 Gianluigi Laccetta1*
Gianluigi Laccetta1* Simona Fiori2Matteo Giampietri1Annarita Ferrari2Valentina Cetica3Manuela Bernardini1Francesca Chesi1Sara Mazzotti2Elena Parrini3
Simona Fiori2Matteo Giampietri1Annarita Ferrari2Valentina Cetica3Manuela Bernardini1Francesca Chesi1Sara Mazzotti2Elena Parrini3 Massimiliano Ciantelli1
Massimiliano Ciantelli1 Andrea Guzzetta2,4
Andrea Guzzetta2,4 Paolo Ghirri1
Paolo Ghirri1- 1Division of Neonatology and Neonatal Intensive Care Unit, Department of Maternal and Child Health, Santa Chiara Hospital, University of Pisa, Pisa, Italy
- 2Department of Developmental Neuroscience, IRCCS Stella Maris, Pisa, Italy
- 3Pediatric Neurology, Neurogenetics and Neurobiology Unit and Laboratories, Neuroscience Department, Meyer Children's University Hospital, University of Florence, Florence, Italy
- 4Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
Among neonatal epileptic syndromes, benign familial neonatal seizures (BFNS) are often due to autosomal-dominant mutations of the KCNQ2 gene. Seizures are usually characterized by asymmetric tonic posturing with apnea with onset in the first 7 days of life; they may even occur more than 10 times per day or evolve into status epilepticus. The delivery course of our patient was uneventful and family history was negative; on the second day of life the baby became pale, rigid, and apnoic during breastfeeding and appeared jittery and irritable when stimulated or examined. At age 3 days, she experienced clusters of generalized tonic seizures with pallor, desaturation, bradycardia, and partial response to intravenous phenobarbital; during her 4th and 5th days of life, three episodes of tonic seizures were noticed. At age 6 days, the patient experienced about 10 episodes of tonic seizures involving both sides of the body, which gradually responded to intravenous phenytoin. Electroencephalograms revealed abnormalities but brain MRI was normal. The patient is seizure-free since postnatal day 21; she is now 12 months old with cognitive development within normal limits at Bayley III Scale and mild motor delay. The patient is on maintenance therapy with phenobarbital since she was 7 months old. A de novo heterozygous mutation (c.853C>T/p.P285S) in the KCNQ2 gene was identified. We therefore describe a case of de novo KCNQ2-related neonatal convulsions with necessity of multiple anticonvulsants for the control of seizures, mutation occurring in the pore channel of the voltage-gated potassium channel subfamily Q member 2 associated with a likely benign course; furthermore, the same mutation of the KCNQ2 gene and a similar one (c.854C>A/p.P285H) have already been described in association with Ohtahara syndrome. Probably acquired environmental, perinatal and genetic risk factors are very important in determining the different phenotype; we hope that the rapid progress of analysis tools in molecular diagnosis can also be used in the search of an individualized therapeutic approach for these patients.
Introduction
Neonatal seizures are often caused by hypoxic-ischemic encephalopathy, intracranial hemorrhage, hypoglycemia, electrolytes imbalance, and infections; metabolic disorders and neonatal epileptic syndromes occurring in the first week after birth, are rarely involved (1–3).
The KCNQ2 gene encodes for the voltage-gated potassium channel subfamily Q member 2 underlying the M-current; it was firstly described as a cause of autosomal-dominant seizures in 1998 and it is located at 20q13 (4–7). Each KCNQ2 subunit is made up of 6 transmembrane domains (S1–S6) with the voltage-sensor in S4 and the pore region defined by S5, S6, and the intervening linker (4, 8, 9). Mutations of the KCNQ2 gene can result in haploinsufficiency or dominant negative effect (4, 8–11).
Patients carrying KCNQ2 gene mutations will have benign familial neonatal seizures (BFNS), benign familial neonatal-infantile seizures (BFNIS) or benign familial infantile seizures (BFIS); they usually have benign courses even if some affected children will experience recurrent febrile seizures, benign childhood epilepsy with centrotemporal spikes (BCECTS), or photosensitive epilepsy during follow-up (2, 3, 12–15). On the other side, KCNQ2 gene mutations have also been associated with early myoclonic encephalopathy (EME) and early onset epileptic encephalopathy (EOEE), such as Ohtahara syndrome (OS) and intractable partial epilepsy, characterized by poor developmental prognosis (4, 12, 13, 16, 17).
Despite some case series reports and functional studies, the association between the phenotype and the genotype still remains unclear, thus it is not possible to establish prognosis for these patients at the moment of diagnosis (3, 12, 18–20).
Asymmetric tonic posturing and apnea are usually the main features of seizures at onset; they are often followed by unilateral or bilateral clonic jerking (2, 21–23). Seizures may even occur more than 10 times per day or evolve into status epilepticus (2, 21–23). Interictal EEG is usually normal in BFNS but it shows multifocal epileptiform abnormalities, random attenuation, or burst-suppression (BS) pattern in KCNQ2-related encephalopathy: thus, a severely abnormal interictal EEG is important in the differential diagnosis between KCNQ2-related encephalopathy and BFNS (2, 11, 21–23). Patients with KCNQ2-related epilepsies usually present a generalized EEG attenuation or suppression even after brief seizures (2, 17, 22, 23). Brain magnetic resonance imaging (MRI) is usually normal or shows a transient hyperintense globus pallidus (11, 23).
