 Sarah Winter
Sarah Winter Adélaïde Durand
Adélaïde Durand Raja Brauner
Raja Brauner- Fondation Ophtalmologique Adolphe de Rothschild and Université Paris Descartes, Paris, France
Background: Precocious and early puberty are reported findings in children with pre-existing medical conditions including certain syndromes. Series pertaining to such situations are limited.
Methods: A retrospective, single-center study was conducted on children with central precocious puberty (onset before the age of 8 years in girls and 9 years in boys) or early puberty (onset between 8 and 9 years in girls and between 9 and 10.5 years in boys) diagnosed on the background of a known pre-existing chronic significant medical condition. Patients with a CNS tumor and those exposed to cranial irradiation were excluded.
Results: Precocious puberty was diagnosed in 13 patients and early puberty in 12. Mean age at onset of puberty was 6.65 ± 2.3 years in girls (n = 15) and 9.4 ± 0.84 years in boys (n = 10). The most common disorders were psychomotor delay (n = 12), psychiatric disorders (n = 7) and/or epilepsy (n = 5). Precocious or early puberty was among the symptoms experienced by patients with a variety of syndromes including lipofuscinosis (2 siblings), Dravet syndrome and Silver-Russel syndrome. Pituitary stalk interruption with agenesis of olfactory bulbs and optic nerve atrophy was found on imaging in one patient who presented with blindness, epilepsy, and autism spectrum disorder. The other diseases associated with precocious or early puberty are adrenocorticotropic deficiency, dyspraxia and bone abnormalities, glomerulopathy with complete renal failure, and repeated intra-fetal deaths in the mother. Karyotype analysis revealed chromosomal duplication (chromosome 15 in 2 cases; chromosomes 17 and 11 in one case each) in 4 of 8 patients evaluated.
Conclusions: Data from patients with complex disease who experience precocious or early puberty may provide clues regarding the genetic determinants of pubertal development.
Introduction
The onset of puberty is driven by an increase in amplitude and frequency of gonadotropin-releasing hormone (GnRH) pulses after a quiescent period during childhood. The reactivation of pulsatile GnRH secretion leads to increases in the secretion of the gonadotropins, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) by the anterior pituitary gland and consequent activation of gonadal function (1). Patients experiencing pubertal development as a result of the activation of the hypothalamic-pituitary-gonadal axis before the ages of 8 years (girls) or 9 years (boys) are diagnosed with central precocious puberty (CPP) (2). Those experiencing this activation between the ages of 8–9 years (girls) (3) and 9–10.5 years (boys) (4) are known to have early puberty, an entity that is frequently considered as a normal variant and yet may underscore significant disease.
Complex interactions among genetic, nutritional, environmental, and socio-economic factors control the onset of normal puberty (5, 6). In a previous report on 154 patients from 139 families presenting with precocious or early puberty, we were able to demonstrate that the mode of inheritance of either phenotype is predominantly maternal (7). More than half of the families had individuals with either precocious or early puberty, suggesting that both entities may be controlled by similar factors and could likely be studied as a single entity in this particular context (7).
Precocious or early puberty have also been reported in the context of a variety of pre-existing medical conditions including certain syndromes. Series on this particular presentation were absent until the recent presentation of Canton et al. (8) including 26 patients (representing 17% of their idiopathic CPP) presenting as CPP and at least two additional features and/or conditions, characterizing complex phenotypes. The presence of a known, pre-existing, medical diagnosis provides the opportunity to further study potentially contributing factors to precocious or early puberty and shed additional light on the mechanisms of pubertal onset in this population. To this end, we have summarized in this report clinical, laboratory, imaging, and cytogenetic data from 25 patients presenting with this association. Patients with a central nervous system tumor and those exposed to cranial irradiation were excluded from this study.
Subjects and Methods
Ethics Approval and Consent to Participate
The protocol was approved by the Ethical Review Committee Comité de Protection des Personnes Ile de France III (certificate AC 048). All of the clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. Written informed consent for the evaluations was obtained from the children's parents and was included in their hospital medical records. No additional activity to routine patient care was performed. The authors had no direct interaction with the patients enrolled in the study, except for their medical follow-up, which was performed by R. Brauner.
Patients
This retrospective, single-center, cohort study included all patients monitored for precocious or early puberty in the context of a pre-existing chronic significant medical condition by a senior pediatric endocrinologist (R. Brauner) in a university pediatric hospital between 2004 and 2014. In girls, CPP was diagnosed by breast development before the age of 8 years accompanied by the presence of pubic or axillary hair, a growth rate >2 SD scores (SDS) the year before clinical evaluation and/or a bone age (BA) >2 years above chronological age (6). In boys, CPP was diagnosed by testicular enlargement (testicular volume index >3.5–4 cm2) before the age of 9 years (2). Early puberty was diagnosed by the appearance of these signs between the ages of 8 and 9 years in girls (3) and 9 and 10.5 years in boys (4). A total of 15 girls and 10 boys fulfilled the diagnostic criteria to be included in this study.
Methods
The initial evaluation included the determination of birth data, height expressed as SDS, growth rate, weight, body mass index (BMI) (expressed as SDS), pubertal stage rated according to Marshall and Tanner and BA assessed using the Greulich and Pyle method (9–11) except in 7 (28%) patients. Age at the first pubertal sign was reported by the parents.
