 Corinne Summers
Corinne Summers Vipul S. Sheth1
Vipul S. Sheth1 Marie Bleakley
Marie Bleakley- 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, United States
- 2Department of Pediatrics, University of Washington, Seattle, WA, United States
Minor Histocompatibility (H) antigens are major histocompatibility complex (MHC)/Human Leukocyte Antigen (HLA)-bound peptides that differ between allogeneic hematopoietic stem cell transplantation (HCT) recipients and their donors as a result of genetic polymorphisms. Some minor H antigens can be used as therapeutic T cell targets to augment the graft-vs.-leukemia (GVL) effect in order to prevent or manage leukemia relapse after HCT. Graft engineering and post-HCT immunotherapies are being developed to optimize delivery of T cells specific for selected minor H antigens. These strategies have the potential to reduce relapse risk and thereby permit implementation of HCT approaches that are associated with less toxicity and fewer late effects, which is particularly important in the growing and developing pediatric patient. Most minor H antigens are expressed ubiquitously, including on epithelial tissues, and can be recognized by donor T cells following HCT, leading to graft-vs.-host disease (GVHD) as well as GVL. However, those minor H antigens that are expressed predominantly on hematopoietic cells can be targeted for selective GVL. Once full donor hematopoietic chimerism is achieved after HCT, hematopoietic-restricted minor H antigens are present only on residual recipient malignant hematopoietic cells, and these minor H antigens serve as tumor-specific antigens for donor T cells. Minor H antigen-specific T cells that are delivered as part of the donor hematopoietic stem cell graft at the time of HCT contribute to relapse prevention. However, in some cases the minor H antigen-specific T cells delivered with the graft may be quantitatively insufficient or become functionally impaired over time, leading to leukemia relapse. Following HCT, adoptive T cell immunotherapy can be used to treat or prevent relapse by delivering large numbers of donor T cells targeting hematopoietic-restricted minor H antigens. In this review, we discuss minor H antigens as T cell targets for augmenting the GVL effect in engineered HCT grafts and for post-HCT immunotherapy. We will highlight the importance of these developments for pediatric HCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (HCT) is widely employed in the management of very high-risk leukemia in children, adolescents, and young adults (1, 2), because HCT as consolidation therapy is generally associated with a reduced relapse risk compared to chemotherapy alone (3, 4). Although HCT reduces the risk, relapse remains the major cause of death following HCT for leukemia (5). Reported post-HCT relapse rates are variable, with rates of 10–30% for patients transplanted with leukemia in minimal residual disease (MRD) negative remission, 20–70% for those in remission but with MRD, and 50–90% for those in relapse (6, 7). Long-term survival following relapse after HCT is infrequent (8–10).
HCT involves two important elements that confer protection from relapse: first, “conditioning” incorporating chemotherapy and/or radiotherapy as the pre-HCT preparative regimen, and second, donor lymphocytes in the hematopoietic cell graft as cellular therapy. Donor T cells respond to non-donor antigens on recipient cells, including minor histocompatibility (H) antigens, and thereby lead to a graft-vs.-leukemia (GVL) effect when those antigens are presented on leukemic cells. Minor Histocompatibility (H) antigens are Human Leukocyte Antigen (HLA)-presented peptides derived from normal self-proteins that differ in amino acid sequence between donor and recipient due to genetic polymorphisms outside of the chromosome 6 HLA loci (11). The GVL effect and intensive conditioning both substantially contribute to relapse prevention in myeloablative HCT, while the GVL effect is particularly critical with use of less intensive, “non-myeloablative” and “reduced intensity” HCT preparative regimens.
The efficacy of HCT for pediatric leukemia currently depends highly on both the GVL effect and on delivery of myeloablative chemo(radio)therapy in the pre-HCT conditioning regimen. Intense myeloablative conditioning, particularly involving total body irradiation (TBI), causes serious short- and long-term adverse effects, including growth and neurocognitive impairments in pediatric patients (12–16). Thus, there is a critical need to advance HCT strategies that allow reduced conditioning intensity and associated toxicity, while mitigating relapse risk. Furthermore, avoiding severe and chronic graft-vs.-host disease (GVHD) in children is highly desirable due to the morbidity, mortality, disability, and social handicaps, and late effects associated with chronic GVHD (12, 15, 17).
In this review, we discuss minor H antigens as T cell targets for augmenting the GVL effect in engineered HCT grafts and for post-HCT immunotherapy. We will highlight the importance of these developments for pediatric HCT.
Minor Histocompatibility Antigens
The GVL effect in allogeneic HLA-matched related or unrelated donor HCT is largely attributed to T cell responses to recipient minor H antigens. Specifically, when a HCT recipient has a homozygous or heterozygous polymorphism that encodes a minor H antigen and their HLA-matched donor is homozygous for the “negative” allele, the donor may have T cells in their repertoire that recognize the minor H antigen peptide/HLA complex on the recipient's leukemia cell surface, and those cells may eliminate any residual leukemia after HCT (Figure 1).
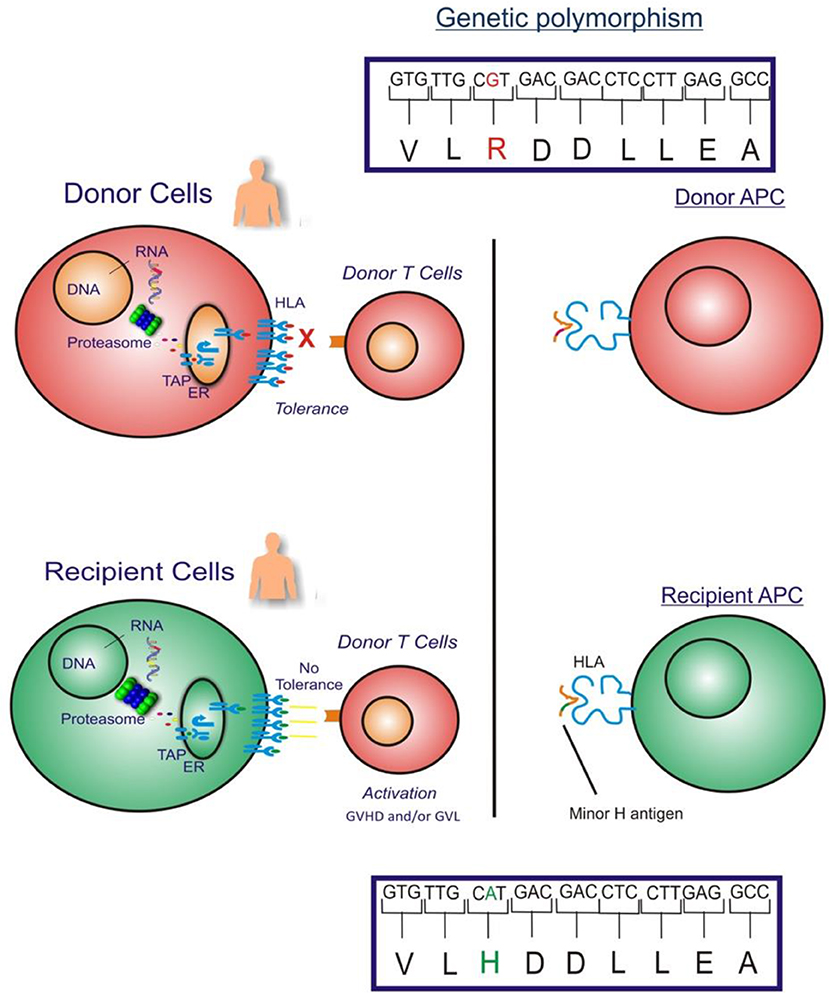
Figure 1. The figure illustrates the GVHD and/or GVL effect in allogeneic HLA-matched donor HCT which is largely attributed to donor T cell responses to recipient cell minor H antigens. When an HCT recipient has a homozygous or heterozygous polymorphism that encodes a minor H antigen and their donor is homozygous negative for the allele, the donor may have T cells in their repertoire that can target the minor H antigen peptide/HLA complex on the recipient's leukemia cell surface leading to an elimination of any residual leukemia following HCT.
From the perspective of the donor T cells, minor H antigens are foreign antigens and consequently donor T cells specific for minor H antigens are not subject to self-tolerance mechanisms, allowing for highly avid minor H antigen-specific T cell responses. Most known minor H antigens arise from single nucleotide substitutions in the coding sequences of homologous donor and recipient genes, which change peptide-HLA binding or T cell receptor (TCR) recognition of the peptide-HLA complex. There are at least 660,000,000 single nucleotide polymorphisms (SNPs) and insertion-deletion polymorphisms (indels) in the human genome (18) although less than 1% of SNPs are non-synonymous limiting the number of potential minor H antigens (19). Moreover, only non-synonymous SNPs that give rise to recipient donor mismatches in the graft-vs.-host direction (recipient homozygous positive or heterozygous, donor homozygous negative for the immunogenic allelic variant) are relevant to the GVL effect. T cell recognition of most minor H antigens is unidirectional, mostly due to the lack of T cell recognition of the allelic variant peptide despite cell surface presentation with HLA molecules (20). Alternatively, the corresponding donor peptide may not be generated (21–25), transported by antigen processing machinery (26), escape proteasomal degradation (27), or stably bind to MHC molecules (28–31).
Minor H Antigens and Selective GVL
HCT outcome data demonstrate the potency of the GVL effect, with reduced relapse rates noted in patient cohorts that develop acute and/or chronic GVHD following allogeneic HCT (32–34). However, the GVL effect is apparently separable from GVHD; reduced relapse rates are still observed in patients who underwent allogeneic HCT and did not develop clinically significant GVHD, as compared to relapse rates in syngeneic HCT recipients (33).
Most minor H antigens are expressed ubiquitously, including on epithelial tissues. Recognition of such minor H antigens by donor T cells following HCT can potentially lead to GVHD as well as GVL (35, 36). However, those minor H antigens that are expressed predominantly on hematopoietic cells can be targets of a selective GVL effect (37, 38) (Figure 2). Once full donor normal hematopoietic chimerism is achieved after HCT, only residual or recurrent recipient malignant hematopoietic cells will present hematopoietic-restricted minor H antigens, and these minor H antigens serve as tumor-specific antigens for donor T cells (Figure 3). Over 100 fully-characterized or candidate human minor H antigens have been identified (22, 25, 28, 39–52). Of these, an important minority are expressed predominantly or exclusively on hematopoietic cells and are of particular interest as targets for GVL-augmenting therapeutic T cells delivered with or following HCT (Table 1). Examples, of well-characterized minor H antigens of high therapeutic interest include HA-1, ACC-1, ACC-2, and LRH1, described below.
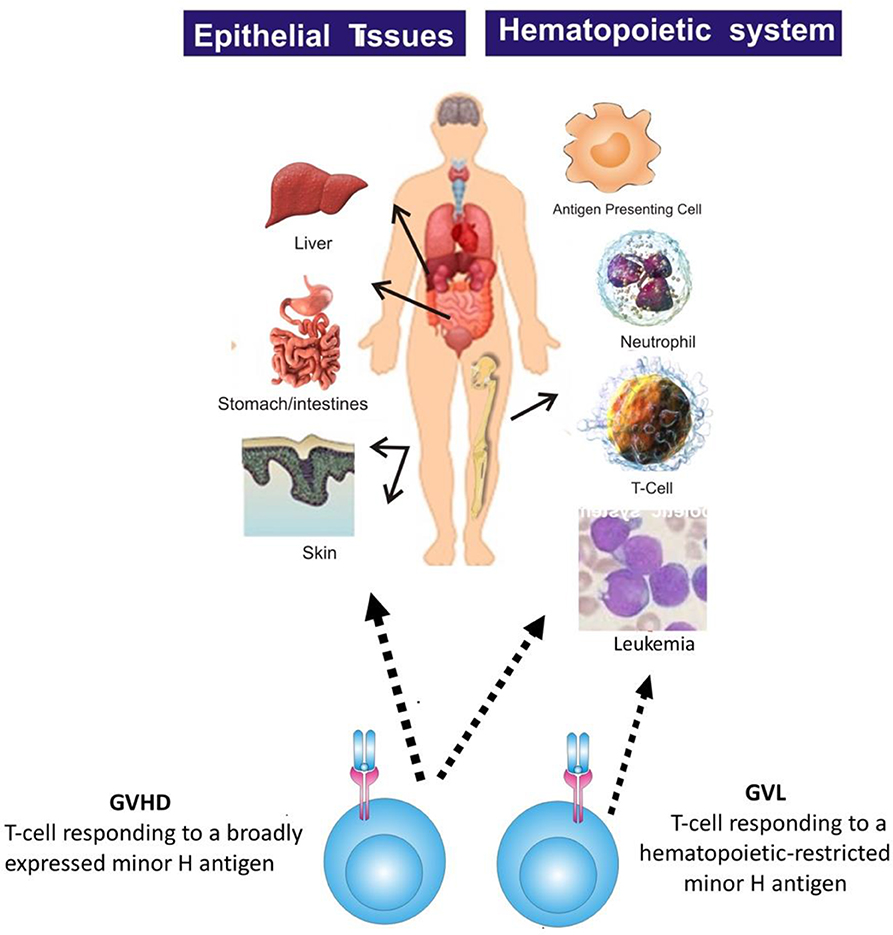
Figure 2. The figure illustrates that most minor H antigens are ubiquitously expressed on epithelial tissues and hematopoietic cells. Donor T cell recognition of such antigens leads to GVHD as well as GVL. However, minor H antigens expressed predominantly on hematopoietic cells can be targets of a selective GVL effect.
