 Meiqiu Wang
Meiqiu Wang Ren Wang2†
Ren Wang2†- 1Department of Pediatrics, Jinling Hospital, The First School of Clinical Medicine, Southern Medical University, Nanjing, China
- 2Department of Pediatrics, Jinling Hospital, Nanjing Medical University, Nanjing, China
- 3Department of Pediatrics, Jinling Hospital, Nanjing, China
- 4Department of Neonatology, Taizhou People's Hospital, Taizhou, China
Background: The phenotypes of TRPC6 mutations have been reported mainly in familial and sporadic focal segmental glomerulosclerosis (FSGS), which can occur in both adults and children. Herein, we report on two children with novel TRPC6 spontaneous missense mutations associated with immune complex-mediated glomerulonephritis and minor glomerular abnormality (MGA) that showed to be resistant to corticosteroids and other immunosuppressants.
Case Presentation: A 9-year-old girl presented with steroid-resistant nephrotic syndrome (SRNS), while another 11-year-old boy developed proteinuria at 7 years old. Treatment with a variety of immunosuppressants had no effect, and the renal biopsy showed immune complex-mediated glomerulonephritis and MGA. No members of their family were clinically affected. Genetic testing was performed in the two patients, revealing two novel spontaneous missense mutations in TRPC6—N110S and P112R. The girl developed end-stage renal disease (ESRD) 5 months after onset while the boy continued to have sub-nephrotic range proteinuria and normal creatinine.
Conclusions: Two novel TRPC6 mutations were associated with the atypical phenotype—immune complex-mediated glomerulonephritis and MGA, rather than FSGS as previously reported. Their rates of disease progression are different. Genetic testing is helpful to identify the etiology and avoid the side effects brought on by immunosuppressants.
Introduction
TRPC6, which encodes the protein transient receptor potential cation channel protein, is a non-selective cation channel, which is expressed in podocytes and interacts with nephrin and podocin to participate in signal transduction, cell polarization, skeletal structure stabilization, and other physiological functions between podocytes (1). In 2005, Winn identified an autosomal dominant family of focal segmental glomerulosclerosis (FSGS) with a point mutation in the TRPC6 gene on chromosome 11q (2). Since then, some new mutations of TRPC6 have been reported. It was confirmed that 24 mutations of the TRPC6 gene were involved in the pathogenesis of FSGS (Table 1). However, other pathological types associated with TRPC6 mutations have been less reported. Herein, we report on two children with novel TRPC6 mutations related to immune complex-mediated glomerulonephritis and minor glomerular abnormality (MGA).
