 Jonathan R. Kerr
Jonathan R. Kerr- Department of Microbiology, West Suffolk Hospital Foundation Trust, Bury St Edmunds, United Kingdom
Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME) is a chronic multisystem disease characterized by a variety of symptoms, and exhibits various features of an autoimmune-like disease. Subtypes are well recognized but to date are difficult to identify objectively. The disease may be triggered by infection with a variety of micro-organisms, including Epstein-Barr virus (EBV). A subset of CFS/ME patients exhibit up regulation of EBV virus induced gene 2 (EBI2) mRNA in peripheral blood mononuclear cells (PBMC), and these patients appear to have a more severe disease phenotype and lower levels of EBNA1 IgG. EBI2 is induced by EBV infection and has been found to be upregulated in a variety of autoimmune diseases. EBI2 is a critical gene in immunity and central nervous system function; it is a negative regulator of the innate immune response in monocytes. Its heterogeneous expression in CFS/ME could explain the variable occurrence of a variety of immune and neurological abnormalities which are encountered in patients with CFS/ME. The EBI2 subtype occurred in 38–55% CFS/ME patients in our studies. Further work is required to confirm the role of EBV and of EBI2 and its oxysterol ligands in CFS/ME, and to identify the most practical means to identify patients of the EBI subtype. There are two EBI2 antagonists currently in development, and these may hold promise in the treatment of CFS/ME patients of the EBI subtype.
Introduction
Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME) is a chronic multisystem disease characterized by at least 6 months of fatigue and a variety of other symptoms, including headache, sore throat, muscle pain, joint pain, muscle weakness, post-exertional malaise, sleep abnormalities, and secondary anxiety and depression (1). It is most likely that heterogeneity in CFS/ME is the reason that although there have been many immune and other abnormalities found in patients, none are universal and so there are currently no biomarkers of CFS/ME per se. This is used as evidence against a biological pathogenesis of the disease, however, the most plausible explanation is that CFS/ME is a heterogeneous autoimmune-like disease with a variety of subtypes, a phenomenon typical of autoimmune disease.
Particular problems are, first, that the CFS/ME is unique as a chronic autoimmune-like disease, in that there is no objective means to confirm the diagnosis. Secondly, there are a variety of names and diagnostic criteria some of which do not exclude major depression, as is required by the CDC criteria (1) and Canadian criteria (2). Thirdly, there has been a push to combine biological and unrelated psychological approaches in CFS/ME research. Of course, psychological aspects must be included, but only in terms of what we know is relevant to current knowledge of CFS/ME. Namely, that psychological stress can trigger the disease, and that anxiety and depression are secondary phenomena in CFS/ME (1, 2).
By analogy to autoimmune diseases, psychological aspects are known to be almost universal, for example, anxiety in ulcerative colitis, rheumatoid arthritis, psoriasis, asthma, etc. And the anxiety is believed to underlie relapses and flare-ups which may precipitate hospital admission and use of immune-modifying treatments in a variety of autoimmune diseases.
In this paper, I will review the heterogeneity of CFS/ME, its clinical presentation and triggering factors including Epstein-Barr Virus (EBV) reactivation, CFS/ME as an autoimmune-like disease, Epstein-Barr virus induced (EBI) gene 2 (EBI2) dysregulation in CFS/ME, the “EBI subtype” and the future possibility for therapeutic pharmacological EBI2 antagonism in CFS/ME patients.
Heterogeneity of CFS/ME
Heterogeneity among CFS/ME patients is well recognized. There are clear differences in type of onset (sudden vs. gradual), duration of illness, and different types of illness (predominant pain, predominant flu-like illness, predominant neurological type illness, etc.). In a comparative review of systemic and neurological symptoms in 12 outbreaks of CFS/ME, epidemic neuromyasthenia and ME, marked heterogeneity in the range of neurological symptoms was observed (3), and outbreaks could be grouped into four levels of increasing neurological involvement. Janal et al. (4) subtyped CFS/ME patients according to “minor” symptoms, and identified three subtypes; neurological, musculoskeletal, and infectious. Extreme scores in one or more of these factors accounted for 66% of the sample. Depression and anxiety were not more prevalent in any particular subtype. Jason et al. (5) analyzed data from 18,675 CFS/ME patients and found strong evidence for the existence of a variety of subtypes based on sociodemographic status and disability. Jason et al. (6) have outlined the importance of subtyping of CFS/ME patients, both for the study of pathogenesis and for response to available treatments. These authors identified CFS/ME subtypes based on level of disability, viral, immune, neuroendocrine, neurological, autonomic, and genetic aspects. Proper identification and study of CFS/ME subtypes has been hampered by the lack of concensus as to how to diagnose the disease.
CFS/ME as a Chronic flu-Like Illness Triggered by Virus Infection
At its most simple, CFS/ME can be considered to be a chronic flu-like illness triggered by virus infection, and that the symptoms of CFS/ME are those of a resulting flu-like illness (fatigue, impairment in short term memory or concentration, sore throat, muscle pain, joint pain without swelling or redness, headaches, unrefreshing sleep, and post-exertional malaise) (7). Psychological symptoms (anxiety and depression) are common during flu-like illnesses, and are secondary in otherwise healthy persons. There is a biological basis for such secondary psychological symptoms in that circulating proinflammatory cytokines result in activation of glial cells in the brain and secretion of proinflammatory cytokines by these cells, causing the symptoms of depression, lethargy and anxiety (8). We are all familiar with such short term illness and its symptoms, including the secondary psychological symptoms. We are familiar with the sickness behavior we exhibit during these short term illnesses. For example, we prefer to go to bed early rather than stay out late (8). The principal difference between a short term flu-like illness secondary to virus infection, and CFS/ME, is the duration, severity and the global effect on the lives of patients. In the case of CFS/ME, generally 6 months of symptoms are required (1), while a short term flu-like illness would typically last < 2 weeks.
Psychological Stress is Key to Virus Transmission, Infection, and CFS/ME
For any given virus infection, outcomes vary according to many factors. However, for the purposes of CFS/ME, the key factor is psychological stress. It has been shown for a variety of viruses that psychological stress is necessary for successful virus transmission from one person to another (9). Furthermore, it has been shown that psychological stress is necessary for symptoms to develop after successful transmission, as opposed to asymptomatic infection (9). It is well known that psychological stress is key in the reactivation of herpes viruses, and this precedes the recurrence of cold sores (herpes simplex virus) (10), shingles (varicella-zoster virus) (11), and Epstein-Barr virus (EBV) (12, 13). Psychological stress has been shown to be important in triggering a large proportion of cases of CFS/ME, and this fits perfectly with a viral pathogenesis. Psychological stress is universal and is expected under various circumstances, for example, student examinations, loss of a parent or partner, etc.
Important Microbial Triggers of CFS/ME
Micro-organisms which have been documented to trigger CFS/ME include EBV, enteroviruses, cytomegalovirus (CMV), human herpesvirus-6 (HHV-6), human parvovirus B19, hepatitis C, Chlamydia pneumonia, and Coxiella burnetii (Table 1). EBV is known to infect 90% humans, the majority of which become infected in childhood due to transmission in oral secretions. Following acute infection, EBV persists life-long. It is not clear what percentage of CFS/ME patients are infected with EBV, but in one UK study, it was 90% (34). It is recognized that for each microbial trigger of CFS/ME, that there are a variety of possible clinical outcomes of acute infection, including CFS/ME. It is also recognized, that of those patients who suffered an acute microbial infection which led to development of CFS/ME, there are a number of possible resulting CFS/ME phenotypes, and that this varies depending on other, as yet unknown factors. Therefore, of those patients who developed CFS/ME following parvovirus B19 infection, for example, some will have a CFS/ME phenotype with predominant musculoskeletal pain, while others will have less predominant pain, and more problems with sleep, memory, and concentration, for example. Therefore, there is a lack of correlation between the particular microbial or other trigger and the resulting CFS/ME phenotype.