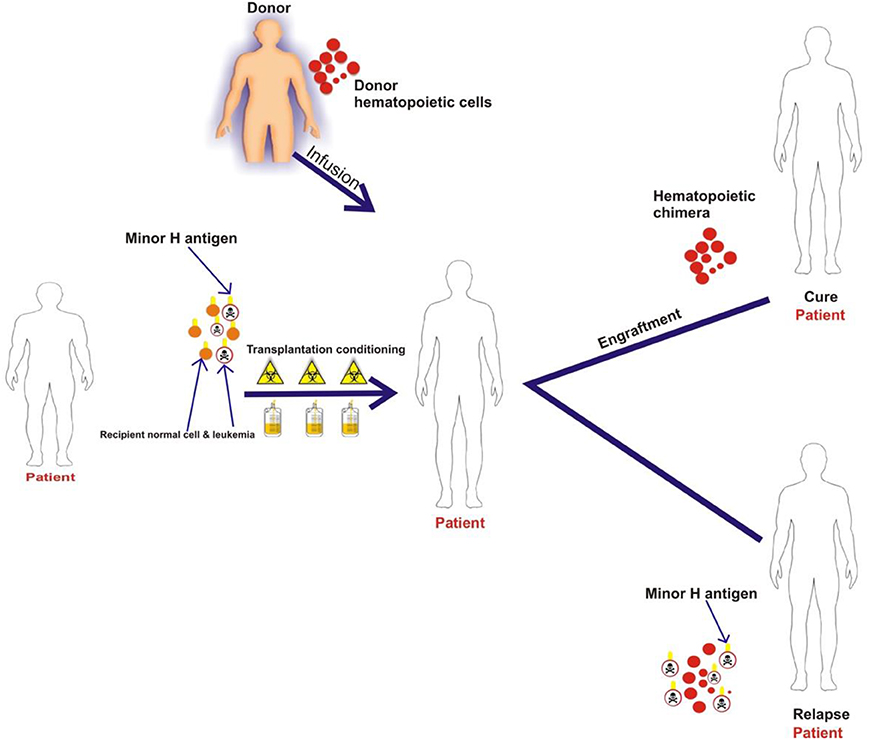
Figure 3. The schematic illustrates that patient normal hematopoietic and leukemia cells with minor H antigens are eliminated by the transplant conditioning regimen and replaced with donor hematopoietic cells. Once full donor normal hematopoietic cell chimerism is achieved only recipient malignant hematopoietic cells present hematopoietic-restricted minor H antigens if disease persists or recurs following HCT. These minor H antigens serve as tumor-specific antigens for donor T cells.
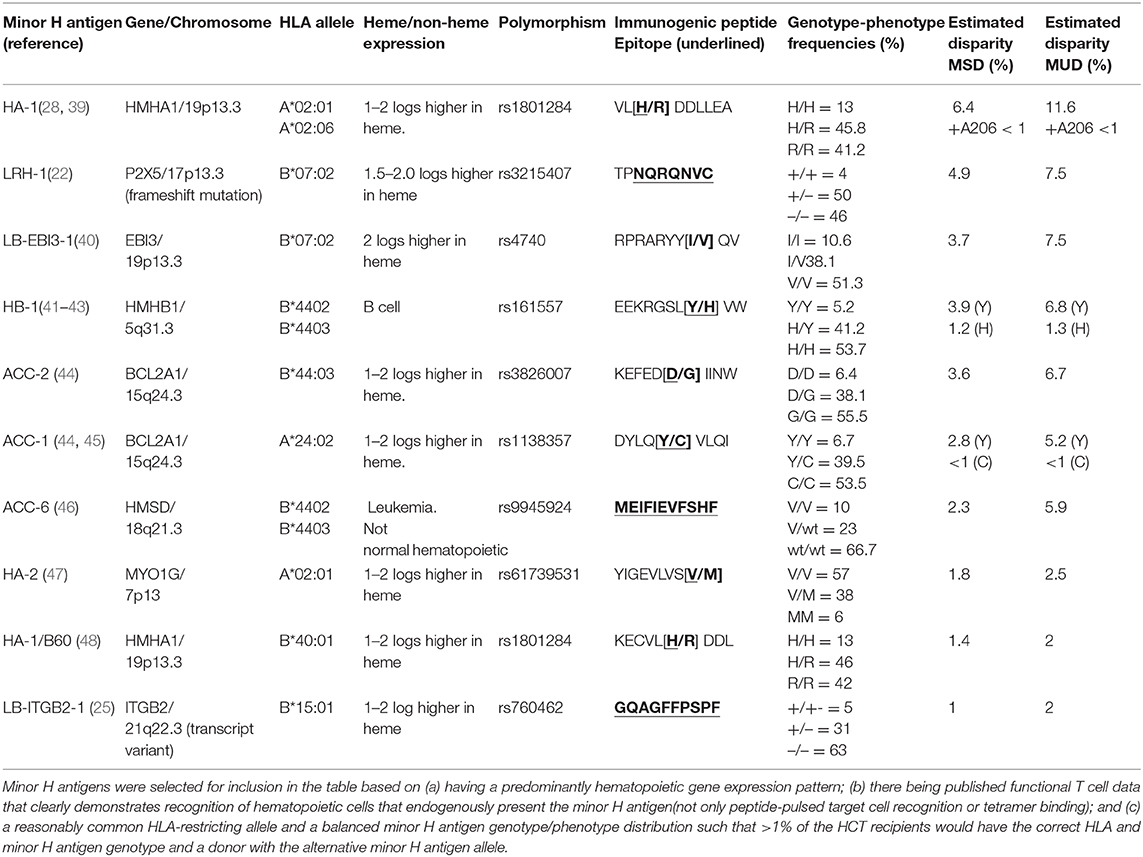
Table 1. Selected hematopoietic-restricted minor H antigens of interest for therapeutic targeting.
There is direct evidence for the anti-leukemic activity of minor H antigen-specific T cells. In humans, donor-derived CD4+ and CD8+ T cells that have been activated and expanded in vivo following recognition of minor H antigens on recipient cells can be isolated and grown in vitro and evaluated for anti-leukemic activity (38). Additionally, minor H antigen-specific T cells can be generated by primary in vitro stimulation (53). Minor H antigen-specific CD8+ T cell clones can inhibit acute myelogenous leukemia (AML) colony growth and lyse primary AML and acute lymphoblastic leukemia (ALL) cells in vitro (38, 53–55). Furthermore, minor H antigen-targeting T cells prevent the engraftment of AML in immunodeficient murine models, supporting the hypothesis that early leukemic progenitors are targets of these cells (56).
In vivo anti-leukemic efficacy of minor H antigen-specific T cells has also been demonstrated in murine models of HCT and GVL. Perreault and colleagues demonstrated that adoptive transfer of T cells specific for a single immunodominant murine minor H antigen (B6dom1, also known as H7a) can eradicate leukemia and has anti-cancer activity in solid tumor models (57–60). Shlomchik and colleagues demonstrated antigen-specific memory T cell (TM)-mediated GVL against chronic phase and blast crisis chronic myeloid leukemia (CML) when they transferred CD8+ TM from murine donors vaccinated against the H60 minor H antigen (61). In both the Perreault and Shlomchik studies, little to no GVHD was observed when the transferred T cells were specific for a single minor H antigen, even if expression of the minor H antigen was not restricted to the hematopoietic system. However, the anti-tumor efficacy was improved if the minor H antigen was not ubiquitously expressed (57, 59–61). Better efficacy of T cells specific for minor H antigens with hematopoietic-restricted vs. ubiquitous expression can be explained by less activation-induced cell death and T cell exhaustion, and better expansion of T cells targeting hematopoietic-restricted minor H antigens (61).
Focusing the T cell response on a limited number of minor H antigens may favor GVL over GVHD. In mice, leukemia was eradicated following adoptive transfer of CD8+ T cells specific for a single broadly-expressed minor H antigen (B6dom/H7a), without the development of GVHD. However, GVHD occurred if B6dom-specific T cells from vaccinated donors were delivered with naïve T (TN) cells specific for other minor H antigens (57). Earlier experiments by Korngold and colleagues, using numerous combinations of congenic mouse strains, also did not reveal GVHD in any experiment where donors and recipients were incompatible for single minor H antigens (62). Research by the Falkenburg group using human cells also demonstrated that the magnitude and diversity of the immune response influences the balance between GVHD and GVL (51). They characterized alloreactive CD8+ T cell responses in recipients of T cell-depleted (TCD) HLA-matched HCT who achieved a clinical complete response and/or full donor chimerism after donor lymphocyte infusion (DLI). Minor H antigen-specific T cell frequency and diversity was lower in patients with who cleared residual leukemia but did not develop GVHD (i.e., selective GVL) compared to those who developed GVHD and although, patients who developed selective GVL had predominantly hematopoietic-restricted minor H antigen-specific T cells, some did also have T cells that recognized more broadly expressed minor H antigens.
Together, these studies imply that minor H antigens targeted with T cells to augment GVL may not need to be absolutely hematopoietic-restricted from a safety perspective, particularly if the T cell infusion occurs beyond the pro-inflammatory period immediately post-HCT and if a limited number of minor H antigens are targeted. However, targeting minor H antigens that are predominantly expressed on hematopoietic tissue may still be safer, and may be more effective as T cells specific for broadly-expressed antigens tend to become exhausted and dysfunctional (60, 61).
Minor H Antigen Discovery
A major challenge for developing minor H antigen-targeting, GVL-augmenting strategies is to create therapeutics for all patients who may benefit from them, which will require the identification of multiple minor H antigens presented by various HLA types. Techniques used for minor H antigen discovery have been reviewed and include forward and reverse immunology approaches. Forward immunology approaches involve several combinations of different component methods of isolation of activated T cells from HCT recipients, primary in vitro stimulation of normal donor T cells, haplotype mapped (HapMap) cell line screening, genetic linkage analysis, genome-wide association studies and cDNA library screening. Combinations of in silico analysis, mass spectrometry, HLA/multimer and functional T cell screening have been used in reverse immunology approaches (63, 64).
As there are numerous non-synonymous SNPs (ns-SNPs) with a variant allele frequency between 0.1 and 0.9 across the human genome and a significant minority are encoded by genes that are predominantly expressed in hematopoietic cells, it is anticipated that there remain many minor H antigens that will be suitable targets for therapeutic T cell yet to be discovered. An in silico analysis was performed by Lansford et al. to predict minor H antigens. Analysis of recurrent SNPs among 101 HLA-matched HCT recipient donor pairs resulted in the identification of 102 peptides with desirable properties for public, leukemia-associated minor H antigens, specifically with: (a) predicted high binding affinity to a common HLA molecule; (b) RNA expression in AML, but not in GVHD target organs; and (c) optimal allele frequencies to give rise to common minor H antigen mismatches and therefore feasible T cell targeting (52). A proportion of these candidate minor H antigens would be expected to be naturally processed and presented on HLA molecules in leukemic cells, and to elicit a T cell response.
