 Hui-Jun Tan
Hui-Jun Tan Mei Deng1
Mei Deng1- 1Department of Pediatrics, The First Affiliated Hospital of Jinan University, Guangzhou, China
- 2Department of Infectious Diseases, Quanzhou Women's and Children's Hospital, Quanzhou, China
Sodium taurocholate cotransporting polypeptide (NTCP) is a carrier protein encoded by the human SLC10A1 gene, acting as the principal transporter of conjugated bile salts from the plasma into hepatocytes. Although NTCP was cloned as early as in 1994 and its function has been studied extensively, clinical description of NTCP deficiency remains rather limited thus far. The patients in this paper were 2 female monozygotic twins, who were referred to our hospital at the age 2 years with the complaint of persistently-raised total bile acids (TBA) for 21 months. At age 3 months, they were both diagnosed to have cholestatic liver disease due to raised serum TBA and direct bilirubin (DBIL) with the fraction >20% of the elevated total bilirubin (TBIL). Thereafter, their jaundice subsided and the DBIL levels recovered gradually, while serum TBA remained raised persistently. In view of their refractory hypercholanemia but negative symptoms and signs, SLC10A1 genetic analysis was performed for all family members to evaluate the possibility of NTCP deficiency. As a result, the twins were both homozygotes, while the parents, carriers, of the reportedly pathogenic variant c.800C>T (p.Ser267Phe). These findings suggested that NTCP deficiency may be a unique genetic factor causing transient cholestasis in early infancy, as well as, persistent hypercholanemia in pediatric patients.
Introduction
Deficiency of sodium taurocholate cotransporting polypeptide (NTCP) is an inborn error of bile acid metabolism caused by biallelic SLC10A1 variants, which impairs the NTCP function as the primary transporter of conjugated bile salts from the plasma into hepatocytes (1–4). NTCP was cloned as early as in the year 1994 (5), and thereafter, its function has been studied extensively (6, 7), while a number of SLC10A1 genetic variants have been identified in humans (1, 7, 8). However, the first patient with NTCP deficiency was just reported very recently by Vaz et al. (9). Since then, several papers about patients with NTCP deficiency have been published (4, 10–14), but the patient number was limited and the laboratory and clinical features of this new disorder still remained open for further investigation.
Herein, we reported two monozygotic twins suffering from NTCP deficiency, who presented with persistent hypercholanemia and transient cholestatic jaundice in their early infancy.
Case Description
Patient 1. A 2-years-old female infant was referred to the First Affiliated Hospital, Jinan University, due to elevated serum total bile acids (TBA) discovered for 21 months. At the age 3 months, she was admitted to Women and Children's Hospital of Quanzhou because of jaundice for 2 months. Laboratory test revealed that the serum levels of TBA, direct bilirubin (DBIL), aspartate transaminase (AST) and alanine transaminase (ALT) were all elevated (Table 1), and she was thus diagnosed to have cholestatic liver disease. After being treated with intravenous reduced glutathione and ademetionine-1,4-butanedisulfonate for 9 days, which were commonly used in cholestatic patients with elevated alanine transaminase levels, her jaundice was alleviated, but the liver function indices remained abnormal (Table 1). Then oral ursodesoxycholic acid was given and she was discharged at the age 3.3 months. During the subsequent follow-up over 20 months, she showed normal anthropometric and neurobehavioral development without abnormal appearance, and her cholestasis resolved completely since the age 7 months. However, persistently raised TBA levels were observed on repeated biochemical analyses (Table 1). So she was referred to our hospital for further investigation and management when aged 2 years.
