 Alejandra Aird1Macarena Lagos2,3Alexander Vargas-Hernández4Jennifer E. Posey5Zeynep Coban-Akdemir5Shalini Jhangiani5,6
Alejandra Aird1Macarena Lagos2,3Alexander Vargas-Hernández4Jennifer E. Posey5Zeynep Coban-Akdemir5Shalini Jhangiani5,6 Emily M. Mace7
Emily M. Mace7 Anaid Reyes4Alejandra King1
Anaid Reyes4Alejandra King1 Felipe Cavagnaro1Lisa R. Forbes4
Felipe Cavagnaro1Lisa R. Forbes4 Ivan K. Chinn4James R. Lupski4,5,6
Ivan K. Chinn4James R. Lupski4,5,6 Jordan S. Orange7
Jordan S. Orange7 Maria Cecilia Poli1,4*
Maria Cecilia Poli1,4*- 1Clínica Alemana de Santiago, Facultad de Medicina Clínica Alemana-Universidad del Desarrollo, Santiago, Chile
- 2Clínica Las Condes, Santiago, Chile
- 3Hospital Padre Hurtado, Santiago, Chile
- 4Section of Immunology, Allergy and Rheumatology, Department of Pediatrics, Center for Human Immunobiology, Baylor College of Medicine, Texas Children's Hospital, Houston, TX, United States
- 5Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, United States
- 6Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, United States
- 7Division of Immunogenetics, Department of Pediatrics, Morgan Stanley Children's Hospital of New York Presbyterian, Columbia University Irving Medical Center, New York, NY, United States
Nuclear factor kappa-B subunit 2 (NF-κB2/p100/p52), encoded by NFKB2 (MIM: 164012) belongs to the NF-κB family of transcription factors that play a critical role in inflammation, immunity, cell proliferation, differentiation and survival. Heterozygous C-terminal mutations in NFKB2 have been associated with early-onset common variable immunodeficiency (CVID), central adrenal insufficiency and ectodermal dysplasia. Only two previously reported cases have documented decreased natural killer (NK) cell cytotoxicity, and little is known about the role of NF-κB2 in NK cell maturation and function. Here we report a 13-year-old female that presented at 6 years of age with a history of early onset recurrent sinopulmonary infections, progressive hair loss, and hypogamaglobulinemia consistent with a clinical diagnosis of CVID. At 9 years of age she had cytomegalovirus (CMV) pneumonia that responded to ganciclovir treatment. Functional NK cell testing demonstrated decreased NK cell cytotoxicity despite normal NK cell numbers, consistent with a greater susceptibility to systemic CMV infection. Research exome sequencing (ES) was performed and revealed a novel de novo heterozygous nonsense mutation in NFKB2 (c.2611C>T, p.Gln871*) that was not carried by either of her parents. The variant was Sanger sequenced and confirmed to be de novo in the patient. At age 12, she presented with a reactivation of the systemic CMV infection that was associated with severe and progressive nephrotic syndrome with histologic evidence of pedicellar effacement and negative immunofluorescence. To our knowledge, this is the third NF-κB2 deficient patient in which an abnormal NK cell function has been observed, suggesting a role for non-canonical NF-κB2 signaling in NK cell cytotoxicity. NK cell function should be assessed in patients with mutations in the non-canonical NF-κB pathway to explore the risk for systemic viral infections that may lead to severe complications and impact patient survival. Similarly NF-κB2 should be considered in patients with combined immunodeficiency who have aberrant NK cell function. Further studies are needed to characterize the role of NF-κB2 in NK cell cytotoxic function.
Background
Common variable immunodeficiency (CVID) is one of the most frequently diagnosed symptomatic primary immunodeficiencies. Clinical and immunophenotypic manifestations are highly heterogeneous and it most typically presents clinically as recurrent infections, pulmonary inflammation with or without bronchiectasis, and sometimes associated with autoimmune manifestations such as granulomatous disease and lymphoproliferation (1). Characteristically, there is B cell dysfunction that results in impaired antibody production and hypogammaglobulinemia; and up to one third of the patients may have an associated T cell defect. The underlying genetic mechanisms for CVID have been elucidated in <10% of cases and more than 20 genetic causes for CVID have been described (1–3). NF-κB transcription factors are essential for the immune response, and mutations in NF-κB transcription factors and their regulators result in primary immunodeficiency and autoimmunity (4, 5). The non-canonical NF-κB pathway is critical for B cell survival, differentiation into plasma cells and isotype class switching as well as dendritic cell activation (6, 7). A number of mono- and biallelic mutations in this pathway have been identified among CVID patients (i.e., BAFFR, TACI, TNFSF12, TRAF3, NIK, NFKB2). Interestingly, in the case of TACI/Taci locus, the association of both heterozygous and biallelic variations with AD/AR CVID disease trait, suggest subtle gene dosage perturbations might underlie this phenotype (8).
Nuclear factor kappa-B subunit 2 (NF-κB2) is composed of p100 protein, a central component of the non-canonical NF-κB pathway that also serves as an inhibitor of the canonical pathway. The p100 protein is activated by phosphorylation of its C-terminal serine residues (Ser 866, Ser 870), triggering its breakdown to p52, that is then translocated into the nucleus (7). The association between CVID and central adrenal insufficiency had been previously recognized as Deficient Anterior pituitary with Variable Immune Deficiency (DAVID) syndrome (9). Later, heterozygous C-terminal mutations in NFKB2 (MIM 164012) were determined to cause early onset CVID with variable association with central adrenal insufficiency, ectodermal dysplasia, and autoimmunity (10–16). The immune phenotype of these patients is characterized by profound B cell deficiency and defects in peripheral T cell proliferation and differentiation (12, 17). Decreased natural killer (NK) cell function has also been reported (15, 18).