In a second example of the potential for additional minor H antigen discovery, Granados et al. focused on identifying HLA-A*02:01 or -B*44:03-restricted polymorphic peptides using a mass spectrometry-based proteogenomic approach and cells from 13 volunteer donors and found thousands of candidate minor H antigens (50). Of the nearly 6,773 candidate minor H antigens generated by ns-SNPs, the authors identified 100 relatively common candidates with a minor allele frequency of >0.05, including a set of 39 putative minor H antigens with RNA expression at least two times higher in bone marrow cells than in skin cells. Two of the 39 were tested and induced a T cell response in vitro. A proportion of the 39 candidate minor H antigens with predominantly hematopoietic expression would be expected to be both adequately hematopoietically-restricted for therapeutic targeting and immunogenic to have therapeutic utility.
Examples of HLA Class I Minor H Antigens With Potential for Therapeutic Targeting
HA-1
HA-1 is the most comprehensively investigated human minor H antigen and is selectively expressed on normal and malignant hematopoietic cells, including AML, myelodysplastic syndromes (MDS), B lineage ALL, and T lineage ALL. The HA-1 peptide epitope is presented on the cell surface in association with HLA-A*02:01 and is recognized by HA-1-specific T cells.
The HA-1H peptide (VLHDDLLEA) is encoded by a nucleotide sequence spanning a single nucleotide polymorphism (RS_1801284) within the HMHA1 gene (28). Individuals with a rs1801284 A/A or A/G genotype express the histidine variant (HA-1H, also referred to as HA-1) and are considered HA-1 “positive.” Conversely, HA-1 “negative” people with the G/G genotype express only the arginine allelic variant (HA-1R, VLRDDLLEA), and may have T cells in their repertoire that can respond to HA-1H. Both HA-1H and HA-1R undergo similar intracellular processing from the HMHA1 protein, with appropriate proteasomal cleavage and transporter associated with antigen processing (TAP) (29, 30). However, the presence of an arginine at position three of the peptide reduces the affinity of VLRDDLLEA binding to HLA-A*02:01, relative to VLHDDLLEA (29, 30). HA-1H-specific T cells do not recognize VLRDDLLEA at peptide concentrations that are naturally presented on cells. Therefore, HA-1-specific T cells can be employed following HCT to selectively target residual leukemic cells from a HA-1 positive patient, without damaging normal hematopoietic cells of HA-1 negative donor origin.
Approximately 50% of the population presents HLA-A*02:01, and HA-1 allelic variants are phenotypically balanced in the population (rs1801284 A/A 16%, A/G 36%, G/G 48%; HA-1 positive 52%, HA-1 negative 48%). This means that ~10–15% of the HCT population will express both HA-1H and the HLA-A*02:01-restricting allele and also have a suitably mismatched HA-1 negative or HLA-A*02:01 negative donor, making HA-1 a relatively feasible minor H antigen target for T cell immunotherapy.
Multiple publications have documented that HMHA1 gene expression is very low to absent in non-hematopoietic cells (65–67), and that HA-1 genotypically-positive hematopoietic cells but not non-hematopoietic cells are recognized by HA-1 -specific T cells in vitro (37). Furthermore, HA-1-specific T cells induce little or no tissue damage when co-cultured with HA-1 positive skin biopsy specimens (68). Following HCT from an HA-1 negative donor to an HLA-A*02:01 HA-1 positive recipient, HA-1-specific T cells can be identified in approximately one-third of patients (69). One study reported a temporal relationship between the presence of HA-1-specific T cells and GHVD early post-HCT (70), but HA-1-specific T cells have been identified in patients without GVHD following DLI therapy (51, 71–73). Additionally, associations between HA-1 donor-recipient genotypic disparity and GVHD have been observed in some (74–77) but not all (78–81) HCT studies. One explanation for this apparent inconsistency is that patient hematopoietic cells remain in the tissues for several months before being replaced by donor-derived cells (72). Thus, HA-1-specific T cells generated by in vivo priming following HCT may contribute to GVHD early post-HCT but are less likely to do so in the context of DLI or delayed HA-1 targeted T cell immunotherapy.
Circumstantial evidence suggests that HA-1 can serve as a therapeutic target in the context of HCT and unmanipulated DLI. Specifically, there have been several reports in which patients with hematological malignancies responded DLI to treat recurrent disease following HCT and coincident emergence of HA-1-specific T cells in vivo was documented using peptide/HLA tetramer analysis and/or isolation of the HA-1-specific T cells from the peripheral blood (29, 51, 71–73). In some of these reports HA-1-specific T cell clones isolated from the patients were further evaluated and demonstrated specific killing of HLA-A*02 positive+ HA-1H -pulsed target cells and primary leukemic cells (71–73). There have also been reports of HA-1 positive patients with multiple myeloma who were treated with dendritic cell vaccines loaded with minor H antigen peptides and adjuvant followed by DLI. Several patients developed a detectable HA-1-specific T cell responses, without adverse effects and two achieved disease control for 6–7 months (82, 83). Together, these observations suggest HA-1 will be a safe and effective target for T cell immunotherapy. HA-1-directed T cell immunotherapy is currently in development, as discussed below.
BCL2A1/ACC-1 and ACC-2
BCL2A1 is a member of the Bcl-2 family of anti-apoptotic genes. Two minor H antigens, ACC-1Y and ACC-2D, result from distinct nucleotide polymorphisms in the BCL2A1 gene and are presented by HLA-A*24:02 and HLA-B*44:03, respectively (44). BCL2A1 is frequently highly expressed in hematologic malignancies and may contribute to the malignant phenotype, making ACC-1 and ACC-2 attractive targets for T cell immunotherapy (84, 85).
The polymorphisms, rs1138357 and rs3826007, lead to single amino acid substitutions in exon 1 of BCL2A1, creating the immunogenic HLA-A*24:02-restricted ACC-1Y [DYLQYVLQI, rs1138357 AA (6%) or AG (39%)] and B*44:03-restricted ACC-2D [KEFEDDIINW, rs3826007 AA (4%) or AG (50%)] (44). ACC-1-specific T cells can be generated from HLA-A*24:02 positive ACC-1 “homozygous negative” donors (rs1138357, GG encoding DYLQCVLQI) and distinguish the single amino acid difference (tyrosine vs. cysteine in ACC-1). Similarly, ACC-2-specific T cells from homozygous negative donors (rs3826007, GG encoding KEFEDGIINW) can distinguish the aspartic acid from the glycine.
ACC-1 and ACC-2 are feasible targets for T cell immunotherapy. Based on the prevalence of the HLA-A*24:02 and HLA-B*44:03 restricting alleles and the frequency of distribution of immunogenic and non-immunogenic variants of ACC-1 and ACC-2, the calculated estimate of finding a polymorphism-discrepant, matched related and unrelated donor is 2.8 and 5.2% for ACC-1, and 3.6 and 6.7% for ACC-2, respectively (86).
There has been controversy regarding how selectively BCL2A1 is expressed in hematopoietic cells. Gene expression analysis by Northern blot (44), quantitative polymerase chain reaction (67), and database microarray (http://biogps.gnf.org) suggests hematopoietic-restricted expression. Although the Goulmy group showed that BCL2A1 expression could be upregulated in non-hematopoietic cells (mesenchymal stromal cells) by simultaneous exposure to interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα), but not to IFNγ alone (87), Akatsuka's research group subsequently demonstrated comparable up-regulation of BCL2A1 and HMHA1 after administration of IFNγ and TNFα (67). In any case, the clinical relevance of upregulation of minor H antigen-encoding genes after exposure to high doses of cytokines in vitro has not been established. ACC-1Y and ACC-2D genotypically positive non-hematopoietic cells are not recognized in cellular assays without exogenous cytokines (44, 67, 87). Moreover, an analysis of HCT outcomes in 320 patients expressing HLA-A*24:02 did not show an increase in GVHD in recipients with an immunogenic BCL2A1 allele and an antigen negative donor (88). Together these data suggest that ACC-1Y is likely to be a safe target.
ACC-1- and ACC-2-specific T cells lysed primary leukemic cells in vitro after isolation from HCT recipients (44). In a subsequent study, direct tetramer analysis was used to identify ACC-1-specific T cells in the bone marrow and peripheral blood in a patient who received an HLA-matched, ACC-1-disparate HCT for CML 14 months earlier (89). The bone marrow ACC-1-specific T cells proliferated in response to ACC-1 peptide stimulation and demonstrated cytotoxicity against a cell line endogenously presenting ACC-1, demonstrating the presence of functional ACC-1 specific T cells that may contribute to protection against post-HCT relapse. Gene expression data suggests that BCL2A1 is not expressed at high levels in all leukemia cells. Although high-level expression is not necessarily required to render leukemia susceptible to lysis by high-avidity minor H antigen-specific T cells, functional assays demonstrating lysis of numerous ACC-1 and ACC-1 genetically positive leukemia will be necessary before ACC-1 and 2 directed T cell immunotherapy is advanced to the clinic.
P2X5/P2RX5/LRH-1
The P2RX5 gene is a member of the P2X purinergic ATP-gated non-selective cation channel protein group and has been recognized as promoting cancer growth and aggression (90–93). A frameshift polymorphism (rs3215407) in P2RX5 leads to major differences in the P2RX5-transcribed sequence, including the immunogenic HLA-B*07:02-restricted LRH-1 minor H antigen TPNQRQNVC (22). On the basis of the frequency of the HLA-B*07:02 restricting allele and 46% prevalence of the homozygous cytosine deletion, the calculated estimate of finding discrepant matched related and unrelated donors is 4.9 and 7.5%, respectively (86). If HCT donors have the rs3215407 polymorphic sequence with a homozygous deletion of a cytosine at position 732, they may generate T cells that recognize the TPNQRQNVC minor H antigen generated in individuals without the cytosine deletion.
The P2RX5 gene is expressed in normal lymphocytes, B and T lineage ALL, a range of lymphoma and multiple myeloma cases, the CD34+ fractions of CML and AML, and possibly at low levels in brain and skeletal muscle, but there is minimal expression in GVHD target tissues (intestine, liver, lung, skin) (22). LRH-1 genotypically-positive fibroblasts, a representative non-hematopoietic tissue, are not recognized in cellular assays (22).
LRH-1-specific T cells kill ALL CD34+, AML CD34+, CML CD34+, and multiple myeloma CD138+ cells in vitro (22, 94, 95). Moreover, LRH-1-specific T cells have been detected in the peripheral blood of patients responding to DLI (22, 95, 96). Dolstra's group studied seven HLA-B*07:02+ LRH-1 positive patients who received HCT and DLI from a HLA-B*07:02 positive LRH-1 negative donor, and detected LRH-1-specific T responses coinciding with declines in detectable leukemia in three of the seven patients, including two patients with CML and one with AML (22, 95). Gene expression data indicates that LRH-1 is not expressed at high levels in all leukemia blasts or progenitors, although progenitors with relatively low levels of LRH-1 expression can be inhibited by LRH-1-specific T cells in functional assays (22, 95). Further functional assays demonstrating activity of LRH-1-specific T cells against multiple LRH-1 genetically positive leukemia cell targets in vitro, and ideally in patient-derived murine xenograft models, are required before translation of LRH-1-specific T cell immunotherapy to the clinic.
HLA Class II Minor H Antigens
The primary focus of minor H antigen discovery has been on HLA class I-restricted minor H antigens as targets for CD8+ T cells. However, class II-restricted minor H antigens are also of interest particularly given that HLA class II molecules are generally expressed at relatively low levels on non-hematopoietic cells under non-inflammatory conditions. As such, class II-restricted minor H antigens may be more likely to induce a selective GVL response even if the gene encoding the minor H antigen is relatively broadly expressed. In a recent study of CD4+ enriched DLI from HLA-identical sibling donors, GVL reactivity without GVHD was associated with CD4+ T cells targeting HLA class II-restricted minor H antigens, some of which were associated with genes expressed in non-hematopoietic cells (97). However, HLA class II gene expression is often downregulated on leukemic cells after HCT (98, 99), which implies that while class II-restricted minor H antigen-specific T cells may make a major contribution to GVL after HCT and drive the HLA class II downregulation, class II-restricted minor H antigens may not be optimal targets for T cell immunotherapy to treat post-HCT relapse.