Thus, the suspicion of a KCNQ2-related disorder is mainly based on familial anamnesis, clinical presentation, and EEG pattern; the diagnosis is finally confirmed by direct sequencing of the KCNQ2 gene or targeted next generation sequencing (NGS) panel of epilepsy genes (3, 8).
As regards therapy of KCNQ2-related epilepsies, a recent review states that both phenobarbital and sodium channel blockers (carbamazepine and phenytoin) lead to seizure disappearance in patients with a benign course and patients with EOEE usually achieve seizure freedom when receiving sodium channel blockers (6, 14, 24, 25).
We report a 12-months-old female with a de novo KCNQ2 gene mutation presumably associated with non-familial benign neonatal seizures (nfBNS) and investigate the correlation between the genotype and the phenotype revisioning the data of literature. Written informed consent for the publication of this case report was obtained from both the parents of the patient.
Case Report
This third child of non-consanguineous parents was born at full term by spontaneous vaginal delivery without asphyxia to a 43-year-old woman. The pregnancy course was uneventful; Apgar score was 9 at 5 min and umbilical cord blood gas analysis was normal. Her birth weight was 3,040 g, her body length was 50 cm, and her head circumference was 35 cm. After birth, her condition was good, and she started breastfeeding regularly. Family history was negative for epilepsies, mental retardation and hereditary diseases.
On the second day of life, the infant was noted to become pale, rigid and apnoeic for some seconds while breastfeeding; furthermore she appeared jittery and irritable when stimulated or examined.
At age 3 days, clusters of generalized tonic seizures associated with pallor, desaturation, and bradycardia, were first noticed. The infant was subsequently admitted to the Neonatal Intensive Care Unit (NICU). At first examination, the infant appeared in good general conditions, with normal cardiorespiratory and abdominal examinations. Arterial blood gas and head ultrasound performed upon admission to the NICU were unremarkable. Bedside Amplitude-Integrated EEG (aEEG) monitoring was promptly started and showed a discontinuous normal voltage; during seizures the aEEG trace registered an immediate rise and a subsequent depression of its amplitude. The patient was therefore loaded with intravenous phenobarbital (20 mg/kg), and a maintenance dose of 2.5 mg/kg was administered every 12 h with reduced frequency of seizures but persistent subclinical seizure patterns at aEEG registration. During her 4 and 5th days of life the infant experienced three episodes of tonic seizures with hyperextension of the trunk, right, or left head deviation, involvement of both lower and upper limbs, oral automatisms, and flushing.
In the meantime, laboratory investigations (complete blood count, electrolytes, glucose, plasma lactate and ammonia, plasma amino acids, acyl-carnitines and urinary organic acids, blood culture, viral serology and genomes, markers of infection) and diagnostic procedures (Holter ECG, echocardiogram and abdomen ultrasound scan) yielded no significant results.
At age 6 days the patient had about 10 episodes of tonic seizures which involved both lower and upper limbs and both sides of the body alternately; seizures were accompanied by apnea and turning of head and eyes to one side. Each episode lasted about 30–40 s and persisted despite the administration of intravenous phenobarbital, midazolam (0.2 mg/kg) and pyridoxine (100 mg). Intravenous phenytoin (8 mg/kg/day) was therefore administered from the same day with gradual disappearance of clinically evident seizures on the subsequent days and improvement of the EEG pattern.
First video electroencephalogram (EEG) at 7 days of age showed discontinuous pattern during sleep and spike waves over bilateral frontal and centrotemporal areas with secondary diffusion, predominantly during sleep. The second and third video EEGs (9 and 16 days of age) indicated an improvement of the sleep pattern and the persistence of the spike waves over bilateral frontal and centrotemporal areas during sleep with secondary diffusion.
Brain MRI at 10 days of age was normal as regards ventricular morphology, cortical folding, and white matter intensity; MR spectroscopy was also normal.
Midazolam and pyridoxine were ineffective, thus they were discontinued at 11 days of age. Because of the baby's good general conditions, treatment with oral phenobarbital (2.5 mg/kg every 12 h) and phenytoin (2 mg/kg every 8 h) was started from postnatal day 15 with no further seizures until postnatal day 21 when the infant presented two episodes of generalized tonic seizures involving her limbs with hyperextension of the trunk, left head, and eye deviation and pallor; both episodes were preceded by a high-pitched cry and each lasted about 30–40 s.
Seizures definitively stopped after the administration of intravenous phenobarbital and phenytoin; the patient was seizure free even after the second attempt of switching to oral phenobarbital (2.5 mg/kg every 12 h) and phenytoin (2 mg/kg every 8 h), which was done at 25 days of age.
The fourth video EEG (23 days of age) showed a predominant discontinuous pattern over the right hemisphere during sleep and the persistence of the spike waves over bilateral frontal and centrotemporal areas during sleep with secondary diffusion. The fifth video EEG (30 days of age) indicated a predominant normal background even during sleep with spike waves over bilateral frontal and centrotemporal areas with secondary diffusion more evident on the right side and during sleep.