Here we describe a female patient with a novel heterozygous nonsense mutation in NFKB2 presenting with early onset CVID, ectodermal dysplasia, subclinical adrenal insufficiency and functional NK cell deficiency with overwhelming systemic CMV infection, uniquely associated with severe nephrotic syndrome. This case report highlights the relevance of NK cell function in prognosis and suggests that functional NK cell evaluation could impact treatment strategies in these patients.
Case Presentation
A female patient born to non-consanguineous parents, with no family history of immunodeficiency or endocrine disorders was asymptomatic until 2 years of age when she presented with hair loss progressing to alopecia universalis, trachyonychia (sandpapered, rough nails with accentuated linear ridges), psoriatic-like dermatitis and atopic dermatitis. Facial and dental anomalies were not detected (Figure 1A). Subsequently, she developed recurrent bacterial upper and lower respiratory infections and immunologic evaluation at 6 years of age showed hypogammaglobulinemia (IgG 180 mg/dl, IgA< 4 mg/dl, IgM 4 mg/dl), low B cells with absent memory B cells and non-protective antibody titers to tetanus and pneumococcal vaccines consistent with CVID with absent B cells. T and NK cell numbers were normal, but T-cell proliferation to phytohemagglutinin (PHA) was decreased (Table 1).
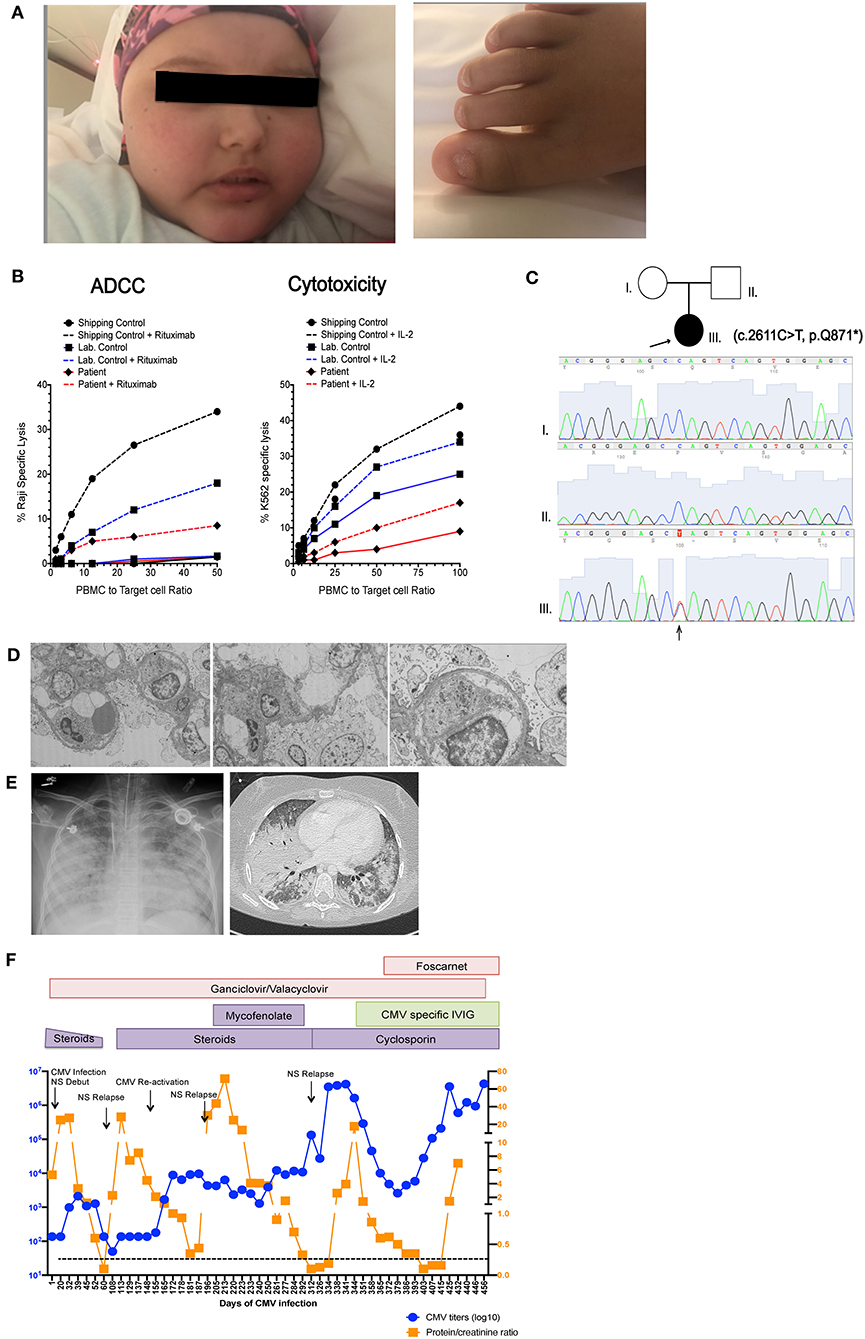
Figure 1. Patient clinical presentation. (A) Left picture shows the face of the patient with alopecia totalis, edema and Cushingoid facies due to prolonged steroid treatment. Right picture shows trachyonychia of the toenails. (B) Pedigree of the family showing two healthy non-consanguineous parents and the patient (arrow) with c.2611C>T mutation in NFKB2. Sanger tracings of each individual in the family are presented. (C) NK cell cytotoxic function measured by 51Cr release assay. Left graph shows antibody dependent cellular cytotoxicity (ADCC) measured against Raji B cell targets in in the presence (dashed line) and absence (solid line) of rituximab. Right shows natural cytotoxicity against K562 targets in the presence (dashed line) and absence (solid line) of IL-2 stimulation. Due to sample availability and transport limitations this demonstrates a single assay performed in triplicate. (D) Electron microscopy of renal biopsy showing pedicellar effacement and few mesangial deposits (E) Left picture shows a chest X ray and chest CT scan taken during the patient's final stage of disease showing multiple bilateral foci of alveolar compromise with extensive consolidation of bilateral basal lobes, superior left lobe and medial lobe. (F) Graphic representation showing variations in CMV titers in blood (in blue) and proteinuria (in orange), Black arrows indicate nephrotic syndrome (NS) reactivations. Immunosuppressive and antiviral treatments are indicated in colored boxes as they were administered during the course of disease.