T Cell Immunotherapy Targeting Minor H Antigens (Figure 4)
Minor H antigens have several advantages as targets for therapeutic T cells aimed to prevent or manage relapse. First, minor H antigens arise from germline variants and are therefore likely to be expressed, at least initially, in all leukemia cells in an individual, unlike neoantigens that are often subclonal, permitting escape from neoantigen-specific T cells. Second, minor H antigens are foreign to donor T cells, like neoantigens and unlike overexpressed non-polymorphic leukemia-associated antigens. High-affinity minor H antigen-specific TCRs can be relatively readily found in the repertoire of normal donors and exploited as therapeutics (100). Lastly, as described above, minor H antigen expression is inherently specific to HCT-recipient cells, and therefore specific to recipient leukemia after myeloablative HCT. This inherent specificity avoids many of the challenges for chimeric antigen receptor T cells (CAR-T) that target cell surface antigens that are shared with normal recipient or donor hematopoietic cells, leading to problems with prolonged marrow aplasia in the case of AML CAR-T, for example.
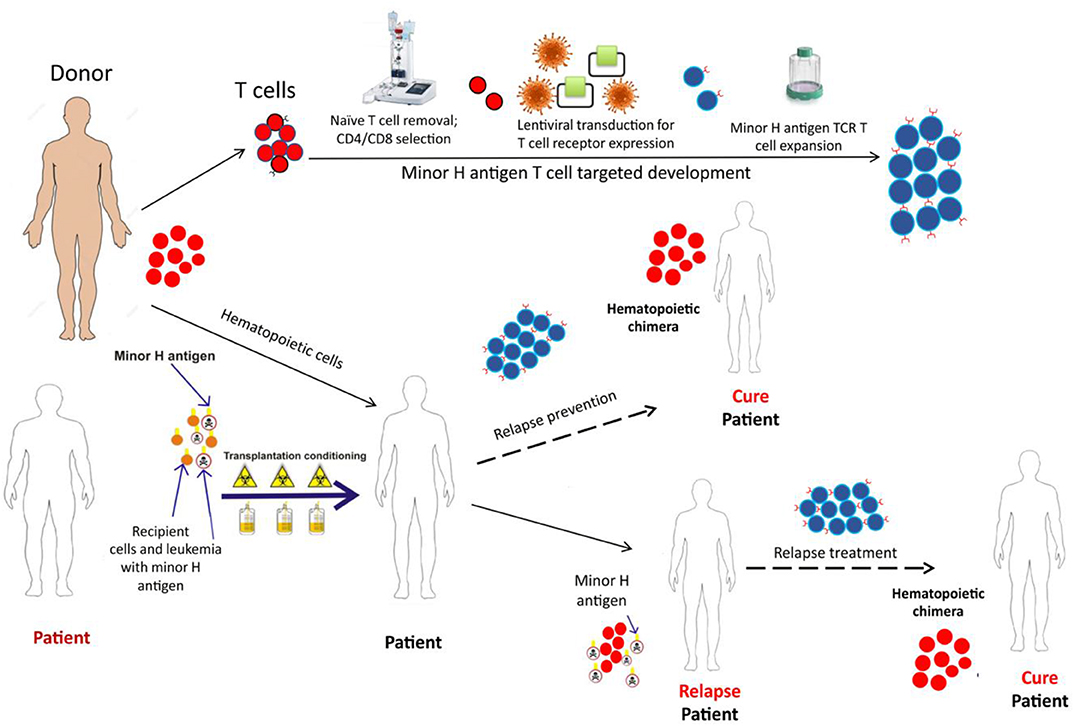
Figure 4. The schematic depicts the development and potential therapeutic use of donor derived minor H antigen targeting T cell therapy. A patient, with the hematopoietically-restricted minor H antigen target, would undergo allogeneic HCT. The donor, lacking the minor H target, would donate hematopoietic cells for the HCT graft and T cells for the immunotherapy product. The T cells would undergo depletion of the naïve T cell fraction (to reduce GVHD risk) and then undergo modification using a lentiviral construct containing a potent TCR targeting the minor H antigen of interest. Following this modification, the cell product would be expanded to develop a therapeutically active cell dose which could be infused soon after the HCT as a relapse prevention strategy or following relapse detection to treat post-HCT relapse.
Immunotherapy employing T cells genetically modified with transgenic TCR alpha and beta chains is a promising strategy for treating hematologic malignancies and solid tumors (101, 102). Genetic TCR transfer into a selected T cell subset facilitates relatively rapid production of a T cell immunotherapy product with high potential for expansion, function and persistence after infusion into the patient. We developed T cell immunotherapy employing donor TM cells transduced with a lentiviral vector encoding a TCR specific for HA-1 and are currently evaluating this novel therapeutic in a phase I clinical trial for the treatment of post-HCT MRD or relapse (NCT03326921) (100). The cell product incorporates multiple elements designed to optimize efficacy and safety: (1) a high-avidity HA-1 specific TCR with potent anti-leukemic activity; (2) a CD8+ co-receptor to promote function of the class I-restricted TCR in CD4+ T cells; (3) an inducible caspase 9 safety switch that can be triggered by the dimerizer AP1903/rimiducid in the event of unexpected side effects; (4) a CD34-CD20 epitope to facilitate selection and tracking of the engineered T cells; and (5) predominantly central memory T cells to promote persistence after infusion and to avoid infusing GVHD-inducing TN cells (100, 103, 104). This is the currently the only minor H antigen targeting clinical trial enrolling pediatric patients that we are aware of. Another HA-1 TCR T cell immunotherapy trial (MDG1021) will open in Europe this year, employing a HA-1 TCR T cell product developed in Leiden (105, 106).
Of note, while current clinical trials targeting minor H antigens are evaluating the safety profile of minor H antigen-specific T cells as treatment for post-HCT leukemia recurrence, a longer-term goal is to deliver hematopoietic-restricted minor H antigen-specific T cells soon after the HCT graft to augment GVL and prevent relapse.
Graft Engineering to Augment the T Cell Response to Selected Minor H Antigens
The term “graft engineering” refers to manipulation of the composition of cells collected from an allogenic HCT donor prior to infusion into the patient. To date, graft engineering has primarily involved selections and/or depletions of particular cell subsets for the purpose of mitigating the risk of GVHD. However, more complex manipulations can be considered with emerging technology, including enrichment for rare antigen-specific T cells, genetic modification of cells with CAR-T or TCR-T, and/or knockdown of genes in order to protect certain cells or augment their function. Here we will discuss recent progress in graft engineering to create an improved platform for delivery of antigen-specific T cells, and to develop strategies for enhanced delivery of leukemia-associated minor H antigen-specific T cells.
Minimizing the risk of serious GVHD is a critical first step to enabling the effective delivery and function of hematopoietic-restricted, minor H antigen-specific T cells, since the management of GVHD requires pharmacologic immunosuppression that is not conducive to T cell expansion and function. Non-selective removal of T cells from the donor graft can be achieved through pre-infusion physical depletion of T cells or enrichment of CD34+ stem cells using immunomagnetic beads, or by in vivo depletion using T cell antibody-directed therapy. Non-selective T cell depletion strategies have led to reduced GVHD rates but also increased infection risk due to prolonged immune reconstitution (107–111). Selective T cell depletion strategies, including CD45RA+ TN depletion and αβ TCR T cell depletion, are aimed at reducing GVHD but retaining lymphocytes with activity against pathogens and malignant cells and are being evaluated in children and adults.
TN-Depletion
Mature αβ TCR T cells can be divided into categories based on differentiation: naïve, T memory stem cells, central memory, effector memory, and effectors (112). Naïve T cells (TN) which are antigen inexperienced, include cells that can react to minor H antigens expressed on epithelial tissues following recipient infusion, resulting in GVHD. Murine modeling using MHC matched and mismatched mice have demonstrated that antigen experienced TM cells cause less or no GVHD compared to TN (113–121). They do, however, show appropriate antigen-specific T cell responses and beneficial GVL (114, 120). Additionally, in vitro studies found that human minor H antigen-specific T cells were more prevalent among TN than TM cells (53). Bleakley et al. developed a technique to engineer CD34+ cell-enriched, TN-depleted peripheral blood stem cells (PBSC) for HCT (122). Initial published experience has demonstrated remarkably low levels of chronic GVHD without increases in relapse or infection rates following HCT of TN-depleted HLA-matched related donor PBSC in patients with leukemia (104). Additional unpublished data shows comparable results in a larger cohort of pediatric and adult recipients of TN-depleted PBSC from HLA-matched related and unrelated donors. This approach is currently being further studied in randomized prospective trials comparing the TN-depleted PBSC to conventional bone marrow stem cells for pediatric patients (NCT03779854) and to alternative strategies for GVHD reduction in adults (NCT03970096).
αβ TCR T Cell Depletion (αβ-TCD)
HCT strategies that deplete the donor stem cell graft of all αβ TCR T cells to remove alloreactive T cells and CD19 B cells to avoid EBV post-transplant lymphoproliferative disorder, while retaining γδ TCR T cells and NK cells, are also being investigated. HCT with αβ-TCD grafts shows promise as a platform to reduce GVHD, especially in pediatric patients (123–128). γδ TCR T cells and NK cells both have activity against pathogens and malignant cells, so their retention in the graft may protect patients against infection and relapse, respectively (128–134). In long-term analysis of 98 pediatric patients with leukemia, the outcomes of patients who received haploidentical αβ-TCD grafts were encouraging with 3-year DFS, relapse, severe acute and extensive chronic GVHD rates of 62, 29, 0, and 1%, respectively (126, 127). Moreover, in a large retrospective analysis comparing outcomes of unmanipulated HLA matched or mismatched unrelated donor HCT (MUD; MMURD) with haploidentical αβ-TCD HCT, 5-year chronic GVHD free, relapse free survival (GRFS) was superior in the haploidentical αβ-TCD compared to the MMURD (135).
Assuming continued success in reducing GVHD by graft engineering, the next challenge is to augment HCT grafts for enhanced anti-leukemic activity, including but not limited to enriching for hematopoietic-restricted minor H antigen-specific T cells. The primary aim of this approach would be to overcome quantitative deficiencies in minor H antigen-specific T cells in the donor HCT graft. Minor H antigen-specific T cells will be numerically deficient in grafts that have been depleted of TN or of all αβ TCR T cells. Additionally, minor H antigen-specific T cells are rare in the donor T cell repertoire even in the absence of any T cell depletion (53) and may not consistently adequately expand, migrate to the bone marrow, and persist with durable anti-leukemic activity after HCT. Antigen-specific T cells are effective at controlling and eliminating cancer cells only when the T cells are present in sufficient numbers relative to the cancer cells. Therefore, particularly for patients with residual disease at the time of HCT, increasing the number of hematopoietic-restricted minor H antigen-specific T cells delivered with the graft should facilitate the GVL effect. Delayed sequential infusion of antigen-specific T cells after infusion of the HCT graft (i.e., a “split graft”) may also be considered to allow the pro-inflammatory state resulting from chemo/radiotherapy conditioning to abate and the recipient tissue-resident normal hematopoietic cells to be largely replaced by cells of donor origin, in order to mitigate the risk of inducing GVHD. Delayed sequential T cell infusions should also circumvent the antigen-induced T cell death that may occur when hematopoietic-restricted minor H antigen-specific T cells encounter residual recipient hematopoietic cells immediately post-HCT, improving persistence. Multiple infusions of cells will also avoid the functional limitation of the donor T cells that results from progressive expression of inhibitory molecules over time (99, 136).