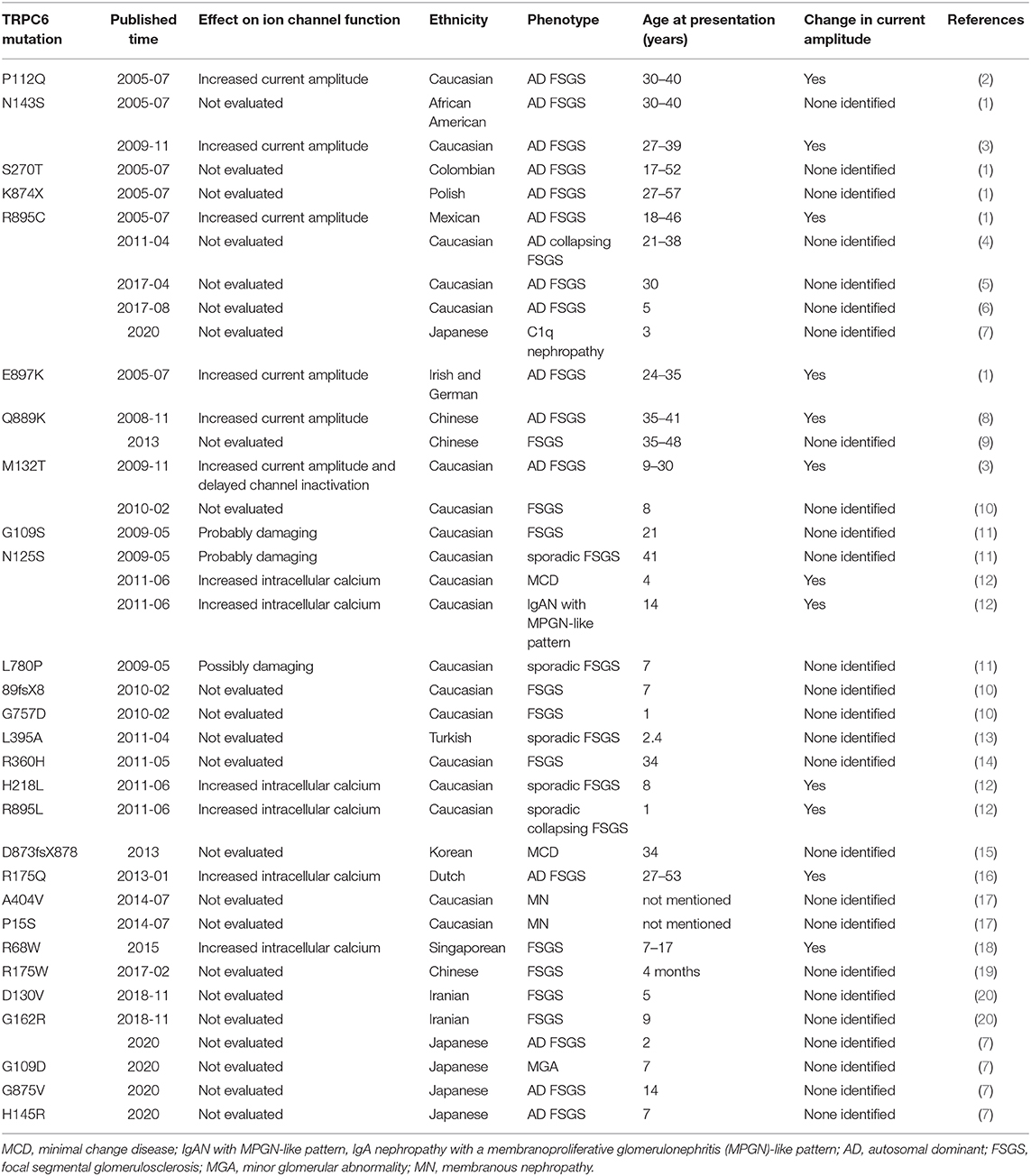
Table 1. Mutations in TRPC6 protein currently identified to cause kidney disease.
Case Presentation
Case 1
A 10-year-old Chinese girl presented at the age of 9 years with steroid-resistant nephrotic syndrome (SRNS). The patient tested serologically negative for hepatitis B virus, human immunodeficiency virus, antinuclear antibodies (ANA) spectrum, and cytoplasmic and perinuclear antineutrophil cytoplasmic autoantibodies. She had periorbital edema and all other physical examinations were normal. She had a normal cardiac exam, normal nails and patellae, and no rashes or arthritis. Her percutaneous kidney biopsy demonstrated 6/26 showing as globally obsolete, 3/26 showing segmental sclerosis, and 1 having a cellular crescent. Mesangial cellularity was mildly increased, and tubules showed a patchy atrophy. Immunofluorescence staining showed that 1+ for C3 and C1q, IgG and IgM were 2+. Electron microscopy revealed that the distribution of medium electron density was observed in the mesangial region and subendothelial basement membrane. No electron density was found in the epithelium of the glomerular basement membrane. The podocyte processes were widely fused (Figure 2). Her treatment included atorvastatin 10 mg daily, and furosemide as needed, titrated to symptoms. Tacrolimus and cyclophosphamide had been added to steroids at different times, without success in achieving remission. No members of the family (parents and a sister) were clinically affected. To identify the pathogeny, the child and her family underwent the Whole Exome sequencing. High-throughput sequencing was performed with an Illumina NovaSeq 6000 series sequencer, and not less than 99% of the target sequence was sequenced. The detected mutations were checked in various databases (e.g., dbSNP, ExAC, ESP, OMIM, HGMD, ClinVar, etc.). It showed that a new missense change (c.329A>G) was identified in exon 2 causing an asparagine-to-serine substitution(N110S) within the first ankyrin repeat of the TRPC6 protein (Figure 1). At the onset of 5 months, the child developed chronic renal insufficiency and began hemodialysis and peritoneal dialysis.