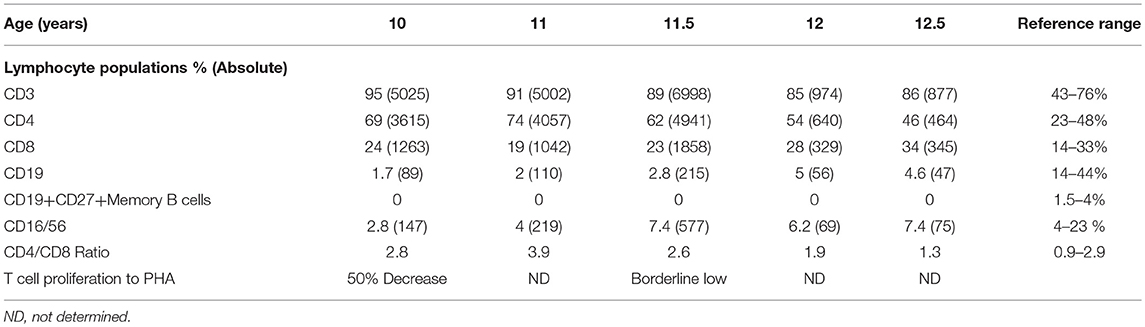
Table 1. Patient immunophenotyping.
The patient remained free of significant infections on monthly intravenous immunoglobulin (IVIG) replacement until 9 years of age, when she developed CMV pneumonia that was successfully treated with intravenous ganciclovir, achieving negative CMV polymerase chain reaction (PCR) testing on follow up. After this first CMV infection she developed recurrent upper and lower respiratory infections despite immunoglobulin replacement and required antibiotic prophylaxis. CMV infection could have been a feature of her depressed T cell function but also raised the possibility for a functional NK cell abnormality. Hence, NK cell function was assessed by chromium release assay using K562 target cells, which demonstrated decreased NK cell cytotoxicity with poor improvement with IL-2, as well as decreased antibody-dependent cell cytotoxicity (Figure 1B).
After obtaining informed assent by the patient and informed consent from her parents, trio Whole Exome sequencing (WES) was performed and identified a de novo heterozygous c.2611C>T, (variant to total reads 123/221 = 0.55) p.Gln871* variant in NFKB2 (NM_001077494) leading to a premature stop codon in the C-terminal portion of NF-κB2. WES results at this locus and the de novo nature of the variant allele were confirmed by orthogonal Sanger sequencing (Figure 1C). NMDEscPredictor (19) was used to determine if this variant would be predicted to escape nonsense mediated decay (NMD). Similar to other C-terminal NFKB2 variants (Table 2) Gln871* is predicted to escape NMD and likely result in translation of a truncated protein. This variant is not present in ExAC, gnomAD, the 1000 Genomes Project or other personal genomes from ~9,000 subject samples studies by WES within our internal Center for Mendelian Genomics (CMG) database; therefore we are interpreting it as a novel disease-causing rare variant presumably loss of function (LoF) allele. The ExAC calculated probability of loss of function intolerance (pLi) for this gene is 1, meaning that it is unlikely to tolerate LoF. No other known primary immunodeficiency mutations or potential candidate gene were identified in this patient. With this result, pituitary function was assessed and studied by objective laboratory analyses that demonstrated reduced serum cortisol and adrenocorticotropic hormone (ACTH) levels; thus glucocorticoid replacement was initiated. Other pituitary hormones were within normal limits. Autoantibody testing, including antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibodies (ANCA), anti-adrenal and anti-thyroid antibodies were all negative.
