Abstract
Celiac disease (CD) is a systemic autoimmune disease due to a dysregulated mucosal immune response to gluten and related prolamines in genetically predisposed individuals. It is a common disorder affecting ~1% of the general population, its incidence is steadily increasing. Changes in the clinical presentation have become evident since the 80s with the recognition of extra-intestinal symptoms like short stature, iron deficiency anemia, altered bone metabolism, elevation of liver enzymes, neurological problems. Recent studies have shown that the overall prevalence of extra-intestinal manifestations is similar between pediatric and adult population; however, the prevalence of specific manifestations and rate of improvement differ in the two age groups. For instance, clinical response in children occurs much faster than in adults. Moreover, an early diagnosis is decisive for a better prognosis. The pathogenesis of extra-intestinal manifestations has not been fully elucidated yet. Two main mechanisms have been advanced: the first related to the malabsorption consequent to mucosal damage, the latter associated with a sustained autoimmune response. Importantly, since extra-intestinal manifestations dominate the clinical presentation of over half of patients, a careful case-finding strategy, together with a more liberal use of serological tools, is crucial to improve the detection rate of CD.
Introduction
Celiac disease (CD) is a systemic autoimmune disease due to a dysregulated mucosal immune response to gluten and related prolamins, characterized by a remodeling of the small intestinal mucosa leading to villous atrophy (1), that recedes upon a gluten free diet (GFD). It occurs in genetically susceptible individuals carrying the HLA-DQ2 and/or -DQ8 and affects ~1% of the general population in Europe, North America, North Africa, India and Middle East (2). There is evidence that its incidence is steadily increasing (3), not only because of improved awareness and more extensive use of specific diagnostic tools. Genetic factors cannot explain such a rapid increment, hence it seems to be mainly attributable to environmental factors (4).
Concomitantly with the increase in CD incidence, changes in its clinical presentation have been described since the 80s. In contrast to the “typical” presentation of CD with gastrointestinal symptoms, a higher number of asymptomatic cases has been detected by targeted screening of at-risk groups (5), as well as an increase in the number of “atypical” presentations, including extra-intestinal symptoms such as iron deficiency anemia, altered bone metabolism, short stature, and elevation of liver enzymes. A recent American study showed that non-intestinal symptoms were the most commonly represented in 43% of pediatric CD patients (6). Importantly, the use of terms such as “typical” and “atypical” to describe the clinical presentation of CD, which reflects the historical background, is currently discouraged in favor of the use of “classical” when the gastrointestinal symptoms (such as weight loss, diarrhea, distended abdomen) are prominent and “not classical” indicating cases with a predominance of extra-intestinal symptoms (7).
The overall clinical picture of CD at diagnosis became less severe (8) and the average age at diagnosis increased from below 3 years to the scholar age (9). However, most of these changes seem to have recently reached a plateau, at least as reported in Finland (9).
Recent studies showed that the prevalence of extra-intestinal manifestations is similar between the pediatric and adult population: 60 and 62%, respectively (10). However, clinical manifestations and rate of improvement differ in the two age groups. Short stature was found to be the most common feature in children, while iron deficiency anemia dominates the clinical picture in adults, furthermore, clinical symptoms in children seem to recede much faster than in adults (10, 11).
Here we reviewed the current knowledge about the extra-intestinal manifestations of CD, operating a clear distinction between extra-intestinal symptoms and CD-associated conditions. Emphasis has been given to the mechanisms underlying these pleomorphic manifestations, even though in part still unclear. Finally, we analyzed the impact of the GFD on these extra-intestinal manifestations.
Associated diseases vs. extra-intestinal manifestations
There is often some confusion between the extra-intestinal symptoms and diseases associated to CD. Indeed, some clinical presentations are sometimes described as CD-associated conditions and other times as extra-intestinal manifestations, one example being dermatitis herpetiformis. The main difference is that extra-intestinal symptoms improve on a GFD, particularly if the diet is started early (12). Contrariwise, CD-associated conditions are not correlated with gluten ingestion, despite being more frequent in the CD population. In line with what observed in many autoimmune disorders, there is an association of CD with other autoimmune conditions. The recent literature reports a risk of having another autoimmune disease, in the celiac population is from 3 to 10 times more frequent than in the general population (13, 14). The most frequent associated disease is type 1 diabetes (15), that shares with CD a combination of genetic factors and common pathogenetic mechanisms. The association with other autoimmune disorders is also attributed to shared genetic risk factors, particularly the HLA genes. In addition, a possible role of gluten in triggering autoimmunity has been suggested, based on its pro-inflammatory properties and the increased risk of developing other autoimmune disorders reported in relation to the duration of gluten containing diet (16). On the contrary, the pathogenic link of CD with chromosomal abnormalities such as Down's syndrome or Turner's disease is unclear and so is for other associated conditions such as IgA deficiency.
Clinical manifestations
Short Stature
After serological screening was introduced, in the form of anti-gliadin antibodies testing, the first extra-intestinal manifestation to be identified was short stature (17). Affecting ~10–40% of pediatric patients at the time of diagnosis (18), it still remains the most common extra-intestinal presentation of CD in children (19), sometimes being its only clinical sign (20). Up to 8% of the patients investigated for short stature will eventually receive a diagnosis of CD, that overall represents between 19 and 59% of all non-endocrinological causes of short stature (21–24). Poor growth has been more often described in children with a younger age at diagnosis and a more severe disease onset (25).
The pathogenic mechanisms underlying short stature in CD have not been fully clarified yet. Malnutrition due to malabsorption has traditionally been thought to play a major role, but more recently a multifactorial pathogenesis has been advanced. Particularly, dysfunction of the growth hormone (GH)- Insulin-like growth factor (IGF1) axis and in particular a role for ghrelin has been proposed (26). Reduced blood values of GH, IGF1, IGF-binding protein 1 and 3 (IGFBP 1, IGFBP3) and elevated levels of IGFBP2 have been reported (27). Interestingly, the exogenous administration of GH to untreated CD patients did not induce an increase in IGF-1 levels suggesting a dysfunction of growth axis associated to active CD (23). Dysregulation of the GH axis might be sustained by the elevation of pro-inflammatory cytokines such as IL-6, TNF α, interferon γ, IL-1 (28). An “autoimmune hypothesis” for short stature in CD has also been proposed (29), but evidences have been found only in few patients for whom high titers of anti-pituitary antibodies (APA) have been associated to low levels of IGF-1 (28). Hence APA titers might help identifying CD subjects with suspected GH deficiency (GHD).
The early introduction of GFD leads to a rapid growth catch-up, particularly in the first 6 months, with weight catch-up being much faster than height. Catch-up growth is a remarkable phenomenon characterized by an increase in height up to four times the average rate for the corresponding chronological age. Target height is usually reached within 3 years after diagnosis. However, sometimes CD patients do not reach their target height, possibly because a rapid catch-up growth can associate with accelerated bone maturation (27, 30, 31). When no catch-up growth can be observed despite a strict GFD, an endocrinological evaluation is mandatory to exclude GHD, condition that has been observed in ~0.23% of CD patients (32). Other comorbidities such as inflammatory bowel diseases (IBD), food aversion, Turner syndrome, have been reported in children with persistent short stature despite strict adherence to the GFD and should be ruled out in those cases. An early diagnosis and proper dietary regimen continuation minimize the risk of a compromised final height. Accordingly, Comba et al. (33) observed that patients who received the diagnosis of CD after the age of 6 had a significantly lower z-score for BMI, height and weight, as compared to children diagnosed at a younger age, indicating that when CD diagnosis is posed after puberty, the chances for growth catch-up are lower.