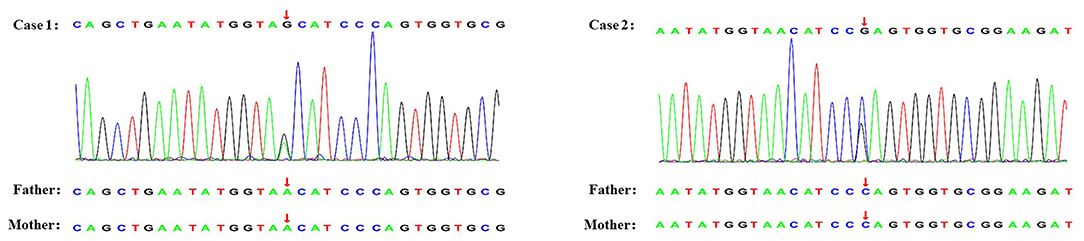
Figure 1. Genetic testing of two novel TRPC6 mutations. The arrow shows the position where base substitution results in changes in amino acids.
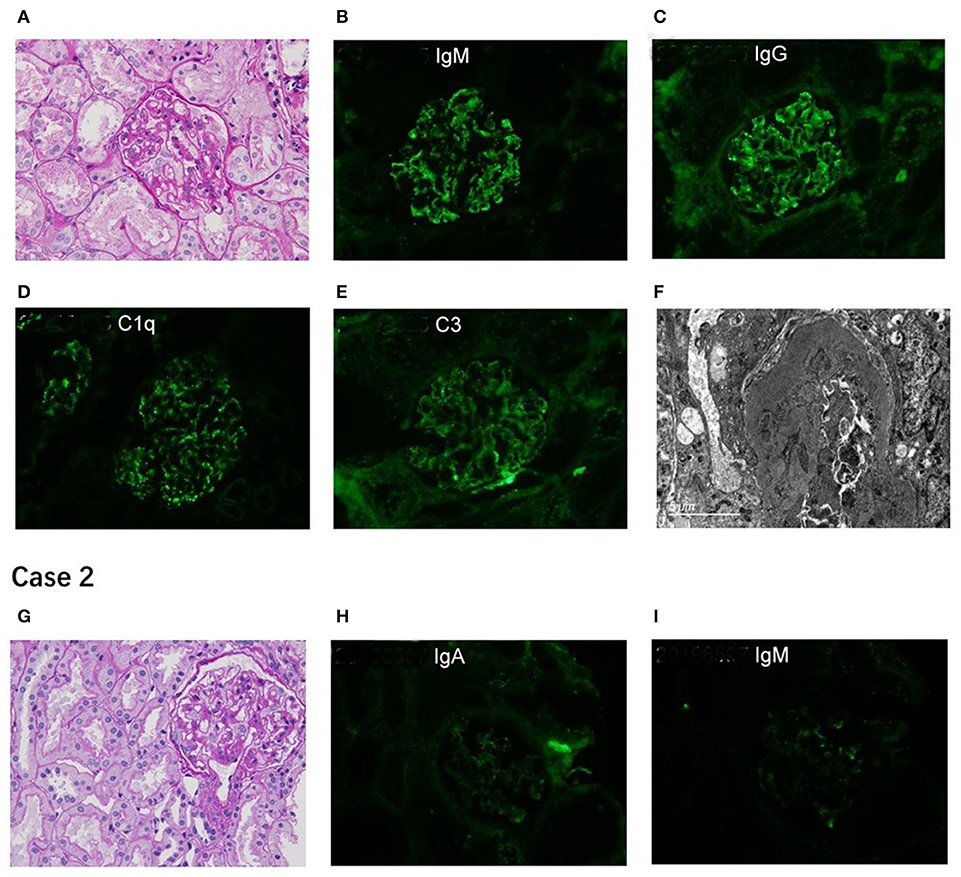
Figure 2. Renal pathological presentation of two proband. (A,G) for light microscopy (PAS×400); (B–E,H,I) for immunofluorescence; (F) for electron microscope. The figure of Case 2 is for subsequent biopsy.
