 Jose Antonio Tavares de Albuquerque1,2
Jose Antonio Tavares de Albuquerque1,2 Alessandra Miramontes Lima2
Alessandra Miramontes Lima2 Edgar Borges de Oliveira Junior1,2
Edgar Borges de Oliveira Junior1,2 Edson Kiyotaka Ishizuka2
Edson Kiyotaka Ishizuka2 Walmir Cutrim Aragão-Filho2
Walmir Cutrim Aragão-Filho2 Nuria Bengala Zurro3
Nuria Bengala Zurro3 Sônia Mayumi Chiba4,5
Sônia Mayumi Chiba4,5 Fátima Rodrigues Fernandes2
Fátima Rodrigues Fernandes2 Antonio Condino-Neto3*
Antonio Condino-Neto3*- 1Immunogenic Inc, São Paulo, Brazil
- 2PENSI Institute - Jose Luiz Egydio Setubal Foundation, Sabará Hospital, São Paulo, Brazil
- 3Department of Immunology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil
- 4Sabará Hospital, São Paulo, Brazil
- 5Department of Pediatrics, Federal University of São Paulo, São Paulo, Brazil
Chronic granulomatous disease (CGD) is an inherited, genetically heterogeneous disease characterized by defective phagocytic cell microbicidal function, leading to increased susceptibility to bacterial and fungal infections. CGD is caused by mutations in components of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system, which is responsible for reactive oxygen species production during phagocytosis. Mutations in the neutrophil cytosolic factor 2 (NCF2) gene account for <5% of all cases. Here, we report a case of a 2-year-old female with persistent recurrent pneumopathy, even under trimethoprim-sulfamethoxazole (TMP-SMX) and itraconazole prophylaxis combined with IFNγ treatment. Genetic analysis revealed a novel homozygous mutation in NCF2, sequence depletion in a splicing region (c.256_257+2delAAGT NM_000433), leading to a K86Ifs*2 residue change in the p67−phox protein.
Introduction
Chronic granulomatous disease (CGD) is a severe immunodeficiency syndrome caused by functional impairment of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex in neutrophilic granulocytes and monocytes. Patients with CGD are generally diagnosed in early childhood after developing recurrent and severe infections, dysregulated granulomatous inflammation, or autoimmunity (1). Mutations in NADPH oxidase affect the generation of reactive oxygen species (ROS) within phagosomes and is essential for the normal killing of bacteria and fungi (2).
Subunits of NADPH oxidase are located at the membrane and in the cytoplasm. For example, the catalytic glycoprotein gp91−phox and the non-glycosylated protein p22−phox are located at the cell membrane and together form the heterodimer cytochrome b558. Upon induction, the cytosolic proteins p47−phox, p67−phox, and p40−phox migrate toward cytochrome b558 to activate the enzyme complex and produce ROS (1, 2).
Mutations in all five structural genes of NADPH oxidase have been implicated in CGD, affecting 1 in 250,000 individuals. In addition, the X-linked CGD caused by cytochrome b-245 beta chain (CYBB) gene mutations is the most common form affecting gp91−phox and accounts for 74% of all CGD patients in Latin America (3). Defects in the neutrophil cytosolic factor 1 (NCF1) gene encoding the p47−phox subunit represent the most common form of autosomal recessive (AR) CGD, accounting for 20% of all cases. In contrast, mutations in the genes encoding p22−phox (cytochrome b-245 alpha chain, CYBA) and p67−phox (neutrophil cytosolic factor 2, NCF2) are rare and account for <5% of all cases of CGD (1, 4).
As a result of the antimicrobial activity defect in CGD, these patients are susceptible to infections such as pneumonia, lymphadenitis, cutaneous and hepatic abscesses, osteomyelitis and, septicemia (3, 5). Pneumonia is the most common pulmonary infection, and patients may also have complications such as lung abscesses, empyema, and hilar lymphadenopathy.