The GnRH test, consisting of the intravenous injection of GnRH (100 μg/m2) and measurements of LH and FSH peaks at 0, 30, 60, and 90 min, was performed in 9 girls and 9 boys. Plasma LH, FSH, estradiol and testosterone concentrations were measured using radioimmunoassay. The following values were considered to be pubertal: LH/FSH peak ratio after GnRH test ≥ 0.66 in girls and ≥ 2 in boys (12), plasma estradiol concentrations ≥ 15 pg/mL in girls and testosterone levels ≥ 0.5 ng/mL in boys. Pelvic ultrasound evaluation was performed in 7 girls to evaluate the estrogenic stimulation of the uterus and to determine GnRH analog treatment. A uterus length value ≥35 mm was considered to be pubertal (13). Magnetic resonance imaging (MRI) was performed to exclude hypothalamic-pituitary lesion. It was not performed in 3 girls with CPP because they had a normal neurological evaluation, were >6 years of age at the onset of puberty and had low plasma estradiol concentration (< 15 pg/mL, cases 6 and 8) or because MRI has already been performed in the brother (case 9). Plasma 17-hydroxyprogesterone and testosterone concentrations were measured in all of the boys and in the girls with pubic or axillary hair development as the first pubertal sign to exclude abnormal androgen secretion and congenital adrenal hyperplasia (14). Plasma thyroxin and thyroid-stimulating hormone concentrations were measured to exclude hypothyroidism, and 24-h urinary cortisol levels were measured to exclude hypercortisolism in those who were overweight or had a rapid weight increase; all of the values were normal. Karyotype analysis was performed in 6 girls and 2 boys in the medical follow-up of their pre-existing medical condition.
The results are expressed as the means ± SD.
Results
Characteristics of the Patients
The characteristics of the 25 patients at the initial evaluation are shown in Table 1. Fifteen girls and 10 boys were included in this study, corresponding to a sex ratio at 1.5. Mean age at onset of puberty was 6.65 ± 2.3 years in girls and 9.4 ± 0.84 years in boys. In girls, the age at onset of puberty was < 3 years in 2 patients (13.3%), 3–7 years in 4 patients (26.6%), 7–8 years in 4 patients (26.6%), and 8–9 years in 5 patients (33.3%). In boys, the age at onset of puberty was 8–9 years in 3 patients (30.0%) and 9–10.5 years in 7 patients (70.0%). Thus, 66.5% of the girls and 30% of the boys have CPP, the others having early puberty. LH/FSH peak ratio was pubertal in 6 girls (66.6% of those evaluated) and in 7 boys (77.8%). Bone age was advanced over chronological age by 0.5 ± 0 years in girls and 0.85 ± 0.9 years in boys. One girl (case 1) experienced the onset of breast and pubic hair development at the age of 0.5 years. Pubertal development remained stable without treatment until the age of 8 years when breast and plasma estradiol concentration increased. Five girls (30.0%) and six boys (60.0%) received GnRH agonist treatment.
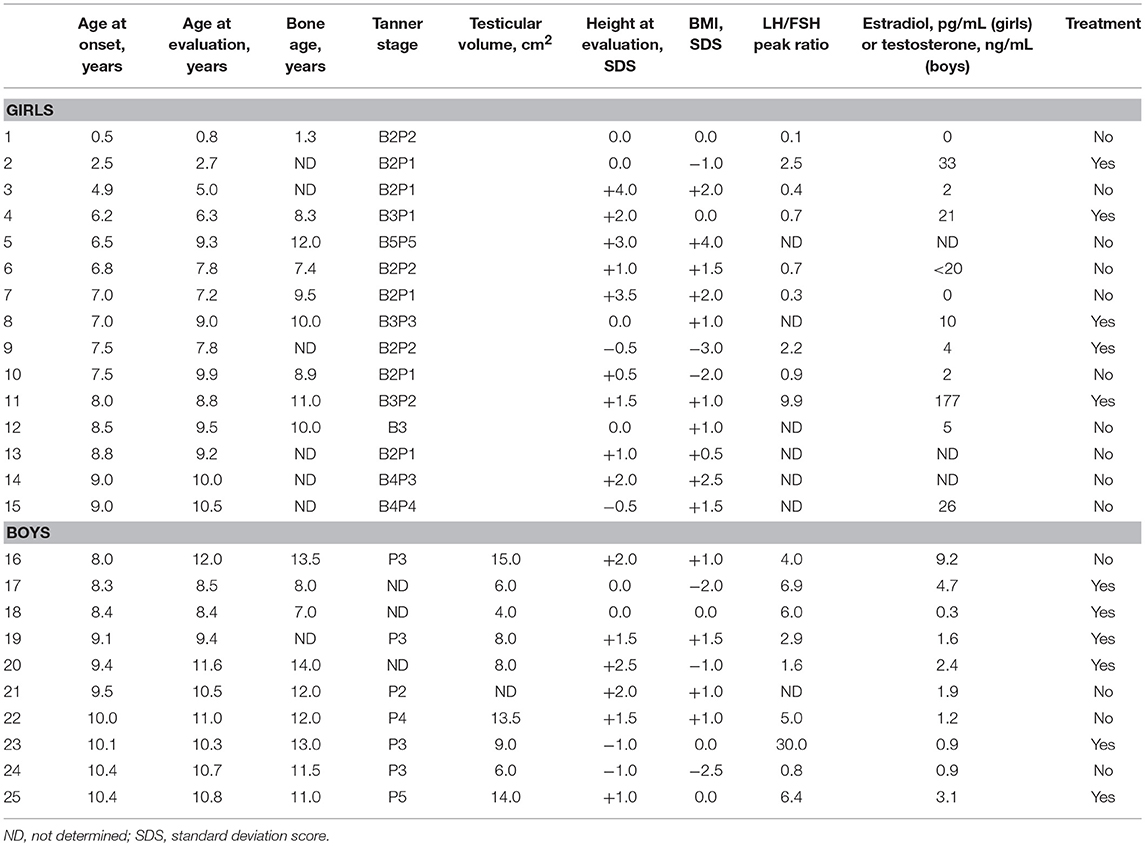
Table 1. Characteristics of the patients with precocious or early puberty associated with other disorders.