The following video EEG (37 days of age) showed centro-temporal isolated spikes predominantly on the right hemisphere, with a normal background activity.
At the age of 38 days the patient was discharged from hospital; her general conditions were satisfactory and she was seizure free with oral phenobarbital (5 mg/kg/day in two doses) and phenytoin (6 mg/kg/day in three doses). Neurologic examination before discharge revealed mild generalized hypotonia and poor writhing movements. Based on these findings, a genetically-determined epilepsy of neonatal onset was strongly suspected, therefore genetic testing, including epilepsy NGS panel for 109 genes involved in EOEE (Table 1), was ordered on postnatal day 8.
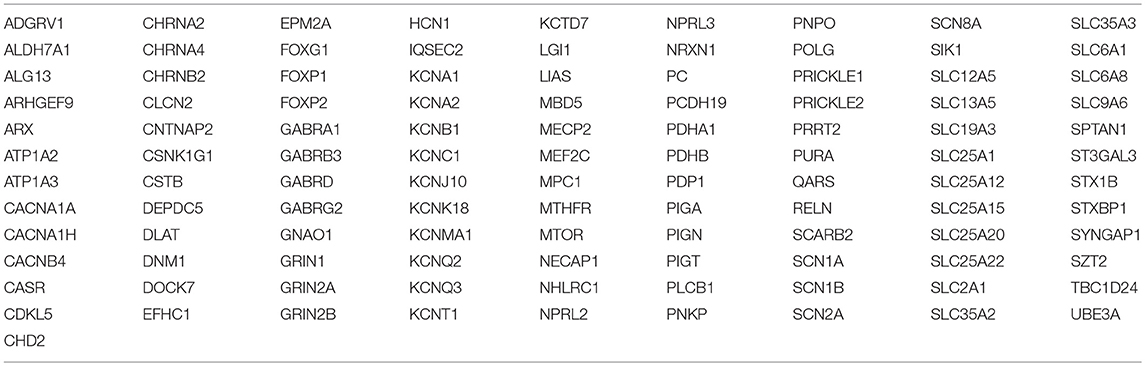
Table 1. Epilepsy NGS panel for 109 genes involved in early epileptic encephalopathies.
After discharge the patient has been followed closely by the Infant Neurology Section at Stella Maris Scientific Institute. At 12 months of age she is seizure-free with progressively reducing doses of phenytoin; from the age of 7 months she is on monotherapy with phenobarbital. Her follow-up neurological examinations reveal a slightly delayed trunk control, sitting position acquired at 8 months of age and mild hypotonia; at 12 months of age she can stand with support, but no walking is possible yet. Video EEG performed at 12 months of age shows spike waves in the frontal-central, centro-parietal and centro-parieto-temporal regions with left predominance. The cognitive subscale of Bayley Scale of Infant Development III performed at 12 months of age shows a normal cognitive development. Communication abilities consist of varied babbling and first words; pointing is present.
DNA samples from the patient and both her parents were extracted from peripheral whole blood by means of a genomic DNA purification kit (Gentra Systems, Minneapolis, MN) after written informed consents were obtained. For the proband, all 17 exons of the KCNQ2 gene were amplified by polymerase chain reaction and sequenced using NexSeq Illumina platform from Illumina (San Diego, CA, USA); the variant detection software used were BWA v0.7.7-r441, Picard v1.109, and GATK v3.1.
A de novo heterozygous mutation (c.853C>T, p.P285S) in the KCNQ2 gene was identified in the patient. No mosaicism for the normal variant of the KCNQ2 gene was found in the proband; similarly, no mosaicism for the KCNQ2 variant was identified in the parents. Thus, the family was referred to a genetic counselor: the estimated risk of recurrence was <1% for the parents and 50% for the proband.
Discussion
In this patient, a heterozygous mutation (c.853C>T, p.P285S) in the KCNQ2 gene was identified, with the consequence of a proline substitution to serine.
The voltage-gated potassium channel Kv7.2, encoded by the KCNQ2 gene, assembles with Kv7.3, encoded by the KCNQ3 gene, to form heteromultimeric channels which regulate neuronal excitability through the M-current (3, 5, 9, 20, 26).
KCNQ2-related epilepsy spectrum ranges from KCNQ2-related BFNS to KCNQ2-related EOEE; KCNQ2 gene mutations have also been linked with other phenotypes such as myokymia associated with neonatal or early infantile epilepsy, peripheral nerve excitability without known epilepsy, fever sensitive epilepsy forms of the generalized epilepsy with febrile seizure plus syndrome and Dravet syndrome (1, 3, 6, 11, 27–31).
Age of onset of seizures strongly correlates with underlying genetics, as most of the non-lesional patients with BFNS/BFNIS show KCNQ2 mutations and those with BFIS commonly show PRRT2 mutations (32).
The baby experienced some episodes of tonic seizures, each one involving one side of the body; these seizure attacks were also accompanied by involvement of both lower and upper limbs, apnea, and turning of head and eyes to one side: these features have already been described in a child with epilepsy of infancy with migrating focal seizures (EIMFS) which evolved to infantile spasms (28). Interestingly, this infant presented a mutation, p.881C>T (p.A294V), in the pore region of the KCNQ2 gene (28).
Furthermore, tonic seizures are more common than generalized tonic-clonic seizures among newborns because of immature myelination (1, 2, 33).