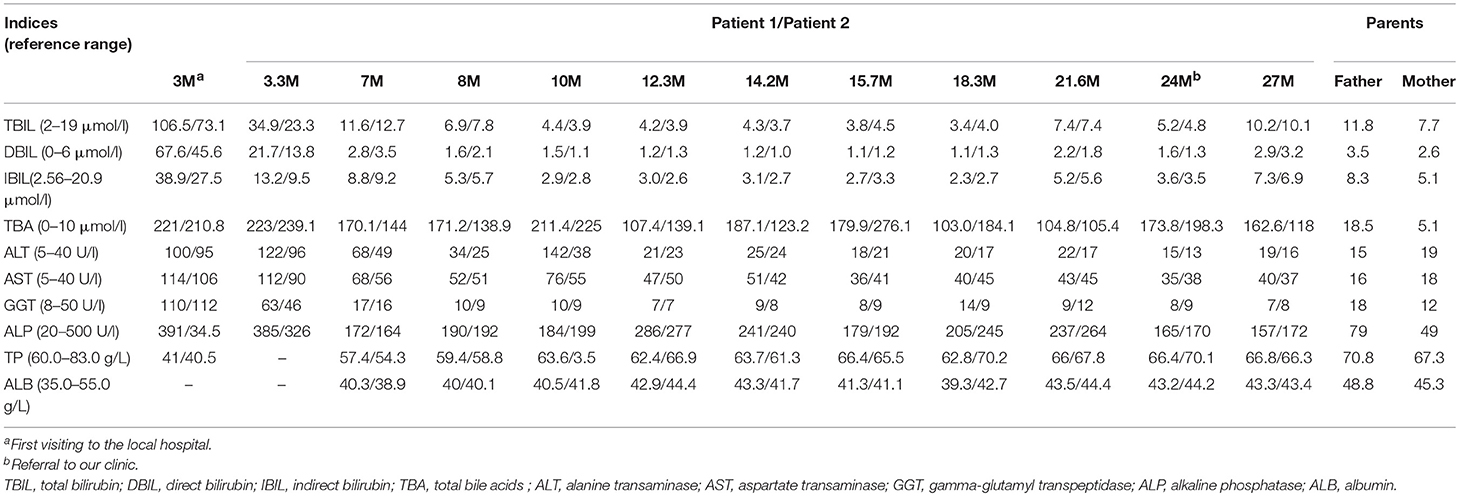
Table 1. Biochemical indices of the monozygotic twins and their parents.
The patient was the elder sister of two monochorionic diamniotic twins who was delivered by cesarean section at the gestation age of 37 weeks and 2 days with the birth weight 2.25 kg and body length 45.0 cm. Her father is a hepatitis B virus (HBV) carrier who was clinically healthy but with slightly elevated serum TBA level on biochemistry analysis, and her mother was physically and biochemically healthy (Table 1). There was no family history of any genetic disease.
Physical examination revealed a body weight of 12.0 kg, height 83.5 cm and head circumference 46.0 cm. No jaundice was observed in the skin and sclera. No stridor, crackles or crepitus was heard in the two lungs, and the heart sound was normal without any murmurs. There was no abdominal distention, and the liver and spleen were non-palpable. Physiological reflexes were normal and no pathological reflexes could be found on nervous system examination. On biochemical analysis, the TBA level reached 173.8 μmol/L (0–10 μmol/L) as other indices were normal (Table 1).
Patient 2 was the younger sister of patient 1, who also experienced transient cholestatic jaundice in early infancy and was referred to our hospital with the same complaint of persistently elevated serum TBA levels. As the younger one of two monozygotic twins, her birth weight was 2.30 kg and body length, 46.0 cm. On physical examination at referral, the body weight was 13.0 kg, height 85.0 cm, and head circumference 46.0 cm. No jaundiced skin or sclera was observed. Examinations of the lungs, the heart, the abdomen and nervous system were all normal. Her TBA level was 198.3 μmol/L (0–10 μmol/L), with otherwise normal biochemistry indices (Table 1).
Considering their refractory hypercholanemia whist lack of clinical symptoms and signs on physical examination, NTCP deficiency was highly suspected and hence Sanger sequencing of the SLC10A1 gene was carried out for all family members. As a result, the twins were both homozygotes of the variant c.800C>T (p.Ser267Phe), and their parents were both carriers of the same variant (Figure 1A). Electrophoresis of the PCR-RFLP products showed that the twins both harbored two additional bands of 164 bp and 65 bp rising from the aforementioned SLC10A1 variant, which further confirmed the SLC10A1 genotypes of all family members (Figure 1B).