Consanguinity was reported in one case (case 2) without any additional details. A history of familial precocious or early puberty was reported in 8 cases (5 girls and 3 boys); it affected the mother in 6 cases and an older sister in 2 cases (Table 2).
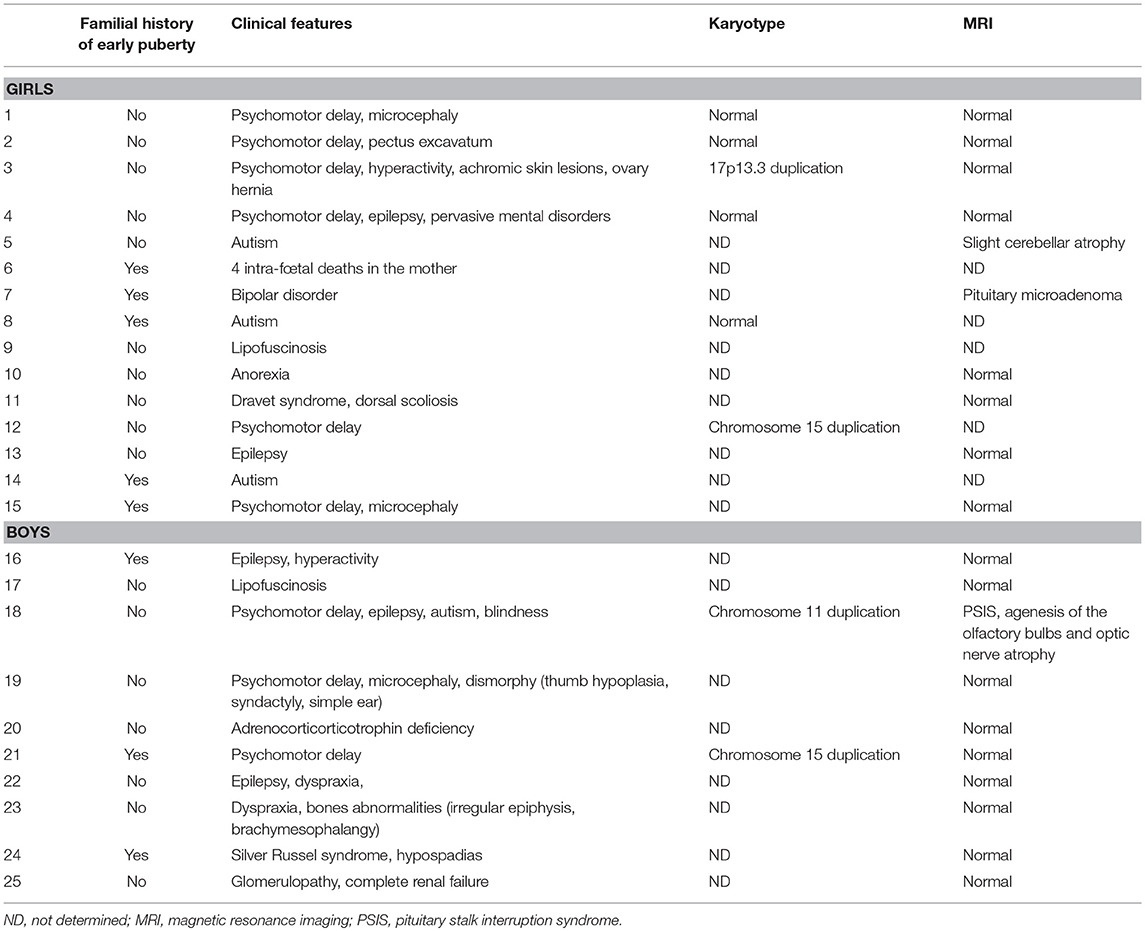
Table 2. Clinical, karyotype, and MRI findings associated with precocious or early puberty.
Pre-existing Medical Diagnoses Associated With Precocious or Early Puberty
In all of the patients, the diagnosis of the associated disorder was made before that of precocious or early puberty, except in case 25 with glomerulopathy and complete renal failure that required renal transplantation, who was diagnosed during the evaluation of early puberty by systematic urinary analysis.