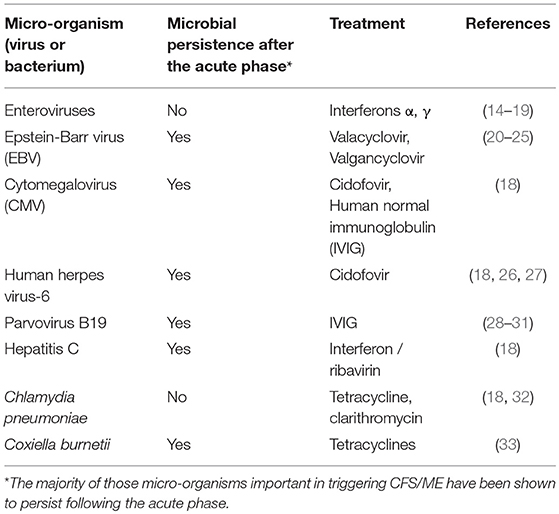
Table 1. Microbial infections which have been shown to trigger CFS/ME.
CFS/ME Exhibits Features of an Autoimmune Disease
A variety of features suggest that CFS/ME may be an autoimmune-like disease. CFS/ME may be triggered by virus infection, and its course characterized by a typical “viral” flu-like illness (7). These observations have led to the recognition that the immune response plays a large and significant role in the pathogenesis of the disease. There are striking similarities between CFS/ME and various autoimmune diseases, for example, Multiple Sclerosis (MS). And, the existence of subtypes of CFS/ME is a further parallel to autoimmune diseases, in which subtypes are well recognized. There are various examples of subtypes of autoimmune diseases exhibiting specific pathogenetic mechanisms, such that particular subtypes of particular autoimmune diseases may be amenable to specific treatments while other subtypes of the same autoimmune disease are not. Studies have demonstrated a variety of immune abnormalities in CFS/ME patients (Table 2), many of which are also found in patients with autoimmune disease. A variety of autoantibodies have been demonstrated in serum of CFS/ME patients including those against nuclear and membrane structures, neurotransmitters and their receptors, cytoplasmic intermediate filaments, EBV dUTPase, and neoepitopes resulting from oxidative or nitrosative damage (53). There is considerable co-morbidity of CFS/ME with other immune or autoimmune diseases, including fibromyalgia (30–77%), postural orthostatic tachycardia syndrome (POTS) (11–40%), Hashimoto's thyroiditis (17–20%), and a family history of an autoimmune disease (18–41%) (53). We have also found upregulated EBI2 mRNA expression in a subset of CFS/ME patients which also occurs in autoimmune diseases (see below).

Table 2. Various immunological abnormalities which have been reported in CFS/ME patients.
Two clinical trials of monoclonal anti-CD20 antibody, rituximab, in CFS/ME patients demonstrate partial or complete benefit in 60%, and in some of these the remission was sustained. This treatment, in both trials, exhibited a delayed onset of response of ~4 months, suggesting that benefit was not directly mediated by CD20 depletion, but by plasma cell depletion followed by washout of short-lived autoantibodies (54, 55).
EBV Reactivation is a Model for Psychological Stress and Triggering of CFS/ME
Acute EBV infection has been shown to be an important virus trigger of CFS/ME (20–22). Study of acute EBV infection in medical students shows that EBV preferentially reactivates during the psychologically stressful time of examinations as compared with other less stressful periods of the academic year (56). This has also been shown for military recruits in training at examination times (57). This reactivation is also paralleled by changes in a large variety of immune markers of cellular immunity which are important in the long-term control and suppression of replication of persistent and asymptomatic EBV in the normal person (12, 13). These studies elegantly document the importance of the balance between persistent EBV and cellular immune system competency which can be disrupted by psychological stress, leading to reactivation and replication of EBV, and subsequent manifestation of symptoms of EBV infection, which if the stress is maintained, can become prolonged and lead to CFS/ME and other diseases, such as nasopharyngeal cancer and post-transplant lymphoproliferative disorder (PTLD) (58). The various types of stress that can adversely affect the efficacy of the cellular immune response include marital stress (59), student examination stress (56), attachment anxiety or fear of abandonment and rejection (60), loneliness (61), etc. But physical stress by itself does not have this effect (62).
Psychological stress triggers release of glucocorticoids which activate EBV lytic infection through the upregulation of the immediate early BZLF1 gene expression (63). The cause of the pro-inflammatory state with EBV reactivation is the EBV-encoded deoxyuridine triphosphate nucleotidohydrolase (dUTPase) which modulates innate immunity in human primary monocyte-derived macrophages through toll-like receptor (TLR)-2 signaling leading to NF-κB activation and the production of pro-inflammatory cytokines. EBV dUTPase induces sickness responses in mice (64). Restraint stress (unavoidable stress which causes autonomic and behavioral changes) results in impairment of learning and memory which is due to expression of EBV dUTPase (65).
CFS/ME patients exhibit prolonged raised antibody titers against EBV dUTPase and EBV DNApol which are neutralizing, and may be used to identify CFS patients in which their disease pathogenesis is due to ongoing EBV reactivation (66). However, global screening of serum antibody responses to an EBV peptide array in serum of CFS/ME patients compared with controls revealed strikingly similar patterns (67).
Epstein-Barr virus (EBV) Induced GENE 2 (EBI2)
Epstein-Barr Virus (EBV) induced gene 2 (EBI2) is a G-Protein Coupled Receptor (GPCR), also known as GPR183, which was originally identified as the main induced gene in Burkitt's Lymphoma cells upon infection with EBV (68). EBI2 has been found to be highly expressed in peripheral blood mononuclear cells (PBMC) (B, T, NK, monocytes, and granulocytes) during EBV reactivation (68–70), is a regulator of B cell partitioning in tissues of the lymphoid system and is critical for T-cell mediated antibody responses (71–74) and inflammation (71, 72, 75). EBI2 has also been found in dendritic cells and monocytes (76). EBI2 is activated by oxysterols and pertussis toxin-sensitive heterotrimeric G proteins, resulting in decreased cyclic AMP, mobilization of calcium and activation of the extracellular signal related kinase (ERK) pathway (69, 77). High affinity EBI2 agonists are the oxysterol, 7α 25-dihydroxycholesterol (7α25HC) and related compounds (78, 79). Activation of EBI2 with 7α25HC results in a wide range of functional responses including cell migration and calcium mobilization (78, 79). 7α25HC is synthesized from cholesterol (80). Other oxysterols also activate EBI2 but with lower potency (78).