Current hurdles to augmenting HCT grafts for hematopoietic-restricted minor H antigen-specific T cells include the relatively limited number of suitable target minor H antigens that have been discovered and characterized (Table 1) and technical issues largely related to inadequate options for clinical-grade sorting of rare cells. Streptamer technology is being evaluated for isolating antigen-specific T cells (137–140). The technology is based on the direct labeling of CD8+ T cells with HLA I-Streptamers composed of peptide-loaded HLA I-Strep-tag fusion proteins reversibly multimerized on magnetically labeled Strep-tactin. After separation, the HLA I-Streptamers can be dissociated from the positively selected cells by the addition of D-Biotin, allowing for rapid enrichment of unlabeled antigen-specific T cells under GMP conditions. However, in a clinical trial of Streptamer-enriched multi-antigen-specific T cells to prevent complications early after T cell-depleted HCT, neither tumor associated antigen or minor H antigen-specific T cells could be confirmed in the majority of antigen-specific T cell products or after HCT, although EBV and CMV-specific T cells were readily detected in the products and sometimes after HCT (140). The greater success in isolating virus-specific T cells compared to minor H antigen- or other tumor-associated antigen-specific T cells, may be due to the relatively high frequencies of virus-specific T cells in the repertoire of normal viral antigen-experienced donors.
Next-generation high-speed cell sorting technology may solve the challenge of isolating very rare cells from donor cell collections. For example, a novel cell sorting technology called OrcaSort™ is being developed by OrcaBio. OrcaSort™ uses fluorescent markers for identification of target cells and high-speed laser pulses to sort cells in fully-enclosed, sterile, disposable cassettes (141). The technology will first be evaluated in clinical trials that require sorting of multiple cell populations for GVHD reduction (NCT03802695, NCT01660607) but could also be adapted to positive selection of antigen-specific T cells.
An alternative strategy for enriching selected minor H antigen-specific T cells in donor cell collections is to first vaccinate the HCT donor against minor H antigens to generate a memory T cell response and increase the frequency of the minor H antigen-specific T cells (61). Shlomchik et al. demonstrated that donor vaccination with recipient minor H antigens, and subsequent transplantation of donor T memory T cells, transferred leukemia- and pathogen-specific immunity in murine bone marrow transplantation (BMT) recipients (61). The transferred memory T cells expanded after BMT and augmented GVL. This approach could be translated to humans by vaccinating donors months before HCT or intended post-HCT infusion and then specifically selecting the minor H antigen T cells using a immunomagnetic bead-based technique, such as Streptamer selection (137–140) or the Miltenyi Biotec CliniMACs Cytokine Capture System (142, 143). Alternatively, after donor vaccination with hematopoietic-restricted minor H antigens, donor T cells could be collected and depleted of naïve T cells to avoid GVHD (104), and the minor H antigen-specific T cell enriched memory T cells could be delivered with or following the stem cell graft. Minor H antigen vaccination of donors is likely to be safe, given that clinical trials of vaccination of HCT recipients against minor H antigens have been completed without excess toxicity (82, 83). Donor vaccination could also be employed to improve the efficacy of DLI to prevent or treat post-HCT relapse by producing a product enriched for particular minor H antigen-specific TM cells. The DLI product could be further engineered by depletion of TN.
Hematopoietic Cell Transplantation for Pediatric Leukemia in the Current Era and Future
Allogeneic HCT is the current standard of care for pediatric patients with very high-risk hematologic malignancies (1, 2). The overall success of HCT as a treatment for pediatric leukemia has improved, with reduced non-relapse mortality rates (144) and leukemia-free survival rates in the range of 60–80% (145, 146) in the current era. However, conditioning regimens and GVHD have long lasting adverse effects, particularly for the pediatric population who undergo significant neurologic and physical development. Long-term effects of HCT, particularly of conditioning and especially related to TBI, include growth disturbance, hormone deficiencies, cataracts, seizures, cerebrovascular events, dyslipidemia, and secondary malignancies (12–16). GVHD can also lead to long-lasting morbidity involving multiple organs, most commonly the skin, gut, and liver, but also lungs, mouth, eyes and joints in the chronic setting (15, 17).
The mortality and morbidity of HCT may be improved by graft engineering and adjunctive T cell immunotherapy to reduce GVHD and to augment GVL. Because T cell immunotherapy has the potential to prevent relapse, it may permit de-escalation of conditioning intensity, which will be critically important for the youngest HCT recipients in whom myeloablative HCT can be associated with devastating neurodevelopmental complications (13). In infants, leukemia is often refractory to chemotherapy and relapse occurs frequently (147). HCT is indicated for infants with ALL in first remission with the highest risk of relapse, and for those who achieve a second remission after relapse. Unfortunately, the youngest infants with ALL generally have the highest risk of relapse with chemotherapy alone, but also the greatest risk of late effects of HCT conditioning. Because of this combination of high risk from both disease and complications of therapy, there is an urgent need to develop reduced toxicity HCT strategies for infants, supplemented by add-back of minor H antigen-specific T cells and/or post-HCT T cell immunotherapy to prevent relapse.
The presence of detectable disease at the time of HCT is consistently and strongly associated with an increased risk of relapse post-HCT (6, 7). As such, other important recent developments that enable reduction of conditioning intensity include sensitive techniques for detecting measurable residual disease (MRD) and therapies for reducing MRD prior to HCT. These advances are particularly important when one contemplates HCT strategies that rely on the T cell-dependent GVL effect, which is relatively delayed compared to the immediate anti-leukemic effect of intensive conditioning. MRD detection has moved from morphologic evaluation to the use of flow cytometry or PCR, which have sensitivities of 0.1% for AML and 0.01% for ALL (148, 149). Technologies to detect even lower levels of disease have been developed and the clinical implications are being studied. Next-generation sequencing (NGS) measuring immunoglobulin heavy chain (variable, diversity and joining) have been developed for ALL; patients who are NGS negative prior to HCT have been shown to have a reduced risk of relapse (150). NGS is also currently being evaluated for AML (151, 152) and may prove useful for risk stratification and perhaps monitoring, but is likely to be complex (153). Highly sensitive MRD evaluation in the pre-HCT period may be used to guide which pediatric patients can undergo HCT with reduced intensity/toxicity conditioning without an excessive risk of relapse, and which patients do have a high risk of relapse and may benefit from the addition of novel relapse prophylaxis, such as minor H antigen-specific T cells. In cases where MRD is detected pre-HCT, new targeted strategies, particularly CAR-T cell immunotherapy and bispecific T cell engagers, can also produce deep MRD negative remissions prior to HCT and thereby may improve post-HCT prognosis (154–158).
Significant success has been achieved using CAR-T cell immunotherapy targeting lineage-specific antigens to treat pediatric and adult patients with acute leukemia. This is highlighted by the efficacy of some CAR-T cell products targeting CD19, an antigen expressed on normal and leukemic B cells in some acute leukemias and lymphomas (154, 156). Patients who have received effective CD19 CAR-T cell therapy develop B cell aplasia and hypogammaglobulinemia as a result. Given hypogammaglobulinemia can be supported with intravenous immunoglobulin administration, and patients do not have significant infectious complications (159), B cell aplasia is not a major barrier for CD19 CAR-T cell immunotherapy. Targeting myeloid-lineage antigens, such as CD33 or CD123, is likely to be more problematic due to the risk of marrow suppression or aplasia placing patients at risk for severe associated complications including infection (160, 161). Minor H antigen-targeted therapies are not suitable for use in patients who have not received allogeneic HCT and therefore have a recipient hematopoietic system. However, following HCT the presence of a normal donor hematopoietic system lacking the minor H antigen target limits the therapeutic targets to neoplastic cells or residual recipient normal cells that are no longer necessary, and should protect the recipient from marrow suppression, B cell aplasia, and other hematopoietic complications. Additionally, many CAR constructs incorporate non-human components, frequently using murine derived svFc, which can be immunogenic leading to a rejection response, whereas the TCR in minor H antigen-specific T cells and TCR-T cell products are of human origin and are inherently less immunogenic favoring in vivo persistence (162). Lastly, although minor H antigen-targeted therapy is restricted to patients with the restricting HLA-allele and appropriate recipient-donor mismatch for the polymorphism, minor H antigen-targeted therapy is not limited to one leukemia subtype, in contrast to most CAR-T cell immunotherapy.
Ultimately, superior survival with reduced morbidity after pediatric HCT could be achieved by a combination of (a) therapies to achieve remission without any detectable disease, potentially including CAR-T cell therapy or other immunotherapy (b) a reduced-intensity/toxicity conditioning regimen, (c) a HCT graft selectively depleted of GVHD-inducing T cells and augmented with T cells specific for a limited number of hematopoietic restricted minor H antigens, and (d) post-HCT minor H antigen-targeted T cell immunotherapy. Although this vision has not yet been reached, and minor H antigen-targeted T cell therapies are currently in early phase clinical trials without proven benefit for pediatric or other patients, steady progress is being made in the fields of antigen discovery, graft engineering and genetically-modified T cell therapy. We anticipate that these advances will continue and come together to benefit children with very high-risk leukemia.
Conclusions
Donor-derived T cell responses to minor H antigens with hematopoietic-restricted expression are a key component of effective GVL. Targeting leukemia-associated minor H antigens with T cells in hematopoietic grafts and/or with post-HCT T cell immunotherapy are potentially low-toxicity, high-efficacy prophylactic and therapeutic strategies for pediatric patients. Pre-clinical studies and clinical trials of T cell immunotherapies to enhance the response to hematopoietic-restricted minor H antigens are underway. These advancing technologies should enable a reduction in HCT conditioning intensity without sacrificing protection from post-HCT relapse, thereby mitigating dangerous late effects of HCT for pediatric patients with leukemia.
Author Contributions
CS, VS, and MB reviewed the literature, wrote, and edited the manuscript.
Funding
This work was supported by National Cancer Institute (NCI CA018029-42) Alex's Lemonade Stand Foundation, and by a Stand Up To Cancer Innovative Research Grant, Grant Number SU2C-AACR-IRG 14-17. Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Conflict of Interest
MB is a Founder and Scientific Advisory Board member of HighPassBio, a Scientific Advisory Board member of Orca Bio, and has also received compensation from Miltenyi Biotec for presentations at conferences and corporate symposia.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Oliansky DM, Camitta B, Gaynon P, Nieder ML, Parsons SK, Pulsipher MA, et al. Role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of pediatric acute lymphoblastic leukemia: update of the 2005 evidence-based review. Biol Blood Marrow Transplant. (2012) 18:505–22. doi: 10.1016/j.bbmt.2011.12.585
2. Taraseviciute A, Pulsipher MA. Advances in hematopoietic cell transplant for the treatment of hematologic malignancies. Curr Opin Pediatr. (2019) 31:3–13. doi: 10.1097/MOP.0000000000000729
3. Cornelissen JJ, van Putten WL, Verdonck LF, Theobald M, Jacky E, Daenen SM, et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: benefits for whom? Blood. (2007) 109:3658–66. doi: 10.1182/blood-2006-06-025627
4. Cornelissen JJ, van der Holt B, Verhoef GE, van't Veer MB, van Oers MH, Schouten HC, et al. Myeloablative allogeneic versus autologous stem cell transplantation in adult patients with acute lymphoblastic leukemia in first remission: a prospective sibling donor versus no-donor comparison. Blood. (2009) 113:1375–82. doi: 10.1182/blood-2008-07-168625
5. Shouval R, Fein JA, Labopin M, Kroger N, Duarte RF, Bader P, et al. Outcomes of allogeneic haematopoietic stem cell transplantation from HLA-matched and alternative donors: a European society for blood and marrow transplantation registry retrospective analysis. Lancet Haematol. (2019) 6:e573–e584. doi: 10.1016/S2352-3026(19)30158-9
6. Bar M, Wood BL, Radich JP, Doney KC, Woolfrey AE, Delaney C, et al. Impact of minimal residual disease, detected by flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute lymphoblastic leukemia. Leuk Res Treat. (2014) 2014:421723. doi: 10.1155/2014/421723
7. Leung W, Pui CH, Coustan-Smith E, Yang J, Pei D, Gan K, et al. Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood. (2012) 120:468–72. doi: 10.1182/blood-2012-02-409813
8. Dahlberg A, Leisenring W, Bleakley M, Meshinchi S, Baker KS, Summers C, et al. Prognosis of relapse after hematopoietic cell transplant (HCT) for treatment of leukemia or myelodysplastic syndrome (MDS) in children. Bone Marrow Transplant. (2019) 54:1337–45. doi: 10.1038/s41409-019-0438-z
9. Uden T, Bertaina A, Abrahamsson J, Ansari M, Balduzzi A, Bourquin JP, et al. Outcome of children relapsing after first allogeneic haematopoietic stem cell transplantation for acute myeloid leukaemia: a retrospective I-BFM analysis of 333 children. Br J Haematol. (2020) 189:745–50. doi: 10.1111/bjh.16441
10. Shah NN, Borowitz MJ, Robey NC, Gamper CJ, Symons HJ, Loeb DM, et al. Feasibility of treating post-transplantation minimal residual disease in children with acute leukemia. Biol Blood Marrow Transplant. (2014) 20:1000–7. doi: 10.1016/j.bbmt.2014.03.021
11. Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer. (2004) 4:371–80. doi: 10.1038/nrc1365
12. Nieder ML, McDonald GB, Kida A, Hingorani S, Armenian SH, Cooke KR, et al. National cancer institute-national heart, lung and blood institute/pediatric blood and marrow transplant consortium first international consensus conference on late effects after pediatric hematopoietic cell transplantation: long-term organ damage and dysfunction. Biol Blood Marrow Transplant. (2011) 17:1573–84. doi: 10.1016/j.bbmt.2011.09.013
13. Vrooman LM, Millard HR, Brazauskas R, Majhail NS, Battiwalla M, Flowers ME, et al. Survival and late effects after allogeneic hematopoietic cell transplantation for hematologic malignancy at less than three years of age. Biol Blood Marrow Transplant. (2017) 23:1327–34. doi: 10.1016/j.bbmt.2017.04.017
14. Baker KS, Leisenring WM, Goodman PJ, Ermoian RP, Flowers ME, Schoch G, et al. Total body irradiation dose and risk of subsequent neoplasms following allogeneic hematopoietic cell transplantation. Blood. (2019) 133:2790–9. doi: 10.1182/blood.2018874115
15. Lee CJ, Kim S, Tecca HR, Bo-Subait S, Phelan R, Brazauskas R, et al. Late effects after ablative allogeneic stem cell transplantation for adolescent and young adult acute myeloid leukemia. Blood Adv. (2020) 4:983–92. doi: 10.1182/bloodadvances.2019001126
16. Saglio F, Zecca M, Pagliara D, Giorgiani G, Balduzzi A, Calore E, et al. Occurrence of long-term effects after hematopoietic stem cell transplantation in children affected by acute leukemia receiving either busulfan or total body irradiation: results of an AIEOP (Associazione Italiana Ematologia Oncologia Pediatrica) retrospective study. Bone Marrow Transplant. (2020). doi: 10.1038/s41409-020-0806-8. [Epub ahead of print].