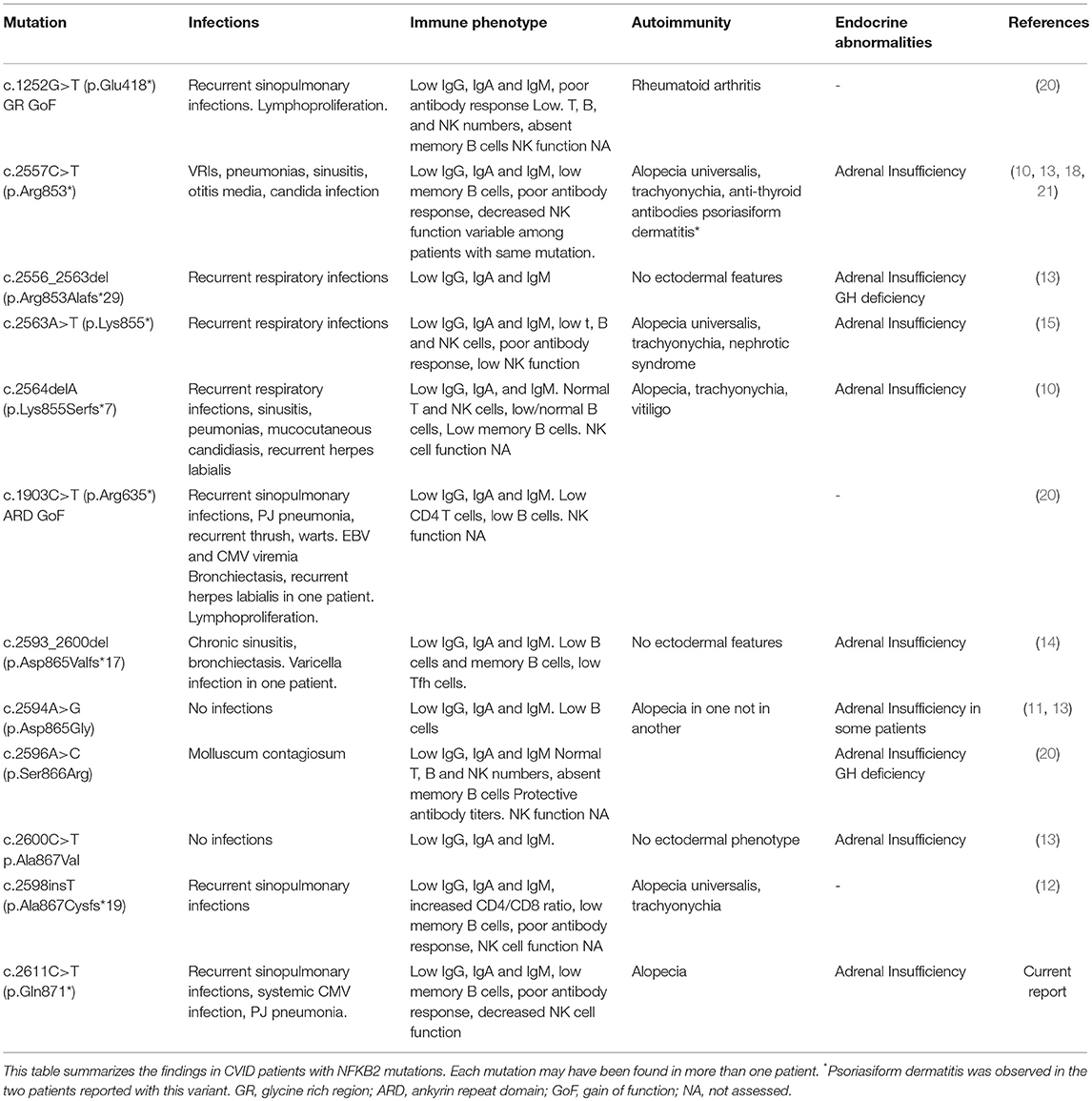
Table 2. NFKB2 mutations.
At 12 years of age the patient was admitted with gastroenteritis, generalized intestinal edema and severe nephrotic syndrome progressing to acute renal failure. Quantitative CMV PCR was positive >2 × 106 copies/ml in blood and positive in a renal biopsy (not quantified in tissue). Renal histology showed focal-segmental glomerulosclerosis with extracapillary hypercellularity and mild tubulointerstitial atrophy. Ultrastructural findings showed complete pedicellar effacement characteristic of minimal change nephropathy and no intracellular inclusions or antibody deposits were demonstrated on cytopathology (Figure 1D). Immunofluorescence was faintly positive for C3 and IgG and negative for C1q, IgA, IgM, and fibrin. Exome files were screened for pathogenic podocin mutations that can be associated with nephrotic syndrome, and no pathogenic variants were identified. Interestingly, elevations in CMV titers in blood seemed to precede nephrotic syndrome relapses and correlate with increases in proteinuria (Figure 1F). Due to chronic systemic CMV infection and severe nephrotic syndrome with acute renal failure, she required intermittent critical level care and treatment with systemic steroids. She also received IVIG, broad-spectrum antibiotics, Pneumocystis jiroveci prophylaxis with cotrimoxazole, ganciclovir progressing to foscarnet due to proven resistant CMV strains and hyperimmune CMV-specific immunoglobulin (Cytogam®). Despite aggressive anti-viral therapy, it was impossible to fully control the CMV infection or the nephrotic syndrome that fluctuated in accordance with blood CMV titers that remained between 1 × 106 and 4 × 106 copies/ml despite aggressive treatment. Immunosuppressive therapy, first mycophenolate mofetil (MMF) later cyclosporine were initiated. She had an initial response to this treatment achieving reduction of proteinuria. Unfortunately, CMV infection could not be cleared and the nephrotic syndrome finally became unresponsive to treatment. In this clinical context stem cell transplant was not feasible. During this final end stage disease, she developed Pneumocytis jiroveci pneumonia and fungal lung infection (Figure 1E). Bronchoalveolar lavage was positive for Pneumocystis jiroveci, CMV and galactomannan. She progressed to multi-organ failure and died at 13 years of age, 1 year after her initial admission for nephrotic syndrome associated with systemic CMV infection.
Heterogenic immune abnormalities may have contributed to patient clinical manifestations and susceptibility to chronic CMV infection. In order to understand these abnormalities, further phenotypic characterization of patient T cells and NK cells was performed post mortem from cryopreserved PBMC. In T cells a reduction in central memory and effector memory T cells in both CD4+ and CD8+ T cell compartments was observed (Figure 2A). Comprehensive NK cell phenotyping was performed as previously described (22). NK cell numbers and the proportion of CD56bright and CD56dim NK cells were within normal limits and similar to a healthy donor control (Figure 2B). There were no detected abnormalities in expression of NK cell receptors associated with adhesion, activation and inhibition, or NK cell development, and despite chronic CMV infection we did not observe expansion of NKG2C+ or CD57+ subsets as has previously been reported (23). Cytokine production was assessed after 5-h stimulation with PMA and ionomycin. While interferon gamma production was similar to control, a reduction in production of GM-CSF and TNFa was observed. CD107a mobilization, and perforin levels were comparable to control (Figure 2C).

