone disease in the United States: an epidemiologic investigation. Urology (2009) 74(1):104–7. doi:10.1016/j.urology.2008.12.079
6. van’t Hoff WG. Aetiological factors in paediatric urolithiasis. Nephron Clin Pract (2004) 98(2):c45–8. doi:10.1159/000080251
7. Habbig S, Beck BB, Hoppe B. Nephrocalcinosis and urolithiasis in children. Kidney Int (2011) 80(12):1278–91. doi:10.1038/ki.2011.336
8. Sas DJ, Hulsey TC, Shatat IF, Orak JK. Increasing incidence of kidney stones in children evaluated in the emergency department. J Pediatr (2010) 157(1):132–7. doi:10.1016/j.jpeds.2010.02.004
9. Kovesdy CP, Furth SL, Zoccali C; World Kidney Day Steering Committee. Obesity and kidney disease: hidden consequences of the epidemic. Nephron (2017) 135(4):243–51. doi:10.1159/000455698
10. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol (2004) 15(12):3225–32. doi:10.1097/01.ASN.0000146012.44570.20
11. Hernandez JD, Ellison JS, Lendvay TS. Current trends, evaluation, and management of pediatric nephrolithiasis. JAMA Pediatr (2015) 169(10):964–70. doi:10.1001/jamapediatrics.2015.1419
12. Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med (1993) 328(12):833–8. doi:10.1056/NEJM199303253281203
13. Waller S, Ridout D, Rees L. Bone mineral density in children with chronic renal failure. Pediatr Nephrol (2007) 22(1):121–7. doi:10.1007/s00467-006-0292-2
14. Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol (2009) 20(10):2253–9. doi:10.1681/ASN.2009030276
15. Carvalho-Salemi J, Moreno L, Michael M. Medical nutrition therapy for pediatric kidney stone prevention, part one. J Ren Nutr (2017) 27(1):e5–8. doi:10.1053/j.jrn.2016.09.004
16. Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol (1993) 7(2):207–11. doi:10.1007/BF00864405
17. Laufer J, Boichis H. Urolithiasis in children: current medical management. Pediatr Nephrol (1989) 3(3):317–31. doi:10.1007/BF00858542
18. Spivacow FR, Negri AL, del Valle EE, Calviño I, Zanchetta JR. Clinical and metabolic risk factor evaluation in young adults with kidney stones. Int Urol Nephrol (2010) 42(2):471–5. doi:10.1007/s11255-009-9623-0
19. Gambaro G, Vezzoli G, Casari G, Rampoldi L, D’Angelo A, Borghi L. Genetics of hypercalciuria and calcium nephrolithiasis: from the rare monogenic to the common polygenic forms. Am J Kidney Dis (2004) 44(6):963–86. doi:10.1053/j.ajkd.2004.06.030
20. Roszko KL, Bi RD, Mannstadt M. Autosomal dominant hypocalcemia (hypoparathyroidism) types 1 and 2. Front Physiol (2016) 7:458. doi:10.3389/fphys.2016.00458
21. Ludwig M, Levtchenko E, Bökenkamp A. Clinical utility gene card for: dent disease (dent-1 and dent-2). Eur J Hum Genet (2014) 22(11):1338. doi:10.1038/ejhg.2014.33
22. Hoppe B, Hesse A, Neuhaus T, Fanconi S, Forster I, Blau N, et al. Urinary saturation and nephrocalcinosis in preterm infants: effect of parenteral nutrition. Arch Dis Child (1993) 69(3 Spec No):299–303. doi:10.1136/adc.69.3_Spec_No.299
23. Bergsland KJ, Worcester EM, Coe FL. Role of proximal tubule in the hypocalciuric response to thiazide of patients with idiopathic hypercalciuria. Am J Physiol Renal Physiol (2013) 305(4):F592–9. doi:10.1152/ajprenal.00116.2013
24. Arrabal-Polo MA, Arias-Santiago S, de Haro-Munoz T, Lopez-Ruiz A, Orgaz-Molina J, Gonzalez-Torres S, et al. Effects of aminobisphosphonates and thiazides in patients with osteopenia/osteoporosis, hypercalciuria, and recurring renal calcium lithiasis. Urology (2013) 81(4):731–7. doi:10.1016/j.urology.2012.12.013
25. Blanchard A, Vargas-Poussou R, Peyrard S, Mogenet A, Baudouin V, Boudailliez B, et al. Effect of hydrochlorothiazide on urinary calcium excretion in dent disease: an uncontrolled trial. Am J Kidney Dis (2008) 52(6):1084–95. doi:10.1053/j.ajkd.2008.08.021