Delayed Puberty
In the pediatric untreated CD population delayed puberty is a common finding, due to hypogonadism in girls and to androgen resistance in boys (34, 35). The prevalence of delayed puberty in CD is about 11–20% (36). The pathogenesis is unclear, however a combination of nutritional deficiencies and autoimmune antibodies against hormones, their receptors and/or endocrine organs, seem to play a role (34, 35, 37). The prognosis is favorable with puberty development occurring within 6–8 months from the introduction of GFD. If the delay in puberty persists, the patient should be referred to the endocrinologist for further evaluation.
Anemia
Anemia is the most common extra-intestinal manifestation in the CD adult population, but roughly present in 15% of the CD pediatric population (10, 11, 19, 38), probably because in adults the diagnosis is delayed. The anemia is correlated with the severity of CD (39). In a recent meta-analysis, Mahadev et al. (40) observed that iron deficiency anemia is frequent in CD irrespectively of patient demographics (age and gender). Iron absorption occurs mainly in the duodenal mucosa, hence the small intestinal damage typical of active CD may lead to its malabsorption. However, the observation that anemia could be found also in children with potential celiac disease (41), seems to suggest a multifactorial pathogenesis. Anemia is most frequently due to iron deficiency, nevertheless vitamin B12 and folate deficiencies may also be responsible. Up to 84% of CD children presenting with mild anemia that strictly follow the GFD and receive iron supplementation show a complete recovery of their iron storages by 12–24 months, when their hemoglobin levels are not very low (10, 39). Oral iron formulations are often preferred over IV formulations. Recently, new oral iron formulations, sucrosomial iron, has been proposed for patients who are intolerant to iron sulfate.
Liver Abnormalities
Hypertransaminasemia has been reported as the most frequent hepatic manifestation in CD patients. Recent studies report its prevalence at about 9–14% (42). Most times the liver damage is not severe and reversible, but in rare cases it can lead to liver failure (43). The grade of hypertransaminasemia is correlated to the duodenal mucosal damage, malabsorption, and serum levels of anti-endomysial and anti-tissue transglutaminase2 (TG2) antibodies. It has been hypothesized that the altered gut permeability can determine an increased exposure to hepatotoxins in the portal circulation leading to inflammation and liver damage (19, 44, 45). Nevertheless, in line with the other extra-intestinal manifestations, also in this case autoimmune factors may play a role, as indicated by the presence of anti-TG2 antibodies deposits in the liver (46).
The response of hypertransaminasemia to a strict GFD is excellent, with a 75–95% rate of complete normalization of liver enzymes in 12–24 months (47). An early diagnosis of CD may prevent future hepatic problems and a strict compliance of GFD can make unnecessary a routine control of liver function (42). A link between thyroid and liver disease has been reported, also based on the observation that usually CD patients with elevated ALT had also a higher TSH. The liver, in fact, performs a central role in the transport, metabolism, and deiodination of thyroid hormones (42).
As far as hepatological conditions associated with CD, primary biliary cholangitis, primary sclerosing cholangitis and autoimmune hepatitis have been found overrepresented in CD patients, but in contrast to the so called “coeliac hepatitis” (elevation of liver enzymes) their course is not modified by GFD.
Bone Disease
Bone manifestations in CD patients are mainly osteopenia, defined as low bone mineral density (BMD), and osteoporosis, defined as low bone density leading to brittleness of the bones. Approximately 75% of pediatric patients have osteopenia and 10–30% osteoporosis (48). The damage of the duodenal mucosa, where both vitamin D and almost 90% of the calcium are absorbed, leads to decreased blood levels of calcium and vitamin D and consequent increased secretion of parathyroid hormone. The hyperparathyroidism is common in celiac children (12–54%) and it determines an increased bone turnover (49). Moreover, the inflammatory milieu including IL-1, TNFα, and IL-6, together with the activation of the RANK-L/RANK/osteoprotegerin pathway stimulates the bone metabolism (50, 51). The trabecular bone is usually the most involved, since it is the most metabolically active. Evaluation of the BMD is important in short-statured CD children, also in relation with height, gender, and age in order not to misinterpret BMD values. However, there is no demonstration of increased risk of fractures during childhood and youth in CD patients.
It is possible to have osteopenia also in the very early phases of the disease. A lower BMD has been observed in screening-detected CD children compared to controls; in the former, lower levels of 1-25-OH-Vitamin D and raised levels of PTH have also been measured. These values returned to the normal range after GFD was started, and that occurred already in the first year of GFD, emphasizing the importance of an early diagnosis (52, 53). Osteopenia may be further worsened by inadequate calcium intake, hence the need to supplement the GFD with Calcium-fortified foods and vitamin D metabolites (54, 55).
Joint and Musculoskeletal Disorders
The most common joint and musculoskeletal disorders in CD include myopathy, arthralgia and non-erosive arthritis that can be silent in the early stages of disease (56). The most common finding in CD pediatric population is a subclinical synovitis, while arthralgia becomes evident with age (>12 years). The incidence of these manifestations is ~5–10% and the most commonly involved joint is the knee, followed by the hip and the ankle (19). Ultrasonography is important for the diagnosis, particularly in those patients whose symptoms are mild. The pathogenesis of the rheumatological manifestations remains obscure (56) and similarly their response to the GFD. Iqbal and colleagues reported an improvement only in 30% of patients (57) upon gluten withdrawal, suggesting that GFD may lead to improvement of symptoms at least in a subset of patients.
It is important to remember that CD can be associated with autoimmune diseases involving joints and muscles such as Sjogren's syndrome, juvenile idiopathic arthritis, rheumatoid arthritis and systemic lupus erythematosus (LES). A two-fold risk of LES has been recently reported in the pediatric celiac population (58).
Neurological Manifestations
Several neurological manifestations are significantly associated with CD in the pediatric population. The most common being headache, that is present in up to one-fifth of the cases. Rarer conditions in the pediatric population are ataxia and neuropathy, ranging from 0.1 to 7.4%. A prevalence of 0.7–2%, not significantly different from the general population, has been described in some studies (59, 60), however other authors reported a 1.4 fold increase of epilepsy in CD children (61, 62). Thus, the link between epilepsy and CD remains still uncertain. The most common seizures patterns are the complex partial, followed by tonic-clonic seizures. A particular type of epilepsy characterized by the presence of occipital calcifications has been specifically reported in association to CD (63, 64)—Figure 1.