Figure 2. (A) Flow cytometry dot plots showing T cell memory subsets in total CD3+ T cells as well as in CD4+ and CD8+ subsets. (B) Flow cytometry dot plots showing patient and control NK CD56+ NK cells and gating on CD56Dim and CD56Bright NK cells. (C) 5 panels showing mean fluorescence intensity and percent of positive cells for different NK cell markers and receptors within CD56 Dim and CD56 Bright NK cells. Low percentages of cells compared to healthy donor are marked in red.
Written informed consent was obtained from the parents of the patient for the publication of this case report and any potentially-identifying information/images.
Discussion
Two members of the NF-κB transcription factor family, NF-κB1 and NF-κB2, are produced as precursor proteins, NF-κB1 p105 and NF-κB2 p100. Transcriptional regulation by NF-κB proteins involves canonical and alternative pathways that result in the nuclear translocation of the activated form of these proteins, p50 and p52 respectively. In the non-canonical pathway, the NFKB2 transcripts are first translated into a p100 precursor protein that undergoes activation-dependent cytoplasmic proteolytic processing through NF-κB-inducing kinase (NIK) into the smaller active transcription factor p52. NF-κB2/p100 processing is stimulated by a subset of NF-κB inducers (lymphotoxin-β, B-cell activating factor and CD40 ligand) to regulate peripheral lymphoid organogenesis, B-lymphocyte differentiation and adaptive humoral immunity (24, 25).
As previously reported, mutations at the 3'end of the NFKB2 gene, encoding the C-terminal region of the protein, both perturb immune and endocrine function resulting in central adrenocorticotropic hormone deficiency and CVID (10–13). Our patient carries a novel C-terminal mutation in NF-κB2 and presented with a clinical and immunological phenotype that very closely resembles other individuals with C-terminal mutations in this gene, including early onset combined immunodeficiency, alopecia and trachyonychia. Adrenal insufficiency was not clinically evident and was only diagnosed after the NFKB2 mutation was identified.
Most of the mutations associated with primary immunodeficiency that have been described for NFKB2 are LoF mutations in the C-terminal domain, with variations at Arg853 being the most frequently reported (Figure 3). In most cases the nonsense/frameshift alleles with premature termination codons (PTC) have transcripts predicted to escape NMD and result in a truncated proteins that loose phosphorylation sites that are important for its interaction with NIK. As a result, p100 is not processed into p52. Two families with gain-of-function (GoF) alleles have been recently described, the mRNA transcribed from these disease-causing nonsense variants also seems to escape NMD and result in truncated proteins that, in contrast to C-terminal nonsense mutations, result in increased accumulation of p52 in the nucleus. Interestingly, for the E418* variant, one asymptomatic family member carries the variant but has no truncated protein expression; suggesting mRNA degradation may be protecting him from disease (20). Our patient's stop gain mutation is also predicted to not render the mRNA unstable and subjected to NMD and thus the mutant transcript is likely to be translated and encode a C-terminal-aberrant protein due to the PTC (19). The Gln871 residue is immediately adjacent to the S870 phosphorylation site of NF-κB2 where p100 interacts with NIK to induce its processing into p52. Hence, it is likely that Gln871* would interfere with p100 processing to p52 resulting in decreased p52 in the nucleus, thus resembling Nfkb2 mutant mice and previously described patients with C-terminal missense mutations that compromise the NIK interaction region as well as nonsense and frameshift mutations that result in PTCs (Table 2, Figure 3) (13, 26).
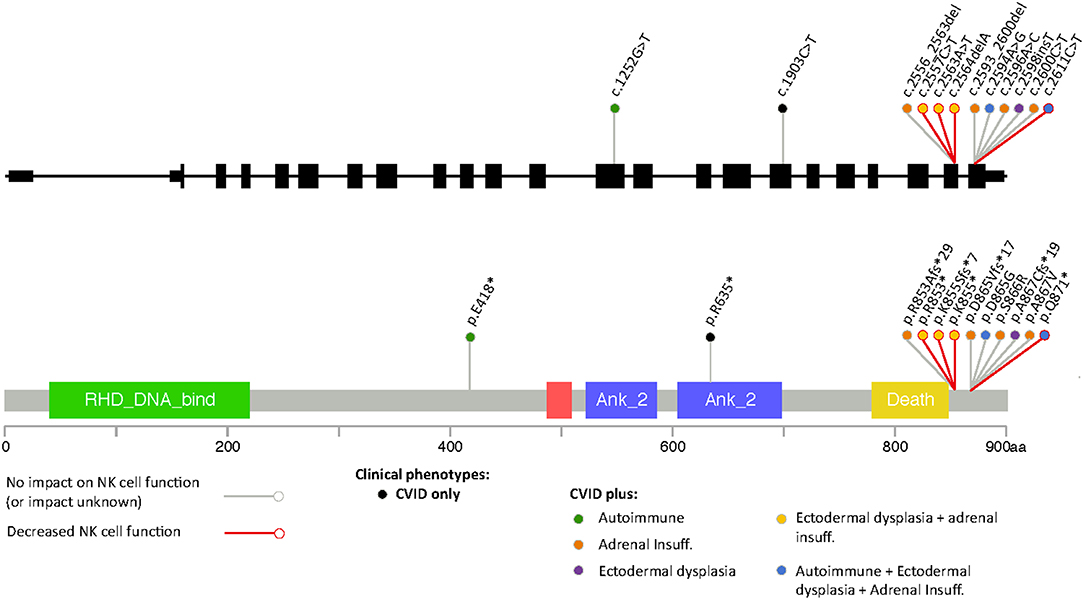
Figure 3. Representation of different variants in NFKB2 that have been associated to disease color-coded with the different clinical manifestations including defects in NK cell function.