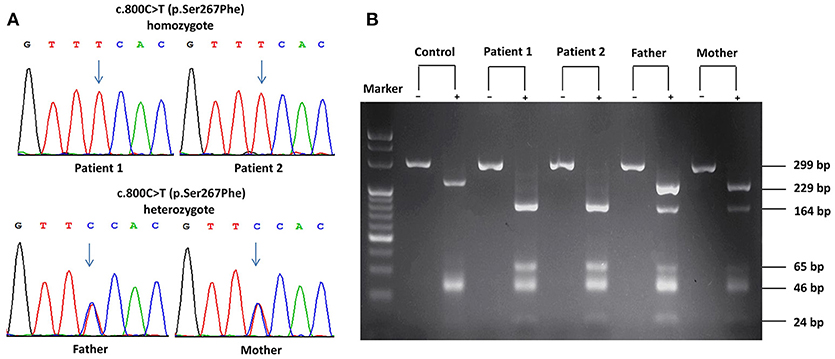
Figure 1. SLC10A1 genotypes of all family members on Sanger sequencing and electrophoresis of the PCR-RFLP products. (A) The two patients were both homozygotes while their parents, carriers, of the c.800C>T (p.Ser267Phe) variant. (B) PCR-RFLP analysis confirmed the SLC10A1 genotypic findings as in (A). In (B), the symbols “+” and “–” denoted with and without digestion of the restriction enzyme HphI, respectively.
NTCP deficiency was thus definitely diagnosed, and the twins were followed-up in the local hospital, and no specific medication was given. Thus far, the twins were both healthy but the serum TBA levels were still raised (Table 1), and their long-term outcomes needed to be observed.
Discussion
In this paper, SLC10A1 genetic analysis provided reliable evidences for the definite diagnosis of NTCP deficiency for the monozygotic twins. The p.Ser267Phe variant in SLC10A1 gene had proven pathogenic by functional, bioinformatic, and clinical evidences (4, 10–12). Of note, this SLC10A1 variant was prevalent in East Asian population, with an allele frequency of 8% in Southern Han Chinese, 12% in Chinese Dai and 11% in Vietnam (http://grch37.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=14:70244693-70245693;v=rs2296651;vdb=variation;vf=1699086) suggesting that this type of hypercholanemia might affect 0.64% of the Southern Han, 1.44% of the Dai Chinese, and 1.21% of the Vietnamese population (11). In addition, other non-synonymous SLC10A1 variants have been identified previously in different ethnic populations as well, such as p.Ile223Thr and p.Ala64Thr, with an allele frequency of 5% in African Americans and 1% in Korean, respectively (1, 7). Therefore, although patients of NTCP deficiency were rarely reported in the past over 20 years, NTCP deficiency may not be rare worldwide, especially in East Asian population.
Persistent hypercholanemia was the prominent feature of the patients with NTCP deficiency in this paper. Bile acids are synthesized from cholesterol in the liver, secreted into bile which is stored in the gallbladder, and then enter the small intestine during a meal. The majority (>90%) of bile acids are reabsorbed from the intestine and returned to the liver via the portal venous circulation. This circling of bile salts is well-known as enterohepatic circulation (15). NTCP plays a key role in the enterohepatic circulation of bile salts as the major transporter of conjugated bile salts from the plasma compartment into the hepatocyte (5). The p.Ser267Phe variant had been reported to result in an almost complete loss of the function for bile acid uptake, rendering NTCP without taurocholate transporting activity (6, 7, 10). Although the hepatocyte sinusoidal membrane also expresses other sodium-independent bile acid transporters, these transporters play limited roles in bile acid clearance (16). As such, it was not strange for the twins in this paper to present with refractory hypercholanemia.
Of particular note, the father of the twins, who was a carrier of the p.Ser267Phe variant and HBV as well, also exhibited slight hypercholanemia (Table 1). Since the reported patients with NTCP deficiency thus far were all compound heterozygotes or homozygotes of SLC10A1 biallelic variants (4, 9–14), indicating an autosomal recessive disorder, the slightly elevated TBA level in the father could not be explained by his carrier status of the p.Ser267Phe variant. Recently, Yan et al. found that NTCP also functions as a cellular receptor for viral entry of HBV through a specific interaction between NTCP and the pre-S1 domain of HBV large envelope protein (17), and showed that pre-S1 lipopeptide binding blocked the physiological function of NTCP in bile salt transport (6). As such, it is reasonable to speculate that the HBV carrier status of the father might affect the NTCP function to uptake bile acid at the basolateral membrane of the hepatocyte, being responsible for his slight hypercholanemia, as shown in Table 1.