BFNS are usually hereditary, autosomal-dominant seizures occurring in the neonatal age: our patient likely represents a rare case of nfBNS (see Table 2 for further cases).
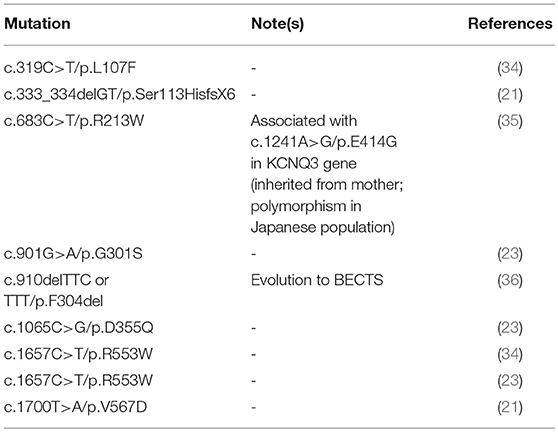
Table 2. KCNQ2 gene mutations associated with non-familial benign neonatal seizures (nf-BNS).
In the present case, benign neonatal seizures (BNS) were mainly a clinical diagnosis: analysis of the KCNQ2 gene pointed out a de novo heterozygous mutation in our patient (c.853C>T, p.P285S), furthermore it was not possible to accurately predict outcome because the association between mutations of the KCNQ2 gene and subsequent phenotype is unclear and the follow-up of our patient is still short (3).
We firstly hypothesized our patient would have suffered from an epileptic encephalopathy (EE) because of her negative family history for epilepsies, mental retardation and hereditary diseases and because of her seizure semiology at 6 days of age, which was similar to that of EIMFS evolving into epileptic spasms (28). Furthermore, the same mutation of the KCNQ2 gene has already been described in a 8-month-old child with OS (17). This patient experienced tonic and simple partial seizures since his first day of life which persisted despite the administration of phenobarbital, topiramate and valproic acid; his EEG showed a burst-suppression pattern and brain MRI was normal (17). No other data are available about the phenotype of the child. In addition, a similar mutation of the KCNQ2 gene (c.854C>A, p.P285H) has already been described as associated with OS (12). In this case a proline is changed to histidine; the patient experienced irritability with hypoxia since 2 days of age and tonic seizures from 8 days of age (12). Seizures persisted despite the administration of phenobarbital and pyridoxal 5′ phosphate but were controlled by valproic acid; electroencephalograms showed burst-suppression and MRI showed T1- and T2-high signal on globus pallidus at postnatal day 12 and T2-high signal on globus pallidus at 3 months of age (12). The patient was seizure-free since 3 months of age but developed moderate mental retardation and hypotonic quadriplegia; the mutation was maternally inherited and the mother of the patient only suffered from idiopathic epilepsy since neonatal age and she was on treatment with valproic acid (12). Probably acquired environmental, perinatal, and genetic risk factors are very important in determining the different phenotype; as proof of this hypothesis, in one report, a patient with a mutation of c.G1620A (p.K526N) and family history of BNS, developed resistant epilepsy and mental retardation (1, 2, 37). Furthermore, Grinton et al. reported some families with KCNQ2 mutations and family members with the same genotype but different phenotypes (1, 3, 15). Thus, the same gene mutation contributes to different phenotypes; this possibility is valid for many genetic diseases and also for KCNQ2-related diseases (2, 6).
The basal ganglia are susceptible to different stressors, and even more the neonatal brain (3, 6, 11, 38, 39). Basal ganglia are also vulnerable to excitotoxicity caused by frequent neonatal seizures; this might explain the lesions seen in patients with KCNQ2-related epilepsies (3, 6, 11, 40). These lesions are highly specific since they have never been described in association with other severe neonatal epileptic syndromes (OS, EME, or pyridoxine-dependent epilepsy) (6, 11, 24, 27). Our patient had a normal brain MRI at 10 days of age but this result did not exclude EE since many patients with negative brain MRI and EE have already been described (34).
Al Yazidi et al. noted an association between severe neonatal EE phenotype and de novo missense mutations of the KCNQ2 gene; even patients with BFNS and missense mutations in KCNQ2, had less benign clinical courses than it is usually predicted, for example they required multiple anticonvulsants for the control of their seizures (3, 11–13, 26). Furthermore, though precise correlation between KCNQ2 gene mutation and phenotype is difficult, BFNS phenotype is usually associated with inherited heterozygous loss-of-function mutations, and EE is usually associated with de novo missense mutations (1, 2, 26).
While BFNS-associated variants are distributed throughout the channel protein, currently known KCNQ2 encephalopathy mutations are located in four functionally important protein domains: the voltage sensor domain, the pore, the C-terminus proximal region, and the calmodulin-binding B helix region (2, 7, 8, 11, 41, 42).
Our patient presented the missense mutation c.853C>T (p.P285S) of the KCNQ2 gene, in which a proline is substituted by a serine; this change, presumably leading to a reduction of current potassium or other abnormal functional properties/subcellular localization of Kv7.2 channel, is responsible for the phenotype of our patient in association with additional genetic, perinatal and environmental factors (2, 9, 10).