EBI2 also plays an important role in the central nervous system (69). Astrocytes are the macrophages of the brain and protect it against invading pathogens and astrocyte abnormalities are implicated in multiple sclerosis (MS), Parkinsons Disease (PD), Amyotrophic Lateral Sclerosis (ALS), and Alzheimer Disease (AD). Oligodendrocytes are the main cells involved in myelination of nerve fibers of the brain and are implicated in leukodystrophies and leukoencephalopathies. Cholesterol is a crucial component of myelin and cholesterol deficiency results in motor symptoms (81). Abnormal levels of oxysterols have been found in AD, MS and Experimental Allergic Encephalomyelitis (EAE). Mutations in CYP7B1 gene have been demonstrated in Spastic Paraplegia Gene 5 (SPG5) and lead to lesions of upper motor neurones, periventricular areas and subcortical white matter (82, 83). Brain cholesterol is synthesized in the brain as it can't traverse the blood brain barrier (BBB) (84, 85). The cholesterol metabolite, 24(S)-hydroxycholesterol (24(S)HC) is a brain-specific oxysterol which is thought to be synthesized exclusively in the brain and secreted into the circulation via the BBB to maintain steady levels of brain cholesterol (85). Circulating 24(S)HC is believed to be a biomarker for brain cholesterol homeostasis and neurodegenerative disease (MS, PD, AD) (85–87). Oxysterols also have detrimental effects on myelin and oligodendrocyte viability (88, 89).
Dysregulation of EBI2 expression has been demonstrated in EBV infection (68, 69, 73, 74, 76), melanoma metastasis, lymphoblastic leukemia, glioblastoma, bone cancer metastasis, systemic lupus erythematous, chronic rhino sinusitis with nasal polyps, Type 1 Diabetes (69), and CFS/ME (90) (see below). Aberrant oxysterol signaling has been demonstrated in Multiple Sclerosis, Experimental Allergic Encephalomyelitis (EAE), Alzheimer's disease, Parkinsons Disease, Motor Neurone Disease, Cerebrotendinous Xanthomatosis, Hereditary spastic paraplegia type 5 (SPG5), Huntingdon Disease, Age related macular degeneration, atherosclerosis, Inflammatory bowel disease, and osteoporosis (69). EBI2 regulates several genes, important in monocyte function, which are important in the pathogenesis of glioblastoma multiforme and Type 1 Diabetes Mellitus. Knock down of the EBI2 gene in rat monocytes, results in upregulated IRF7 expression. As IRF7 is a critical regulator of the type 1 interferon response, this suggests that EBI2 is a negative regulator of the innate immune response in macrophages (91). However, the precise role played by EBI2 in EBV infection remains to be clarified (68, 69).
Expression of EBI2 in CFS/ME
We have previously found that CFS/ME patients exhibit significantly upregulated expression of EBI2 in PBMC as compared with normal controls, in gene expression arrays and reverse-transcriptase polymerase chain reaction (RT-PCR) confirmation assays (90). EBI2 mRNA (NM_004951) expression was found to be significantly upregulated by a factor of 1.3 in microarray experiments using PBMC from 25 CFS patients vs. 50 normal human controls matched 2:1. This upregulation was confirmed using RT-PCR in 55 CFS/ME patients vs. 75 normal human controls at a fold difference of 3.44 (P = 0.0012) using ABI assay number Hs00270639_s1 (90). In this study, EBI2 was found to be unregulated in 55% CFS/ME patients, one of whom was a 26 year old woman whose CFS/ME had been triggered by laboratory documented EBV infection 10 years prior (90). All those with raised expression of EBI2 were also positive for serum anti-VCA IgG.
Microarray experiments identified 88 human genes which were upregulated in CFS/ME, and the RT-PCR expression data on these 88 human genes in 52 CFS patients was then clustered and this identified seven gene expression subtypes. These gene expression derived subtypes differed significantly in measures of clinical symptomatology and neurocognitive functioning (90). Of these 88 human genes which were differentially expressed in CFS patients, they could be divided into two groups each with 44 genes, one group of which showed quite predictable up regulation across most CFS/ME patients and in the other group, the expression was much more variable. EBI2 was one of those which was more variably upregulated in PBMC of CFS/ME patients. This is illustrated in Figure 1, in which 12 of 31 (38%) patients exhibited EBI2 up regulation as compared with none of 40 normal controls.
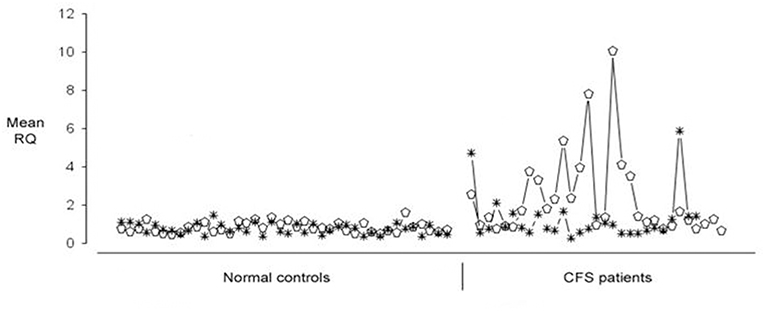
Figure 1. Expression of Epstein-Barr Virus (EBV) induced gene 2 (EBI2) (open ellipse) and Neuropathy Target Esterase (NTE) (asterisk) genes in 40 healthy blood donors (shown on the Left) and 31 Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME) patients (shown on the Right). Upregulated EBI2 mRNA expression was demonstrated in 12 of 31 CFS/ME patients, and in none of the controls.
Although it was known in 2008 that EBI2 was upregulated in PBMC of CFS/ME patients (90), the significance was not understood at the time as little was known about the gene and its function. However, with the recent identification of EBI2 as a critical regulator of the immune response with importance for a variety of autoimmune diseases and cancer, its significance in the pathogenesis of CFS/ME has been recognized.
Possible Link Between EBI2 Expression, EBV Latency, and Clinical Severity in CFS/ME
As part of a separate study of microbial infections in CFS/ME, we analyzed EBV antibody markers in 117 CFS/ME patients which had been grouped into eight subtypes (A-H) based on clustering of RT-PCR expression data for 88 CFS/ME-associated genes (34). These 117 CFS/ME patients included 55 CFS/ME patients who had been included in the previous study (90), 56 CFS/ME patients who had not previously been studied and six whose disease had been triggered by acute Q fever (QFS) (34). The CFS/ME patients exhibited 90% EBV seropositivity which is to be expected (34).
Subtype D was the most interesting in terms of EBV infection markers and clinical phenotype (34). Subtype D consisted only of females and had the most severe clinical phenotype, with the lowest functional level on SF-36 scoring (physical role, vitality, general health, bodily pain, and total score) and a high frequency of muscle pain and sleep problems (Figures 2A,B). EBI2 was expressed at the highest level in PBMC of subtype D patients [fold difference (CFSME/Normal), 14.93] as compared with the other subtypes [Mean fold difference (CFSME/Normal), 3.004] (34). There were no discernible differences in EBV antibody markers [viral capsid antigen (VCA) IgM and IgG, early antigen (EA) IgG, and Epstein-Barr nuclear antigen (EBNA) IgG] between subtypes, except that in Subtype D, CFS/ME patients had a markedly reduced titer of EBNA IgG (Figure 2D).