In patients with C-terminal NFKB2 mutations, the defect in the non-canonical NF-κB pathway invariably impairs B-cell function, resulting in hypogammaglobulinemia and poor antigen-specific antibody responses and presenting as CVID in early childhood (Table 2). Defects in peripheral T cells (12) as well as decreased natural killer (NK) cell function have also been described (15, 18).
NK cells are critically important for anti-tumoral and anti-viral defense and primary immunodeficiencies in which the main immunological defect is in NK cells (NK cell deficiencies) are clinically characterized by disseminated or recurrent viral infections due to CMV, herpes simplex virus (HSV), human papilloma virus (HPV), virus varicella zoster (VVZ) and Epstein Barr virus (EBV) (27). Five out of 6 NK cell deficiencies involve a defect in NK cell maturation.
NF-κB1 plays a role in NK cell maturation and effector function and patients with monoallelic mutations in NF-κB1 have functional NK cell defects that correlate with a susceptibility to viral infections mainly with viruses from the herpes family, including Epstein-Barr virus (EBV), varicella zoster and CMV (28). The role of NF-κB2 in NK cell function has not been clearly defined and an aberrant NK cell function does not seem to be a consistent feature of patients with C-terminal mutations (Figure 3). Thus far, it has been demonstrated in two previously published cases and ruled out in three additional patients (15, 18, 29). Some patients with C-terminal NFKB2 mutations have presented with recurrent herpes labialis and systemic CMV or EBV infection but it remains unclear if these clinical manifestations occurred in the context of functional NK cell defects, although some had low NK cell numbers (Table 2) (10, 14, 20). Similar to our findings Lougaris et al. describe a patient with a C-terminal NFKB2 mutation and functional NK cell deficiency in which NK cell phenotyping suggested intact NK cell maturation. In contrast to our observations, they report an increased expression of CD69 on CD56dimCD16+ NK cells, suggesting an activated steady state, however the viral status of this patient with regards to CMV and EBV that could have an impact in the expression of these markers was not reported (18). In healthy subjects, CMV infection drives an expansion of NKG2C+ NK cells (30); interestingly we did not observe an expansion of NKG2C+ or CD57+ NK cell subsets in our patient, despite chronic CMV infection.
The cytotoxic NK cell defect despite normal NK cell numbers identified in our patient is consistent and likely contributes to her susceptibility to invasive CMV infection, while further suggesting a possible role for NF-κB2 in NK-cell cytotoxic activity. A possible explanation for a functional NK cell defect could be the accumulation of unprocessed p100 in the cytoplasm that could potentially impair the canonical NF-κB pathway, but further investigation is needed to define the mechanism by which NF-κB2 deficiency may impact NK cell development, activation and function. The NK cell abnormality in patients with NIK deficiency had previously raised this question but those patients had defective canonical as well as non-canonical NF-κB activation (31). Functional T cell abnormalities identified in our patient, namely decreased proliferation to PHA and a reduction in CD8+CD45RO+ memory cells are similar to what has been described in one previously reported patient (12). We did not perform further T cell functional studies and it is unclear if these T cell abnormalities are a consistent feature of this disease. It is likely that both T cell and NK cell abnormalities contributed to an overwhelming CMV infection in this patient. Anti-cytokine antibodies were not measured in our patient but they have also been described in patients with mutations in NFKB2, these may be directed to type 1 interferons and also contribute to an impaired antiviral immunity (32).
The outcome and complications of patients with NFKB2 mutations with or without functional perturbations of NK cells has not been compared. Importantly, this case highlights the risk of invasive viral infections including CMV in this disorder, and suggests that functional NK deficiency may play a role in this susceptibility and result in severe complications. We suggest that NK cell function should be assessed in all patients with mutations in the non-canonical NF-κB pathway. In those cases where NK function is affected, hematopoietic stem cell transplant may be appropriate despite the fact that NF-κB2 expression is not restricted to hematopoietic cells, since once CMV infection settles in, it becomes very difficult to clear and therefore reduces the chances for future successful stem cell transplantation.
Endocrine manifestations are more variable and expression of clinically overt ACTH deficiency involves about two-thirds of the cases and is usually an isolated form of hypopituitarism. However, growth hormone (GH) deficiency and hypothyroidism have also been described (15). Alopecia, trachyonychia and psoriatic dermatitis are also disease-associated features. Both endocrine and cutaneous manifestations are thought to be autoimmune mediated as NF-κB2 has been shown to play a role in central tolerance through an Aire-dependent pathway and in regulatory T cell homeostasis and suppressive function (15, 17, 33, 34). The underlying mechanism for the organ specificity of these autoimmune manifestations has not been fully elucidated.
Only one patient with NFKB2 mutation has been previously reported to be associated with a benign nephrotic syndrome not related to CMV infection (15). The nephrotic syndrome in our patient was severe and unresponsive to immunosuppressive treatment. This is the first time an NFKB2 variant has presented with long-lasting renal disease. The precise trigger for this manifestation is unknown. Interestingly, CMV PCR of the kidney biopsy was positive and her nephrotic range proteinuria fluctuated along with blood CMV titers. In periods of overwhelming CMV infection her nephrotic syndrome was most severe and unresponsive to treatment. CMV infection has been previously associated with nephrotic syndrome (35–38) supporting the role of CMV in the persistent proteinuria of our patient. Other possible causes of nephrotic syndrome described in patients with CVID are amyloidosis (39) and membranous nephropathy (40). An underlying autoimmune process cannot be ruled out, especially in the context of an NFKB2 mutation, however the absence of autoantibodies and immune complex deposition in the kidneys argues against this possibility.
In conclusion, we report a patient with a novel mutation C-terminal in NFKB2, presenting as early onset CVID, ectodermal dysplasia and alopecia who had a functional NK cell defect and succumbed to overwhelming CMV infection and an associated nephrotic syndrome.