Case 2
A 7-year-old Chinese boy presented 2 days after the onset of eyelid swelling and tonsillitis. Urine dipstick tested 2+ positive for protein with no blood and 24-h protein excretion was 0.88 g. Initial serum concentrations were normal except for a slightly low albumin 3.3 g/dl and a slightly high total cholesterol 238 mg/dl. Other laboratory results showed that ANA, complement, antistreptolysin O, and serum creatinine were normal, with an initial biopsy showing IgA nephropathy (IgAN) (Lee II, M1E1S0T0). He was not edematous and did not have hypertension. He had normal nails and patellae, a normal cardiac exam, and no rashes or arthritis. His medications included losartan potassium 50 mg daily and fosinopril 10 mg daily. Cyclosporine A, mycophenolate mofetil had been added to steroids at different times, without success in achieving remission. At age 11, a subsequent biopsy was performed. Light microscopy demonstrated 1 out of 14 segmental sclerotic glomeruli with no other abnormalities. Immunofluorescence microscopy tested slightly positive for IgA and IgM. Staining for IgG, C3, and C1q was negative (Figure 2). Electron microscopy showed no glomerulus. The histopathologic diagnosis was MGA. He had no family history of kidney disease. Next, the Whole Exome sequencing was performed on the patient and his parents (case 2 used the same detection method as case 1). It showed that a new missense change (c.335C>G) was identified in exon 2 causing a proline-to-arginine substitution (P112R) within the first ankyrin repeat of the TRPC6 protein (Figure 1). He had been taking angiotensin-converting enzyme inhibitor (ACEI) as his main treatment. Current serum creatinine was 56 μmol/L, and 24-h urine showed proteinuria 1 g/day.
Discussion
TRPC6 interacts with nephrin and podocin and is an important component of the podocyte slit diaphragm (3). Mutations in genes encoding the podocyte structural proteins lead to the development of proteinuria, bringing about progressive kidney failure. To date, a total of 28 novel TRPC6 mutations have been associated with FSGS, SRNS, and proteinuria (Table 1). Here we first identified two novel TRPC6 mutations (N110S, P112R) that manifest as diverse types of renal pathology with early-onset, multidrug resistance, and different clinical outcomes.
Winn identified an autosomal dominant family of FSGS with a point mutation in the gene TRPC6 (2). As shown in Table 1, we found that the incidence of TRPC6 mutation was more familial and less sporadic. The pathological type of these two children was not FSGS and the parents were not clinically affected. But the two children were resistant to immunosuppressants, which we suspected may be caused by gene spontaneous mutations. As a result, the two children tested negative for NPHS2, NPHS1, WT1, ACTN4, COL4A5, and CD2AP genes but positive for TRPC6. Their parents' genetic testing was normal. It shows the importance of environmental or other factors for mutations.
Several autoimmune diseases sporadically cause immune-complex deposition nephropathy. Circulating immune complexes because of autoimmune disease have been assumed to activate the classical complement pathway, which leads to glomerular injury (21). In case 1, the girl's response to treatment and laboratory tests would suggest that it was a non-immune-complex deposition disease. Previous studies have reported cases of IgAN and C1q nephropathy associated with TRPC6 mutations (7, 22), but there is no further explanation for the deposition of immune complexes in the kidney. It has been reported that several candidate genes relating to IgAN are involved in cytokine pathways, immunoregulation, and glycosylation (23, 24). Cox et al. identified 23 genes with candidate pathogenic variants functionally related to a large network of immune-related pathways. They speculated that the IgAN disease status may be affected by a series of mutations that affect immune-related networks (25). Milillo et al. identified the SPRY2 gene related to IgAN, which is part of the MAPK/ERK pathway (26). So, we hypothesized that variants in some pathways caused by TRPC6 mutations are risk factors for the development of immune-complex deposition nephropathy.
According to earlier studies, it has been identified that heterozygous mutations of TRPC6 cause late-onset autosomal-dominant FSGS. Heeringa et al. first reported that an M132T mutation in the TRPC6 gene can lead to early-onset FSGS in 2009 (3). Later, more and more TRPC6 mutations associated with kidney disease have been reported in children. The onset-age of the two children we reported are 7 and 9 years. Heeringa et al. believed that both a direct increase in calcium influx and an impaired channel inactivation may result in a more severe or earlier disease onset (3). It is shown in animal experiments that transient overexpression of wild-type TRPC6 in murine glomeruli causes rapid onset proteinuria (27).