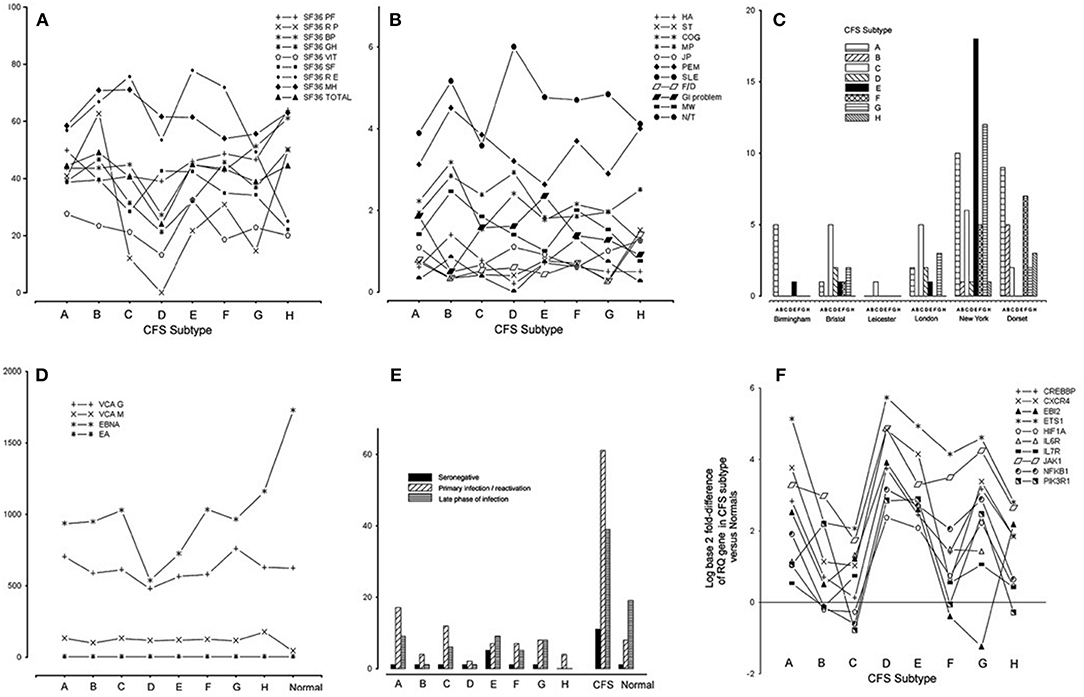
Figure 2. (A) Medical Outcomes Survey Short Form-36 (SF36) domain and total scores for each chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) subtype: physical function (PF), physical role (RP), bodily pain (BP), general health (GH), vitality (VIT), social functioning (SF), emotional role (RE), mental health (MH), and total score (Total). (B) Scores indicating occurrence and severity of 11 clinical symptoms for each CFS/ME subtype: headache (HA), sore throat (ST), swollen glands (GLA), cognitive defect (COG), muscle pain (MP), joint pain (JP), muscle weakness (MW), post-exertional malaise (PEM), sleep problems (SLE), fainting/dizziness (F/D), gastrointestinal complaints (GI), numbness/tingling (N/T), spatial span (SSP), verbal recognition memory (VRM). (C) Histogram showing the numbers of CFS/ME patients of each subtype occurring in each of the six geographical locations. (D) Epstein-Barr virus (EBV) antibody titers [viral capsid antigen (VCA) IgM, VCA IgG, early antigen (EA) IgG, Epstein-Barr nuclear antigen (EBNA) IgG] in each CFS/ME subtype and the normal comparison group. (E) Distribution of categories of EBV serostatus (seronegative, primary/re-activation, late phase of infection) in the CFS/ME subtypes, A-H, in CFS/ME (all subtypes combined) and in normal controls. (F) Log (base 2) of fold-difference values of 10 human genes known to be important in EBV infection, in eight CFS subtypes (A-H). Reproduced from Figure 1 of reference no. 34 with permission from BMJ Publishing Group Ltd. (License number 4413611406714).
Epstein-Barr virus nuclear antigen 1 gene (EBNA1) is important in establishing and maintaining the altered state that cells undergo during EBV infection, and is the only EBV protein found in all EBV-associated malignancies. EBNA1 has a glycine-alanine repeat which stabilizes the protein, prevents its breakdown, impairs antigen processing, and MHC class I-restricted antigen presentation, resulting in inhibition of the CD8-restricted cytotoxic T lymphocyte response against EBV infected cells, thus favoring latency (92). The finding that CFS/ME patients of subtype D with EBI2 mRNA upregulation had lower titers of EBNA IgG than the other subtypes (Figure 2D), supports the idea that subtype D is associated with a higher prevalence of EBV latency, as lytic infection is required to expose this antigen to circulating lymphocytes, a necessary step in developing serum EBNA IgG positivity.
In one study, it was shown that in 10% CFS/ME patients, EBNA IgG titers were low or absent (93). Multicolor flow cytometry revealed that the frequencies of EBNA-1-specific triple TNF-α/IFN-γ/IL-2 producing CD4(+) and CD8(+) T-cell subsets were significantly diminished in CFS/ME patients (93).
Within the CFS/ME-associated gene signature of 88 human genes, 12 had recognized associations with EBV infection. One of these was EBI2, as discussed, and the others were NFKB1, EGR1, ETS1, GABPA, CREBBP, CXCR4, HIF1A, JAK1, IL6R, IL7R, and PIK3R1. Striking associations were found for these 12 genes across subtypes, and subtype D had the highest levels of all of them in PBMC (Figure 2F).
Heterogeneous EBI2 Upregulation May Contribute to the Variability of Immune and Neurological Abnormalities in CFS/ME
The following have been variably been found in CFS/ME patients; increase in the number of circulating B lymphocytes, increase in activated T lymphocytes, reduction in NK cell numbers and/or function, deficiency in antibody-mediated cellular cytotoxicity (ADCC), Th2 profile of helper T cell responsiveness, reduced TGF1 expression, increased neutrophil apoptosis, and deficiencies in particular IgG subsets (Table 2). During the normal humoral immune response, activated B cells upregulate EBI2 which mediates their journey to the outer follicle where they interact with T helper cells. After CD40 engagement, EBI2 expression results in cells moving away from the B-T boundary toward the outer and inner areas of the follicle (73), differentiation into plasmablasts, and mounting of a rapid antibody response. Some B cells move to the central follicle, differentiate into germinal center B cells, to later exhibit antibody affinity maturation (71–75). A higher than normal expression of EBI2 could result in both increased numbers of B cells in the circulation, and in reduced T cell help and therefore deficiencies in particular IgG subsets, and reduced antibody affinity maturation. EBV reactivation is associated with expansion of differentiated and activated CD4 and CD8 T lymphocytes and later with decline in these cells as exhaustion takes over (94), and so the timing of EBV infection in CFS/ME will affect research studies on immune abnormalities. NK cells are important in defense during the early stages of primary EBV infection (94). Innate immune control of lytic EBV infection by early differentiated NK cells was found to attenuate infectious mononucleosis (IM) (95). It has been proposed that NK cells are important in the long-term control of EBV (96), which may account for the variable findings related to NK numbers and function in CFS/ME, as not all CFS/ME patients will have EBV reactivation at the time of sampling.
Abnormalities in white matter, gray matter and in cerebral perfusion have been found in CFS/ME patients, and these occur in a similar presentation to those of MS patients. Both CFS/ME and MS patients have reduced cerebral perfusion, gray matter reduction and white matter hyper intensities, although individual patients are variably affected (97). As EBI2 expression and oxysterol dysregulation have been linked with the pathogenesis of MS (69), and as MS is characterized by a relapsing and remitting course, in which subtypes exist and in which EBI2 is variably upregulated in some of these, it is logical to suggest that heterogeneous EBI2 expression may similarly play a role in the neurological abnormalities found in CFS/ME.
Although we know that there are a variety of immune abnormalities which occur with regularity in CFS/ME patients, these do not occur invariably, and none can be used as a marker for the presence of the disease. However, the upregulation of EBI2 in a subset of CFS/ME patients may contribute to this phenomenon.
EBI2 Modulators
EBI2 is a key receptor in B, T and dendritic cells, modulating the T and B cell response to blood borne antigens (76). As EBI2 and/or its oxysterol ligands are upregulated in B cell malignancies and autoimmune diseases (Type 1 Diabetes, rheumatoid arthritis, systemic lupus erythematosus, and multiple sclerosis), two EBI2 modulators are being developed. GSK682753A is a small molecule, potent EBI2 antagonist which blocks 7a25HC stimulation of the EBI2 receptor in a recombinant system (98). NIBR189 is a potent selective antagonist of EBI2, which has been developed in paradigms relevant to cardiovascular disease (99).