17. Baird K, Cooke K, Schultz KR. Chronic graft-versus-host disease (GVHD) in children. Pediatr Clin North Am. (2010) 57:297–322. doi: 10.1016/j.pcl.2009.11.003
18. Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell. (2019) 177:70–84. doi: 10.1016/j.cell.2019.02.032
19. Burke DF, Worth CL, Priego EM, Cheng T, Smink LJ, Todd JA, et al. Genome bioinformatic analysis of nonsynonymous SNPs. BMC Bioinformatics. (2007) 8:301. doi: 10.1186/1471-2105-8-301
20. Bijen HM, Hassan C, Kester MGD, Janssen GMC, Hombrink P, de Ru AH, et al. Specific T cell responses against minor histocompatibility antigens cannot generally be explained by absence of their allelic counterparts on the cell surface. Proteomics. (2018) 18:e1700250. doi: 10.1002/pmic.201700250
21. Murata M, Warren EH, Riddell SR. A human minor histocompatibility antigen resulting from differential expression due to a gene deletion. J Exp Med. (2003) 197:1279–89. doi: 10.1084/jem.20030044
22. de Rijke B, van Horssen-Zoetbrood A, Beekman JM, Otterud B, Maas F, Woestenenk R, et al. A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. J Clin Invest. (2005) 115:3506–16. doi: 10.1172/JCI24832
23. Brickner AG, Evans AM, Mito JK, Xuereb SM, Feng X, Nishida T, et al. The PANE1 gene encodes a novel human minor histocompatibility antigen that is selectively expressed in B-lymphoid cells and B-CLL. Blood. (2006) 107:3779–86. doi: 10.1182/blood-2005-08-3501
24. Terakura S, Murata M, Warren EH, Sette A, Sidney J, Naoe T, et al. A single minor histocompatibility antigen encoded by UGT2B17 and presented by human leukocyte antigen-A*2902 and -B*4403. Transplantation. (2007) 83:1242–8. doi: 10.1097/01.tp.0000259931.72622.d1
25. Pont MJ, van der Lee DI, van der Meijden ED, van Bergen CA, Kester MG, Honders MW, et al. Integrated whole genome and transcriptome analysis identified a therapeutic minor histocompatibility antigen in a splice variant of ITGB2. Clin Cancer Res. (2016) 22:4185–96. doi: 10.1158/1078-0432.CCR-15-2307
26. Brickner AG, Warren EH, Caldwell JA, Akatsuka Y, Golovina TN, Zarling AL, et al. The immunogenicity of a new human minor histocompatibility antigen results from differential antigen processing. J Exp Med. (2001) 193:195–206. doi: 10.1084/jem.193.2.195
27. Spierings E, Brickner AG, Caldwell JA, Zegveld S, Tatsis N, Blokland E, et al. The minor histocompatibility antigen HA-3 arises from differential proteasome-mediated cleavage of the lymphoid blast crisis (Lbc) oncoprotein. Blood. (2003) 102:621–9. doi: 10.1182/blood-2003-01-0260
28. den Haan JM, Meadows LM, Wang W, Pool J, Blokland E, Bishop TL, et al. The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science. (1998) 279:1054–7. doi: 10.1126/science.279.5353.1054
29. Nicholls S, Piper KP, Mohammed F, Dafforn TR, Tenzer S, Salim M, et al. Secondary anchor polymorphism in the HA-1 minor histocompatibility antigen critically affects MHC stability and TCR recognition. Proc Natl Acad Sci USA. (2009) 106:3889–94. doi: 10.1073/pnas.0900411106
30. Spierings E, Gras S, Reiser JB, Mommaas B, Almekinders M, Kester MG, et al. Steric hindrance and fast dissociation explain the lack of immunogenicity of the minor histocompatibility HA-1Arg null allele. J Immunol. (2009) 182:4809–16. doi: 10.4049/jimmunol.0803911
31. Torikai H, Akatsuka Y, Miyazaki M, Tsujimura A, Yatabe Y, Kawase T, et al. The human cathepsin H gene encodes two novel minor histocompatibility antigen epitopes restricted by HLA-A*3101 and -A*3303. Br J Haematol. (2006) 134:406–16. doi: 10.1111/j.1365-2141.2006.06205.x
32. Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. (1979) 300:1068–73. doi: 10.1056/NEJM197905103001902
33. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. (1990) 75:555–62. doi: 10.1182/blood.V75.3.555.555
34. Inamoto Y, Flowers ME, Lee SJ, Carpenter PA, Warren EH, Deeg HJ, et al. Influence of immunosuppressive treatment on risk of recurrent malignancy after allogeneic hematopoietic cell transplantation. Blood. (2011) 118:456–63. doi: 10.1182/blood-2011-01-330217
35. Korngold R, Sprent J. Lethal GVHD across minor histocompatibility barriers: nature of the effector cells and role of the H-2 complex. Immunol Rev. (1983) 71:5–29. doi: 10.1111/j.1600-065X.1983.tb01066.x
36. Korngold R, Sprent J. Features of T cells causing H-2-restricted lethal graft-vs.-host disease across minor histocompatibility barriers. J Exp Med. (1982) 155:872–83. doi: 10.1084/jem.155.3.872
37. de Bueger M, Bakker A, Van Rood JJ, Van der Woude F, Goulmy E. Tissue distribution of human minor histocompatibility antigens. Ubiquitous versus restricted tissue distribution indicates heterogeneity among human cytotoxic T lymphocyte-defined non-MHC antigens. J Immunol. (1992) 149:1788–94.
38. Warren EH, Greenberg PD, Riddell SR. Cytotoxic T-lymphocyte-defined human minor histocompatibility antigens with a restricted tissue distribution. Blood. (1998) 91:2197–207. doi: 10.1182/blood.V91.6.2197
39. Torikai H, Akatsuka Y, Miyauchi H, Terakura S, Onizuka M, Tsujimura K, et al. The HLA-A*0201-restricted minor histocompatibility antigen HA-1H peptide can also be presented by another HLA-A2 subtype, A*0206. Bone Marrow Transplant. (2007) 40:165–74. doi: 10.1038/sj.bmt.1705689
40. Van Bergen CA, Rutten CE, Van Der Meijden ED, Van Luxemburg-Heijs SA, Lurvink EG, Houwing-Duistermaat JJ, et al. High-throughput characterization of 10 new minor histocompatibility antigens by whole genome association scanning. Cancer Res. (2010) 70:9073–83. doi: 10.1158/0008-5472.CAN-10-1832
41. Dolstra H, Fredrix H, Preijers F, Goulmy E, Figdor CG, de Witte TM, et al. Recognition of a B cell leukemia-associated minor histocompatibility antigen by CTL. J Immunol. (1997) 158:560–5.