Figure 1
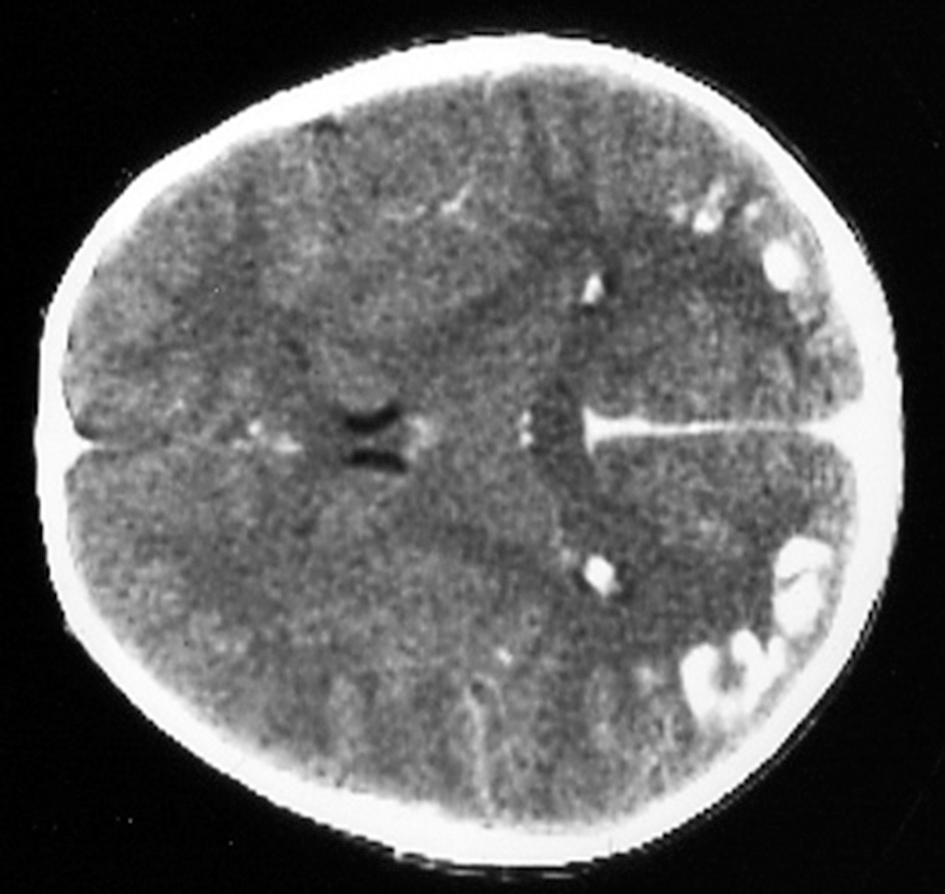
Bilateral occipital calcifications in celiac disease.
Cerebellar ataxia is more common in adults, with a median age of onset around 20 years. Chronic, symmetric distal neuropathy is the most common form of neuropathy described in CD patients, but its precise prevalence is unclear. A possible pathogenetic mechanism behind the neuropathy involves the anti-ganglioside antibodies, but nutritional deficiencies may also have a role. Particularly, some neurological manifestations are correlated with the deficit of vitamins such as E, B12 and D, or micronutrients like magnesium. Nevertheless, these manifestations may be present even in children with no enteropathy, excluding a role for malabsorption and suggesting that other mechanism might be responsible, for example a cross reaction between anti-gliadin antibodies and synapsin has been postulated. The pathogenesis of gluten ataxia can be related to the presence of anti-TG6 antibodies that might be directed against the cerebellar cells. However, their pathogenic role remains unclear since these autoantibodies can also be present in celiac children who are not affected by neurological disorders. The GFD leads to the complete recovery of headache in 76% of celiac children (65, 66) and it can be also responsible of the low prevalence of gluten ataxia and distal neuropathy when started early.
Psychiatric Disorders
An association between CD and psychiatric disorders, including attention deficit and hyperactive disorder (ADHD), autism spectrum disorders (ASD), mood disorders, anxiety, eating disorders and depression, has been reported (67). In a large cohort of CD children, an increased risk of psychiatric disorders development (1.4-fold increase) has been observed (68). There is evidence supporting an association of CD with depression and, although to a less extent, with eating disorders (67). For panic disorder, autism and ADHD there are few reports indicating an association but further studies are necessary. Finally, the association between CD and schizophrenia or other anxiety disorders is still debated. As far as pathogenic mechanisms involved, a direct effect of CD, perhaps based on inflammation and immunological dysregulation has been proposed (69), but another possible concomitant cause could be the psychosocial discomfort associated with a chronic condition for CD children.
Enamel Defects
The exact prevalence of enamel defects in CD is not known. However, in a recent meta-analysis it was observed that CD patients had significantly higher prevalence of enamel defects compared to controls (70). Some reports indicate that it involves up to 40–50% of CD patients at diagnosis (71), but in more recent reports the percentage results to be below 15% (72) probably because clinical presentations have become less severe over time. The enamel defects are characterized by pitting and sometimes by complete loss of enamel; they include discoloration and structural changes. Aine described enamel defects as detectable in all quadrants of the dentition, involving deciduous teeth (incisors and molars are the more frequently involved teeth) and most importantly symmetrical, the latter feature being more specific for CD (73). He proposed a grading, detailed in Table 1. Usually defects in dental enamel occur when CD affects children during dental development (before 7 years of age). When the defect affects permanent teeth, there is no improvement upon GFD.
Table 1
| Grade 0 | No defect |
|---|---|
| Grade 1 | Enamel discoloration with yellow, cream or brown opacities and loss of normal enamel glaze. |
| Grade 2 | Structural defect with some horizontal grooves. Change of color can be find. |
| Grade 3 | Important structural defects with deep horizontal grooves. Discoloration may be present. |
| Grade 4 | Destruction of tooth shape and structure. The material of enamel is fragile. |
Classification of systemic and chronologic enamel defects [modified from Aine (74)].
The defect of amelogenesis, malnutrition, in particular hypocalcemia, and immunological disturbances have been proposed as causes of enamel defects in CD. However, some reports deny a correlation between the degree of enamel defects and the severity of small bowel mucosal damage (75). Patients with HLA-DR3 genotype have been reported to have a higher risk of enamel lesions, pointing to a genetic contribution (76) (Figure 2).
Figure 2
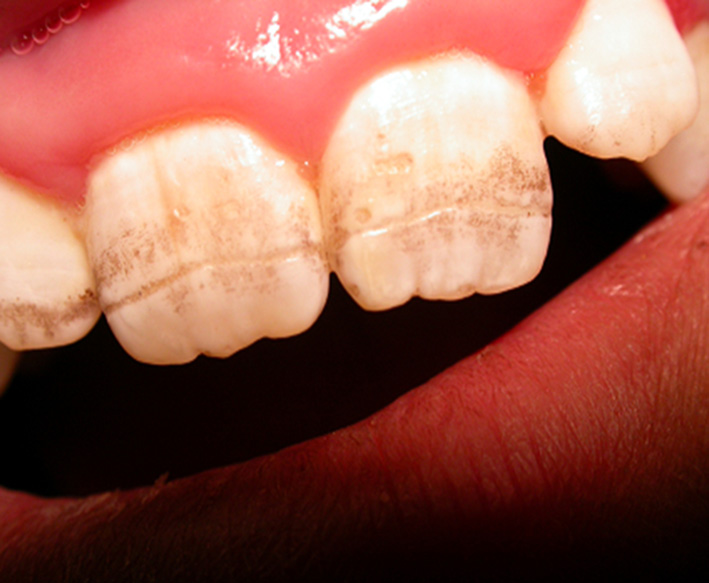
Model of enamel defects in celiac pediatric patient; these lesions have a symmetrical distribution.