Besides the persistent hypercholanemia, the elevated DBIL (67.6 μmol/l and 45.6 μmol/l) fraction >20% of TBIL (106.5 μmol/l and 73.1 μmol/l) in the twins at 3 months (Table 1) clearly indicated cholestatic jaundice in early infancy (18), and lent support to the concept that NTCP deficiency might work as one of the contributing factors that affect the bilirubin homeostasis, particularly in early infants who have very rich bilirubin sources but immature liver function to uptake, conjugate, and excrete bilirubin (19). Actually, hepatic uptake of bile salts is mediated by sodium-dependent and sodium-independent transporters (20). Sodium-independent uptake of bile salts is principally accomplished by the activity of multiple organic anion transporting polypeptides 1B1 and 1B3(OATP1B1/1B3), which work as an dimer protein to uptake plasma bilirubin, especially the direct bilirubin, as well as, bile acids into the hepatocyte (21, 22). NTCP is the primary sodium-dependent transporter of conjugated bile salts from the plasma compartment into the hepatocyte (5). Therefore, the elevated plasma bile acids caused by the deficiency of NTCP might increase the burden of the OATP1B1/1B3 to uptake bile acids into the hepatocyte, competitively inhibit its' function to uptake bilirubin, and result in secondary elevation of bilirubin, especially direct bilirubin, giving rise to cholestatic jaundice in infants as in the twins in this study, as well as, in NTCP-deficient patients reported previously (4, 10, 14).
Conclusion
In summary, this study reported the clinical and genetic findings of two monozygotic twins with NTCP deficiency. Our findings lent support to the important role of NTCP in bile salt homeostasis, and suggested that NTCP deficiency may be a unique genetic factor causing cholestasis in early infancy, as well as, persistent hypercholanemia at pediatric age.
Informed Consent
The authors declare that this study was performed after written informed consent had been obtained from the parents of the twins, which permitted publication of this case report.
Ethics Statement
This study has been approved by the Committee for Medical Ethics, the First Affiliated Hospital, Jinan University.
Author Contributions
H-JT performed data collection and drafted the initial manuscript. Y-ZS conceptualized and designed the study, critically reviewed, and revised the manuscript. MD and J-WQ carried out the genetic analyses and reviewed the manuscript. J-FW treated and followed up the patients and reviewed the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The present study was supported by the Fund for the construction of high-level Universities in Guangdong province (No.11615470) and National Natural Science Foundation (NSFC) of China (No. 81570793).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are sincerely thankful for the twins and their parents for their cooperation. We thank the support of National Natural Science Foundation (NSFC) of China (No. 81741080) as well as the fund for the construction of high-level universities in Guangdong province (No. 11615470).
References
1. Pan W, Song IS, Shin HJ, Kim MH, Choi YL, Lim SJ, et al. Genetic polymorphisms in Na+-taurocholate co-transporting polypeptide (NTCP) and ileal apical sodium-dependent bile acid transporter (ASBT) and ethnic comparisons of functional variants of NTCP among Asian populations. Xenobiotica (2011) 41:501–10. doi: 10.3109/00498254.2011.555567
2. Hagenbuch B, Dawson P. The sodium bile salt cotransport family SLC10. Pflugers Arch. (2004) 447:566–70. doi: 10.1007/s00424-003-1130-z
3. Anwer MS, Stieger B. Sodium-dependent bile salt transporters of the SLC10A transporter family: more than solute transporters. Pflugers Arch. (2014) 466:77–89. doi: 10.1007/s00424-013-1367-0