The microorganisms implicated in CGD patient infections include Staphylococcus aureus, Aspergillus spp., the Burkholderia cepacia complex, Candida spp., enteric gram-negative bacteria, Mycobacterium tuberculosis, and Serratia marcescens (3, 5). Overall, treatment with lifelong antibiotic and antifungal prophylaxis and interferon-gamma (IFNγ) have a clear benefit, with a reduction in both the number and severity of infections in most CGD patients. Regardless, the only curative option is allogenic hematopoietic stem cell transplantation (HSCT), which shows very promising results in well-defined circumstances (6, 7).
Here, we describe a case of autosomal recessive CGD (AR-CGD) diagnosed in a 3-year-old female. The patient carried a novel mutation in NCF2 leading to a K86Ifs*2 residue change in the p67−phox protein. She presented with recurrent pneumonia, even under trimethoprim-sulfamethoxazole (TMP-SMX) and itraconazole prophylaxis plus IFNγ therapy.
Ethical Approval
This study was approved by the Ethics and Research Committees in Humans of the Sabará Hospital and Institute of Biomedical Sciences, University of São Paulo, in accordance with the Declaration of Helsinki. The patient's parents provided written informed consent before the investigation.
Case Report
The patient was a 3-year-old girl, the only child of Lebanese parents without direct consanguinity, with early onset of severe, recurrent pulmonary infections. Her first clinical manifestation occurred at 3 months of life, when she presented with fever without localizing signs. At that time, she was treated with intramuscular ceftriaxone for 3 days until she was admitted at an Intensive Care Unit due to bilateral pneumonia and pleural effusion. Then, she was submitted to pleural drainage and treatment with polymyxin, meropenem, vancomycin, and linezolid for 14 days. No etiological agents were identified in blood cultures and pleural fluids. At discharge, cefuroxime was prescribed for 10 days due to the persistent infection that was identified by radiological images.
At 4 months of age, she presented with oral moniliasis and Bacillus Calmette-Guérin (BCG) scar suppuration with slow healing. She was hospitalized for the second time at 6 months of age when she was diagnosed with bronchiolitis, with identification of Haemophilus parainfluenzae biotype I. She developed bilateral pneumonia, which did not respond adequately to ceftriaxone treatment and evolved to necrotizing pneumonia (Figure 1A). At that time, the patient was transferred to our hospital for evaluation by pulmonologists and immunologists. The patient was diagnosed with necrotizing pneumonia; left lung abscess; anemia, most likely secondary to infection; and stomatitis. She received a transfusion of red blood cell concentrate and was treated with ceftriaxone and linezolid and subsequently treated with meropenem and amphotericin B. Culture of material collected from bronchoscopy and bronchoalveolar lavage showed no growth of aerobic or anaerobic bacteria.
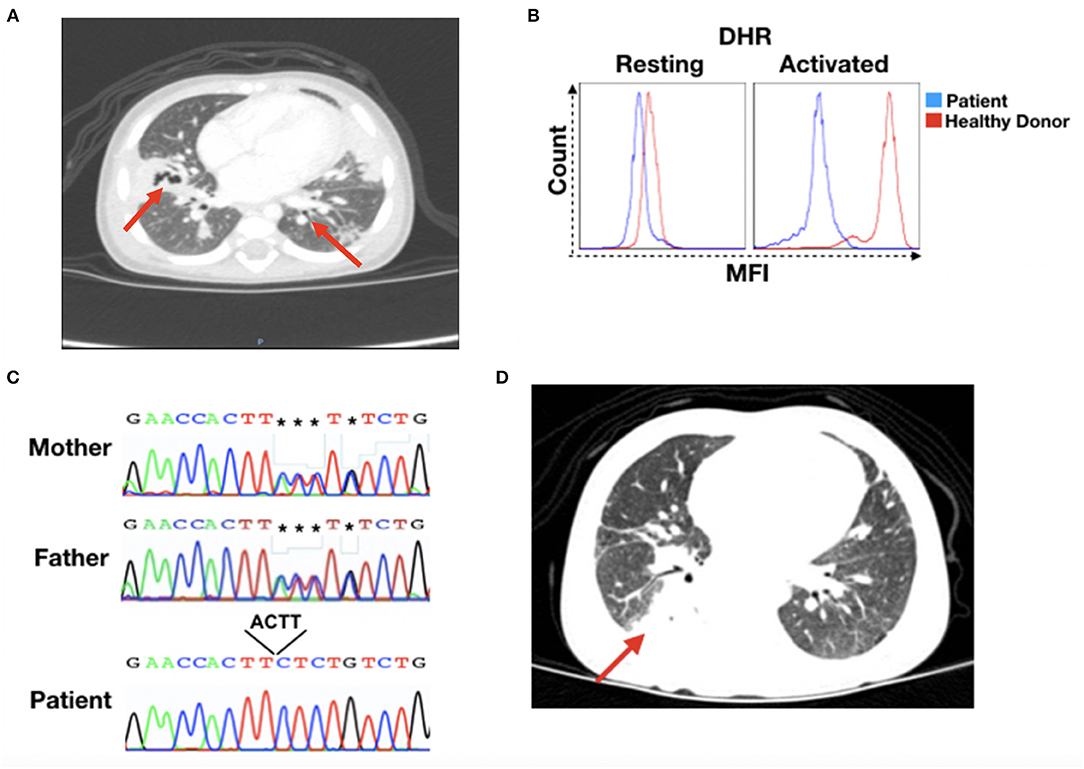