Aphthous Stomatitis
Other oral disorders have been related to CD including delayed teeth eruption, lichen planus, cheilosis, atrophic glossitis, glossodinia. Aphthous stomatitis is an inflammatory ulcerative condition characterized by multiple recurrent small, round or ovoid ulcers with circumscribed margins appearing in the oral cavity. It usually manifests in the non-keratinized oral mucosa and can cause considerable pain. Up to 46% of CD patients have been reported to be affected by aphthous stomatitis (77). The mechanisms underlying this manifestation remain still obscure. It is unclear if there is any relation with malabsorption. Disturbances of the oral ecosystem (saliva, leukocytes, microbioma) have been hypothesized. Usually patients remit completely on GFD (10).
Alopecia
Alopecia has been observed in ~1% of recently diagnosed CD children. Depending on the extension of the lesion it is possible to distinguish alopecia areata, totalis, and universalis. Autoimmune mechanisms are thought to be involved in its pathogenesis, nevertheless GFD can lead to the total regrowth of hair by 12–24 months in half of the cases (78).
Dermatitis Herpetiformis
Dermatitis herpetiformis (DH) is considered an extraintestinal manifestation of CD. In children it is relatively rare (in Finland only 4% of all DH cases are children) (79), but in some series from other countries it seems to be more frequent in the pediatric age (80). It is particularly interesting to note that, in contrast to CD, the annual incidence rate is decreasing. It has been hypothesized that subclinical CD may predispose to DH, but what renders some individuals more prone to the development of skin lesions is unknown. The disease starts in the gut and evolves with the deposition in the papillary dermis of immune complexes of TG3 and high avidity anti-TG3 IgA antibodies (79).
From a clinical standpoint it presents with itchy papules and small blisters, often crusted because of the intense itch and consequent scratching, located on the extensor surfaces of elbows, knees and on the buttocks. Upper back, abdomen, scalp, and face may also be affected. Gastrointestinal symptoms in patients with DH are rare, but up to 72% of patients have a silent enteropathy (81). The diagnosis is made on a biopsy of unaffected skin showing by direct immunofluorescence pathognomonic granular IgA deposits at the dermo-epidermal junction. The rash recedes upon a strict GFD, with almost 100% resolution rate in children. Some patients may need additional medical therapy with dapsone, but with time the lesions are well-controlled by a GFD alone. In children the long-term prognosis on exclusion diet is excellent.
Mechanisms underlying extra-intestinal manifestations
The pathogenesis of extra-intestinal manifestations in many respects is still unclear. There are two main mechanisms probably involved, one related to the mucosal damage and the consequent malabsorption, the second sustained by the autoimmune response. In line with the first, some extra-intestinal manifestations are clearly correlated with the severity of intestinal damage. Indeed, patients with extra-intestinal manifestations at diagnosis have a more severe grade of intestinal mucosal atrophy as compared to patients presenting only with gastrointestinal symptoms (11). Anemia is associated with malabsorption of iron, vitamin B12, and folate (82). Stunted growth is likely caused by nutrients malabsorption (25, 83). Finally, osteopenia may be due to malabsorption of calcium and vitamin D, leading to secondary hyperparathyroidism and subsequently a high bone turnover (84).
Extra-intestinal manifestations may also be the consequence of autoimmune phenomena, although in many cases this hypothesis needs to be supported by more evidences. Tissue-transglutaminase 2 (TG2) is the main, but not the only autoantigen involved in CD. Whether anti-TG2 autoantibodies play a role in CD pathogenesis has not been definitely proven. They bind to several epitopes including the enzymatic core and can then interfere with bioactivity of TG2 (85). The presence of IgA deposits co-localizing with TG2 in liver, lymphnodes, muscle, thyroid, bone and brain (86) indicate that the autoantibodies, probably originated in the gut, can access to TG2 throughout the body and cause pathogenic effects. Consistently with this hypothesis, data in mice showed that the injection of anti-TG2 antibodies in the lateral ventricle of the brain caused deficits in motor coordination (87). Other members of transglutaminase family possibly involved in the pathogenesis of extra-intestinal manifestations in CD are TG6 and TG3. The latter is mainly expressed in the cornified layer of epidermis and for this reason also defined epidermal TG. Antibodies anti-TG3 have been found in almost 95% of DH patients representing a useful diagnostic marker for DH in both pediatric and adult patients (88). They are present also in areas located far away from the skin lesions, suggesting that other factors, besides the mere presence of the antibodies, are necessary to provoke the lesions. The TG6 is mainly expressed in neurons, playing an important role in neurogenesis (89). An association between neurological manifestation in CD and the presence of anti-TG6 has been suggested. In fact, anti-TG6 is elevated in the serum of patients with gluten neuropathy (90). Furthermore, TG6 is the target autoantigen in gluten ataxia (91). However, both specificity of these autoantibodies and gluten-dependence of their production have not been definitely proven (92).
As said, tissue transglutaminase is not the only autoantigen in CD. Antibodies to gangliosides have been reported in immune mediated peripheral neuropathies (93, 94); in particular, they have been described in CD patients in conjunction with neurological symptoms (95), their titers responding to the exclusion of gluten from the diet (96). Neutralizing autoantibodies against osteoprotegerin have also been detected in CD patients (97), but their role in development of osteoporosis is still uncertain (98). Finally, autoantibodies to cardiolipin, enolase alfa, ATP synthase beta chain, and also IgA to collagen type I, III, V, VI have been reported in CD (99), but at present there is no association/correlation between these and extra-intestinal manifestations of CD.
We reported in Table 2 the possible pathogenetic mechanisms for each EIM in CD.
Table 2
| Manifestation | Probable cause(s) |
|---|---|
| CUTANEOUS | |
| Edema | Hypoproteinemia |
| Dermatitis herpetiformis | Epidermal (type 3) tTG autoimmunity |
| ENDOCRINOLOGIC | |
| Amenorrhea, delayed puberty | Malnutrition, hypothalamic-pituitary dysfunction, immune dysfunction |
| Secondary hyperparathyroidism | Calcium and/or vitamin D malabsorption with hypocalcemia |
| HEMATOLOGIC | |
| Anemia | Iron, folate, vitamin B12, or pyridoxine deficiency |
| Hemorrhage | Vitamin K deficiency |
| HEPATIC | |
| Elevated liver biochemical test levels | Celiac hepatitis |
| Autoimmune hepatitis | Autoimmunity |
| MUSCULAR | |
| Atrophy | Malnutrition due to malabsorption |
| Tetany | Calcium, vitamin D, and/or magnesium malabsorption |
| Weakness | Generalized muscle atrophy, hypokalemia |
| NEUROLOGIC | |
| Peripheral neuropathy | Deficiencies of vitamin B12 and thiamine; immune-based neurologic dysfunction |
| Ataxia | Cerebellar and posterior column damage |
| Demyelinating central nervous system lesions | Immune-based neurologic dysfunction |
| Seizures | Unknown |
| SKELETAL | |
| Osteopenia, osteomalacia, and osteoporosis | Malabsorption of calcium and vitamin D, secondary hyperparathyroidism, chronic inflammation |
| Pathologic fractures | Osteopenia and osteoporosis |
| ORAL DISEASES | |
| Enamel hypoplasia | Vitamin D, calcium malabsorption |
| Aphthous stomatitis | Unknown |
Possible pathogenetic mechanisms for each extra-intestinal manifestations in celiac disease.