42. Dolstra H, Fredrix H, Maas F, Coulie PG, Brasseur F, Mensink E, et al. A human minor histocompatibility antigen specific for B cell acute lymphoblastic leukemia. J Exp Med. (1999) 189:301–8. doi: 10.1084/jem.189.2.301
43. Dolstra H, de Rijke B, Fredrix H, Balas A, Maas F, Scherpen F, et al. Bi-directional allelic recognition of the human minor histocompatibility antigen HB-1 by cytotoxic T lymphocytes. Eur J Immunol. (2002) 32:2748–58. doi: 10.1002/1521-4141(2002010)32:10<2748::AID-IMMU2748>3.0.CO2-T
44. Akatsuka Y, Nishida T, Kondo E, Miyazaki M, Taji H, Iida H, et al. Identification of a polymorphic gene, BCL2A1, encoding two novel hematopoietic lineage-specific minor histocompatibility antigens. J Exp Med. (2003) 197:1489–500. doi: 10.1084/jem.20021925
45. Kawase T, Nannya Y, Torikai H, Yamamoto G, Onizuka M, Morishima S, et al. Identification of human minor histocompatibility antigens based on genetic association with highly parallel genotyping of pooled DNA. Blood. (2008) 111:3286–94. doi: 10.1182/blood-2007-10-118950
46. Kawase T, Akatsuka Y, Torikai H, Morishima S, Oka A, Tsujimura A, et al. Alternative splicing due to an intronic SNP in HMSD generates a novel minor histocompatibility antigen. Blood. (2007) 110:1055–63. doi: 10.1182/blood-2007-02-075911
47. Pierce RA, Field ED, Mutis T, Golovina TN, Von Kap-Herr C, Wilke M, et al. The HA-2 minor histocompatibility antigen is derived from a diallelic gene encoding a novel human class I myosin protein. J Immunol. (2001) 167:3223–30. doi: 10.4049/jimmunol.167.6.3223
48. Mommaas B, Kamp J, Drijfhout JW, Beekman N, Ossendorp F, Van Veelen P, et al. Identification of a novel HLA-B60-restricted T cell epitope of the minor histocompatibility antigen HA-1 locus. J Immunol. (2002) 169:3131–6. doi: 10.4049/jimmunol.169.6.3131
49. Griffioen M, van Bergen CA, Falkenburg JH. Autosomal minor histocompatibility antigens: how genetic variants create diversity in immune targets. Front Immunol. (2016) 7:100. doi: 10.3389/fimmu.2016.00100
50. Granados DP, Rodenbrock A, Laverdure JP, Cote C, Caron-Lizotte O, Carli C, et al. Proteogenomic-based discovery of minor histocompatibility antigens with suitable features for immunotherapy of hematologic cancers. Leukemia. (2016) 30:1344–54. doi: 10.1038/leu.2016.22
51. van Bergen CA, van Luxemburg-Heijs SA, de Wreede LC, Eefting M, von dem Borne PA, van Balen P, et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J Clin Invest. (2017) 127:517–29. doi: 10.1172/JCI86175
52. Lansford JL, Dharmasiri U, Chai S, Hunsucker SA, Bortone DS, Keating JE, et al. Computational modeling and confirmation of leukemia-associated minor histocompatibility antigens. Blood Adv. (2018) 2:2052–62. doi: 10.1182/bloodadvances.2018022475
53. Bleakley M, Otterud BE, Richardt JL, Mollerup AD, Hudecek M, Nishida T, et al. Leukemia-associated minor histocompatibility antigen discovery using T-cell clones isolated by in vitro stimulation of naive CD8+ T cells. Blood. (2010) 115:4923–33. doi: 10.1182/blood-2009-12-260539
54. Falkenburg JH, Goselink HM, van der Harst D, van Luxemburg-Heijs SA, Kooy-Winkelaar YM, Faber LM, et al. Growth inhibition of clonogenic leukemic precursor cells by minor histocompatibility antigen-specific cytotoxic T lymphocytes. J Exp Med. (1991) 174:27–33. doi: 10.1084/jem.174.1.27
55. van der Harst D, Goulmy E, Falkenburg JH, Kooij-Winkelaar YM, van Luxemburg-Heijs SA, Goselink HM, et al. Recognition of minor histocompatibility antigens on lymphocytic and myeloid leukemic cells by cytotoxic T-cell clones. Blood. (1994) 83:1060–6. doi: 10.1182/blood.V83.4.1060.1060
56. Bonnet D, Warren EH, Greenberg PD, Dick JE, Riddell SR. CD8(+) minor histocompatibility antigen-specific cytotoxic T lymphocyte clones eliminate human acute myeloid leukemia stem cells. Proc Natl Acad Sci USA. (1999) 96:8639–44. doi: 10.1073/pnas.96.15.8639
57. Fontaine P, Roy-Proulx G, Knafo L, Baron C, Roy DC, Perreault C. Adoptive transfer of minor histocompatibility antigen-specific T lymphocytes eradicates leukemia cells without causing graft-versus-host disease. Nat Med. (2001) 7:789–94. doi: 10.1038/89907
58. Meunier MC, Delisle JS, Bergeron J, Rineau V, Baron C, Perreault C. T cells targeted against a single minor histocompatibility antigen can cure solid tumors. Nat Med. (2005) 11:1222–9. doi: 10.1038/nm1311
59. Meunier MC, Roy-Proulx G, Labrecque N, Perreault C. Tissue distribution of target antigen has a decisive influence on the outcome of adoptive cancer immunotherapy. Blood. (2003) 101:766–70. doi: 10.1182/blood-2002-04-1032
60. Meunier MC, Baron C, Perreault C. Two host factors regulate persistence of H7-specific T cells injected in tumor-bearing mice. PLoS ONE. (2009) 4:e4116. doi: 10.1371/journal.pone.0004116
61. Li N, Matte-Martone C, Zheng H, Cui W, Venkatesan S, Tan HS, et al. Memory T cells from minor histocompatibility antigen-vaccinated and virus-immune donors improve GVL and immune reconstitution. Blood. (2011) 118:5965–76. doi: 10.1182/blood-2011-07-367011
62. Korngold R, Leighton C, Mobraaten LE, Berger MA. Inter-strain graft-vs.-host disease T-cell responses to immunodominant minor histocompatibility antigens. Biol Blood Marrow Transplant. (1997) 3:57–64.
63. Zilberberg J, Feinman R, Korngold R. Strategies for the identification of T cell-recognized tumor antigens in hematological malignancies for improved graft-versus-tumor responses after allogeneic blood and marrow transplantation. Biol Blood Marrow Transplant. (2015) 21:1000–7. doi: 10.1016/j.bbmt.2014.11.001
64. Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. (2011) 89:396–407. doi: 10.1038/icb.2010.124
65. Wilke M, Dolstra H, Maas F, Pool J, Brouwer R, Falkenburg JH, et al. Quantification of the HA-1 gene product at the RNA level; relevance for immunotherapy of hematological malignancies. Hematol J. (2003) 4:315–20. doi: 10.1038/sj.thj.6200318
66. Fujii N, Hiraki A, Ikeda K, Ohmura Y, Nozaki I, Shinagawa K, et al. Expression of minor histocompatibility antigen, HA-1, in solid tumor cells. Transplantation. (2002) 73:1137–41. doi: 10.1097/00007890-200204150-00022
67. Torikai H, Akatsuka Y, Yatabe Y, Morishima Y, Kodera Y, Kuzushima K, et al. Aberrant expression of BCL2A1-restricted minor histocompatibility antigens in melanoma cells: application for allogeneic transplantation. Int J Hematol. (2008) 87:467–73. doi: 10.1007/s12185-008-0076-5
68. Dickinson AM, Wang XN, Sviland L, Vyth-Dreese FA, Jackson GH, Schumacher TN, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. (2002) 8:410–4. doi: 10.1038/nm0402-410
69. Hobo W, Broen K, van der Velden WJ, Greupink-Draaisma A, Adisty N, Wouters Y, et al. Association of disparities in known minor histocompatibility antigens with relapse-free survival and graft-versus-host disease after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. (2013) 19:274–82. doi: 10.1016/j.bbmt.2012.09.008
70. Mutis T, Gillespie G, Schrama E, Falkenburg JH, Moss P, Goulmy E. Tetrameric HLA class I-minor histocompatibility antigen peptide complexes demonstrate minor histocompatibility antigen-specific cytotoxic T lymphocytes in patients with graft-versus-host disease. Nat Med. (1999) 5:839–42. doi: 10.1038/10563
71. Kircher B, Stevanovic S, Urbanek M, Mitterschiffthaler A, Rammensee HG, Grunewald K, et al. Induction of HA-1-specific cytotoxic T-cell clones parallels the therapeutic effect of donor lymphocyte infusion. Br J Haematol. (2002) 117:935–9. doi: 10.1046/j.1365-2141.2002.03536.x
72. Marijt WA, Heemskerk MH, Kloosterboer FM, Goulmy E, Kester MG, van der Hoorn MA, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci USA. (2003) 100:2742–7. doi: 10.1073/pnas.0530192100
73. Kloosterboer FM, van Luxemburg-Heijs SA, van Soest RA, Barbui AM, van Egmond HM, Strijbosch MP, et al. Direct cloning of leukemia-reactive T cells from patients treated with donor lymphocyte infusion shows a relative dominance of hematopoiesis-restricted minor histocompatibility antigen HA-1 and HA-2 specific T cells. Leukemia. (2004) 18:798–808. doi: 10.1038/sj.leu.2403297
74. Goulmy E, Schipper R, Pool J, Blokland E, Falkenburg JH, Vossen J, et al. Mismatches of minor histocompatibility antigens between HLA-identical donors and recipients and the development of graft-versus-host disease after bone marrow transplantation. N Engl J Med. (1996) 334:281–5. doi: 10.1056/NEJM199602013340501
75. Tseng LH, Lin MT, Hansen JA, Gooley T, Pei J, Smith AG, et al. Correlation between disparity for the minor histocompatibility antigen HA-1 and the development of acute graft-versus-host disease after allogeneic marrow transplantation. Blood. (1999) 94:2911–4. doi: 10.1182/blood.V94.8.2911.420k21_2911_2914
76. Socie G, Loiseau P, Tamouza R, Janin A, Busson M, Gluckman E, et al. Both genetic and clinical factors predict the development of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Transplantation. (2001) 72:699–706. doi: 10.1097/00007890-200108270-00024
77. Gallardo D, Arostegui JI, Balas A, Torres A, Caballero D, Carreras E, et al. Disparity for the minor histocompatibility antigen HA-1 is associated with an increased risk of acute graft-versus-host disease (GvHD) but it does not affect chronic GvHD incidence, disease-free survival or overall survival after allogeneic human leucocyte antigen-identical sibling donor transplantation. Br J Haematol. (2001) 114:931–6. doi: 10.1046/j.1365-2141.2001.03013.x
78. Murata M, Emi N, Hirabayashi N, Hamaguchi M, Goto S, Wakita A, et al. No significant association between HA-1 incompatibility and incidence of acute graft-versus-host disease after HLA-identical sibling bone marrow transplantation in Japanese patients. Int J Hematol. (2000) 72:371–5.
79. Heinemann FM, Ferencik S, Ottinger HD, Beelen DW, Grosse-Wilde H. Impact of disparity of minor histocompatibility antigens HA-1, CD31, and CD49b in hematopoietic stem cell transplantation of patients with chronic myeloid leukemia with sibling and unrelated donors. Transplantation. (2004) 77:1103–6. doi: 10.1097/01.TP.0000120175.25116.CB
80. Lin MT, Gooley T, Hansen JA, Tseng LH, Martin EG, Singleton K, et al. Absence of statistically significant correlation between disparity for the minor histocompatibility antigen-HA-1 and outcome after allogeneic hematopoietic cell transplantation. Blood. (2001) 98:3172–3. doi: 10.1182/blood.V98.10.3172
81. Spellman S, Warden MB, Haagenson M, Pietz BC, Goulmy E, Warren EH, et al. Effects of mismatching for minor histocompatibility antigens on clinical outcomes in HLA-matched, unrelated hematopoietic stem cell transplants. Biol Blood Marrow Transplant. (2009) 15:856–63. doi: 10.1016/j.bbmt.2009.03.018
82. Franssen LE, Roeven MWH, Hobo W, Doorn R, Oostvogels R, Falkenburg JHF, et al. A phase I/II minor histocompatibility antigen-loaded dendritic cell vaccination trial to safely improve the efficacy of donor lymphocyte infusions in myeloma. Bone Marrow Transplant. (2017) 52:1378–83. doi: 10.1038/bmt.2017.118
83. Oostvogels R, Kneppers E, Minnema MC, Doorn RC, Franssen LE, Aarts T, et al. Efficacy of host-dendritic cell vaccinations with or without minor histocompatibility antigen loading, combined with donor lymphocyte infusion in multiple myeloma patients. Bone Marrow Transplant. (2017) 52:228–37. doi: 10.1038/bmt.2016.250
84. Nagy B, Lundan T, Larramendy ML, Aalto Y, Zhu Y, Niini T, et al. Abnormal expression of apoptosis-related genes in haematological malignancies: overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br J Haematol. (2003) 120:434–41. doi: 10.1046/j.1365-2141.2003.04121.x
85. Sochalska M, Schuler F, Weiss JG, Prchal-Murphy M, Sexl V, Villunger A. MYC selects against reduced BCL2A1/A1 protein expression during B cell lymphomagenesis. Oncogene. (2017) 36:2066–73. doi: 10.1038/onc.2016.362
86. Spierings E, Drabbels J, Hendriks M, Pool J, Spruyt-Gerritse M, Claas F, et al. A uniform genomic minor histocompatibility antigen typing methodology and database designed to facilitate clinical applications. PLoS ONE. (2006) 1:e42. doi: 10.1371/journal.pone.0000042