The PolyPhen program predicted the TRPC6P112R(c.335C>G) mutation in case 2 to be “probably damaging” and the SIFT program anticipated this mutation to be “Deleterious” (Table 2). The mutation was not found in any public database of SNP. Winn indicated that Pro112 is highly conserved in TRPC protein homologs from multiple species and the peak intracellular concentrations were considerably higher in cells expressing TRPC6P112Q(c.335C>A) as compared with WT controls (2). The mutation in case 2 is also in Pro112, but it causes a proline-to-arginine substitution. We speculated that P112R may increase the current amplitude. The clinical manifestations and pathological type caused by P112R in our report are less severe than P112Q. The mechanism needs to be further explored. Additionally, other genetic and/or environmental factors may have an important impact on disease expressivity and penetrance. Two renal biopsies of case 2 had different outcomes, possibly because of immunofluorescence trapping. Nagano et al. found that 62% were FSGS and 28% were MGA in the histopathological diagnosis of 230 Japanese patients with proteinuria (7). MGA is a common pathological type of proteinuria, but only 1 case of MGA with normal renal function related to TRPC6 has been reported. MGA associated with TRPC6 mutation may progress slowly.
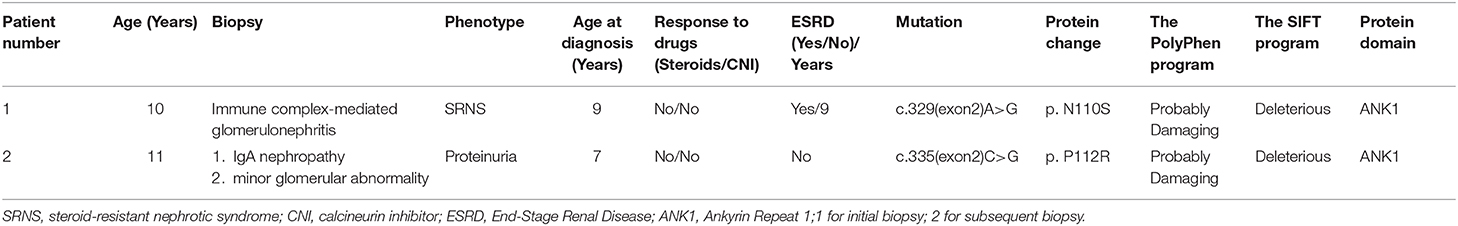
Table 2. Basic characteristics and clinical manifestations of 2 children.
The PolyPhen program predicted the TRPC6N110S(c.329A>G) mutation in case 1 to be “probably damaging” and the SIFT program anticipated this mutation to be “Deleterious” (Table 2). The N110S mutation, which was not found in any public database of SNP, is located very close to P112Q, within the same ankyrin repeat. Considering their close location, we speculated that the channel kinetics of both mutants would be similar. The girl in case 1 entered end-stage renal disease (ESRD) only 5 months after the onset of the disease. One patient with IgAN associated with TRPC6 mutation had normal renal function (12), while another patient with C1q nephropathy had developed ESRD in the previous report (7). They have different rates of disease progression, and IgAN may progress more slowly. As it was a novel mutation that had not been previously reported on, and because we did not enter the molecular level to learn about the mechanism, the possible explanation we can offer is that the children may have proteinuria at an early stage, but it is not detected in time.
The girl in case 1 presented with SRNS and the boy in case 2 presented with proteinuria. After the failure of steroid therapy, they added immunosuppressants to relieve proteinuria. But it still did not work. A German study showed that 81% of patients with genetic SRNS did not respond to calcineurin inhibitor (CNI)—cyclosporin A (28). There are 28 TRPC6 mutations shown in Table 1; eleven (P112Q, N143S, R895C, E897K, Q889K, M132T, N125S, H218L, R895L, R175Q, R68W mutation) were gain-of-function mutations that resulted in increasing calcium channel activity (1–3, 8, 12, 16, 18). CNIs may affect the CaN-NFAT signaling pathway, which may promote cell apoptosis and destroy the podocyte actin cytoskeleton, thereby improving proteinuria (29). However, two of our patients were resistant to CNI. To avoid the side effects of immunosuppressants, they have stopped using the drugs. At present, the treatment goal in both patients is to protect kidney function, delay the progression of renal decline, and promote symptomatic management. Currently, the girl in case 1 receives regular peritoneal dialysis, while the boy in case 2 only takes ACEI orally.