Figure 1. CGD patient's clinical recordings. (A) Patient's chest X-ray at 15 months of age. Red arrows indicate lesion sites. (B) Dihydrorhodamine (DHR) for patient and healthy donor granulocytes in the resting or activated state. (C) DNA sequencing results for the NCF2 gene. Chromatographs for the mother (top), father (middle), and patient (bottom). The black box shows the sequence depletion in a splicing region (c.256_257+2delAAGT NM_000433), resulting in a K86Ifs*2 residue change in the p67−phox protein. The reverse nucleotide sequences are shown. (D) Patient's thorax computed tomography at 17 months of age. The red arrow indicates the lesion site. MFI, mean fluorescence intensity.
At this time, the patient was submitted to an immunological investigation, which revealed normal immunoglobulin levels, lymphocyte subset numbers, and complement system. Nevertheless, a dihydrorhodamine (DHR) test showed that the patient's stimulated granulocytes presented abnormal ROS production, a characteristic of CGD patients (Figure 1B). Thus, CGD prophylaxis was initiated with the administration of Bactrim, itraconazole, and IFNγ.
Despite prophylactic treatment, the patient continued to need hospitalization for pneumonia at 8 and 11 months of age that required systemic antibiotics. The first genetic analysis was performed on one of these occasions, although Sanger sequencing revealed no mutations in the NCF1 gene (data not shown). At this time, the patient's mother reported that one of the patient's female cousin had received two transplants for an unidentified immunological problem.
At 23 months of age, the patient presented with adenitis and cervical abscess, which were treated with ceftriaxone, clindamycin, and surgical drainage. She was discharged with cefuroxima treatment; however, 1 week later, she required medical assistance after developing right lobe pneumonia and atelectasis. These last conditions were treated with cefepime and prednisolone.
Whole-exome sequencing was performed to confirm or discard a CGD diagnosis, and sequence analysis revealed a mutation (c.256_257+2delAAGT NM_000433) in the NCF2 gene that leads to a K86Ifs*2 residue change in the p67−phox protein, which was confirmed by Sanger sequencing (Figure 1C). Sanger analysis also showed that the parents were heterozygous for the mutation. The sequence data were analyzed using the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov) and Ensembl SNP (www.ensembl.org) databases, confirming this novel mutation in NCF2.
Prophylactic medications were maintained during patient follow-up. However, at 2 years and 7 months of age, she was readmitted with pneumonia that was treated with ceftazidima. Thorax computed tomography revealed pulmonary consolidation in the upper segment of the right lower lobe and areas of retractable stretch marks, with a bilateral sparse sequential aspect that was more evident in the posterior segment of the right upper lobe, the middle lobe, and the lingula (Figure 1D). Bronchoscopy showed non-purulent laryngotracheobronchitis, with negative results for acid-alcohol resistant bacilli, fungi, cytomegalovirus or Pneumocystis jirovecii, as well as normal serum galactomannan levels.