26. Dong B, Endo I, Ohnishi Y, Kondo T, Hasegawa T, Amizuka N, et al. Calcilytic ameliorates abnormalities of mutant calcium-sensing receptor (CaSR) knock-in mice mimicking autosomal dominant hypocalcemia (ADH). J Bone Miner Res (2015) 30(11):1980–93. doi:10.1002/jbmr.2551
27. Gabriel SS, Belge H, Gassama A, Debaix H, Luciani A, Fehr T, et al. Bone marrow transplantation improves proximal tubule dysfunction in a mouse model of dent disease. Kidney Int (2017) 91(4):842–55. doi:10.1016/j.kint.2016.11.016
28. Rendu J, Montjean R, Coutton C, Suri M, Chicanne G, Petiot A, et al. Functional characterization and rescue of a deep intronic mutation in OCRL gene responsible for Lowe syndrome. Hum Mutat (2017) 38(2):152–9. doi:10.1002/humu.23139
29. Hawkes CP, Li D, Hakonarson H, Meyers KE, Thummel KE, Levine MA. CYP3A4 induction by rifampin: an alternative pathway for vitamin D inactivation in patients with CYP24A1 mutations. J Clin Endocrinol Metab (2017) 102(5):1440–6. doi:10.1210/jc.2016-4048
30. Mandrile G, van Woerden CS, Berchialla P, Beck BB, Acquaviva Bourdain C, Hulton S-A, et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int (2014) 86(6):1197–204. doi:10.1038/ki.2014.222
31. Cregeen DP, Williams EL, Hulton S, Rumsby G. Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat (2003) 22(6):497. doi:10.1002/humu.9200
32. Beck BB, Baasner A, Buescher A, Habbig S, Reintjes N, Kemper MJ, et al. Novel findings in patients with primary hyperoxaluria type III and implications for advanced molecular testing strategies. Eur J Hum Genet (2013) 21(2):162–72. doi:10.1038/ejhg.2012.139
33. Williams HE, Wandzilak TR. Oxalate synthesis, transport and the hyperoxaluric syndromes. J Urol (1989) 141(3 Pt 2):742–9. doi:10.1016/S0022-5347(17)40999-2
35. Dhondup T, Lorenz EC, Milliner DS, Lieske JC. Combined liver-kidney transplantation for primary hyperoxaluria type 2: a case report. Am J Transplant (2017) 18(1):253–7. doi:10.1111/ajt.14418
36. Zhao F, Bergstralh EJ, Mehta RA, Vaughan LE, Olson JB, Seide BM, et al. Predictors of incident ESRD among patients with primary hyperoxaluria presenting prior to kidney failure. Clin J Am Soc Nephrol (2016) 11(1):119–26. doi:10.2215/CJN.02810315
37. Mulay SR, Eberhard JN, Desai J, Marschner JA, Kumar SV, Weidenbusch M, et al. Hyperoxaluria requires TNF receptors to initiate crystal adhesion and kidney stone disease. J Am Soc Nephrol (2017) 28(3):761–8. doi:10.1681/ASN.2016040486
38. Ermer T, Eckardt K-U, Aronson PS, Knauf F. Oxalate, inflammasome, and progression of kidney disease. Curr Opin Nephrol Hypertens (2016) 25(4):363–71. doi:10.1097/MNH.0000000000000229
39. Ludwig-Portugall I, Bartok E, Dhana E, Evers BD, Primiano MJ, Hall JP, et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int (2016) 90(3):525–39. doi:10.1016/j.kint.2016.03.035
40. Miyata N, Steffen J, Johnson ME, Fargue S, Danpure CJ, Koehler CM. Pharmacologic rescue of an enzyme-trafficking defect in primary hyperoxaluria 1. Proc Natl Acad Sci U S A (2014) 111(40):14406–11. doi:10.1073/pnas.1408401111
41. Kelly JP, Curhan GC, Cave DR, Anderson TE, Kaufman DW. Factors related to colonization with Oxalobacter formigenes in U.S. adults. J Endourol (2011) 25(4):673–9. doi:10.1089/end.2010.0462
42. Hoppe B, Beck B, Gatter N, von Unruh G, Tischer A, Hesse A, et al. Oxalobacter formigenes: a potential tool for the treatment of primary hyperoxaluria type 1. Kidney Int (2006) 70(7):1305–11. doi:10.1038/sj.ki.5001707
43. Hoppe B, Groothoff JW, Hulton S-A, Cochat P, Niaudet P, Kemper MJ, et al. Efficacy and safety of Oxalobacter formigenes to reduce urinary oxalate in primary hyperoxaluria. Nephrol Dial Transplant (2011) 26(11):3609–15. doi:10.1093/ndt/gfr107