Clinical, MRI and cytogenetic (karyotype) findings are detailed in Table 2. The most common associated medical diagnoses were psychomotor delay (12 cases) and/or epilepsy (5 cases). Syndromes included lipofuscinosis (diagnosed in one brother and his sister; cases 9 and 17), Dravet syndrome (case 11), and Silver Russel syndrome (case 24). Seven patients had psychiatric diagnoses including autism spectrum disorder (cases 5, 8, 14, and 18), pervasive developmental disorders (case 4), bipolar disease (case 7), and eating disorder (case 10). In one patient with blindness, psychomotor delay, epilepsy, and autism (case 18), the cerebral MRI performed because of multiple hypothalamic-pituitary deficiencies showed pituitary stalk interruption syndrome with agenesis of the olfactory bulbs and optic nerve atrophy. The other conditions associated with precocious or early puberty were adrenocorticotropic hormone deficiency (case 20), dyspraxia and bone abnormalities (case 23), and glomerulopathy with complete renal failure (case 25). The remaining patient (case 6) had no known pre-existing medical diagnosis but her mother experienced 4 pregnancies complicated by 4 intra-fetal deaths in the mother.
Karyotype analysis revealed abnormalities in 4 of the 8 patients in whom these data were available. These were chromosomal duplications (chromosome 15 in 2 cases, 17 and 11 in one case each). All patients with chromosomal abnormalities had psychomotor delay.
Discussion
Despite the data suggesting that age at the onset of puberty is primarily driven by genetic factors, the genetic determinants of the timing of pubertal development and particularly of precocious or early puberty remain unknown in a majority of cases (15–17).
In our best knowledge, only one multicentric series of 26 patients presenting as CPP and at least two additional features and/or conditions has been reported in 2018 as an Abstract by Canto et al. (8). They represent 17% of their idiopathic CPP, while the girls of the present study represent 4.5% (10/219) of those seen for CPP in the same conditions. Their most clinical features were: overweight or obesity (n = 15), born small for gestational age (n = 10), learning difficulties/intellectual disability/autistic spectrum (n = 7), short stature (n = 7), motor and/or speech delay (n = 6), high palate (n = 6), acanthosis nigricans (n = 5), and hyperinsulinemia (n = 5). Five patients were previously diagnosed with Temple syndrome (uniparental disomy of chromosome 14), Williams syndrome (7q11.23 microdeletion) or deletions in SHOX (n = 3).
Our study findings suggest that the association between precocious or early puberty and a variety of pre-existing medical conditions may provide additional insight. Indeed, out of 8 patients presenting with psychomotor delay on whom cytogenetic data were available, 50% had chromosomal duplications. Extensive efforts have been made to elucidate the mechanisms that reactivate pulsatile GnRH secretion at the time of puberty, and several genes have been identified in the complex network of inhibitory, stimulatory and permissive neuroendocrine factors involved in the control of puberty onset (Table 3). In the last decade, the kisspeptin system was revealed as a major gatekeeper of puberty onset (55). Several loss-of-function mutations in KISS1R were described in patients with congenital isolated hypogonadotropic hypogonadism (56). Only two mutations related to this pathway—one mutation in the gene encoding kisspeptin-1 and one in the gene encoding its receptor—have been associated with CPP (18, 19). In 2012, Tusset et al. identified a variant in the TAC3 gene in a Brazilian girl with CPP (20). In 2013, applying whole exome sequencing, Abreu et al. were able to identify 4 loss-of-function mutations in MKRN3 (encoding makorin RING-finger protein 3), an imprinted gene located on the long arm of chromosome 15q11.2, in five of 15 families with CPP (21). Later, Macedo et al. and Settas et al. reported novel MKRN3 heterozygous mutations, and currently 10 different loss-of-function mutations of MKRN3 have been described (22, 57). Interestingly, this gene is located in the Prader-Willi syndrome critical region; patients with Prader-Willi syndrome typically have incomplete sexual development; however, few cases of children with CPP have been reported (31–34).