Treatment
Lifelong gluten-free diet (GFD) is the unique, effective therapy for CD (100). None of the pharmacological alternatives nowadays available or under investigation seems to be capable to replace the GFD (101).
In children GFD can lead to a complete recovery and a faster remission of extra-intestinal manifestations as compared to adults (10). The prognosis in children is very good, if adequately treated with GFD. However, timing of diagnosis is crucial. It is important to start the GFD as soon as possible. That means that an early diagnosis is decisive for a good prognosis. That is particularly true for bone diseases or for short stature. Patients diagnosed early during childhood did not have an increased risk of later development of osteoporotic fractures (102) and showed a very satisfactory catch up growth with a good final height (22). Nevertheless, sometimes the diet alone is not sufficient. This is the case of severe anemia, when it is advised to complement the dietary regimen with iron supplementation, or of severe DH, when adding a medical therapy with dapsone could be necessary in some cases (103), or even of severe osteopenia, when supplement the diet with calcium and vitamin D is important (54, 55). Problems arise when the GFD is not properly followed. This occurs mostly in adolescents (104), the pediatric population affected also by the highest prevalence of extra-intestinal manifestations and complications. When extra-intestinal manifestations show no improvement despite the GFD, patients' compliance to the dietary regimen should be questioned. In recent years a non-invasive, specific and novel approach to assess compliance with GFD has been reported, based on the detection of gluten immunogenic peptides (GIP) in the stools or in the urine by ELISA (105).
In case of no clinical improvement despite a strict GFD, additional diagnosis or pathogenic mechanism should be investigated. For instance, CD children presenting with short stature, but failing to show a satisfactory catch-up growth despite a verified compliance to the GFD, should be investigated for other co-existing conditions (e.g., growth hormone deficiency) (32) and consult the endocrinologist.
Conclusions
With extra-intestinal manifestations dominating the clinical presentation of over half of the patients, also in children CD may be considered a systemic disease. These proteiform clinical features may complicate the diagnosis. With still too many cases being undetected, education becomes very important. Given the availability of very efficient non-invasive diagnostic tools, such as measurement of serum anti-TG2 antibodies, it is necessary to increase the awareness among general pediatricians, but also other specialists (hematologists, neurologists, rheumatologists, endocrinologists). A more careful case finding strategy, together with a more liberal use of CD-specific serological tests, will improve the detection rate. Furthermore, an early diagnosis is particularly important for a faster remission of symptoms, better prognosis as well as to prevent long-term complications, especially those that are no more correctable after a certain age (e.g., osteopenia, short stature). Finally, extra-intestinal manifestations may help understanding the pathogenic mechanisms of CD, in particular the role played by autoimmunity in the different clinical presentations.
Statements
Author contributions
SN: first draft of the manuscript, manuscript revision, final approval of the version to be published, and agreement to be accountable for all aspects of the work. RT: corresponding author, primary responsibility for communication with the journal during the manuscript submission, drafting the work, manuscript revision, final approval of the version to be published, agreement to be accountable for all aspects of the work. RA and VD: critical revision of the article, final approval of the version to be published, agreement to be accountable for all aspects of the work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
LundinKEQiaoSWSnirOSollidLM. Coeliac disease-from genetic and immunological studies to clinical applications. Scand J Gastroenterol. (2015) 50:708–17. 10.3109/00365521.2015.1030766
2.
LionettiEGattiSPulvirentiACatassiC. Celiac disease from a global perspective. Best Practice Res Clin Gastroenterol. (2015) 29:365–79. 10.1016/j.bpg.2015.05.004
3.
LiuEDongFBarónAETakiINorrisJMFrohnertBIet al. High incidence of celiac disease in a long-term study of adolescents with susceptibility genotypes. Gastroenterology. (2017) 152:1329–36. 10.1053/j.gastro.2017.02.002
4.
AbadieVSollidLMBarreiroLBJabriB. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol. (2011) 29:493–525. 10.1146/annurev-immunol-040210-092915
5.
McGowanKECastiglioneDAButznerJD. The changing face of childhood celiac disease in North America: Impact of serological testing. Pediatrics. (2009) 124:1572–8. 10.1542/peds.2008-2373
6.
AlmallouhiEKingKSBhavishaPChungWJunhnJYMurrayJAet al. Increasing incidence and altered presentation in a population-based study of pediatric celiac disease in North America. J Pediatr Gastroenterol Nutr. (2017) 65:432–7. 10.1097/MPG.0000000000001532
7.
LudvigssonJFLefflerDABaiJCBiagiFFasanoAGreenPHet al. The Oslo definitions for coeliac disease and related terms. Gut. (2013) 62:43–52. 10.1136/gutjnl-2011-301346
8.
TapsasDHollénEStenhammarLFalth-MagnussonK. The clinical presentation of coeliac disease in 1030 Swedish children: changing features over the past four decades. Dig Liver Dis. (2016) 48:16–22. 10.1016/j.dld.2015.09.018
9.
KivelaLKaukinenKLahdeahoMLHuhtalaHAshornMRuuskaTet al. Presentation of celiac disease in Finnish children is no longer changing: A 50-year perspective. J Pediatr. (2015) 167:1109–15. 10.1016/j.jpeds.2015.07.057
10.
JerichoHSansottaNGuandaliniS. Extraintestinal manifestations of celiac disease: effectiveness of the gluten-free diet. J Pediatr Gastroenterol Nutr. (2017) 65:75–9. 10.1097/MPG.0000000000001420
11.
NurminenSKiveläLHuhtalaHKaukinenKKurppaK. Extraintestinal manifestations were common in children with coeliac disease and were more prevalent in patients with more severe clinical and histological presentation. Acta Paediatr. (2018). [Epub ahead of print]. 10.1111/apa.14324
12.
LauretERodrigoL. Celiac disease and autoimmune-associated conditions. Biomed Res Int. (2013) 2013:127589. 10.1155/2013/127589
13.
KahalyGJFrommerLSchuppanD. Celiac disease and glandular autoimmunity. Nutrients. (2018) 10:814. 10.3390/nu10070814
14.
AssaAFrenkel-NirYTzurDKatzLHShamirR. Large population study shows that adolescents with celiac disease have an increased risk of multiple autoimmune and nonautoimmune comorbidities. Acta Paediatr. (2017) 106:967–72. 10.1111/apa.13808
15.
AkirovAPinhas-HamielO. Co-occurrence of type 1 diabetes mellitus and celiac disease. World J Diabetes. (2015) 6: 707–14. 10.4239/wjd.v6.i5.707
16.
VenturaAMagazzùGGrecoL. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. SIGEP Study Group for Autoimmune disorders in Celiac Disease. Gastroenterology. (1999) 117:297–303. 10.1053/gast.1999.0029900297
17.
CacciariESalardiSLazzariRCicognaniACollinaAPirazzoliPet al. Short stature and celiac disease: a relationship to consider even in patients with no gastrointestinal tract symptoms. J Pediatr. (1983) 103:708–11. 10.1016/S0022-3476(83)80462-4
18.