44. Hoppe B, Niaudet P, Salomon R, Harambat J, Hulton S-A, Van’t Hoff W, et al. A randomised phase I/II trial to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Pediatr Nephrol (2017) 32(5):781–90. doi:10.1007/s00467-016-3553-8
45. Langman CB, Grujic D, Pease RM, Easter L, Nezzer J, Margolin A, et al. A double-blind, placebo controlled, randomized phase 1 cross-over study with ALLN-177, an orally administered oxalate degrading enzyme. Am J Nephrol (2016) 44(2):150–8. doi:10.1159/000448766
46. Vaishnaw AK, Gollob J, Gamba-Vitalo C, Hutabarat R, Sah D, Meyers R, et al. A status report on RNAi therapeutics. Silence (2010) 1(1):14. doi:10.1186/1758-907X-1-14
47. Liebow A, Li X, Racie T, Hettinger J, Bettencourt BR, Najafian N, et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J Am Soc Nephrol (2017) 28(2):494–503. doi:10.1681/ASN.2016030338
48. Alnylam Pharmaceuticals. Alnylam Reports Positive Initial Results from Ongoing Phase 1/2 Study of ALN-GO1, an Investigational RNAi Therapeutic for the Treatment of Primary Hyperoxaluria Type 1: Press Release. Available from: http://investors.alnylam.com/news-releases/news-release-details/alnylam-reports-positive-initial-results-ongoing-phase-12-study (Accessed: September 21, 2017).
49. Dicerna Pharmaceuticals. Dicerna Announces Dosing of First Patient in Phase 1 Clinical Trial of DCR-PH1 in Patients with Primary Hyperoxaluria Type 1 (PH1): Press Release. Available from: http://investors.dicerna.com/news-releases/news-release-details/dicerna-announces-dosing-first-patient-phase-1-clinical-trial (Accessed: September 22, 2017).
50. Dicerna Pharmaceuticals. Dicerna Files Clinical Trial Application for DCR-PHXC, the Company’s Most Advanced GalXC™ Product Candidate, for Phase 1 Study in Primary Hyperoxaluria (PH): Press Release. Available from: http://investors.dicerna.com/news-releases/news-release-details/dicerna-files-clinical-trial-application-dcr-phxc-companys-most (Accessed: September 22, 2017).
51. Knoll T, Zöllner A, Wendt-Nordahl G, Michel MS, Alken P. Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr Nephrol (2005) 20(1):19–24. doi:10.1007/s00467-004-1663-1
52. Dent CE, Senior B. Studies on the treatment of cystinuria. Br J Urol (1955) 27(4):317–32. doi:10.1111/j.1464-410X.1955.tb03486.x
53. Chow GK, Streem SB. Medical treatment of cystinuria: results of contemporary clinical practice. J Urol (1996) 156(5):1576–8. doi:10.1016/S0022-5347(01)65451-X
54. Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J Urol (1995) 154(1):164–6. doi:10.1016/S0022-5347(01)67256-2
55. Moe OW, Pearle MS, Sakhaee K. Pharmacotherapy of urolithiasis: evidence from clinical trials. Kidney Int (2011) 79(4):385–92. doi:10.1038/ki.2010.389
56. Birwe H, Schneeberger W, Hesse A. Investigations of the efficacy of ascorbic acid therapy in cystinuria. Urol Res (1991) 19(3):199–201. doi:10.1007/BF00303750
57. Sahota A, Parihar JS, Capaccione KM, Yang M, Noll K, Gordon D, et al. Novel cystine ester mimics for the treatment of cystinuria-induced urolithiasis in a knockout mouse model. Urology (2014) 84(5):1249.e9–15. doi:10.1016/j.urology.2014.07.043
58. Hu L, Yang Y, Aloysius H, Albanyan H, Yang M, Liang J-J, et al. L-cystine diamides as L-cystine crystallization inhibitors for cystinuria. J Med Chem (2016) 59(15):7293–8. doi:10.1021/acs.jmedchem.6b00647
59. Zee T, Bose N, Zee J, Beck JN, Yang S, Parihar J, et al. α-Lipoic acid treatment prevents cystine urolithiasis in a mouse model of cystinuria. Nat Med (2017) 23(3):288–90. doi:10.1038/nm.4280
60. University of California, San Francisco. Lipoic Acid Supplement for Cystine Stone (ALA). Available from: https://clinicaltrials.gov/ct2/show/NCT02910531?cond=Cystinuria&draw=2&rank=4 (Accessed: September 16, 2017).