The mutation has been classified as pathogenic according to the American College of Medical Genetics (ACMG) Scoring System, deleterious at SIFT analysis and damaging at Mutation Taster analysis, thus confirming previous findings (17).
The mutation might be responsible for the phenotype of the child even because no other pathogenic or likely pathogenic mutations have been found in the other 108 genes included in the NGS panel for epilepsy. The phenotype of our patient (presumably BNS and not OS) is not explained by an underlying mosaicism for the normal variant of the KCNQ2 gene. The lack of a functional experiment is a limit of the present case report; Miceli et al. drew a genotype-phenotype correlation based on distinct functional properties of Kv7.2 channels carrying different variants on the same residue found in two patients affected by widely divergent epileptic syndromes (43).
Both c.854C>A and c.853C>T are mutations occurring in the portion of the KCNQ2 gene encoding for the pore channel of the voltage-gated potassium channel subfamily Q member 2; other mutations involving the pore region have already been described (Table 3).
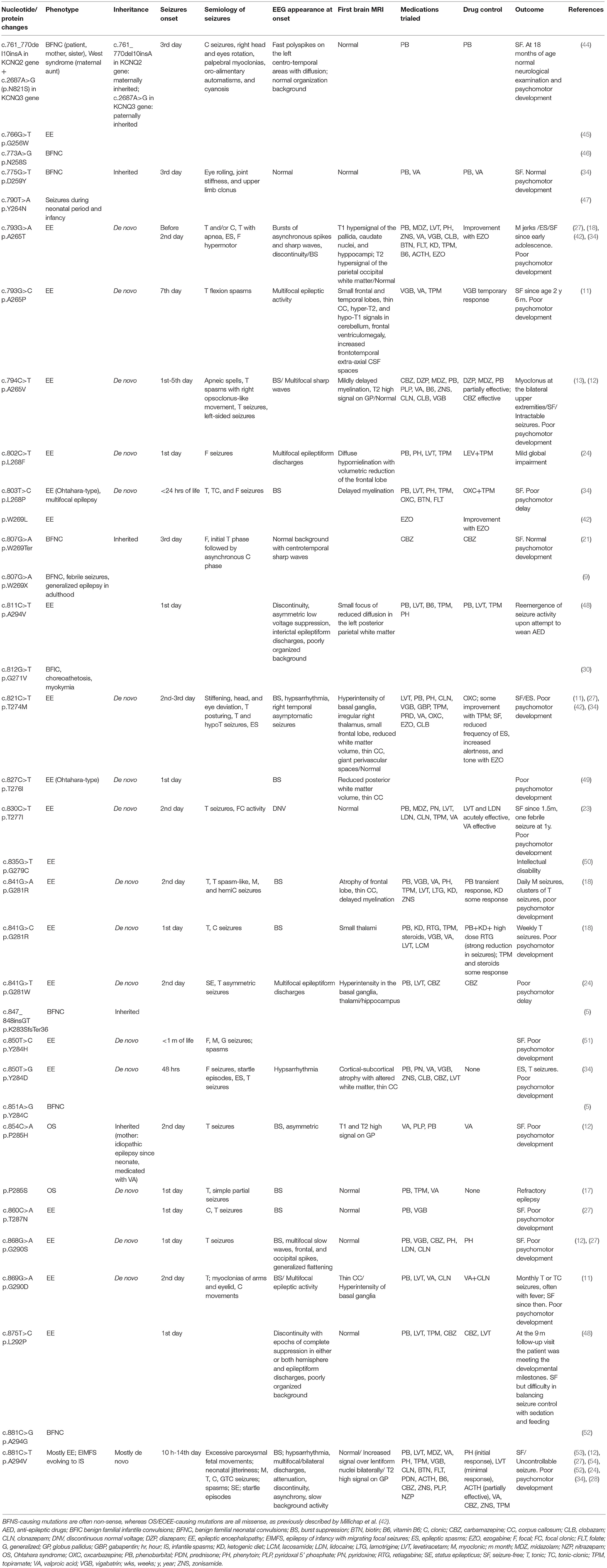
Table 3. Review of the KCNQ2 gene mutations involving the pore region of the voltage-gated potassium channel subfamily Q member 2.
Even though neonatal convulsions constitute a “benign” epileptic syndrome, our patient showed clusters of seizures with apnea, desaturation and bradycardia, which required second-line drugs to achieve seizures control. After the administration of intravenous phenobarbital, seizures showed reduced frequency; at age 6 days the patient experienced about ten episodes of tonic seizures which persisted despite the administration of intravenous phenobarbital, midazolam and pyridoxine, and gradually disappeared after the administration of intravenous phenytoin. At age 21 days, while on treatment with oral phenobarbital and phenytoin, the infant experienced two episodes of generalized tonic seizures which stopped after the administration of intravenous phenobarbital and phenytoin. At 25 days of age, the patient was therefore switched to oral phenobarbital and phenytoin with no further recurrence of seizures; she is now seizure-free on oral treatment with phenobarbital.