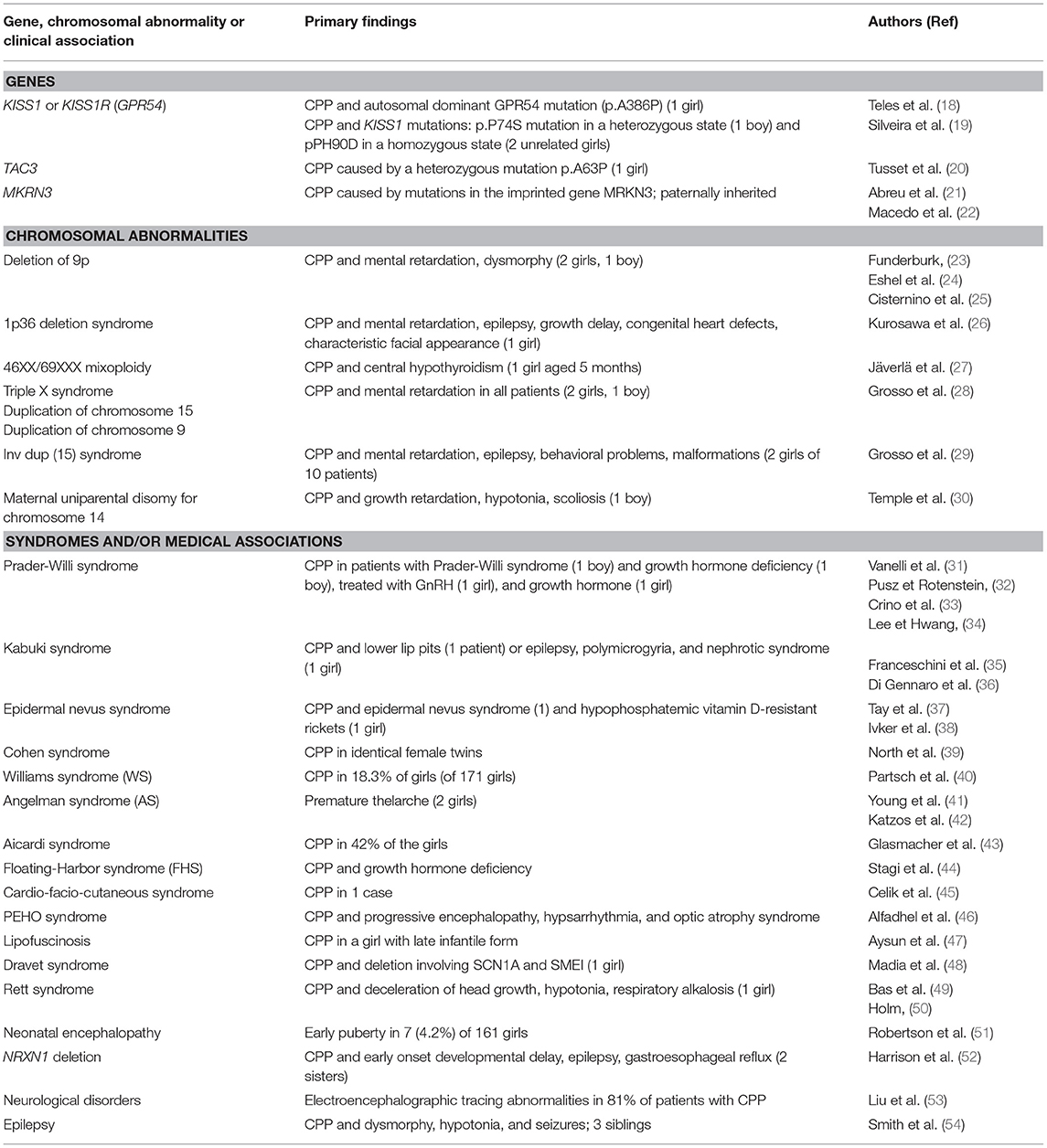
Table 3. Reported data related to genetic factors and various disorders associated with precocious or early puberty.
Chromosomal abnormalities were found in 4 patients (among 8 evaluated) of 25 (16%) of the present series: chromosome duplication of 11, 15 (n = 2) and 17. Cases of precocious or early puberty associated with chromosomal abnormalities have been reported (Table 3): deletion (23–25) or duplication (28) of 9p, deletion of 1p36 (26), 46,XX/69XXX mixoploidy (27), triple X syndrome (28), duplication or inversion duplication of 15 (29), or maternal uniparental disomy for chromosome 14 (30). These data suggested that performing a karyotype analysis should be recommended for patients with precocious or early puberty and associated disorders without any obvious etiology (primarily tumor or neurofibromatosis).
The association of precocious or early puberty with syndromes and/or medical history is not uncommon and, in most of the cases, the causative factor for this association is unknown. It has been reported in patients with Kabuki syndrome, epidermal nevus syndrome, Cohen syndrome, Williams syndrome, Angelman syndrome, Aicardi syndrome, Floating-Harbor syndrome, cardio-facio-cutaneous syndrome or PEHO syndrome (35–46). In our series, two siblings diagnosed with lipofuscinosis developed CPP. In the literature, this association has been described in one case report (47) in which the authors suggested that the early onset of puberty could be attributed to selective destruction of inhibitory tracts within the hypothalamus, chronic disruption of GnRH containing neurons or stimulation of gonadotropin release from the pituitary by the accumulation of lipofuscin-like material. CPP has also been associated with neurologic disorders, such as Dravet syndrome, Rett syndrome, neonatal encephalopathy or epilepsy (48–54). In our series, neurological and/or psychiatric disorders affected most of the patients. This observation is consistent with the findings reported in the literature; in nearly all of the chromosomal abnormalities or the syndromes mentioned above, the patients suffered from neurological disorders, including psychomotor delay, epilepsy, and encephalopathy. Epilepsy and CPP may also co-occur in the context of a central nervous system tumor or insult (58) which should be ruled out via appropriate imaging (59). There have been studies suggesting that valproic acid or other antiepileptic agents, such as clonazepam, could contribute to precocious or early puberty; so far, no evidence could be found in the literature (60–62). A lot of patients in the present study received various seizure medications.
Limitations and Strengths
The 25 patients constitute a small series. However, they were collected over 10 years by one senior pediatric endocrinologist, and no other series had been reported until that of the Sao Paulo University hospital group who reported recently 26 patients (8). We included patients with precocious or early puberty because, as reported in the familial forms, including our series (7), the families frequently include both entities suggesting that they may be controlled by similar factors. Only a small number of patients had a karyotype. The patients of this series are followed by other pediatric specialist, mainly neurologist who posed the indication of the karyotype and sent the patients to us because their pubertal development began early. The Sao Paulo group reported only two patients evaluated for karyotype (See above) (8). Furthermore, as most of the patients were referred to us by a pediatric neurologist, that could explain why so many of the patients had psychomotor delay and/or epilepsy. This could affect the generalizability of the findings of the present study.