61. Revive Therapeutics. Revive Therapeutics Announces Initiation of a Phase 2 Study of REV-004 (Bucillamine) in Cystinuria: Press Release. Available from:http://www.marketwired.com/revive-therapeutics-announces-initiation-phase-2-study-rev-004-bucillamine-cystinuria-tsx-venture-rvv-2193865.htm (Accessed: September 6, 2017).
62. Nelson CP. TCUPS-Tolvaptan Use in Cyctinuria and Urolithiasis: A Pilot Study (TCUPS). Available from: https://clinicaltrials.gov/ct2/show/NCT02538016?cond=Cystinuria&draw=2&rank=3 (Accessed: September 6, 2017).
63. Ceballos-Picot I, Perignon JL, Hamet M, Daudon M, Kamoun P. 2,8-Dihydroxyadenine urolithiasis, an underdiagnosed disease. Lancet (1992) 339(8800):1050–1. doi:10.1016/0140-6736(92)90569-O
64. Arikyants N, Sarkissian A, Hesse A, Eggermann T, Leumann E, Steinmann B. Xanthinuria type I: a rare cause of urolithiasis. Pediatr Nephrol (2007) 22(2):310–4. doi:10.1007/s00467-006-0267-3
65. Abou-Elela A. Epidemiology, pathophysiology, and management of uric acid urolithiasis: a narrative review. J Adv Res (2017) 8(5):513–27. doi:10.1016/j.jare.2017.04.005
66. Nanmoku K, Kurosawa A, Shinzato T, Shimizu T, Kimura T, Yagisawa T.Febuxostat for the prevention of recurrent 2,8-dihydroxyadenine nephropathy due to adenine phosphoribosyltransferase deficiency following kidney transplantation. Intern Med (2017) 56(11):1387–91. doi:10.2169/internalmedicine.56.8142
68. Sikora P, Roth B, Kribs A, Michalk DV, Hesse A, Hoppe B. Hypocitraturia is one of the major risk factors for nephrocalcinosis in very low birth weight (VLBW) infants. Kidney Int (2003) 63(6):2194–9. doi:10.1046/j.1523-1755.2003.t01-4-00001.x
69. López M, Hoppe B. History, epidemiology and regional diversities of urolithiasis. Pediatr Nephrol (2010) 25(1):49–59. doi:10.1007/s00467-008-0960-5
70. Karsli O, Izol V, Aridogan IA, Borekoglu A, Satar N. Metabolic risk factors and the effect of metaphylaxis in pediatric stone disease with hypocitraturia. Urolithiasis (2013) 41(1):9–13. doi:10.1007/s00240-012-0539-2
72. van Rossum AM, Dieleman JP, Fraaij PL, Cransberg K, Hartwig NG, Gyssens IC, et al. Indinavir-associated asymptomatic nephrolithiasis and renal cortex atrophy in two HIV-1 infected children. AIDS (2001) 15(13):1745–7. doi:10.1097/00002030-200109070-00025
73. Chang H-Y, Hsu C-H, Tsai J-D, Li S-T, Hung H-Y, Kao H-A, et al. Renal calcification in very low birth weight infants. Pediatr Neonatol (2011) 52(3):145–9. doi:10.1016/j.pedneo.2011.03.004
74. Gil-Peña H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr (2014) 164(4):691–8.e1. doi:10.1016/j.jpeds.2013.10.085
75. Advicenne Pharmaceuticals. Advicenne Announces Positive 6-Month Extension Study Data from Pivotal Phase III Study of ADV7103 Phase III Study of ADV7103 in Adults and Children Suffering from Distal Renal Tubular Acidosis (dRTA): Press Release. Available from: http://advicenne.com/web/wp-content/uploads/2017/11/171101_ADV7103_6-month_data_presented_at_ASN.pdf (Accessed: October 2, 2017).
76. Advicenne Pharmaceuticals. Our Current Clinical and Pre-Clinical Portfolio of Paediatric Friendly Therapeutics for Renal Disease. Available from: http://advicenne.com/pipeline/ (Accessed: October 2, 2017).