Ethics Statement
This study was carried out in accordance with the recommendations of Institutional review board for Baylor college of Medicine and affiliated Hospitals with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by The Baylor College of Medicine Institutional Review Board.
Author Contributions
AA and MP wrote the manuscript. AV-H performed experiments and analyzed the data. EM and LF supervised analysis of NK cell studies. AA, ML, AK, and FC participated in the patient's clinical care. ZC-A, SJ, and IC performed and analyzed ES results. AR participated in coordination of sample analysis and Sanger sequencing confirmation. JL and JP supervised data interpretation of genomic studies. JL and JO were involved in study design and critically revised the manuscript. All authors reviewed the manuscript.
Funding
This work was supported in part by the NIH National Institute of Allergy and Infectious Diseases (R01AI120989 to JO), the Jeffrey Modell Foundation (to JO), the NIH National Institute of Neurological Disorders and Stroke (R35NS105078 to JL), the National Human Genome Research Institute and National Heart, Lung, and Blood Institute (UM1HG006542 to the Baylor Hopkins Center for Mendelian Genomics). FONDECYT #11181222 provided support to MP and JP was supported by the National Human Genome Research Institute (NHGRI K08 HG008986).
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patient and her family for facilitating this work.
References
1. Ameratunga R, Lehnert K, Woon ST, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol. (2018) 54:261–8. doi: 10.1007/s12016-017-8645-0
2. Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
3. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The (2017) IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
4. Schipp C, Nabhani S, Bienemann K, Simanovsky N, Kfir-Erenfeld S, Assayag-Asherie N, et al. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica. (2016) 101:e392–6. doi: 10.3324/haematol.2016.145136
5. Aksentijevich I, Zhou Q. NF-kappaB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. doi: 10.3389/fimmu.2017.00399
6. Brightbill HD, Jackman JK, Suto E, Kennedy H, Jones C III, Chalasani S, et al. Conditional deletion of NF-κB-inducing kinase (NIK) in adult mice disrupts mature B cell survival and activation. J Immunol. (2015) 195:953–64. doi: 10.4049/jimmunol.1401514
7. Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. (2011) 21:71–85. doi: 10.1038/cr.2010.177
8. Chinen J, Martinez-Gallo M, Gu W, Cols M, Cerutti A, Radigan L, et al. Transmembrane activator and CAML interactor (TACI) haploinsufficiency results in B-cell dysfunction in patients with Smith-Magenis syndrome. J Allergy Clin Immunol. (2011) 127:1579–86. doi: 10.1016/j.jaci.2011.02.046
9. Quentien MH, Delemer B, Papadimitriou DT, Souchon PF, Jaussaud R, Pagnier A, et al. Deficit in anterior pituitary function and variable immune deficiency (DAVID) in children presenting with adrenocorticotropin deficiency and severe infections. J Clin Endocrinol Metab. (2012) 97:E121–8. doi: 10.1210/jc.2011-0407
10. Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet. (2013) 93:812–24. doi: 10.1016/j.ajhg.2013.09.009
11. Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood. (2014) 124:2964–72. doi: 10.1182/blood-2014-06-578542
12. Lindsley AW, Qian Y, Valencia CA, Shah K, Zhang K, Assa'ad A. Combined immune deficiency in a patient with a novel NFKB2 mutation. J Clin Immunol. (2014) 34:910–15. doi: 10.1007/s10875-014-0095-3
13. Brue T, Quentien MH, Khetchoumian K, Bensa M, Capo-Chichi JM, Delemer B, et al. Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Med Genet. (2014) 15:139. doi: 10.1186/s12881-014-0139-9
14. Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MA. Novel NFKB2 mutation in early-onset CVID. J Clin Immuno. (2014) 34:686–90. doi: 10.1007/s10875-014-0064-x
15. Shi C, Wang F, Tong A, Zhang XQ, Song HM, Liu ZY, et al. NFKB2 mutation in common variable immunodeficiency and isolated adrenocorticotropic hormone deficiency: a case report and review of literature. Medicine. (2016) 95:e5081. doi: 10.1097/MD.0000000000005081
16. Tovo PA, Lala R, Martino S, Pastorelli G, De Sanctis C. Isolated adrenocorticotropic hormone deficiency associated with common variable immunodeficiency. Eur J Pediatr. (1991) 150:400–2. doi: 10.1007/BF02093717
17. Grinberg-Bleyer Y, Caron R, Seeley JJ, De Silva NS, Schindler CW, Hayden MS, et al. The alternative NF-κB Pathway in Regulatory T cell homeostasis and suppressive function. J Immunol. (2018) 200:2362–71. doi: 10.4049/jimmunol.1800042
18. Lougaris V, Tabellini G, Vitali M, Baronio M, Patrizi O, Tampella G, et al. Defective natural killer-cell cytotoxic activity in NFKB2-mutated CVID-like disease. J Allergy Clin Immunol. (2015) 135:1641–3. doi: 10.1016/j.jaci.2014.11.038
19. Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. (2018) 103:171–87. doi: 10.1016/j.ajhg.2018.06.009
20. Kuehn HS, Niemela JE, Sreedhara K, Stoddard JL, Grossman J, Wysocki CA, et al. Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype. Blood. (2017) 130:1553–64. doi: 10.1182/blood-2017-05-782177