Conclusion
Although our cohort included a small number of patients, this study is the first, to our knowledge, to describe in detail a series of patients with precocious or early puberty occurring in the context of a pre-existing medical diagnosis. The majority of patients included in this study presented neurological and/or psychiatric disorders, some fulfilling the diagnostic criteria of a syndrome, and others not. Surprisingly high rates of chromosomal abnormalities were found in assessed patients. Additional studies are needed to investigate the mechanisms through which precocious or early puberty is triggered in patients with complex medical histories.
Author Contributions
SW collected and analyzed the data and prepared the manuscript. AD collected and analyzed the data. RB directed the work, participated to the preparation and edited the manuscript according to the questions of the Reviewers. All authors discussed the results, commented on the manuscript, and approved the final version to be submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Wassim Chemaitilly for his editorial help.
Abbreviations
BMI, body mass index; BA, bone age; CPP, central precocious puberty; FSH, follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; MRI, magnetic resonance imaging; SDS, standard deviation score.
References
1. Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev. (2001) 22:111–51. doi: 10.1210/er.22.1.111
2. Carel J-C, Eugster EA, Rogol A, Ghizzoni L, Palmert MR, ESPE-LWPES, GnRH Analogs Consensus Conference Group, et al. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics (2009) 123:e752–62. doi: 10.1542/peds.2008-1783
3. Lazar L, Kauli R, Pertzelan A, Phillip M. Gonadotropin-suppressive therapy in girls with early and fast puberty affects the pace of puberty but not total pubertal growth or final height. J Clin Endocrinol Metab. (2002) 87:2090–4. doi: 10.1210/jcem.87.5.8481
4. Lazar L, Pertzelan A, Weintrob N, Phillip M, Kauli R. Sexual precocity in boys: accelerated versus slowly progressive puberty gonadotropin-suppressive therapy and final height. J Clin Endocrinol Metab. (2001) 86:4127–32. doi: 10.1210/jcem.86.9.7852
5. Gajdos ZK, Hirschhorn JN, Palmert MR. What controls the timing of puberty? An update on progress from genetic investigation. Curr Opin Endocrinol Diabetes Obes. (2009) 16:16–24. doi: 10.1097/MED.0b013e328320253c
6. Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. (2001) 86:2364–8. doi: 10.1210/jcem.86.6.7603
7. Durand A, Bashamboo A, McElreavey K, Brauner R. Familial early puberty: presentation and inheritance pattern in 139 families. BMC Endocr Disord. (2016) 16:50. doi: 10.1186/s12902-016-0130-x
8. Canton A, Brito V, Montenegro L, Ramos C, Macedo D, Bessa D, et al. Clinical and genetic features of central precocious puberty associated with complexe phenotypes. In: Hormone Research in Paediatrics European Society for Paediatric Endocrinology (ESPE) 57th Annual Meeting. Athens (2018). p. 98.
9. Greulich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and the Wrist, 2nd ed. Stanford, CA: Stanford University Press (1959).
10. Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. (1969) 44:291–303. doi: 10.1136/adc.44.235.291
11. Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. (1970) 45:13–23.
12. Oerter KE, Uriarte MM, Rose SR, Barnes KM, Cutler GB. Gonadotropin secretory dynamics during puberty in normal girls and boys. J Clin Endocrinol Metab. (1990) 71:1251–8. doi: 10.1210/jcem-71-5-1251
13. De Vries L, Horev G, Schwartz M, Phillip M. Ultrasonographic and clinical parameters for early differentiation between precocious puberty and premature thelarche. Eur J Endocrinol Eur Fed Endocr Soc. (2006) 154:891–8. doi: 10.1530/eje.1.02151
14. Armengaud J-B, Charkaluk M-L, Trivin C, Tardy V, Bréart G, Brauner R, et al. Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. J Clin Endocrinol Metab. (2009) 94:2835–40. doi: 10.1210/jc.2009-0314
15. Fischbein S. Onset of puberty in MX and DZ twins. Acta Genet Med Gemellol. (1977) 26:151–8. doi: 10.1017/S0001566000009946
16. Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Mol Genet Metab. (2003) 80:1–10. doi: 10.1016/S1096-7192(03)00107-0
17. Parent A-S, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon J-P. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. (2003) 24:668–93. doi: 10.1210/er.2002-0019
18. Teles MG, Bianco SDC, Brito VN, Trarbach EB, Kuohung W, Xu S, et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. (2008) 358:709–15. doi: 10.1056/NEJMoa073443
19. Silveira LG, Noel SD, Silveira-Neto AP, Abreu AP, Brito VN, Santos MG, et al. Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab. (2010) 95:2276–80. doi: 10.1210/jc.2009-2421
20. Tusset C, Noel SD, Trarbach EB, Silveira LFG, Jorge AAL, Brito VN, et al. Mutational analysis of TAC3 and TACR3 genes in patients with idiopathic central pubertal disorders. Arq Bras Endocrinol Metabol. (2012) 56:646–52. doi: 10.1590/S0004-27302012000900008
21. Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. (2013) 368:2467–75. doi: 10.1056/NEJMoa1302160
22. Macedo DB, Abreu AP, Reis ACS, Montenegro LR, Dauber A, Beneduzzi D, et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J Clin Endocrinol Metab. (2014) 99:E1097–103. doi: 10.1210/jc.2013-3126
23. Funderburk SJ, Sparkes RS, Klisak I. The 9p-syndrome. J Med Genet. (1979) 16:75–9. doi: 10.1136/jmg.16.1.75
24. Eshel G, Lahat E, Reish O, Barr J. Neurodevelopmental and behavioral abnormalities associated with deletion of chromosome 9p. J Child Neurol. (2002) 17:50–1. doi: 10.1177/088307380201700113
25. Cisternino M, Della Mina E, Losa L, Madè A, Rossetti G, Bassi LA, et al. Idiopathic central precocious puberty associated with 11 mb de novo distal deletion of the chromosome 9 short arm. Case Rep Genet. (2013) 2013:978087. doi: 10.1155/2013/978087
26. Kurosawa K, Kawame H, Okamoto N, Ochiai Y, Akatsuka A, Kobayashi M, et al. Epilepsy and neurological findings in 11 individuals with 1p36 deletion syndrome. Brain Dev. (2005) 27:378–82. doi: 10.1016/j.braindev.2005.02.004
27. Järvelä IE, Salo MK, Santavuori P, Salonen RK. 46,XX/69,XXX diploid-triploid mixoploidy with hypothyroidism and precocious puberty. J Med Genet. (1993) 30:966–7.