At 3 years and 5 months of age, the child was hospitalized with jugal abscess arising from a dental procedure. The abscess was drained, and she received ceftriaxone, clindamycin and oral steroids for 5 days. Once she presented an anaphylactic reaction to ceftriaxone, the patient received epinephrine and continued treatment with clindamycin and steroids for an additional 3 days. Thereafter, she was discharged under treatment with sulfamethoxazole and trimethoprim.
Discussion
CGD comprises a group of genetic disorders of the NADPH oxidase complex of phagocytes that results in defective microbicidal activity due to dysregulated ROS generation. Patients with CGD suffer from recurrent life-threatening bacterial and fungal infections and from uncontrolled inflammatory responses leading to granuloma formation in multiple organs. These infections are caused by catalase-positive microorganisms, Staphylococcus spp., Serratia spp., Aspergillus spp., and Burkholderia cepacia, as well as by BCG and M. tuberculosis in developing countries (1, 3, 5).
Here, we report the case of a patient who developed recurrent pneumonia beginning at 3 months of age. At this age, she was admitted to an intensive care unit due to bilateral pneumonia and pleural effusion. At 6 months of age, she developed bilateral pneumonia that evolved to necrotizing pneumonia, presenting with left lung abscess, anemia, and stomatitis. These clinical manifestations are present in CGD patients in which the lungs, followed by the gastrointestinal tract, lymph nodes, and skin are the most prevalent infection sites. Recurrent pneumonia is the most frequent clinical condition associated with CGD and may be complicated by abscess formation or pleural effusion (1, 3, 5, 8). Moreover, respiratory complications are among the signs and symptoms that constitute significant causes of mortality and morbidity in CGD.
In a retrospective analysis involving 71 patients with CGD and mycobacterial infection (9), 75% of these patients (53 individuals) presented adverse effects from BCG vaccination. This finding brings up the necessity of a CGD investigation when such adverse effects are observed. Considering the occurrence of BCG scar suppuration with slow healing when our patient was 4 months old and the characteristics of her infections with evolution to empyema and pulmonary abscess, we investigated the hypothesis for primary immunodeficiency in phagocytes. Then, a DHR test was performed, and the results were suggestive of CGD.
Genetic investigations confirmed that the patient has AR-CGD due to a novel homozygous mutation in the NCF2 gene. This mutation leads to sequence depletion in a splicing region (c.256_257+2delAAGT NM_000433), resulting in a K86Ifs*2 residue change in the p67−phox protein. The occurrence of AR forms of CGD is well-correlated with the high frequency of consanguineous marriages in Western Asian countries, which includes the Lebanese Republic, the birth place of our patient. Indeed, in these countries, AR-CGD is more common (10–13) than the X-linked CGD form, which predominates in the United States (14), Europe (15–18), and Japan (19) (60% of cases). Nevertheless, our patient had Lebanese parents without direct consanguinity, and Sanger analysis showed that they were heterozygous for the same NCF2 mutation probably inherited from a progenitor many generations ago.
The current prophylactic therapy for CGD is daily doses of TMP-SMX and itraconazole, which results in reduced bacterial and fungal infections among CGD patients. Indeed, lifelong prophylaxis with TMP-SMX (5 mg/kg/d up to 320 mg trimethoprim a day) has been used routinely since the 1970s, and it has reduced the incidence of serious non-fungal infections from 7.1 to 2.4 per 100 patient-months in patients with AR-CGD (20, 21). Moreover, prophylaxis with itraconazole (5 mg/kg/d up to 200 mg daily) can decrease fungal infection mortality and morbidity (22). The addition of IFNγ to CGD prophylactic therapy is still under debate, and this decision should be made on a case-by-case basis. For example, an Italian study did not find evidence of increased protection from infections in CGD patients treated with IFNγ (16). By contrast, another report showed that CGD patients treated with IFNγ (50 μg/m2 subcutaneously three times weekly) exhibited a clear reduction in severe infections without exacerbation of granulomatous or inflammatory complications in 67% of patients (23).