The pattern of BNS involves apnea, cyanosis, automatisms, focal clonic seizures and, rarely, generalized tonic-clonic seizures; some patients experience recurrence of seizures after 1 year of age (most commonly febrile seizures, generalized seizures with febrile seizures-plus, or BCECTS) (1, 2). For all these reasons, the diagnosis of neonatal seizures can be difficult and genetic testing becomes mandatory; furthermore, seizures semiology made clinical diagnosis more difficult for our patient. We would have preferred to start therapy with oral carbamazepine immediately because it is safe and highly effective for BNS and it permits to avoid sedation, hypotonia and delayed oral feeding associated with intravenous phenobarbital and phenytoin but it requires prompt recognition of the electroclinical phenotype; furthermore, our patient presented clusters of seizures with apnea, desaturation, and bradycardia requiring a rapidly effective intravenous therapy (21).
Our patient will stop oral therapy with phenobarbital within a few months because the risk of recurrence of seizures is minimum when medications are weaned between 12 and 18 months of life (7, 21, 55). Unfortunately, up to 25% of neonates with BNS may experience other forms of epilepsy later in childhood and mental retardation occurs in rare cases, therefore our infant is strictly followed up (2, 7, 8, 15, 21, 37).
Data Availability
The datasets generated for this study can be found in UO Neonatologia AOUP.
Ethics Statement
The work involves a human subject and it has been conducted in conformity with ethical standards of the field and with consent of both parents of the child. Written informed consent for the publication of this case report was obtained from both the parents of the patient.
Author Contributions
Study concept and design: GL, PG, SF, and AG. Patient collection: PG, FC, MB, MG, MC, AF, SF, AG, and SM. Mutation screening: VC and EP. Data analysis and review: GL and PG. Drafting of the manuscript: GL and PG. Critical revision of the manuscript for important intellectual content: PG, SF, and AG. Obtained funding: PG.
Funding
Publication fees: Pisa University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Herlenius E, Heron SE, Grinton BE, Keay D, Scheffer IE, Mulley JC, et al. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia. (2007) 48:1138–42. doi: 10.1111/j.1528-1167.2007.01049.x
2. Lee I-C, Chen JY, Chen YJ, Yu JS, Su PH. Benign familial neonatal convulsions: novel mutation in a newborn. Pediatr Neurol. (2009) 40:387–91. doi: 10.1016/j.pediatrneurol.2008.12.004
3. Lee I-C, Yang JJ, Liang JS, Chang TM, Li SY. KCNQ2-Associated neonatal epilepsy: phenotype might correlate with genotype. J Child Neurol. (2017) 32:704–11. doi: 10.1177/0883073817701873
4. Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, et al. A potassium channel mutation in neonatal human epilepsy. Science. (1998) 16:279:403–6. doi: 10.1126/science.279.5349.403
5. Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. (1998) 18:25–9. doi: 10.1038/ng0198-25
6. Hewson S, Puka K, Mercimek-Mahmutoglu S. Variable expressivity of a likely pathogenic variant in KCNQ2 in a three-generation pedigree presenting with intellectual disability with childhood onset seizures. Am J Med Genet A. (2017) 173:2226–30. doi: 10.1002/ajmg.a.38281
7. Coppola G, Castaldo P, Miraglia del Giudice E, Bellini G, Galasso F, et al. A novel KCNQ2 K+ channel mutation in benign neonatal convulsions and centro-temporal spikes. Neurology. (2003) 61:131–4. doi: 10.1212/01.WNL.0000069465.53698.BD
8. Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. (2007) 73:245–49. doi: 10.1016/j.eplepsyres.2006.10.010
9. Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. (2003) 126:2726–37. doi: 10.1093/brain/awg286
10. Claes LR, Ceulemans B, Audenaert D, Deprez L, Jansen A, Hasaerts D, et al. De novo KCNQ2 mutations in patients with benign neonatal seizures. Neurology. (2004) 63:2155–8. doi: 10.1212/01.WNL.0000145629.94338.89
11. Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LR, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. (2012) 71:15–25. doi: 10.1002/ana.22644
12. Kato M, Yamagata T, Kubota M, Arai H, Yamashita S, Nakagawa T, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. (2013) 54:1282–7. doi: 10.1111/epi.12200
13. Saitsu H, Kato M, Koide A, Goto T, Fujita T, Nishiyama K, et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol. (2012) 72:298–300. doi: 10.1002/ana.23620
14. De Haan GJ, Pinto D, Carton D, Bader A, Witte J, Peters E, van Erp G, et al. A novel splicing mutation in KCNQ2 in a multigenerational family with BFNC followed for 25 years. Epilepsia. (2006) 47:851–59. doi: 10.1111/j.1528-1167.2006.00552.x