28. Grosso S, Anichini C, Berardi R, Balestri P, Pucci L, Morgese G. Central precocious puberty and abnormal chromosomal patterns. Endocr Pathol. (2000) 11:69–75. doi: 10.1385/EP:11:1:69
29. Grosso S, Balestri P, Anichini C, Bartalini G, Pucci L, Morgese G, et al. Pubertal disorders in inv dup(15) syndrome. Gynecol Endocrinol Off J Int Soc Gynecol Endocrinol. (2001) 15:165–9. doi: 10.1080/gye.15.3.165.169
30. Temple IK, Cockwell A, Hassold T, Pettay D, Jacobs P. Maternal uniparental disomy for chromosome 14. J Med Genet. (1991) 28:511–4. doi: 10.1136/jmg.28.8.511
31. Vanelli M, Bernasconi S, Caronna N, Virdis R, Terzi C, Giovannelli G. Precocious puberty in a male with Prader-Labhart-Willi syndrome. Helv Paediatr Acta (1984) 39:373–7.
32. Pusz ER, Rotenstein D. Treatment of precocious puberty in a female with Prader-Willi syndrome. J Pediatr Endocrinol Metab. (2008) 21:495–500. doi: 10.1515/JPEM.2008.21.5.495
33. Crinò A, Di Giorgio G, Schiaffini R, Fierabracci A, Spera S, Maggioni A, et al. Central precocious puberty and growth hormone deficiency in a boy with Prader-Willi syndrome. Eur J Pediatr. (2008) 167:1455–8. doi: 10.1007/s00431-008-0679-0
34. Lee HS, Hwang JS. Central precocious puberty in a girl with Prader-Willi syndrome. J Pediatr Endocrinol Metab. (2013) 26:1201–4. doi: 10.1515/jpem-2013-0040
35. Franceschini P, Vardeu MP, Guala A, Franceschini D, Testa A, Corrias A, et al. Lower lip pits and complete idiopathic precocious puberty in a patient with Kabuki make-up (Niikawa-Kuroki) syndrome. Am J Med Genet. (1993) 47:423–5. doi: 10.1002/ajmg.1320470326
36. Di Gennaro G, Condoluci C, Casali C, Ciccarelli O, Albertini G. Epilepsy and polymicrogyria in Kabuki make-up (Niikawa-Kuroki) syndrome. Pediatr Neurol. (1999) 21:566–8. doi: 10.1016/S0887-8994(99)00030-2
37. Tay YK, Weston WL, Ganong CA, Klingensmith G. Epidermal nevus syndrome: association with central precocious puberty and woolly hair nevus. J Am Acad Dermatol. (1996) 35:839–42. doi: 10.1016/S0190-9622(96)90098-5
38. Ivker R, Resnick SD, Skidmore RA. Hypophosphatemic vitamin D-resistant rickets, precocious puberty, and the epidermal nevus syndrome. Arch Dermatol. (1997) 133:1557–61. doi: 10.1001/archderm.1997.03890480077011
39. North C, Patton MA, Baraitser M, Winter RM. The clinical features of the Cohen syndrome: further case reports. J Med Genet. (1985) 22:131–4. doi: 10.1136/jmg.22.2.131
40. Partsch C-J, Japing I, Siebert R, Gosch A, Wessel A, Sippell WG, et al. Central precocious puberty in girls with Williams syndrome. J Pediatr. (2002) 141:441–4. doi: 10.1067/mpd.2002.127280
41. Young C, Wang PJ, Tsai WY, Shen YZ. Precocious puberty in a case with probable Angelman syndrome. Brain Dev. (1994) 16:249–52. doi: 10.1016/0387-7604(94)90080-9
42. Katzos G, Triantafyllou P, Gombakis N, Sofocleous C, Zafeiriou DI. Thelarche variant in a girl with Angelman syndrome. Brain Dev. (2004) 26:339–41. doi: 10.1016/j.braindev.2003.10.005
43. Glasmacher MAK, Sutton VR, Hopkins B, Hopkins B, Eble T, Lewis RA, et al. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children. J Child Neurol. (2007) 22:176–84. doi: 10.1177/0883073807300298
44. Stagi S, Galluzzi F, Bindi G, Lapi E, Cecchi C, Salti R, et al. Precocious puberty in a girl with floating-harbor syndrome. J Pediatr Endocrinol Metab. (2007) 20:1333–7. doi: 10.1515/JPEM.2007.20.12.1333
45. Çelik N, Cinaz P, Bideci A, Yüce O, Emeksiz HC, Döger E, et al. Cardio-facio-cutaneous syndrome with precocious puberty, growth hormone deficiency and hyperprolactinemia. J Clin Res Pediatr Endocrinol. (2014) 6:55–8. doi: 10.4274/Jcrpe.1151
46. Alfadhel M, Yong SL, Lillquist Y, Langlois S. Precocious puberty in two girls with PEHO syndrome: a clinical feature not previously described. J Child Neurol. (2011) 26:851–7. doi: 10.1177/0883073810396582