GokceSArslantasE. Changing face and clinical features of celiac disease in children. Pediatr Int. (2015) 57:107–12. 10.1111/ped.12448
19.
JerichoHGuandaliniS. Extra-Intestinal manifestation of celiac disease in children. Nutrients. (2018) 10:755. 10.3390/nu10060755
20.
IughettiLBulgarelliSForeseRLoriniFBalliSBernasconi. Endocrine aspects of coeliac disease. J Pediatr Endocrinol Metab. (2003) 16:805–18. 10.1515/JPEM.2003.16.6.805
21.
HyerWCotterillAMSavageMO. Common causes of short stature detectable by a height surveillance programme. J Med Screen. (1995) 2:150–3. 10.1177/096914139500200310
22.
SaariAHarjuSMakitieOSahaMTDunkelLSankilampiU. Systematic growth monitoring for the early detection of celiac disease in children. JAMA Pediatr. (2015) 169:e1525. 10.1001/jamapediatrics.2015.25
23.
BonamicoMSciréGMarianiPPasquinoAMTriglionePScacciaSet al. Short stature as the primary manifestation of monosymptomatic celiac disease. J Pediatr Gastroenterol Nutr. (1992) 14:12–6.
24.
SinghPSharmaPKAgnihotriAJyotsnaVPDasPGuptaSDet al. Coeliac disease in patients with short stature: a tertiary care centre experience. Natl Med J India. (2015) 28:176–80.
25.
NurminenSKivelaLTaavelaJHuhtalaHMakiMKaukinenKet al. Factors associated with growth disturbance at celiac disease diagnosis in children: a retrospective cohort study. BMC Gastroenterol. (2015) 15:125. 10.1186/s12876-015-0357-4
26.
MeazzaCPaganiSLaarejKCantoniFCivalleroPBonciminoAet al. Short stature in children with coeliac disease. Pediatr Endocrinol Rev. (2009) 6:457–63.
27.
TronconeRKosovaR. Short stature and catch-up growth in celiac disease. J Pediatr Gastroenterol Nutr. (2010) 51 (Suppl. 3):S137–8. 10.1097/MPG.0b013e3181f1dd66
28.
StreetMEVoltaCZiveriMAZanaccaCBanchiniGVianiIet al. Changes and relationships of IGFS and IGFBPS and cytokines in coeliac disease at diagnosis and on gluten-free diet. Clin Endocrinol. (2008) 68:22–8. 10.1111/j.1365-2265.2007.02992.x
29.
DelvecchioMDe BellisAFrancavillaRRutiglianoVPredieriBIndrioFet al. Anti-pituitary antibodies in children with newly diagnosed celiac disease: a novel finding contributing to linear-growth impairment. Am J Gastroenterol. (2010) 105:691–6. 10.1038/ajg.2009.642
30.
PatwariAKKapurGSatyanarayanaLAnandVKJainAGangilAet al. Catch-up growth in children with late-diagnosed celiac disease. Br J Nutr. (2005) 94:437–42. 10.1079/BJN20051479
31.
PeracchiMMolteniNCantalamessaLBardellaMTPeracchiGOrsattiAet al. Abnormal growth hormone responsiveness to stimuli in women with active celiac sprue. Am J Gastroenterol. (1992) 87:580–3.
32.
GiovenaleDMeazzaCCardinaleGMSpositoMMastrangeloCMessiniBet al. The prevalence of growth hormone deficiency and celiac disease in short children. Clin Med Res. (2006) 4:180–3. 10.3121/cmr.4.3.180
33.
CombaACaltepeGYuceOErenaEKalayciAG. Effects of age of diagnosis and dietary compliance on growth parameters of patients with celiac disease. Arch Argent Pediatr. (2018) 116:248–55. 10.5546/aap.2018.eng.248
34.
LefflerDAGreenHRFasanoA. Extraintestinal manifestations of coeliac disease. Nat Rev Gastroenterol Hepatol. (2015) 12:561–71. 10.1038/nrgastro.2015.131
35.
AbaciAEsenIUnuvarTArslanNBoberE. Two cases presenting with pubertal delay and diagnosed as celiac disease. Clin Pediatr Phila. (2008) 47:607–9. 10.1177/0009922808316185
36.
PhilipRPatidarPSaranSAgarwalPAryaTGuptaK. Endocrine manifestations of celiac disease. Indian J Endocrinol Metab. (2012) 16 (Suppl. 2):S506–8. 10.4103/2230-8210.104149
37.
BonaGMarinelloDOderdaG. Mechanisms of abnormal puberty in coeliac disease. Horm Res. (2002) 57 (Suppl. 2):63–5. 10.1159/000058103
38.
KalayciAGKanberYBirinciAYildizLAlbayrakD. The prevalence of coeliac disease as detected by screening in children with iron deficiency anaemia. Acta Paediatr. (2005) 94:678–81. 10.1080/08035250510025879
39.
RajalahtiTRepoMKiveläLHuhtalaHMäkiMKaukinenKet al. Anemia in pediatric celiac disease: Association with clinical and histological features and response to gluten-free diet. J Pediatr Gastroenterol Nutr. (2017) 64:e1–e6. 10.1097/MPG.0000000000001221
40.
MahadevSLaszkowskaMSundströmJBjörkholmMLebwohlBGreenPHRet al. Prevalence of celiac disease in patients with iron deficiency anemia- A systematic review with meta-analysis. Gastroenterology. (2018) 155:374–382.e1. 10.1053/j.gastro.2018.04.016
41.
RepoMLindforsKMäkiMHuhtalaHLaurilaKLähdeahoMLet al. Anemia and iron deficiency in children with potential celiac disease. J Pediatr Gastroenterol Nutr. (2017) 64:56–62. 10.1097/MPG.0000000000001234
42.
AarelaLNurminenSKivelaLHuhtalaHMäkiMViitasaloAet al. Prevalence and associated factors of abnormal liver values in children with celiac disease. Dig Liver Dis. (2016) 48:1023–9. 10.1016/j.dld.2016.05.022
43.
KaukinenKHalmeLCollinPFärkkiläMMäkiMVehmanenPet al. Celiac disease in patients with severe liver disease: gluten-free diet may reverse hepatic failure. Gastroenterology. (2002) 122:881–8. 10.1053/gast.2002.32416
44.
SchwabeRFSekiEBrennerDA. Toll-like receptor signaling in the liver. Gastroenterology. (2006) 130:1886–900. 10.1053/j.gastro.2006.01.038
45.
AnaniaCDe LucaEDe CastroGChiesaCPacificoL. Liver involvement in pediatric celiac disease. World J Gastroenterol. (2015) 21:5813–22. 10.3748/wjg.v21.i19.5813
46.
Korponay-SzabóIRHalttunenTSzalaiZLaurilaKKirályRKovácsJBet al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. (2004) 53:641–8. 10.1136/gut.2003.024836
47.
LeeGJBoyleBEdigerTHillI. Hypertransaminasemia in newly diagnosed pediatric patients with celiac disease. J Pediatr Gastroenterol Nutr. (2016) 63:340–3. 10.1097/MPG.0000000000001153
48.
PantaleoniSLuchinoMAdrianiAPellicanoRStradellaDRibaldoneDGet al. Bone mineral density at diagnosis of celiac disease and after 1 year of gluten-free diet. Sci World J. (2014) 2014:173082. 10.1155/2014/173082
49.