21. Nagai M, Imai Y, Yamanishi K. Psoriasiform dermatitis associated with common variable immunodeficiency 10 due to an Arg853* mutation in the NFKB2 gene. J Dermatol. (2018) 46:e24–26. doi: 10.1111/1346-8138.14524
22. Mahapatra S, Mace EM, Minard CG, Forbes LR, Vargas-Hernandez A, Duryea TK, et al. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS ONE. (2017) 12:e0181–34. doi: 10.1371/journal.pone.0181134
23. Nerreter T, Zeiss S, Herrmann T, Einsele H, Seggewiss-Bernhardt R. Robust 8-color flow cytometry panel reveals enhanced effector function of NKG2C(+) CD57(+) FcepsilonRgamma(-) NK cells in CMV seropositive human blood donors. Immunobiology. (2017) 222:719–25. doi: 10.1016/j.imbio.2017.01.005
24. Beinke S, Ley SC. Functions of NF-κB1 and NF-κB2 in immune cell biology. Biochem J. (2004) 382:393–409. doi: 10.1042/BJ20040544
25. Sun SC. The noncanonical NF-κB pathway. Immunol Rev. (2012) 246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x
26. Tucker E, O'Donnell K, Fuchsberger M, Hilton AA, Metcalf D, Greig K, et al. A novel mutation in the Nfkb2 gene generates an NF-κB2 “super repressor”. J Immunol. (2007) 179:7514–22. doi: 10.4049/jimmunol.179.11.7514
27. Mace EM, Orange JS. Emerging insights into human health and NK cell biology from the study of NK cell deficiencies. Immunol Rev. (2019) 287:202–25. doi: 10.1111/imr.12725
28. Lougaris V, Patrizi O, Baronio M, Tabellini G, Tampella G, Damiati E, et al. NFKB1 regulates human NK cell maturation and effector functions. Clin Immunol. (2017) 175:99–108. doi: 10.1016/j.clim.2016.11.012
29. Montin D, Licciardi F, Giorgio E, Ciolfi A, Pizzi S, Mussa A, et al. Functional evaluation of natural killer cell cytotoxic activity in NFKB2-mutated patients. Immunol Lett. (2018) 194:40–3. doi: 10.1016/j.imlet.2017.12.006
30. Malmberg KJ, Beziat V, Ljunggren HG. Spotlight on NKG2C and the human NK-cell response to CMV infection. Eur J Immunol. (2012) 42:3141–3145. doi: 10.1002/eji.201243050
31. Willmann KL, Klaver S, Dogu F, Santos-Valente E, Garncarz W, Bilic I, et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. (2014) 5:5360. doi: 10.1038/ncomms6360
32. Ramakrishnan KA, Rae W, Barcenas-Morales G, Gao Y, Pengelly RJ, Patel SV, Kumararatne DS, et al. Anticytokine autoantibodies in a patient with a heterozygous NFKB2 mutation. J Allergy Clin Immunol. (2018) 141:1479–82. doi: 10.1016/j.jaci.2017.11.014
33. Fletcher AL, Seach N, Reiseger JJ, Lowen TE, Hammett MV, Scott HS, et al. Reduced thymic Aire expression and abnormal NF-κB2 signaling in a model of systemic autoimmunity. J Immunol. (2009) 182:2690–99. doi: 10.4049/jimmunol.0801752
34. Zhu M, Chin RK, Christiansen PA, Lo JC, Liu X, Ware C, et al. NF-κB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J Clin Invest. (2006) 116:2964–71. doi: 10.1172/JCI28326
35. Dossier C, Sellier-Leclerc AL, Rousseau A, Michel Y, Gautheret-Dejean A, Englender M, et al. Prevalence of herpesviruses at onset of idiopathic nephrotic syndrome. Pediatr Nephrol. (2014) 29:2325–31. doi: 10.1007/s00467-014-2860-1
36. Rahman H, Begum A, Jahan S, Muinuddin G, Hossain MM. Congenital nephrotic syndrome, an uncommon presentation of cytomegalovirus infection. Mymensingh Med J. (2008) 17:210–3.
37. Besbas N, Bayrakci US, Kale G, Cengiz AB, Akcoren Z, Akinci D, et al. Cytomegalovirus-related congenital nephrotic syndrome with diffuse mesangial sclerosis. Pediatr Nephrol. (2006) 21:740–2. doi: 10.1007/s00467-006-0051-4
38. Hogan J, Fila M, Baudouin V, Peuchmaur M, Deschenes G, Niel O. Cytomegalovirus infection can mimic genetic nephrotic syndrome: a case report. BMC Nephrol. (2015) 16:156. doi: 10.1186/s12882-015-0152-z
39. Aydin Z, Gursu M, Ozturk S, Kilicaslan I, Kazancioglu R. A case of primary immune deficiency presenting with nephrotic syndrome. NDT Plus. (2010) 3:456–8. doi: 10.1093/ndtplus/sfq083
Keywords: primary immunodeficiency, NF-κB2, common variable immunodeficiency (CVID), nephrotic syndrome, systemic cytomegalovirus, pituitary deficiency, NK cell deficiency
Citation: Aird A, Lagos M, Vargas-Hernández A, Posey JE, Coban-Akdemir Z, Jhangiani S, Mace EM, Reyes A, King A, Cavagnaro F, Forbes LR, Chinn IK, Lupski JR, Orange JS and Poli MC (2019) Novel Heterozygous Mutation in NFKB2 Is Associated With Early Onset CVID and a Functional Defect in NK Cells Complicated by Disseminated CMV Infection and Severe Nephrotic Syndrome. Front. Pediatr. 7:303. doi: 10.3389/fped.2019.00303
Received: 04 January 2019; Accepted: 08 July 2019;
Published: 30 July 2019.
Edited by:
Sergio Rosenzweig, National Institutes of Health (NIH), United StatesReviewed by:
Michael Daniel Keller, Children's National Health System, United StatesPietro Merli, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2019 Aird, Lagos, Vargas-Hernández, Posey, Coban-Akdemir, Jhangiani, Mace, Reyes, King, Cavagnaro, Forbes, Chinn, Lupski, Orange and Poli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Cecilia Poli, Y3BvbGlAdWRkLmNs