Limitations of the Hypothesis that EBI2 Upregulation is Important in a Subset of CFS/me Patients
The raw data underpinning the present review were generated by only one research group. Therefore, it would be important that these findings are replicated in additional CFS/ME patients and normal controls by independent research groups.
Elevated levels of antibodies to EBV VCA, EA, and DNase have been reported to occur in CFS/ME patients (20, 21, 66, 100–105) albeit inconsistently (106–108). However, it is important to understand that EBV antibody markers may associate with CFS/ME, but this does not prove that EBV has triggered the disease in those particular cases. The seroprevalence of EBV in the general population and in CFS/ME patients is ~90%. And the proportion of CFS/ME patients with EBI2 upregulation was found to be between 38 and 55% CFS/ME patients, all of whom had IgG to EBV VCA. For a disease in which a variety of microbial triggers are recognized, our findings are consistent with the hypothesis that upregulation of EBI2 is important in the pathogenesis of disease in a subset of CFS/ME patients. However, this hypothesis remains wholly unproven and it is not understood what factors, in addition to EBV infection, are required for upregulation of EBI2 in an individual patient.
Although EBI2 was found to be the most upregulated gene in EBV-infected Burkitt lymphoma cells (68) and has been shown to be important in a variety of autoimmune diseases and cancers, the particular role of EBI2 in the pathogenesis of EBV infection still remains to be elucidated. Therefore, the possible pathogenetic role of EBI2 upregulation in CFS/ME patients remains speculative at present.
Conclusion
CFS/ME is a heterogeneous disease which is frequently triggered by virus infection, including EBV. Subtypes are well recognized but to date are difficult to identify objectively. Evidence is presented to document that a subset of CFS/ME patients exhibit up regulation of EBI2 mRNA in PBMC. EBI2 is a gene which is induced by EBV infection and which has been found to be upregulated in a variety of autoimmune diseases. EBI2 is a critical gene in immunity and central nervous system function; it is a negative regulator of the innate immune response in monocytes. Its heterogeneous expression in CFS/ME may indicate an ongoing host response to EBV reactivation and on this basis, could explain the heterogeneous occurrence of many of the immune and neurological abnormalities reported in CFS/ME patients. The EBI subtype may account for 38–55% CFS/ME patients. EBI2 antagonists may hold promise for the treatment of CFS/ME patients of the EBI subtype. Further work is required to confirm the role of EBV and of EBI2 and its oxysterol ligands in CFS/ME, and to identify the most practical means to identify patients of the EBI subtype.
Data Availability
All relevant data is contained within the manuscript.
Author Contributions
JK conceived the idea for this review, collated the data, and wrote the paper, without assistance from any other person.
Funding
The author acknowledges the kind support of the New Jersey Myalgic Encephalomyelitis / Chronic Fatigue Syndrome Association (NJME/CFSA) with the production costs of this article.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med. (1994) 121:953–9. doi: 10.7326/0003-4819-121-12-199412150-00009
2. Carruthers BM, van de Sande MI, De Meirleir KL, Klimas NG, Broderick G, Mitchell T, et al. Myalgic encephalomyelitis: international consensus criteria. J Intern Med. (2011) 270:327–38. doi: 10.1111/j.1365-2796.2011.02428.x
3. Briggs NC, Levine PH. A comparative review of systemic and neurological symptomatology in 12 outbreaks collectively described as chronic fatigue syndrome, epidemic neuromyasthenia, and myalgic encephalomyelitis. Clin Infect Dis. (1994) 18(Suppl. 1):S32–42. doi: 10.1093/clinids/18.Supplement_1.S32
4. Janal MN, Ciccone DS, Natelson BH. Sub-typing CFS patients on the basis of “minor” symptoms. Biol Psychol. (2006) 73:124–31. doi: 10.1016/j.biopsycho.2006.01.003
5. Jason LA, Taylor RR, Kennedy CL, Jordan KM, Song S, Johnson D, et al. Chronic fatigue syndrome: symptom subtypes in a community based sample. Women Health. (2003) 37:1–13. doi: 10.1300/J013v37n01_01
6. Jason LA, Corradi K, Torres-Harding S, Taylor RR, King C. Chronic fatigue syndrome: the need for subtypes. Neuropsychol Rev. (2005) 15:29–58. doi: 10.1007/s11065-005-3588-2
7. Devanur LD, Kerr JR. Chronic fatigue syndrome. J Clin Virol. (2006) 37:139–50. doi: 10.1016/j.jcv.2006.08.013
8. Dantzer R. Cytokine, sickness behavior, and depression. Neurol Clin. (2006) 24:441–60. doi: 10.1016/j.ncl.2006.03.003
9. Cohen S, Tyrrell DA, Smith AP. Psychological stress and susceptibility to the common cold. N Engl J Med. (1991) 325:606–12. doi: 10.1056/NEJM199108293250903
10. Padgett DA, Sheridan JF, Dorne J, Berntson GG, Candelora J, Glaser R. Social stress and the reactivation of latent herpes simplex virus type 1. Proc Natl Acad Sci USA. (1998) 95:7231–5. doi: 10.1073/pnas.95.12.7231
11. Kim CK, Choi YM, Bae E, Jue MS, So HS, Hwang ES. Reduced NK cell IFN-γ secretion and psychological stress are independently associated with herpes zoster. PLoS ONE. (2018) 13:e0193299. doi: 10.1371/journal.pone.0193299
12. Glaser R, Rice J, Sheridan J, Fertel R, Stout J, Speicher C, et al. Stress-related immune suppression: health implications. Brain Behav Immun. (1987) 1:7–20. doi: 10.1016/0889-1591(87)90002-X
13. Glaser R, Pearson GR, Jones JF, Hillhouse J, Kennedy S, Mao HY, et al. Stress-related activation of Epstein-Barr virus. Brain Behav Immun. (1991) 5:219–32. doi: 10.1016/0889-1591(91)90018-6
14. Yousef GE, Bell EJ, Mann GF, Murugesan V, Smith DG, McCartney RA, et al. Chronic enterovirus infection in patients with postviral fatigue syndrome. Lancet. (1988) 1:146–50. doi: 10.1016/S0140-6736(88)92722-5
15. Gow JW, Behan WM, Clements GB, Woodall C, Riding M, Behan PO. Enteroviral RNA sequences detected by polymerase chain reaction in muscle of patients with postviral fatigue syndrome. BMJ. (1991) 302:692–6. doi: 10.1136/bmj.302.6778.692
16. Clements GB, McGarry F, Nairn C, Galbraith DN. Detection of enterovirus- specific RNA in serum: the relationship to chronic fatigue. J Med Virol. (1995) 45:156–61. doi: 10.1002/jmv.1890450208
17. Chia JK. The role of enterovirus in chronic fatigue syndrome. J Clin Pathol. (2005) 58:1126–32. doi: 10.1136/jcp.2004.020255
18. Chia JK, Chia A. Diverse etiologies for chronic fatigue syndrome. Clin Infect Dis. (2003) 36:671–2. doi: 10.1086/367666
19. Lane RJ, Soteriou BA, Zhang H, Archard LC. Enterovirus related metabolic myopathy: a postviral fatigue syndrome. J Neurol Neurosurg Psychiatry. (2003) 74:1382–6. doi: 10.1136/jnnp.74.10.1382