KeavenyAPFreaneyRMcKennaMJMastersonJO'DonoghueDP. Bone remodeling indices and secondary hyperparathyroidism in celiac disease. Am J Gastroenterol. (1996) 91:1226–31.
50.
WeiSKitauraHZhouPRossFPTeitelbaumSL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. (2005) 115:282–90. 10.1172/JCI23394
51.
FioreCEPennisiPFerroGXimenesBPrivitelliLMangiaficoRAet al. Altered osteoprotegerin/RANKL ratio and low bone mineral density in celiac patients on long-term treatment with gluten-free diet. Horm Metab Res. (2006) 38:417–22. 10.1055/s-2006-944548
52.
MargoniDChouliarasGDuscasGVoskakiIVoutsasNPapadopoulouAet al. Bone health in children with celiac disease assessed by dual x-ray absorptiometry: effect of gluten-free diet and predictive value of serum biochemical indices. J Pediatr Gastroenterol Nutr. (2012) 54:680–64. 10.1097/MPG.0b013e31823f5fc5
53.
BjörckSBrundinCKarlssonMAgardhD. Reduced bone mineral density in children with screening-detected celiac disease. J Pediatr Gastroenterol Nutr. (2017) 65:526–32. 10.3390/nu10081015
54.
Krupa-KozakUDrabinskaN. Calcium in gluten-free life: health-related and nutritional implications. Foods. (2016) 5:E51. 10.3390/foods5030051
55.
Krupa-KozakUMarkiewiczLHLamparskiGJuśkiewiczJ. Administration of inulin-supplemented gluten-free diet modified calcium absorption and caecal microbiota in rats in a calcium-dependent manner. Nutrients. (2017) 9:E702. 10.3390/nu9070702
56.
GargKAgarwalPGuptaRKSitaramanS. Joint involvement in children with celiac disease. Indian Pediatr. (2017) 54:946–948. 10.1007/s13312-017-1188-x
57.
IqbalTZaidiMAWellsGAKarshJ. Celiac disease arthropathy and autoimmunity study. J Gastroenterol Hepatol. (2013) 28:99–105. 10.1111/j.1440-1746.2012.07272.x
58.
LudvigssonJFRubio-TapiaAChowdharyVMurrayJASimardJF. Increased risk of systemic lupus erythematosus in 29.000 patients with biopsy-verified celiac disease. J Rheumatol. (2012) 39:1964–70. 10.3899/jrheum.120493
59.
CasellaGBordoMBSchallingRBiancaMVillanacciVSalemmeMet al. Neurological disorders and celiac disease. Minerva Gastroenterologica e Dietologica. (2016) 62:197–206.
60.
LionettiEFrancavillaRPavonePPavoneLFrancavillaTPulvirentiAet al. The neurology of coeliac disease in childhood: what is the evidence? A systematic review and meta-analysis. Dev Med Child Neurol. (2010) 52:700–7. 10.1111/j.1469-8749.2010.03647.x
61.
LudvigssonJFZingoneFTomsonTEkbomACiacciC. Increased risk of epilepsy in biopsy-verified celiac disease: a population-based cohort study. Neurology. (2012)78:1401–7. 10.1212/WNL.0b013e3182544728
62.
Pengiran TengahDSHolmesGKWillsAJ. The prevalence of epilepsy in patients with celiac disease. Epilepsia. (2004) 45:1291–3. 10.1111/j.0013-9580.2004.54104.x
63.
ZelnikNPachtAObeidRLernerA. Range of neurologic disorders in patients with celiac disease. Pediatrics. (2004) 113:1672–6. 10.1542/peds.113.6.1672
64.
GobbiGBouquetFGrecoLLambertiniATassinariCAVenturaAet al. Celiac disease, epilepsy, and cerebral calcifications. Lancet. (1992) 340:439–43. 10.1016/S0387-7604(12)80275-0
65.
ZisPJulianTHadjivassiliouM. Headache associated with coeliac disease: A systematic review and meta-analysis. Nutrients. (2018) 10:1445. 10.3390/nu10101445
66.
NennaRPetrarcaLVerdecchiaPFlorioMPietropaoliNMastrogiorgioGet al. Celiac disease in a large cohort of children and adolescents with recurrent headache: a retrospective study. Dig Liver Dis. (2016) 48:495–498. 10.1016/j.dld.2015.12.015
67.
SlimMRico-VillademorosFCalandreEP. Psychiatric comorbidity in children and adults with gluten-related disorders: a narrative review. Nutrients. (2018) 10:E875. 10.3390/nu10070875
68.
ButwickaALichtensteinPFrisénLAlmqvistCLarssonHLudvigssonJF. Celiac disease is associated with childhood psychiatric disorders: a population-based study. J Pediatr. (2017) 184:87–93.e1. 10.1016/j.jpeds.2017.01.043
69.
NajjarSPearlmanDMAlperKNajjarADevinskyO. Neuroinflammation and psychiatric illness. J Neuroinflammation. (2013) 10:43. 10.1186/1742-2094-10-43
70.
FarmakisEPuntisJWToumbaKJ. Enamel defects in children with coeliac disease. Eur J Paediatr Dent. (2005) 6:129–32.
71.
MajoranaABardelliniERavelliAPlebaniAPolimeniA. Campus G: Implications of gluten exposure period CD clinical forms and HLA typing in the association between celiac disease and dental enamel defects in children. A case-control study. Int J Paediatr Dent. (2010) 20:119–24. 10.1111/j.1365-263X.2009.01028.x
72.
El-HodhodMAEl-AgouzaIAAbdel-AlHKabilNSBayomiKA. Screening for celiac disease in children with dental enamel defects. Pediatrics. (2012) 2012:763–783. 10.5402/2012/763783
73.
AineLMakiMCollinPKeyrilainenO. Dental enamel defects in celiac disease. J Oral Pathol Med. (1990) 19:241–25. 10.1111/j.1600-0714.1990.tb00834.x
74.
AineL. Dental enamel defects and dental maturity in children and adolescents with coeliac disease. Proc Finn Dent Soc. (1986) 82:1–71.
75.
VenturaAMartelossiS. Dental enamel defects and coeliac disease. Arch Dis Child. (1997) 77:91. 10.1136/adc.77.1.91
76.
MarianiPMazzilliMCMarguttiGLionettiPTriglionePPetronzelliFet al. Coeliac disease, enamel defects and HLA typing. Acta Paediatr. (1994) 83:1272–5.
77.
BucciPCarileFSangianantoniAD'AngiòFSantarelliALo MuzioL. Oral aphthous ulcers and dental enamel defects in children with coeliac disease. Acta Paediatr. (2006) 95:203–7. 10.1080/08035250500355022
78.
FessatouSKostakiMKarpathiosT. Coeliac disease and alopecia areata in childhood. J Paediatr Child Health. (2003) 39:152–4. 10.4103/0019-5154.131468
79.
GrazianoMRossiM. An update on the cutaneous manifestations of coeliac disease and non-coeliac gluten sensitivity. Int Rev Immunol. (2018) 5:1–10. 10.1080/08830185.2018.1533008
80.