15. Grinton BE, Heron SE, Pelekanos JT, Zuberi SM, Kivity S, Afawi Z, et al. Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia. (2015) 56:1071–80. doi: 10.1111/epi.13020
16. Kojima K, Shirai K, Kobayashi M, Miyauchi A, Saitsu H, Matsumoto N, et al. A patient with early myoclonic encephalopathy (EME) with a de novo KCNQ2 mutation. Brain Dev. (2018) 40:69–73. doi: 10.1016/j.braindev.2017.06.004
17. Miao P, Feng J, Guo Y, Wang J, Xu X, Wang Y, et al. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin Genet. (2018) 94:512–20. doi: 10.1111/cge.13441
18. Weckhuysen S, Ivanovic V, Hendrickx R, Van Coster R, Hjalgrim H, Møller RS, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology. (2013) 81:1697–703. doi: 10.1212/01.wnl.0000435296.72400.a1
19. Wuttke TV, Penzien J, Fauler M, Seebohm G, Lehmann-Horn F, Lerche H, et al. Neutralization of a negative charge in the S1-S2 region of the KV7.2 (KCNQ2) channel affects voltage-dependent activation in neonatal epilepsy. J Physiol. (2008) 586:545–55. doi: 10.1113/jphysiol.2007.143826
20. Maljevic S, Wuttke TV, Lerche H. Nervous system KV7 disorders: breakdown of a sub-threshold brake. J Physiol. (2008) 586:1791–801. doi: 10.1113/jphysiol.2008.150656
21. Sands TT, Balestri M, Bellini G, Mulkey SB, Danhaive O, Bakken EH, et al. Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia. (2016) 57:2019–30. doi: 10.1111/epi.13596
22. Numis AL, Angriman M, Sullivan JE, Lewis AJ, Striano P, Nabbout R, et al. KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology. (2014) 82:368–70. doi: 10.1212/WNL.0000000000000060
23. Vilan A, Mendes Ribeiro J, Striano P, Weckhuysen S, Weeke LC, Brilstra E, et al. A distinctive ictal amplitude-integrated electroencephalography pattern in newborns with neonatal epilepsy associated with KCNQ2 mutations. Neonatology. (2017) 112:387–93. doi: 10.1159/000478651
24. Pisano T, Numis AL, Heavin SB, Weckhuysen S, Angriman M, Suls A, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. (2015) 56:685–91. doi: 10.1111/epi.12984
25. Kuersten M, Tacke M, Gerstl L, Hoelz H, Stülpnagel CV, Borggraefe I. Antiepileptic therapy approaches in KCNQ2 related epilepsy: a systematic review. Eur J Med Genet. (2019). doi: 10.1016/j.ejmg.2019.02.001
26. Al Yazidi G, Shevell MI, Srour M. Two novel KCNQ2 mutations in 2 families with benign familial neonatal convulsions. Child Neurol Open. (2017) 4:1–4. doi: 10.1177/2329048X17691396
27. Milh M, Boutry-Kryza N, Sutera-Sardo J, Mignot C, Auvin S, Lacoste C, et al. Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J Rare Dis. (2013) 8:80. doi: 10.1186/1750-1172-8-80
28. Duan H, Peng J, Kessi M, Yin F. De Novo KCNQ2 Mutation in one case of epilepsy of infancy with migrating focal seizures that evolved to infantile spasms. Child Neurol Open. (2018) 5:2329048X18767738. doi: 10.1177/2329048X18767738
29. Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci USA. (2001) 98:12272–7. doi: 10.1073/pnas.211431298
30. Zhou X, Ma A, Liu X, Huang C, Zhang Y, Shi R, et al. Infantile seizures and other epileptic phenotypes in a Chinese family with a missense mutation of KCNQ2. Eur J Pediatr. (2006) 165:691–5. doi: 10.1007/s00431-006-0157-5
31. Wuttke TV, Jurkat-Rott K, Paulus W, Garncarek M, Lehmann-Horn F, Lerche H. Peripheral nerve hyperexcitability due to dominant-negative KCNQ2 mutations. Neurology. (2007) 69:2045–53. doi: 10.1212/01.wnl.0000275523.95103.36
32. Zara F, Specchio N, Striano P, Robbiano A, Gennaro E, Paravidino R, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia. (2013) 54:425–36. doi: 10.1111/epi.12089
33. Serino D, Specchio N, Pontrelli G, Vigevano F, Fusco L. Video/EEG findings in a KCNQ2 epileptic encephalopathy: a case report and revision of literature data. Epileptic Disord. (2013) 15:158–65. doi: 10.1684/epd.2013.0578
34. Hortigüela M, Fernández-Marmiesse A, Cantarín V, Gouveia S, García-Peñas JJ, Fons C, et al. Clinical and genetic features of 13 Spanish patients with KCNQ2 mutations. J Hum Genet. (2017) 62:185–9. doi: 10.1038/jhg.2016.104
35. Sadewa AH, Sasongko TH, Lee MJ, Daikoku K, Yamamoto A, Yamasaki T, et al. Germ-line mutation of KCNQ2, p.R213W, in a Japanese family with benign familial neonatal convulsion. Pediatr Int. (2008) 50:167–71. doi: 10.1111/j.1442-200X.2008.02539.x
36. Ishii A, Miyajima T, Kurahashi H, Wang JW, Yasumoto S, Kaneko S, et al. KCNQ2 abnormality in BECTS: benign childhood epilepsy with centrotemporal spikes following benign neonatal seizures resulting from a mutation of KCNQ2. Epilepsy Res. (2012) 102:122–5. doi: 10.1016/j.eplepsyres.2012.07.011