77. Pronicka E, Ciara E, Halat P, Janiec A, Wójcik M, Rowińska E, et al. Biallelic mutations in CYP24A1 or SLC34A1 as a cause of infantile idiopathic hypercalcemia (IIH) with vitamin D hypersensitivity: molecular study of 11 historical IIH cases. J Appl Genet (2017) 58(3):349–53. doi:10.1007/s13353-017-0397-2
78. Tebben PJ, Singh RJ, Kumar R. Vitamin D-mediated hypercalcemia: mechanisms, diagnosis, and treatment. Endocr Rev (2016) 37(5):521–47. doi:10.1210/er.2016-1070
79. Ghemigian A, Ghemigian M, Popescu I, Vija L, Petrova E, Dumitru N, et al. Familial isolated primary hyperparathyroidism due to HRPT2 mutation. Hormones (Athens) (2013) 12(3):454–60.
80. Recker F, Zaniew M, Bockenhauer D, Miglietti N, Bokenkamp A, Moczulska A, et al. Characterization of 28 novel patients expands the mutational and phenotypic spectrum of Lowe syndrome. Pediatr Nephrol (2015) 30(6):931–43. doi:10.1007/s00467-014-3013-2
81. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, et al. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol (2014) 25(10):2366–75. doi:10.1681/ASN.2013101085
82. Prie D, Huart V, Bakouh N, Planelles G, Dellis O, Gerard B, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med (2002) 347(13):983–91. doi:10.1056/NEJMoa020028
83. Godron A, Harambat J, Boccio V, Mensire A, May A, Rigothier C, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol (2012) 7(5):801–9. doi:10.2215/CJN.12841211
84. Di Stefano V, Lionetti E, Rotolo N, La Rosa M, Leonardi S. Hypercalciuria and nephrocalcinosis as early feature of Wilson disease onset: description of a pediatric case and literature review. Hepat Mon (2012) 12(8):e6233. doi:10.5812/hepatmon.6233
85. Pober BR. Williams-Beuren syndrome. N Engl J Med (2010) 362(3):239–52. doi:10.1056/NEJMra0903074
86. de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis (2013) 8:8. doi:10.1186/1750-1172-8-8
87. Wang L-P, Yang K-Q, Jiang X-J, Wu H-Y, Zhang H-M, Zou Y-B, et al. Prevalence of Liddle syndrome among young hypertension patients of undetermined cause in a Chinese population. J Clin Hypertens (Greenwich) (2015) 17(11):902–7. doi:10.1111/jch.12598
88. Ellison DH. Pseudohypoaldosteronism type II. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, et al., editors. GeneReviews(®). Seattle, WA (1993). Available from: https://www.ncbi.nlm.nih.gov/books/NBK65707/ (Accessed: October 1, 2017).
89. Torres RJ, Puig JG. Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J Rare Dis (2007) 2:48. doi:10.1186/1750-1172-2-48
90. Torres RJ, Prior C, Puig JG. Efficacy and safety of allopurinol in patients with the Lesch-Nyhan syndrome and partial hypoxanthine-phosphoribosyltransferase deficiency: a follow-up study of 18 Spanish patients. Nucleosides Nucleotides Nucleic Acids (2006) 25(9–11):1077–82. doi:10.1080/15257770600893974
91. Cochat P, Pichault V, Bacchetta J, Dubourg L, Sabot J-F, Saban C, et al. Nephrolithiasis related to inborn metabolic diseases. Pediatr Nephrol (2010) 25(3):415–24. doi:10.1007/s00467-008-1085-6
92. Wakida N, Tuyen DG, Adachi M, Miyoshi T, Nonoguchi H, Oka T, et al. Mutations in human urate transporter 1 gene in presecretory reabsorption defect type of familial renal hypouricemia. J Clin Endocrinol Metab (2005) 90(4):2169–74. doi:10.1210/jc.2004-1111
Keywords: nephrolithiasis, urolithiasis, nephrocalcinosis, treatment, therapy
Citation: Weigert A and Hoppe B (2018) Nephrolithiasis and Nephrocalcinosis in Childhood—Risk Factor-Related Current and Future Treatment Options. Front. Pediatr. 6:98. doi: 10.3389/fped.2018.00098
Received: 20 October 2017; Accepted: 26 March 2018;
Published: 12 April 2018
Edited by:
Robert P. Woroniecki, Stony Brook Children’s Hospital, United StatesReviewed by:
Shamir Tuchman, Children’s National Health System, United StatesFrancois Cachat, University Hospital Bern, Switzerland
Copyright: © 2018 Weigert and Hoppe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernd Hoppe, YmVybmQuaG9wcGVAdWtib25uLmRl