20. Jones JF. Epstein-Barr virus and the chronic fatigue syndrome: a short review. Microbiol Sci. (1988) 5:366–9.
21. Straus SE, Tosato G, Armstrong G, Lawley T, Preble OT, Henle W, et al. Persisting illness and fatigue in adults with evidence of Epstein-Barr virus infection. Ann Intern Med. (1985) 102:7–16. doi: 10.7326/0003-4819-102-1-7
22. White PD, Thomas JM, Kangro HO, Bruce-Jones WD, Amess J, Crawford DH, et al. Predictions and associations of fatigue syndromes and mood disorders that occur after infectious mononucleosis. Lancet. (2001) 358:1946–54. doi: 10.1016/S0140-6736(01)06961-6
23. Lerner AM, Beqaj SH, Deeter RG, Dworkin HJ, Zervos M, Chang CH, et al. A 6-month trial of valacyclovir in the Epstein-Barr virus subset of chronic fatigue syndrome: improvement in left ventricular function. Drugs Today. (2002) 38:549–61. doi: 10.1358/dot.2002.38.8.820095
24. Montoya JG, Kogelnik AM, Bhangoo M, Lunn MR, Flamand L, Merrihew LE, et al. Randomized clinical trial to evaluate the efficacy and safety of valganciclovir in a subset of patients with chronic fatigue syndrome. J Med Virol. (2013) 85:2101–9. doi: 10.1002/jmv.23713
25. Watt T, Oberfoell S, Balise R, Lunn MR, Kar AK, Merrihew L, et al. Response to valganciclovir in chronic fatigue syndrome patients with human herpesvirus 6 and Epstein-Barr virus IgG antibody titers. J Med Virol. (2012) 84:1967–74. doi: 10.1002/jmv.23411
26. Ablashi DV, Eastman HB, Owen CB, Roman MM, Friedman J, Zabriskie JB, et al. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J Clin Virol. (2000) 16:179–91. doi: 10.1016/S1386-6532(99)00079-7
27. Nicolson GL, Gan R, Haier J. Multiple co-infections (Mycoplasma, Chlamydia, human herpes virus-6) in blood of chronic fatigue syndrome patients: association with signs and symptoms. APMIS. (2003) 111:557–66. doi: 10.1034/j.1600-0463.2003.1110504.x
28. Jacobson SK, Daly JS, Thorne GM, McIntosh K. Chronic parvovirus B19 infection resulting in chronic fatigue syndrome: case history and review. Clin Infect Dis. (1997) 24:1048–51. doi: 10.1086/513627
29. Kerr JR, Barah F, Mattey DL, Laing I, Hopkins SJ, Hutchinson IV, et al. Circulating tumour necrosis factor-alpha and interferon-gamma are detectable during acute and convalescent parvovirus B19 infection and are associated with prolonged and chronic fatigue. J Gen Virol. (2001) 82:3011–9. doi: 10.1099/0022-1317-82-12-3011
30. Kerr JR, Bracewell J, Laing I, Mattey DL, Bernstein RM, Bruce IN, et al. Chronic fatigue syndrome and arthralgia following parvovirus B19 infection. J Rheumatol. (2002) 29:595–602.
31. Kerr JR, Cunniffe VS, Kelleher P, Bernstein RM, Bruce IN. Successful intravenous immunoglobulin therapy in 3 cases of parvovirus B19-associated chronic fatigue syndrome. Clin Infect Dis. (2003) 36:e100–6. doi: 10.1086/374666
32. Chia JK, Chia LY. Chronic Chlamydia pneumoniae infection: a treatable cause of chronic fatigue syndrome. Clin Infect Dis. (1999) 29:452–3. doi: 10.1086/520239
33. Arashima Y, Kato K, Komiya T, Kumasaka K, Matsukawa Y, Murakami M, et al. Improvement of chronic nonspecific symptoms by long-term minocycline treatment in Japanese patients with Coxiella burnetii infection considered to have post-Q fever fatigue syndrome. Intern Med. (2004) 43:49–54. doi: 10.2169/internalmedicine.43.49
34. Zhang L, Gough J, Christmas D, Mattey DL, Richards SC, Main J, et al. Microbial infections in eight genomic subtypes of chronic fatigue syndrome/myalgic encephalomyelitis. J Clin Pathol. (2010) 63:156–64. doi: 10.1136/jcp.2009.072561
35. Klimas NG, Salvato FR, Morgan R, Fletcher MA. Immunologic abnormalities in chronic fatigue syndrome. J Clin Microbiol. (1990) 28:1403–10.
36. Tirelli U, Marotta G, Improta S, Pinto A. Immunological abnormalities in patients with chronic fatigue syndrome. Scand J Immunol. (1994) 40:601–8. doi: 10.1111/j.1365-3083.1994.tb03511.x
37. Robertson MJ, Schacterle RS, Mackin GA, Wilson SN, Bloomingdale KL, Ritz J, et al. Lymphocyte subset differences in patients with chronic fatigue syndrome, multiple sclerosis and major depression. Clin Exp Immunol. (2005) 141:326–32. doi: 10.1111/j.1365-2249.2005.02833.x
38. Sabath DE, Barcy S, Koelle DM, Zeh J, Ashton S, Buchwald D. Cellular immunity in monozygotic twins discordant for chronic fatigue syndrome. J Infect Dis. (2002) 185:828–32. doi: 10.1086/339194
39. Straus SE, Fritz S, Dale JK, Gould B, Strober W. Lymphocyte phenotype and function in the chronic fatigue syndrome. J Clin Immunol. (1993) 13:30–40. doi: 10.1007/BF00920633
40. Whiteside TL, Friberg D. Natural killer cells and natural killer cell activ- ity in chronic fatigue syndrome. Am J Med. (1998) 105:27–34S. doi: 10.1016/S0002-9343(98)00155-7
41. Caligiuri M, Murray C, Buchwald D, Levine H, Cheney P, Peterson D, et al. Phenotypic and functional deficiency of natural killer cells in patients with chronic fatigue syndrome. J Immunol. (1987) 139:3306–13.