Prophylactic therapy is effective for all genetic types of CGD, and children younger than 10 years old appear to benefit the most. The use of IFNγ in CGD prophylaxis is particularly encouraged for patients experiencing increased infection frequency but should be interrupted in cases of adverse events, such as fever, fatigue, headache, rash, myalgia, abdominal pain, and/or granulomatous colitis (6, 7). Since our patient still presents persistent respiratory complications, particularly in the right lung, even under prophylactic treatment, HSCT is currently in consideration.
In summary, we report a novel mutation in the NCF2 gene that led to AR-CGD in a young patient with recurrent lung infection even under TMP-SMX and itraconazole prophylaxis combined with IFNγ treatment. Our patient's clinical manifestations illustrate the signs that clinicians must take into account to suspect a diagnosis of CGD even in female children and to trigger diligent immunological and genetic investigations to confirm or discard this hypothesis. Further genetic investigations in the patient's family and her transplanted cousin should be performed for a complete correct genetic counseling.
Data Availability Statement
This manuscript contains previously unpublished data. The name of the repository and accession number are not available.
Ethics Statement
This study was approved by the Ethics and Research Committees in Humans of the Sabará Hospital and Institute of Biomedical Sciences, University of São Paulo, in accordance with the Declaration of Helsinki. The patient's parents provided written informed consent before the investigation.
Author Contributions
JA, EO, and NZ designed, conducted the experiments, and wrote the manuscript. EI conducted the experiments. AL, SC, and FF performed clinical and laboratory data collection and analysis. WA-F performed data analysis, wrote, and reviewed the manuscript. AC-N designed the experiments, wrote, and reviewed the manuscript. AL also reviewed the manuscript.
Funding
This work was supported by PENSI Institute, Ministério da Saúde do Brasil (PRONAS/PDC 2015, 25000.077928/2015-06, JATA, AC-N) and the Jeffrey Modell Diagnostic Center São Paulo (AC-N).
Conflict of Interest
JA and EO were employed by Immunogenic Inc.
The remaining authors do not declare any commercial or financial relationships that could lead to any potential conflict of interest.
Abbreviations
CGD, chronic granulomatous disease; TMP-SMX, trimethoprim-sulfamethoxazole; IFNγ, interferon-gamma; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species; AR, autosomal recessive; HSCT, hematopoietic stem cell transplantation; BCG, Bacillus Calmette-Guérin.
References
1. Arnold DE, Heimall JR. A Review of chronic granulomatous disease. Adv Ther. (2017) 34:2543–57. doi: 10.1007/s12325-017-0636-2
2. Gardiner GJ, Deffit SN, McLetchie S, Pérez L, Walline CC, Blum JS. A role for NADPH oxidase in antigen presentation. Front Immunol. (2013) 4:295. doi: 10.3389/fimmu.2013.00295
3. Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke LF, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American Patients: first report from the LASID registry. Pediatr Blood Cancer. (2015) 62:2101–7. doi: 10.1002/pbc.25674
4. Ben-Farhat K, Ben-Mustapha I, Ben-Ali M, Rouault K, Hamami S, Mekki N, et al. A founder effect of c.257 + 2T > C mutation in NCF2 gene underlies severe chronic granulomatous disease in eleven patients. J Clin Immunol. (2016) 36:547–54. doi: 10.1007/s10875-016-0299-9
5. Zhou Q, Hui X, Ying W, Hou J, Wang W, Liu D, et al. A cohort of 169 chronic granulomatous disease patients exposed to BCG vaccination: a retrospective study from a Single Center in Shanghai, China (2004-2017). J Clin Immunol. (2018) 38:260–72. doi: 10.1007/s10875-018-0486-y
6. Marciano BE, Wesley R, De Carlo ES, Anderson VL, Barnhart LA, Darnell D, et al. Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin Infect Dis. (2004) 39:692–9. doi: 10.1086/422993