37. Borgatti R, Zucca C, Cavallini A, Ferrario M, Panzeri C, Castaldo P, et al. A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology. (2004) 63:57–65. doi: 10.1212/01.WNL.0000132979.08394.6D
38. Nishino H, Hida H, Kumazaki M, Shimano Y, Nakajima K, Shimizu H, et al. The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: a hypothesis to explain its specific vulnerability. J Neurotrauma. (2000) 17:251–60. doi: 10.1089/neu.2000.17.251
39. Johnston MV. Excitotoxicity in neonatal hypoxia. Ment Retard Dev Disabil Res Rev. (2001) 7:229–34. doi: 10.1002/mrdd.1032
40. Chen Q, Veenman CL, Reiner A. Cellular expression of ionotropic glutamate receptor subunits on specific striatal neuron types and its implication for striatal vulnerability in glutamate receptor-mediated excitotoxicity. Neuroscience. (1996) 73:715–31. doi: 10.1016/0306-4522(96)00011-5
41. Mulkey SB, Ben-Zeev B, Nicolai J, Carroll JL, Grønborg S, Jiang YH, et al. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia. (2017) 58:436–45. doi: 10.1111/epi.13676
42. Millichap JJ, Park KL, Tsuchida T, Ben-Zeev B, Carmant L, Flamini R, et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. (2016) 2:e96. doi: 10.1212/NXG.0000000000000096
43. Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Migliore M, Cilio MR, et al. Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of Kv7.2 potassium channel subunits. Proc Natl Acad Sci USA. (2013) 110:4386–91. doi: 10.1073/pnas.1216867110
44. Bassi MT, Balottin U, Panzeri C, Piccinelli P, Castaldo P, Barrese V, et al. Functional analysis of novel KCNQ2 and KCNQ3 gene variants found in a large pedigree with benign familial neonatal convulsions (BFNC). Neurogenetics. (2005) 6:185–93. doi: 10.1007/s10048-005-0012-2
45. Lim BC, Kim H, Hwang H, Chae J, Kim KJ, Hwang YS. Targeted massively parallel sequencing of infantile onset epileptic encephalopathy. In: Annual meeting of American Epilepsy Society. (2014). Available online at: https://www.aesnet.org/meetings_events/annual_meeting_abstracts/view/1867824
46. Yalçin O, Caglayan SH, Saltik S, Cokar O, Agan K, Dervent A, et al. A novel missense mutation (N258S) in the KCNQ2 gene in a Turkish family afflicted with benign familial neonatal convulsions (BFNC). Turk J Pediatr. (2007) 49:385–9.
47. Stosser MB, Lindy AS, Butler E, Retterer K, Piccirillo-Stosser CM, Richard G, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med. (2018) 20:403–10. doi: 10.1038/gim.2017.114
48. Chen DY, Chowdhury S, Farnaes L, Friedman JR, Honold J, Dimmock DP, et al. Rapid Diagnosis of KCNQ2-associated early infantile epileptic encephalopathy improved outcome. Pediatr Neurol. (2018) 86:69–70. doi: 10.1016/j.pediatrneurol.2018.06.002
49. Martin HC, Kim GE, Pagnamenta AT, Murakami Y, Carvill GL, Meyer E, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. (2014) 23:3200–11. doi: 10.1093/hmg/ddu030
50. Milh M, Lacoste C, Cacciagli P, Abidi A, Sutera-Sardo J, Tzelepis I, et al. Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am J Med Genet A. (2015) 167A:2314–8. doi: 10.1002/ajmg.a.37152
51. Fung CW, Kwong AK, Wong VC. Gene panel analysis for nonsyndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open. (2017) 2:236–43. doi: 10.1002/epi4.12055
52. Abidi A, Devaux JJ, Molinari F, Alcaraz G, Michon FX, Sutera-Sardo J, et al. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol Dis. (2015) 80:80–92. doi: 10.1016/j.nbd.2015.04.017
53. Lee EH, Yum MS, Jeong MH, Lee KY, Ko TS. A case of malignant migrating partial seizures in infancy as a continuum of infantile epileptic encephalopathy. Brain Dev. (2012) 34:768–72. doi: 10.1016/j.braindev.2011.11.011
54. Allen NM, Mannion M, Conroy J, Lynch SA, Shahwan A, Lynch B, King MD. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. (2014) 55:e99–105. doi: 10.1111/epi.12715
Keywords: KCNQ2, seizures, mutation, benign familial neonatal convulsions, phenobarbital
Citation: Laccetta G, Fiori S, Giampietri M, Ferrari A, Cetica V, Bernardini M, Chesi F, Mazzotti S, Parrini E, Ciantelli M, Guzzetta A and Ghirri P (2019) A de novo KCNQ2 Gene Mutation Associated With Non-familial Early Onset Seizures: Case Report and Revision of Literature Data. Front. Pediatr. 7:348. doi: 10.3389/fped.2019.00348
Received: 02 May 2019; Accepted: 05 August 2019;
Published: 06 September 2019.
Edited by:
Arjan Te Pas, Leiden University, NetherlandsReviewed by:
Maurizio Taglialatela, University of Naples Federico II, ItalyMaria Virginia Soldovieri, University of Molise, Italy
Pasquale Striano, University of Genoa, Italy
Copyright © 2019 Laccetta, Fiori, Giampietri, Ferrari, Cetica, Bernardini, Chesi, Mazzotti, Parrini, Ciantelli, Guzzetta and Ghirri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianluigi Laccetta, Z2lhbmx1aWdpbGFjY2V0dGFAcm9ja2V0bWFpbC5jb20=