42. Levine PH, Whiteside TL, Friberg D, Bryant J, Colclough G, Herberman RB. Dysfunction of natural killer activity in a family with chronic fatigue syndrome. Clin Immunol Immunopathol. (1998) 88:96–104. doi: 10.1006/clin.1998.4554
43. Aoki T, Miyakoshi H, Usuda Y, Herberman RB. Low NK syndrome and its relationship to chronic fatigue syndrome. Clin Immunol Immunopathol. (1993) 69:253–65. doi: 10.1006/clin.1993.1178
44. Visser J, Blauw B, Hinloopen B, Brommer E, de Kloet ER, Kluft C, et al. CD4 T lymphocytes from patients with chronic fatigue syndrome have decreased interferon-gamma production and increased sensitivity to dexamethasone. J Infect Dis. (1998) 177:451–4. doi: 10.1086/517373
45. Skowera A, Cleare A, Blair D, Bevis L, Wessely SC, Peakman M. High levels of type 2 cytokine-producing cells in chronic fatigue syndrome. Clin Exp Immunol. (2004) 135:294–302. doi: 10.1111/j.1365-2249.2004.02354.x
46. Hanson SJ, Gause W, Natelson B. Detection of immunologically significant factors for chronic fatigue syndrome using neural-network classifiers. Clin Diagn Lab Immunol. (2001) 8:658–62. doi: 10.1128/CDLI.8.3.658-662.2001
47. Tomoda A, Joudoi T, Rabab el-M, Matsumoto T, Park TH, Miike T. Cytokine production and modulation: comparison of patients with chronic fatigue syndrome and normal controls. Psychiatry Res. (2005) 134:101–4. doi: 10.1016/j.psychres.2005.01.002
48. Kennedy G, Spence V, Underwood C, Belch JJ. Increased neutrophil apoptosis in chronic fatigue syndrome. J Clin Pathol. (2004) 57:891–3. doi: 10.1136/jcp.2003.015511
49. Read R, Spickett G, Harvey J, Edwards AJ, Larson HE. IgG1 subclass deficiency in patients with chronic fatigue syndrome. Lancet. (1988) 1:241–2. doi: 10.1016/S0140-6736(88)91091-4
50. Natelson BH, Haghighi MH, Ponzio NM. Evidence for the presence of immune dysfunction in chronic fatigue syndrome. Clin Diagn Lab Immunol. (2002) 9:747–52. doi: 10.1128/CDLI.9.4.747-752.2002
51. Komaroff AL, Geiger AM, Wormsely S. IgG subclass deficiencies in chronic fatigue syndrome. Lancet. (1988) 1:1288–9. doi: 10.1016/S0140-6736(88)92109-5
52. Linde A, Hammarstrom L, Smith CI. IgG subclass deficiency and chronic fatigue syndrome. Lancet. (1988) 1:885–6. doi: 10.1016/S0140-6736(88)91633-9
53. Sotzny F, Blanco J, Capelli E, Castro-Marrero J, Steiner S, Murovska M, et al. European network on ME/CFS (EUROMENE). Myalgic encephalomyelitis/chronic fatigue syndrome - evidence for an autoimmune disease. Autoimmun Rev. (2018) 17:601–9. doi: 10.1016/j.autrev.2018.01.009
54. Fluge O, Bruland O, Risa K, Storstein A, Kristoffersen EK, Sapkota D, et al. Benefit from B- lymphocyte depletion using the anti-CD20 antibody rituximab in chronic fatigue syndrome. A double- blind and placebo-controlled study. PLoS ONE. (2011) 6:e26358. doi: 10.1371/journal.pone.0026358
55. Fluge O, Risa K, Lunde S, Alme K, Rekeland IG, Sapkota D, et al. B-lymphocyte depletion in Myalgic encephalopathy/chronic fatigue syndrome. An open-label phase II study with rituximab maintenance treatment. PLoS ONE. (2015) 10:e0129898. doi: 10.1371/journal.pone.0129898
56. Glaser R, Pearl DK, Kiecolt-Glaser JK, Malarkey WB. Plasma cortisol levels and reactivation of latent Epstein-Barr virus in response to examination stress. Psychoneuroendocrinology. (1994) 19:765–72. doi: 10.1016/0306-4530(94)90023-X
57. Glaser R, Friedman SB, Smyth J, Ader R, Bijur P, Brunell P, et al. The differential impact of training stress and final examination stress on herpesvirus latency at the United States Military Academy at West Point. Brain Behav Immun. (1999) 13:240–51. doi: 10.1006/brbi.1999.0566
58. Li H, Liu S, Hu J, Luo X, Li N, M Bode A, et al. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int J Biol Sci. (2016) 12:1309–18. doi: 10.7150/ijbs.16564
59. Jaremka LM, Glaser R, Malarkey WB, Kiecolt-Glaser JK. Marital distress prospectively predicts poorer cellular immune function. Psychoneuroendocrinology. (2013) 38:2713–9. doi: 10.1016/j.psyneuen.2013.06.031
60. Fagundes CP, Jaremka LM, Glaser R, Alfano CM, Povoski SP, Lipari AM, et al. Attachment anxiety is related to Epstein-Barr virus latency. Brain Behav Immun. (2014) 41:232–8. doi: 10.1016/j.bbi.2014.04.002
61. Glaser R, Kiecolt-Glaser JK, Speicher CE, Holliday JE. Stress, loneliness, and changes in herpesvirus latency. J Behav Med. (1985) 8:249–60. doi: 10.1007/BF00870312
62. Aubrecht TG, Weil ZM, Abi Salloum B, Ariza ME, Williams M, Reader B, et al. Chronic physical stress does not interact with Epstein-Barr Virus (EBV)-encoded dutpase to alter the sickness response. J Behav Brain Sci. (2015) 5:513–23. doi: 10.4236/jbbs.2015.511049
63. Yang EV, Webster Marketon JI, Chen M, Lo KW, Kim SJ, Glaser R. Glucocorticoids activate Epstein Barr virus lytic replication through the upregulation of immediate early BZLF1 gene expression. Brain Behav Immun. (2010) 24:1089–96. doi: 10.1016/j.bbi.2010.04.013
64. Padgett DA, Hotchkiss AK, Pyter LM, Nelson RJ, Yang E, Yeh PE, et al. Epstein-Barr virus-encoded dUTPase modulates immune function and induces sickness behavior in mice. J Med Virol. (2004) 74:442–8. doi: 10.1002/jmv.20196
65. Aubrecht TG, Weil ZM, Ariza ME, Williams M, Reader BF, Glaser R, et al. Epstein-Barr virus (EBV)-encoded dUTPase and chronic restraint induce impaired learning and memory and sickness responses. Physiol Behav. (2014) 137:18–24. doi: 10.1016/j.physbeh.2014.07.001
66. Lerner AM, Ariza ME, Williams M, Jason L, Beqaj S, Fitzgerald JT, et al. Antibody to Epstein-Barr virus deoxyuridine triphosphate nucleotidohydrolase and deoxyribonucleotide polymerase in a chronic fatigue syndrome subset. PLoS ONE. (2012) 7:e47891. doi: 10.1371/journal.pone.0047891
67. Loebel M, Eckey M, Sotzny F, Hahn E, Bauer S, Grabowski P, et al. Serological profiling of the EBV immune response in Chronic Fatigue Syndrome using a peptide microarray. PLoS ONE. (2017) 12:e0179124. doi: 10.1371/journal.pone.0179124
68. Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, Kieff E. Epstein-Barr virus-induced genes: first lymphocyte-specific G protein-coupled peptide receptors. J Virol. (1993) 67:2209–20.
69. Rutkowska A, Dev KK, Sailer AW. The role of the Oxysterol/EBI2 pathway in the immune and central nervous systems. Curr Drug Targets. (2016) 17:1851–60. doi: 10.2174/1389450117666160217123042
70. Benned-Jensen T, Norn C, Laurent S, Madsen CM, Larsen HM, Arfelt KN, et al. Molecular characterization of oxysterol binding to the Epstein-Barr virus-induced gene 2 (GPR183). J Biol Chem. (2012) 287:35470–83. doi: 10.1074/jbc.M112.387894
71. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity. (2009) 31:259–69. doi: 10.1016/j.immuni.2009.06.016
72. Gatto D, Wood K, Brink R. EBI2 operates independently of but in cooperation with CXCR5 and CCR7 to direct B cell migration and organization in follicles and the germinal center. J Immunol. (2011) 187:4621–8. doi: 10.4049/jimmunol.1101542
73. Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and nonlymphoid tissues. J Immunol. (2011) 187:3026–32. doi: 10.4049/jimmunol.1101262
74. Pereira JP, Xu Y, Cyster JG. A role for S1P and S1P1 in immature-B cell egress from mouse bone marrow. PLoS ONE. (2010) 5:e9277. doi: 10.1371/journal.pone.0009277
75. Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature. (2009) 460:1122–6. doi: 10.1038/nature08226
76. Sun S, Liu C. 7α, 25-dihydroxycholesterol-mediated activation of EBI2 in immune regulation and diseases. Front Pharmacol. (2015) 6:60. doi: 10.3389/fphar.2015.00060
77. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature. (2011) 475:524–7. doi: 10.1038/nature10280
78. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature. (2011) 475:519–23. doi: 10.1038/nature10226
79. Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity. (2012) 37:535–48. doi: 10.1016/j.immuni.2012.06.015
80. Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC, et al. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. (2005) 8:468–75. doi: 10.1038/nn1426
81. Schüle R, Schöls L. Genetics of hereditary spastic paraplegias. Semin Neurol. (2011) 31:484–93. doi: 10.1055/s-0031-1299787
82. Biancheri R, Ciccolella M, Rossi A, Tessa A, Cassandrini D, Minetti C, et al. White matter lesions in spastic paraplegia with mutations in SPG5/CYP7B1. Neuromuscul Disord. (2009) 19:62–5. doi: 10.1016/j.nmd.2008.10.009
83. Leoni V, Caccia C. Oxysterols as biomarkers in neurodegenerative diseases. Chem Phys Lipids. (2011) 164:515–24. doi: 10.1016/j.chemphyslip.2011.04.002
84. Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. (2004) 24:806–15. doi: 10.1161/01.ATV.0000120374.59826.1b
85. Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD, et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. (2000) 41:195–8.