ReunalaTSalmiTTHervonenKKaukinenKCollinP. Dermatitis herpetiformis: a common extraintestinal manifestation of coeliac disease. Nutrients. (2018) 10:E602. 10.3390/nu10050602
81.
MansikkaEHervonenKKaukinenKCollinPHuhtalaHReunalaTet al. Prognosis of dermatitis herpetiformis patients with and without villous atrophy at diagnosis. Nutrients. (2018) 10:E641. 10.3390/nu10050641
82.
HalfdaransonTRLitzowMRMurrayJA. Hematologic manifestations of celiac disease. Blood. (2007) 109:412–21. 10.1182/blood-2006-07-031104
83.
CorneanREGhebanDSimionescuBMargescuM. Celiac disease among adolescents. Poor growth and delayed puberty. Int J Celiac Dis. (2018) 2:52–7. 10.12691/ijcd-6-2-1
84.
Krupa-KozakU. Pathologic bone alterations in celiac disease: etiology, epidemiology, and treatment. Nutrition. (2014) 30:16–24. 10.1016/j.nut.2013.05.027
85.
HnidaKStamnaesJduPré MFMyslingSJorgensenTJDSollidLMet al. Epitope-dependent functional effects of celiac disease autoantibodies on transglutaminase 2. J Biol Chem. (2016) 291:25542–52. 10.1074/jbc.M116.738161
86.
YuXBUhdeMGreenPHAlaediniA. Review autoantibodies in the extraintestinal manifestations of celiac disease. Nutrients. (2018) 10:1123. 10.3390/nu10081123
87.
Korponay-SzabóIRLaurilaKSzondyZHalttunenTSzalaiZDahlbomIet al. Missing endomysial and reticulin binding of coeliac antibodies in transglutaminase 2 knockout tissues. Gut. (2003) 52:199–204. 10.1136/gut.52.2.199
88.
RoseCArmbrusterFPRuppertJIglBWZillikensDShimanovichI. Autoantibodies against epidermal transglutaminase are a sensitive diagnostic marker in patients with dermatitis herpetiformis on a normal or gluten-free diet. J Am Acad Dermatol. (2009) 61:39–43. 10.1016/j.jaad.2008.12.037
89.
ThomasHBeckKAdamczykMAeschlimannPLangleyMOitaRCet al. Transglutaminase 6: a protein associated with central nervous system development and motor function. Amino Acids. (2013) 44:161–177. 10.1007/s00726-011-1091-z
90.
ZisPRaoDGSarrigiannisPGAeschlimannPAeschlimannDPSandersDet al. Transglutaminase 6 antibodies in gluten neuropathy. Br J Dermatol. (2017) 49:1196–200. 10.1016/j.dld.2017.08.019
91.
HadjivassiliouMAeschlimannPStrigunASandersDDWoodroofeNAeschlimannD. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Br J Dermatol. (2008) 64:332–43. 10.1002/ana.21450
92.
MulderCJJRouvroyeMDvan DamAM. Transglutaminase 6 antibodies are not yet mainstream in neuro-coeliac disease. Dig Liver Dis. (2018) 50:96–7. 10.1016/j.dld.2017.10.001
93.
AlaediniALatovN. A surface plasmon resonance biosensor assay for measurement of anti-GM(1) antibodies in neuropathy. Neurology. (2001) 56:855–60. 10.1212/WNL.56.7.855
94.
AlaediniAGreenPHSanderHWHaysAPGamboaETFasanoAet al. Ganglioside reactive antibodies in the neuropathy associated with celiac disease. J Neuroimmunol. (2002) 127:145–8. 10.1016/S0165-5728(02)00102-9
95.
VoltaUGranitoADe GiorgioR. Antibodies to gangliosides in coeliac disease with neurological manifestations. Aliment Pharmacol Ther. (2005) 21:291–2. 10.1111/j.1365-2036.2005.02291.x
96.
VoltaUDe GiorgioRGranitoAStanghelliniVBarbaraGAvoniPet al. Anti-ganglioside antibodies in coeliac disease with neurological disorders. Dig Liver Dis. (2006). 10:1123. 10.1016/j.dld.2005.11.013
97.
RealAGilbertNHauserBKennedyNShandAGillettHet al. Characterisation of osteoprotegerin autoantibodies in coeliac disease. Calcif Tissue Int. (2015) 97:125–33. 10.1007/s00223-015-0023-4
98.
LarussaTSuraciENazionaleILeoneIMontalciniTAbenavoliLet al. No evidence of circulating autoantibodies against osteoprotegerin in patients with celiac disease. World J Gastroenterol. (2012)18:1622–7. 10.3748/wjg.v18.i14.1622
99.
StulikJHernychovaLPorkertovaSPozlerOTuckovaLSanchezDet al. Identification of new celiac disease autoantigens using proteomic analysis. Proteomics. (2003) 3:951–6. 10.1002/pmic.200300370
100.
LebwohlBLudvigssonJFGreenPH. Celiac disease and non-celiac gluten sensitivity. BMJ. (2015) 351:4347. 10.1136/bmj.h4347
101.
LudvigssonJFCiacciCGreenPHRKaukinenKKorponay-SzaboIRKurppaKet al. Outcome measures in coeliac disease trials: the Tampere recommendations. Gut. (2018) 67:1410–24. 10.1136/gutjnl-2017-314853
102.
CompstonJ. Is fracture risk increased in patients with coeliac disease?Gut. (2003) 52:459–60. 10.1136/gut.52.4.459
103.
SalmiTTHervonenKKurppaKCollinPKaukinenKReunalaT. Celiac disease evolving into dermatitis herpetiformis in patients adhering to normal or gluten-free diet. Scand J Gastroenterol. (2015) 50:387–92. 10.3109/00365521.2014.974204
104.
MeyerSRosenblumS. Activities, Participation and quality of life concepts in children and adolescents with celiac disease: a scoping review. Nutrients. (2017) 9:929. 10.3390/nu9090929
105.
CominoIFernández-Ba-aresFEsteveMOrtigosaLCatillejoGFambuenaBet al. Fecal gluten peptides reveal limitations of serological tests and food questionnaires for monitoring gluten-free diet in celiac disease patients. Am J Gastroenterol. (2016) 111:1456–65. 10.1038/ajg.2016.439
Summary
Keywords
extraintestinal, celiac disease, children, gluten free diet, manifestation, clinical presentation, prognosis
Citation
Nardecchia S, Auricchio R, Discepolo V and Troncone R (2019) Extra-Intestinal Manifestations of Coeliac Disease in Children: Clinical Features and Mechanisms. Front. Pediatr. 7:56. doi: 10.3389/fped.2019.00056
Received
31 December 2018
Accepted
13 February 2019
Published
05 March 2019
Volume
7 - 2019
Edited by
Andrew S. Day, University of Otago, New Zealand
Reviewed by
Tudor Lucian Pop, Iuliu Haţieganu University of Medicine and Pharmacy, Romania; Victor Manuel Navas-López, Hospital Materno-Infantil, Spain
Updates
Copyright
© 2019 Nardecchia, Auricchio, Discepolo and Troncone.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccardo Troncone troncone@unina.it
This article was submitted to Pediatric Gastroenterology, Hepatology and Nutrition, a section of the journal Frontiers in Pediatrics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.