7. Marciano BE, Holland SM. Primary immunodeficiency diseases: current and emerging therapeutics. Front Immunol. (2017) 8:937. doi: 10.3389/fimmu.2017.00937
8. de Albuquerque JAT, de Oliveira Junior EB, Zurro NB, Vendramini P, Ishizuka EK, Borgli DSP, et al. A C126R de novo mutation in CYBB leads to X-linked chronic granulomatous disease with recurrent pneumonia and BCGitis. Front Pediatr. (2018) 6:248. doi: 10.3389/fped.2018.00248
9. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E Jr, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. (2016) 138:241–8.e3. doi: 10.1016/j.jaci.2015.11.041
10. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. (2011) 31:792–801. doi: 10.1007/s10875-011-9567-x
11. Köker MY, Camcioglu Y, van Leeuwen K, Kiliç SS, Barlan I, Yilmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. (2013) 132:1156–63. doi: 10.1016/j.jaci.2013.05.039
12. Tajik S, Badalzadeh M, Fazlollahi MR, Houshmand M, Bazargan N, Movahedi M, et al. Genetic and molecular findings of 38 Iranian patients with chronic granulomatous disease caused by p47-phox defect. Scand J Immunol. (2019) e12767. doi: 10.1111/sji.12767
13. Kutukculer N, Aykut A, Karaca NE, Durmaz A, Aksu G, Genel F, et al. Chronic granulamatous disease: two decades of experience from a pediatric immunology unit in a country with high rate of consanguineous marriages. Scand J Immunol. (2018) 1:e12737. doi: 10.1111/sji.12737
14. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
15. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS ONE. (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
16. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. (2008) 126:155–64. doi: 10.1016/j.clim.2007.09.008
17. Raptaki M, Varela I, Spanou K, Tzanoudaki M, Tantou S, Liatsis M, et al. Chronic granulomatous disease: a 25-year patient registry based on a multistep diagnostic procedure, from the referral center for primary immunodeficiencies in Greece. J Clin Immunol. (2013) 33:1302–9. doi: 10.1007/s10875-013-9940-z
18. Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. (2008) 152:211–8. doi: 10.1111/j.1365-2249.2008.03644.x
19. Kobayashi S, Murayama S, Takanashi S, Takahashi K, Miyatsuka S, Fujita T, et al. Clinical features and prognoses of 23 patients with chronic granulomatous disease followed for 21 years by a single hospital in Japan. Eur J Pediatr. (2008) 167:1389–94. doi: 10.1007/s00431-008-0680-7
20. Margolis DM, Melnick DA, Alling DW, Gallin JI. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. (1990) 162:723–6. doi: 10.1093/infdis/162.3.723
21. Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med. (2003) 348:2416–22. doi: 10.1056/NEJMoa021931
22. Blumental S, Mouy R, Mahlaoui N, Bougnoux ME, Debré M, Beauté J, et al. Invasive mold infections in chronic granulomatous disease: a 25-year retrospective survey. Clin Infect Dis. (2011) 53:e159–69. doi: 10.1093/cid/cir731
Keywords: chronic granulomatous disease, NADPH oxidase, NCF2 gene, novel mutation, interferon-gamma
Citation: de Albuquerque JAT, Lima AM, de Oliveira Junior EB, Ishizuka EK, Aragão-Filho WC, Zurro NB, Chiba SM, Fernandes FR and Condino-Neto A (2019) A Novel Mutation in the NCF2 Gene in a CGD Patient With Chronic Recurrent Pneumopathy. Front. Pediatr. 7:391. doi: 10.3389/fped.2019.00391
Received: 14 February 2019; Accepted: 11 September 2019;
Published: 27 September 2019.
Edited by:
Fabio Candotti, Lausanne University Hospital (CHUV), SwitzerlandReviewed by:
Caterina Cancrini, University of Rome Tor Vergata, ItalyHenner Morbach, University Hospital Würzburg, Germany
Copyright © 2019 de Albuquerque, Lima, de Oliveira Junior, Ishizuka, Aragão-Filho, Zurro, Chiba, Fernandes and Condino-Neto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Condino-Neto, YW50b25pb2NvbmRpbm9AZ21haWwuY29t;Y29uZGlub0B1c3AuYnI=