86. Leoni V, Masterman T, Diczfalusy U, De Luca G, Hillert J, Björkhem I. Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci Lett. (2002) 331:163–6. doi: 10.1016/S0304-3940(02)00887-X
87. Makoukji J, Shackleford G, Meffre D, Grenier J, Liere P, Lobaccaro JM, et al. Interplay between LXR and Wnt/β-catenin signaling in the negative regulation of peripheral myelin genes by oxysterols. J Neurosci. (2011) 31:9620–9. doi: 10.1523/JNEUROSCI.0761-11.2011
88. Trousson A, Bernard S, Petit PX, Liere P, Pianos A, El Hadri K, et al. 25-hydroxycholesterol provokes oligodendrocyte cell line apoptosis and stimuates the secreted phospholipase A2 type IIA via LXR beta and PXR. J Neurochem. (2009) 109:945–58. doi: 10.1111/j.1471-4159.2009.06009.x
89. Moog C, Luu B, Beck JP, Italiano L, Bischoff P. Studies on the immunosuppressive properties of 7,25-dihydroxycholesterol–I. Reduction of interleukin production by treated lymphocytes. Int J Immunopharmacol. (1988) 10:511–8. doi: 10.1016/0192-0561(88)90067-7
90. Kerr JR, Petty R, Burke B, Gough J, Fear D, Sinclair LI, et al. Gene expression subtypes in patients with chronic fatigue syndrome/myalgic encephalomyelitis. J Infect Dis. (2008) 197:1171–84. doi: 10.1086/533453
91. Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. (2010) 467:460–4. doi: 10.1038/nature09386
92. Frappier L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses. (2012) 4:1537–47. doi: 10.3390/v4091537
93. Loebel M, Strohschein K, Giannini C, Koelsch U, Bauer S, Doebis C, et al. Deficient EBV-specific B- and T-cell response in patients with chronic fatigue syndrome. PLoS ONE. (2014) 9:e85387. doi: 10.1371/journal.pone.0085387
94. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin Transl Immunol. (2017) 6:e126. doi: 10.1038/cti.2016.87
95. Chijioke O, Landtwing V, Münz C. NK cell influence on the outcome of primary Epstein-Barr virus infection. Front Immunol. (2016) 7:323. doi: 10.3389/fimmu.2016.00323
96. Münz C. Epstein-Barr virus-specific immune control by innate lymphocytes. Front Immunol. (2017) 8:1658. doi: 10.3389/fimmu.2017.01658
97. Morris G, Berk M, Puri BK. A comparison of neuroimaging abnormalities in multiple sclerosis, major depression and chronic fatigue syndrome (Myalgic Encephalomyelitis): is there a common cause? Mol Neurobiol. (2018) 55:3592–609. doi: 10.1007/s12035-017-0598-z
98. Benned-Jensen T, Madsen CM, Arfelt KN, Smethurts C, Blanchard A, Jepras R, et al. Small molecule antagonism of oxysterol-induced Epstein-Barr virus induced gene 2 (EBI2) activation. FEBS Open Bio. (2013) 3:156–60. doi: 10.1016/j.fob.2013.02.003
99. Gessier F, Preuss I, Yin H, Rosenkilde MM, Laurent S, Endres R, et al. Identification and characterization of small molecule modulators of the Epstein-Barr virus-induced gene 2 (EBI2) receptor. J Med Chem. (2014) 57:3358–68. doi: 10.1021/jm4019355
100. Buchwald D, Sullivan JL, Komaroff AL. Frequency of 'chronic active Epstein-Barr virus infection' in a general medical practice. JAMA. (1987) 257:2303–7.
101. Hellinger WC, Smith TF, Van Scoy RE, Spitzer PG, Forgacs P, Edson RS. Chronic fatigue syndrome and the diagnostic utility of antibody to Epstein-Barr virus early antigen. JAMA. (1988) 260:971–3. doi: 10.1001/jama.1988.03410070099036
102. Holmes GP, Kaplan JE, Stewart JA, Hunt B, Pinsky PF, Schonberger LB. A cluster of patients with a chronic mononucleosis-like syndrome. Is Epstein-Barr virus the cause? JAMA. (1987) 257:2297–302. doi: 10.1001/jama.1987.03390170053027
103. Lerner AM, Beqaj SH, Deeter RG, Fitzgerald JT. IgM serum antibodies to Epstein-Barr virus are uniquely present in a subset of patients with the chronic fatigue syndrome. In Vivo. (2004) 18:101–6.
104. Sairenji T, Yamanishi K, Tachibana Y, Bertoni G, Kurata T. Antibody responses to Epstein-Barr virus, human herpesvirus 6 and human herpesvirus 7 in patients with chronic fatigue syndrome. Intervirology. (1995) 38:269–73. doi: 10.1159/000150450
105. Kawai K, Kawai A. Studies on the relationship between chronic fatigue syndrome and Epstein-Barr virus in Japan. Intern Med. (1992) 31:313–8. doi: 10.2169/internalmedicine.31.313
106. Cameron B, Flamand L, Juwana H, Middeldorp J, Naing Z, Rawlinson W, et al. Serological and virological investigation of the role of the herpesviruses EBV, CMV and HHV-6 in post-infective fatigue syndrome. J Med Virol. (2010) 82:1684–8. doi: 10.1002/jmv.21873
107. Wallace HL II, Natelson B, Gause W, Hay J. Human herpesviruses in chronic fatigue syndrome. Clin Diagn Lab Immunol. (1999) 6:216–23.
Keywords: Chronic Fatigue Syndrome, Myalgic Encephalomyelitis, Epstein-Barr virus, Epstein-Barr virus induced gene 2, autoimmune, microarray, real-time polymerase chain reaction
Citation: Kerr JR (2019) Epstein-Barr Virus Induced Gene-2 Upregulation Identifies a Particular Subtype of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. Front. Pediatr. 7:59. doi: 10.3389/fped.2019.00059
Received: 28 August 2018; Accepted: 13 February 2019;
Published: 13 March 2019.
Edited by:
Zaher Nahle, Arthritis National Research Foundation, United StatesReviewed by:
Jill Edith Cadwgan, Evelina London Children's Hospital, United KingdomVincent C. Lombardi, University of Nevada, United States
Copyright © 2019 Kerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan R. Kerr, am9uYXRoYW5Ac3NsLW1haWwuY29t