Herein, we reported two new mutations in the TRCP6 gene that are related to different types of renal pathology, rather than FSGS as previously reported in the literature. But their diseases progress at different rates. Even though the PolyPhen and the SIFT program are very useful for predicting probable mutations, to date the functional assay is the definitive step to determine if a variant is a mutation. The lack of animal model experiments is a limitation of our research. We hypothesized that mutations in certain pathways caused by TRPC6 mutations may be responsible for the development of immune-complex deposition nephropathy in case 1. However, the exact mechanism of this disease needs to be confirmed by further experiments.
Conclusions
Two novel TRPC6 mutations were associated with atypical phenotype—immune complex-mediated glomerulonephritis and MGA. Their rates of disease progression are different. Genetic testing is helpful to identify the etiology and avoid the side effects brought on by immunosuppressants.
Ethics Statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MW, RW, XH, MY, ZX, and CG drafted the manuscript or revised it critically for important intellectual content, provided final approval of the version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
Funding
Funding was provided by the Clinical Advanced Techniques, Primary Research & Development Plan of Jiangsu Province (BE2017719); the Pediatric Medical Innovation Team of Jiangsu Province (CXTDA2017022); and Effect and Mechanism of Polygonin in Acute Renal Injury in Mice (2018K089B).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This is a short text to acknowledge the contributions of specific colleagues, institutions, or agencies that aided the efforts of the authors.
References
1. Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. (2005) 37:739–44. doi: 10.1038/ng1592
2. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. (2005) 308:1801–4. doi: 10.1126/science.1106215
3. Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, et al. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE. (2009) 4:e7771. doi: 10.1371/journal.pone.0007771
4. Liakopoulos V, Huerta A, Cohen S, Pollak MR, Sirota RA, Superdock K, et al. Familial collapsing focal segmental glomerulosclerosis. Clin Nephrol. (2011) 75:362–8. doi: 10.5414/cn106544
5. Iqbal Z, Sayer JA. Case Report: making a diagnosis of familial renal disease - clinical and patient perspectives. F1000Res. (2017) 6:470. doi: 10.12688/f1000research.11316.1
6. Tan W, Lovric S, Ashraf S, Rao J, Schapiro D, Airik M, et al. Analysis of 24 genes reveals a monogenic cause in 11.1% of cases with steroid-resistant nephrotic syndrome at a single center. Pediatr Nephrol. (2018) 33:305–14. doi: 10.1007/s00467-017-3801-6
7. Nagano C, Yamamura T, Horinouchi T, Aoto Y, Ishiko S, Sakakibara N, et al. Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Sci Rep. (2020) 10:270. doi: 10.1038/s41598-019-57149-5
8. Zhu B, Chen N, Wang ZH, Pan XX, Ren H, Zhang W, et al. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res. (2009) 664:84–90. doi: 10.1016/j.mrfmmm.2008.11.021
9. Zhang Q, Ma J, Xie J, Wang Z, Zhu B, Hao X, et al. Screening of ACTN4 and TRPC6 mutations in a Chinese cohort of patients with adult-onset familial focal segmental glomerulosclerosis. Contrib Nephrol. (2013) 181:91–100. doi: 10.1159/000348471
10. Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2010) 5:2075–84. doi: 10.2215/cjn.01190210
11. Santin S, Ars E, Rossetti S, Salido E, Silva I, Garcia-Maset R, et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2009) 24:3089–96. doi: 10.1093/ndt/gfp229
12. Gigante M, Caridi G, Montemurno E, Soccio M, d'Apolito M, Cerullo G, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. (2011) 6:1626–34. doi: 10.2215/cjn.07830910
13. Mir S, Yavascan O, Berdeli A, Sozeri B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant. (2012) 27:205–9. doi: 10.1093/ndt/gfr202