4. Song YZ, Deng M. Sodium taurocholate cotransporting polypeptide deficiency manifesting as cholestatic jaundice in early infancy: a complicated case study. Zhongguo Dang Dai Er Ke Za Zhi (2017) 19:350–4. doi: 10.7499/j.issn.1008-8830.2017.03.020
5. Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. (1994) 93:1326–31. doi: 10.1172/JCI117091
6. Yan H, Peng B, Liu Y, Xu G, He W, Ren B, et al. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J Virol. (2014) 88:3273–84. doi: 10.1128/JVI.03478-13
7. Ho RH, Leake BF, Roberts RL, Lee W, Kim RB. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem. (2004) 279:7213–22. doi: 10.1074/jbc.M305782200
8. Saito S, Iida A, Sekine A, Oqawa C, Kawauchi S, Higuchi S, et al. Catalog of 238 variations among six human genes encoding solute carriers (hSLCs) in the Japanese population. J Hum Genet. (2002)47:576–84. doi: 10.1007/s100380200088
9. Vaz FM, Paulusma CC, Huidekoper H, de Ru M, Lim C, Koster J, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology (2015) 61:260–7. doi: 10.1002/hep.27240
10. Deng M, Mao M, Guo L, Chen FP, Wen WR, Song YZ. Clinical and molecular study of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Exp Ther Med. (2016) 12:3294–300. doi: 10.3892/etm.2016.3752
11. Liu R, Chen C, Xia X, Liao Q, Wang Q, Newcombe PJ, et al. Homozygous p.Ser267Phe in SLC10A1 is associated with a new type of hypercholanemia and implications for personalized medicine. Sci Rep. (2017) 7:9214. doi: 10.1038/s41598-017-07012-2
12. Van Herpe F, Waterham HR, Adam CJ, Mannens M, Bikker H, Vaz FM, et al. NTCP deficiency and persistently raised bile salts: an adult case. J Inherit Metab Dis. (2017) 40:313–5. doi: 10.1007/s10545-017-0031-9
13. Qiu JW, Deng M, Cheng Y, Atif RM, Lin WX, Song YZ, et al. Sodium taurocholate cotransporting polypeptide (NTCP) defciency: identifcation of a novel SLC10A1 mutation in two unrelated infants presenting with neonatal indirect hyperbilirubinemia and remarkable hypercholanemia. J Oncotarget (2017) 8:106598–607. doi: 10.18632/oncotarget.22503
14. Li H, Qiu JW, Lin GZ, Deng M, Lin WX, Song YZ, et al. Clinical and genetic analysis of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Zhongguo Dang Dai Er Ke Za Zhi (2018) 20:279–84. doi: 10.7499/j.issn.1008-8830.2018.04.005
15. Dawson PA. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol. (2011) 201:169–203. doi: 10.1007/978-3-642-14541-4_4
16. Karpen SJ, Dawson PA. Not all (bile acids) who wander are lost: the frst report of a patient with an isolated NTCP defect. Hepatology (2015) 61:24–7. doi: 10.1002/hep.27294
17. Yan H, Zhong G, Xu G, He W, Jing Z, Li W, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife (2012) 1:e00049. doi: 10.7554/eLife.00049
18. Andrianov MG, Azzam RK. Cholestasis in infancy. Pediatr Ann. (2016) 45:e414–9. doi: 10.3928/19382359-20161118-01
19. Grijalva J, Vakili K. Neonatal liver physiology. Semin Pediatr Surg. (2013) 22:185–9. doi: 10.1053/j.sempedsurg.2013.10.006
20. Van Dyke RW, Stephens JE, Scharschmidt BF. Bile acid transport in cultured rat hepatocytes. Am J Physiol. (1982) 243:G484–92.
21. Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta (2003) 1609:1–18. doi: 10.1016/S0005-2736(02)00633-8
Keywords: sodium taurocholate cotransporting polypeptide deficiency, SLC10A1, bile acid, hypercholanemia, cholestasis
Citation: Tan H-J, Deng M, Qiu J-W, Wu J-F and Song Y-Z (2018) Monozygotic Twins Suffering From Sodium Taurocholate Cotransporting Polypeptide Deficiency: A Case Report. Front. Pediatr. 6:354. doi: 10.3389/fped.2018.00354
Received: 15 August 2018; Accepted: 30 October 2018;
Published: 20 November 2018.
Edited by:
André Hörning, Universitätsklinikum Erlangen, GermanyReviewed by:
Corentin Babakissa, Université de Sherbrooke, CanadaVictor Manuel Navas-López, Hospital Materno-Infantil, Spain
Copyright © 2018 Tan, Deng, Qiu, Wu and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan-Zong Song, c29uZ3l1YW56b25nQHZpcC50b20uY29t