47. Aysun S, Apak RA, Küçükali T. A case of late infantile neuronal ceroid lipofuscinosis associated with precocious puberty. J Child Neurol. (2000) 15:204–5. doi: 10.1177/088307380001500311
48. Madia F, Striano P, Gennaro E, Malacarne M, Paravidino R, Biancheri R, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology (2006) 67:1230–5. doi: 10.1212/01.wnl.0000238513.70878.54
49. Baş, VN, Çetinkaya S, Agladioglu SY, Aksoy A, Gülpinar B, Aycan Z. Report of the first case of precocious puberty in Rett syndrome. J Pediatr Endocrinol Metab. (2013) 26:937–9. doi: 10.1515/jpem-2012-0418
50. Holm VA. Rett syndrome: a case report from an audiovisual program. Brain Dev. (1985) 7:297–9. doi: 10.1016/S0387-7604(85)80031-0
51. Robertson CM, Morrish DW, Wheler GH, Grace MG. Neonatal encephalopathy: an indicator of early sexual maturation in girls. Pediatr Neurol. (1990) 6:102–8. doi: 10.1016/0887-8994(90)90042-Y
52. Harrison V, Connell L, Hayesmoore J, McParland J, Pike MG, Blair E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A (2011) 155A:2826–31. doi: 10.1002/ajmg.a.34255
53. Liu N, Grumbach MM, De Napoli RA, Morishima A. Prevalence of electroencephalographic abnormalities in idiopathic precocious puberty and premature pubarche: bearing on pathogenesis and neuroendocrine regulation of puberty. J Clin Endocrinol Metab. (1965) 25:1296–308. doi: 10.1210/jcem-25-10-1296
54. Smith A, Leask K, Tomlin P, Donnai D. A familial dysmorphic condition with hypotonia, seizures and precocious puberty. Clin Dysmorphol. (2008) 17:161–4. doi: 10.1097/MCD.0b013e328302f0c4
55. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. (2003) 349:1614–27. doi: 10.1056/NEJMoa035322
56. Seminara SB. Mechanisms of disease: the first kiss-a crucial role for kisspeptin-1 and its receptor, G-protein-coupled receptor 54, in puberty and reproduction. Nat Clin Pract Endocrinol Metab. (2006) 2:328–34. doi: 10.1038/ncpendmet0139
57. Settas N, Dacou-Voutetakis C, Karantza M, Kanaka-Gantenbein C, Chrousos GP, Voutetakis A. Central precocious puberty in a girl and early puberty in her brother caused by a novel mutation in the MKRN3 gene. J Clin Endocrinol Metab. (2014) 99:E647–51. doi: 10.1210/jc.2013-4084
58. Taylor M, Couto-Silva A-C, Adan L, Trivin C, Sainte-Rose C, Zerah M, et al. Hypothalamic-pituitary lesions in pediatric patients: endocrine symptoms often precede neuro-ophthalmic presenting symptoms. J Pediatr. (2012) 161:855–63. doi: 10.1016/j.jpeds.2012.05.014
59. Chemaitilly W, Trivin C, Adan L, Gall V, Sainte-Rose C, Brauner R. Central precocious puberty: clinical and laboratory features. Clin Endocrinol. (2001) 54:289–94. doi: 10.1046/j.1365-2265.2001.01229.x
60. Choonara IA, Rosenbloom L, Smith CS. Clonazepam and sexual precocity. N Engl J Med. (1985) 312:185. doi: 10.1056/NEJM198501173120315
61. Rättyä J, Vainionpää L, Knip M, Lanning P, Isojärvi JI. The effects of valproate, carbamazepine, and oxcarbazepine on growth and sexual maturation in girls with epilepsy. Pediatrics (1999) 103:588–93.
Keywords: central precocious puberty, chromosomal duplication, early puberty, epilepsy, hypothalamic-pituitary-gonadal axis, precocious puberty, psychomotor delay, psychiatric disorders
Citation: Winter S, Durand A and Brauner R (2019) Precocious and Early Central Puberty in Children With Pre-existing Medical Conditions: A Single Center Study. Front. Pediatr. 7:35. doi: 10.3389/fped.2019.00035
Received: 01 October 2018; Accepted: 25 January 2019;
Published: 14 February 2019.
Edited by:
Sandro Loche, Ospedale Microcitemico, ItalyReviewed by:
Ronald Cohen, University of Chicago, United StatesCarla Bizzarri, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2019 Winter, Durand and Brauner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raja Brauner, cmFqYS5icmF1bmVyQHdhbmFkb28uZnI=