14. Buscher AK, Konrad M, Nagel M, Witzke O, Kribben A, Hoyer PF, et al. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol. (2012) 78:47–53. doi: 10.5414/cn107320
15. Mottl AK, Lu M, Fine CA, Weck KE. A novel TRPC6 mutation in a family with podocytopathy and clinical variability. BMC Nephrol. (2013) 14:104. doi: 10.1186/1471-2369-14-104
16. Hofstra JM, Lainez S, van Kuijk WH, Schoots J, Baltissen MP, Hoefsloot LH, et al. New TRPC6 gain-of-function mutation in a non-consanguineous dutch family with late-onset focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2013) 28:1830–8. doi: 10.1093/ndt/gfs572
17. Hofstra JM, Coenen MJ, Schijvenaars MM, Berden JH, van der Vlag J, Hoefsloot LH, et al. TRPC6 single nucleotide polymorphisms and progression of idiopathic membranous nephropathy. PLoS ONE. (2014) 9:e102065. doi: 10.1371/journal.pone.0102065
18. Sun ZJ, Ng KH, Liao P, Zhang Y, Ng JL, Liu ID, et al. Genetic interactions between TRPC6 and NPHS1 variants affect posttransplant risk of recurrent focal segmental glomerulosclerosis. Am J Transplant. (2015) 15:3229–38. doi: 10.1111/ajt.13378
19. Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2017) 32:1181–92. doi: 10.1007/s00467-017-3590-y
20. Gheissari A, Meamar R, Kheirollahi M, Rouigari M, Dehbashi M, Dehghani L, et al. TRPC6 mutational analysis in iranian children with focal segmental glomerulosclerosis. Iran J Kidney Dis. (2018) 12:341–9
21. Zand L, Fervenza FC, Nasr SH, Sethi S. Membranoproliferative glomerulonephritis associated with autoimmune diseases. J Nephrol. (2014) 27:165–71. doi: 10.1007/s40620-014-0049-0
22. Magnasco A, Ravani P, Edefonti A, Murer L, Ghio L, Belingheri M, et al. Rituximab in children with resistant idiopathic nephrotic syndrome. J Am Soc Nephrol. (2012) 23:1117–24. doi: 10.1681/asn.2011080775
23. Wang W, Li G, Hong D, Zou Y, Fei D, Wang L. Replication of genome-wide association study identified seven susceptibility genes, affirming the effect of rs2856717 on renal function and poor outcome of IgA nephropathy. Nephrology (Carlton). (2017) 22:811–7. doi: 10.1111/nep.12860
24. Xie J, Shapiro S, Gharavi A. Genetic studies of IgA nephropathy: what have we learned from genome-wide association studies. Contrib Nephrol. (2013) 181:52–64. doi: 10.1159/000348652
25. Cox SN, Pesce F, El-Sayed Moustafa JS, Sallustio F, Serino G, Kkoufou C, et al. Multiple rare genetic variants co-segregating with familial IgA nephropathy all act within a single immune-related network. J Intern Med. (2017) 281:189–205. doi: 10.1111/joim.12565
26. Milillo A, La Carpia F, Costanzi S, D'Urbano V, Martini M, Lanuti P, et al. A SPRY2 mutation leading to MAPK/ERK pathway inhibition is associated with an autosomal dominant form of IgA nephropathy. Eur J Hum Genet. (2015) 23:1673–8. doi: 10.1038/ejhg.2015.52
27. Moller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. (2007) 18:29–36. doi: 10.1681/asn.2006091010
28. Buscher AK, Beck BB, Melk A, Hoefele J, Kranz B, Bamborschke D, et al. Rapid response to cyclosporin a and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2016) 11:245–53. doi: 10.2215/cjn.07370715
Keywords: TRPC6, SRNS, proteinuria, Genetic testing, immune complex-mediated glomerulonephritis
Citation: Wang M, Wang R, He X, Yu M, Xia Z and Gao C (2020) Two Children With Novel TRPC6 Spontaneous Missense Mutations and Atypical Phenotype: A Case Report and Literature Review. Front. Pediatr. 8:269. doi: 10.3389/fped.2020.00269
Received: 23 February 2020; Accepted: 29 April 2020;
Published: 15 May 2020.
Edited by:
Asha Moudgil, Children's National Hospital, United StatesReviewed by:
John William Foreman, School of Medicine Duke University, United StatesBakri Alzarka, University of Maryland, Baltimore, United States
Copyright © 2020 Wang, Wang, He, Yu, Xia and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengkun Xia, bmp4emtAMTI2LmNvbQ==; Chunlin Gao, U2h1YW5nbXUzNEAxNjMuY29t
†These authors have contributed equally to this work