87. Kloosterboer FM, van Luxemburg-Heijs SA, van Soest RA, van Egmond HM, Willemze R, Falkenburg JH. Up-regulated expression in nonhematopoietic tissues of the BCL2A1-derived minor histocompatibility antigens in response to inflammatory cytokines: relevance for allogeneic immunotherapy of leukemia. Blood. (2005) 106:3955–7. doi: 10.1182/blood-2004-09-3749
88. Nishida T, Akatsuka Y, Morishima Y, Hamajima N, Tsujimura K, Kuzushima K, et al. Clinical relevance of a newly identified HLA-A24-restricted minor histocompatibility antigen epitope derived from BCL2A1, ACC-1, in patients receiving HLA genotypically matched unrelated bone marrow transplant. Br J Haematol. (2004) 124:629–35. doi: 10.1111/j.1365-2141.2004.04823.x
89. Akatsuka Y, Torikai H, Inamoto Y, Tsujimura K, Morishima Y, Kodera Y, et al. Bone marrow may be a reservoir of long-lived memory T cells specific for minor histocompatibility antigen. Br J Haematol. (2006) 135:413–4. doi: 10.1111/j.1365-2141.2006.06313.x
90. Nadolny C, Dong X. Liver receptor homolog-1 (LRH-1): a potential therapeutic target for cancer. Cancer Biol Ther. (2015) 16:997–1004. doi: 10.1080/15384047.2015.1045693
91. Pang JB, Molania R, Chand A, Knower K, Takano EA, Byrne DJ, et al. LRH-1 expression patterns in breast cancer tissues are associated with tumour aggressiveness. Oncotarget. (2017) 8:83626–36. doi: 10.18632/oncotarget.18886
92. Xiao L, Wang Y, Liang W, Liu L, Pan N, Deng H, et al. LRH-1 drives hepatocellular carcinoma partially through induction of c-myc and cyclin E1, and suppression of p21. Cancer Manage Res. (2018) 10:2389–400. doi: 10.2147/CMAR.S162887
93. Xiao L, Wang Y, Xu K, Hu H, Xu Z, Wu D, et al. Nuclear receptor LRH-1 functions to promote castration-resistant growth of prostate cancer via its promotion of intratumoral androgen biosynthesis. Cancer Res. (2018) 78:2205–18. doi: 10.1158/0008-5472.CAN-17-2341
94. Overes IM, de Rijke B, van Horssen-Zoetbrood A, Fredrix H, de Graaf AO, Jansen JH, et al. Expression of P2X5 in lymphoid malignancies results in LRH-1-specific cytotoxic T-cell-mediated lysis. Br J Haematol. (2008) 141:799–807. doi: 10.1111/j.1365-2141.2008.07125.x
95. Norde WJ, Overes IM, Maas F, Fredrix H, Vos JC, Kester MG, et al. Myeloid leukemic progenitor cells can be specifically targeted by minor histocompatibility antigen LRH-1-reactive cytotoxic T cells. Blood. (2009) 113:2312–23. doi: 10.1182/blood-2008-04-153825
96. Overes IM, Fredrix H, Kester MG, Falkenburg JH, van der Voort R, de Witte TM, et al. Efficient activation of LRH-1-specific CD8+ T-cell responses from transplanted leukemia patients by stimulation with P2X5 mRNA-electroporated dendritic cells. J Immunother. (2009) 32:539–51. doi: 10.1097/CJI.0b013e3181987c22
97. van Balen P, van Bergen CAM, van Luxemburg-Heijs SAP, de Klerk W, van Egmond EHM, Veld SAJ, et al. CD4 Donor lymphocyte infusion can cause conversion of chimerism without GVHD by inducing immune responses targeting minor histocompatibility antigens in HLA class II. Front Immunol. (2018) 9:3016. doi: 10.3389/fimmu.2018.03016
98. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. (2018) 379:2330–41. doi: 10.1056/NEJMoa1808777
99. Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med. (2019) 25:603–11. doi: 10.1038/s41591-019-0400-z
100. Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. (2018) 131:108–20. doi: 10.1182/blood-2017-07-791608
101. Biernacki MA, Brault M, Bleakley M. T-Cell receptor-based immunotherapy for hematologic malignancies. Cancer J. (2019) 25:179–90. doi: 10.1097/PPO.0000000000000378
102. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. (2019) 290:127–47. doi: 10.1111/imr.12772
103. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. (2008) 118:294–305. doi: 10.1172/JCI32103
104. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. (2015) 125:2677–89. doi: 10.1172/JCI81229
105. van Loenen MM, de Boer R, Hagedoorn RS, van Egmond EH, Falkenburg JH, Heemskerk MH. Optimization of the HA-1-specific T-cell receptor for gene therapy of hematologic malignancies. Haematologica. (2011) 96:477–81. doi: 10.3324/haematol.2010.025916
106. van Loenen MM, de Boer R, van Liempt E, Meij P, Jedema I, Falkenburg JH, et al. A good manufacturing practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica. (2014) 99:759–68. doi: 10.3324/haematol.2013.093690
107. Pasquini MC, Devine S, Mendizabal A, Baden LR, Wingard JR, Lazarus HM, et al. Comparative outcomes of donor graft CD34+ selection and immune suppressive therapy as graft-versus-host disease prophylaxis for patients with acute myeloid leukemia in complete remission undergoing HLA-matched sibling allogeneic hematopoietic cell transplantation. J Clin Oncol. (2012) 30:3194–201. doi: 10.1200/JCO.2012.41.7071
108. Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versus-host disease prophylaxis for allogeneic hematopoietic stem cell transplantation. Blood. (2001) 98:3192–204. doi: 10.1182/blood.V98.12.3192
109. Lee YJ, Chung D, Xiao K, Papadopoulos EB, Barker JN, Small TN, et al. Adenovirus viremia and disease: comparison of T cell-depleted and conventional hematopoietic stem cell transplantation recipients from a single institution. Biol Blood Marrow Transplant. (2013) 19:387–92. doi: 10.1016/j.bbmt.2012.10.014
110. Barker JN, Hough RE, van Burik JA, DeFor TE, MacMillan ML, O'Brien MR, et al. Serious infections after unrelated donor transplantation in 136 children: impact of stem cell source. Biol Blood Marrow Transplant. (2005) 11:362–70. doi: 10.1016/j.bbmt.2005.02.004
111. van Burik JA, Carter SL, Freifeld AG, High KP, Godder KT, Papanicolaou GA, et al. Higher risk of cytomegalovirus and aspergillus infections in recipients of T cell-depleted unrelated bone marrow: analysis of infectious complications in patients treated with T cell depletion versus immunosuppressive therapy to prevent graft-versus-host disease. Biol Blood Marrow Transplant. (2007) 13:1487–98. doi: 10.1016/j.bbmt.2007.08.049
112. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. (2014) 14:24–35. doi: 10.1038/nri3567
113. Anderson BE, Zheng H, Taylor PA, Matte-Martone C, McNiff JM, Jain D, et al. Memory T cells in GVHD and GVL. Biol Blood Marrow Transplant. (2008) 14(1 Suppl. 1):19–20.
114. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. (2003) 112:101–8. doi: 10.1172/JCI17601
115. Zhang Y, Joe G, Zhu J, Carroll R, Levine B, Hexner E, et al. Dendritic cell-activated CD44hiCD8+ T cells are defective in mediating acute graft-versus-host disease but retain graft-versus-leukemia activity. Blood. (2004) 103:3970–8. doi: 10.1182/blood-2003-09-3135
116. Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L- memory T cells without graft-versus-host disease. Blood. (2004) 103:1534–41. doi: 10.1182/blood-2003-08-2987
117. Xystrakis E, Bernard I, Dejean AS, Alsaati T, Druet P, Saoudi A. Alloreactive CD4 T lymphocytes responsible for acute and chronic graft-versus-host disease are contained within the CD45RChigh but not the CD45RClow subset. Eur J Immunol. (2004) 34:408–17. doi: 10.1002/eji.200324528
118. Dutt S, Tseng D, Ermann J, George TI, Liu YP, Davis CR, et al. Naive and memory T cells induce different types of graft-versus-host disease. J Immunol. (2007) 179:6547–54. doi: 10.4049/jimmunol.179.10.6547
119. Chen BJ, Deoliveira D, Cui X, Le NT, Son J, Whitesides JF, et al. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood. (2007) 109:3115–23. doi: 10.1182/blood-2006-04-016410
120. Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood. (2008) 111:2476–84. doi: 10.1182/blood-2007-08-109678
121. Zheng H, Matte-Martone C, Jain D, McNiff J, Shlomchik WD. Central memory CD8+ T cells induce graft-versus-host disease and mediate graft-versus-leukemia. J Immunol. (2009) 182:5938–48. doi: 10.4049/jimmunol.0802212
122. Bleakley M, Heimfeld S, Jones LA, Turtle C, Krause D, Riddell SR, et al. Engineering human peripheral blood stem cell grafts that are depleted of naive T cells and retain functional pathogen-specific memory T cells. Biol Blood Marrow Transplant. (2014) 20:705–16. doi: 10.1016/j.bbmt.2014.01.032
123. Schumm M, Lang P, Bethge W, Faul C, Feuchtinger T, Pfeiffer M, et al. Depletion of T-cell receptor alpha/beta and CD19 positive cells from apheresis products with the CliniMACS device. Cytotherapy. (2013) 15:1253–8. doi: 10.1016/j.jcyt.2013.05.014
124. Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. (2014) 124:822–6. doi: 10.1182/blood-2014-03-563817
125. Lang P, Feuchtinger T, Teltschik HM, Schwinger W, Schlegel P, Pfeiffer M, et al. Improved immune recovery after transplantation of TCRαβ/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transplant. (2015) 50(Suppl. 2):S6–10. doi: 10.1038/bmt.2015.87
126. Airoldi I, Bertaina A, Prigione I, Zorzoli A, Pagliara D, Cocco C, et al. γδ T-cell reconstitution after HLA-haploidentical hematopoietic transplantation depleted of TCR-αβ+/CD19+ lymphocytes. Blood. (2015) 125:2349–58. doi: 10.1182/blood-2014-09-599423
127. Locatelli F, Merli P, Pagliara D, Li Pira G, Falco M, Pende D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood. (2017) 130:677–85. doi: 10.1182/blood-2017-04-779769
128. Handgretinger R, Schilbach K. The potential role of γδ T cells after allogeneic HCT for leukemia. Blood. (2018) 131:1063–72. doi: 10.1182/blood-2017-08-752162
129. Locatelli F, Pende D, Falco M, Della Chiesa M, Moretta A, Moretta L. NK cells mediate a crucial graft-versus-leukemia effect in haploidentical-HSCT to cure high-risk acute leukemia. Trends Immunol. (2018) 39:577–90. doi: 10.1016/j.it.2018.04.009
130. Silva-Santos B, Mensurado S, Coffelt SB. γδ T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat Rev Cancer. (2019) 19:392–404. doi: 10.1038/s41568-019-0153-5
131. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. (2015) 21:938–45. doi: 10.1038/nm.3909
132. Knight A, Madrigal AJ, Grace S, Sivakumaran J, Kottaridis P, Mackinnon S, et al. The role of Vδ2-negative γδ T cells during cytomegalovirus reactivation in recipients of allogeneic stem cell transplantation. Blood. (2010) 116:2164–72. doi: 10.1182/blood-2010-01-255166
133. Scheper W, van Dorp S, Kersting S, Pietersma F, Lindemans C, Hol S, et al. γδT cells elicited by CMV reactivation after allo-SCT cross-recognize CMV and leukemia. Leukemia. (2013) 27:1328–38. doi: 10.1038/leu.2012.374
134. Ravens S, Schultze-Florey C, Raha S, Sandrock I, Drenker M, Oberdorfer L, et al. Human γδ T cells are quickly reconstituted after stem-cell transplantation and show adaptive clonal expansion in response to viral infection. Nat Immunol. (2017) 18:393–401. doi: 10.1038/ni.3686
135. Bertaina A, Zecca M, Buldini B, Sacchi N, Algeri M, Saglio F, et al. Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood. (2018) 132:2594–607. doi: 10.1182/blood-2018-07-861575
136. Noviello M, Manfredi F, Ruggiero E, Perini T, Oliveira G, Cortesi F, et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat Commun. (2019) 10:1065. doi: 10.1038/s41467-019-08871-1
137. Freimuller C, Stemberger J, Artwohl M, Germeroth L, Witt V, Fischer G, et al. Selection of adenovirus-specific and epstein-barr virus-specific T cells with major histocompatibility class I streptamers under good manufacturing practice (GMP)-compliant conditions. Cytotherapy. (2015) 17:989–1007. doi: 10.1016/j.jcyt.2015.03.613
138. Neuenhahn M, Albrecht J, Odendahl M, Schlott F, Dossinger G, Schiemann M, et al. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia. (2017) 31:2161–71. doi: 10.1038/leu.2017.16
139. Roex MCJ, Hageman L, Heemskerk MT, Veld SAJ, van Liempt E, Kester MGD, et al. The simultaneous isolation of multiple high and low frequent T-cell populations from donor peripheral blood mononuclear cells using the major histocompatibility complex I-Streptamer isolation technology. Cytotherapy. (2018) 20:543–55. doi: 10.1016/j.jcyt.2018.01.008
140. Roex MCJ, van Balen P, Germeroth L, Hageman L, van Egmond E, Veld SAJ, et al. Generation and infusion of multi-antigen-specific T cells to prevent complications early after T-cell depleted allogeneic stem cell transplantation-a phase I/II study. Leukemia. (2020) 34:831–44. doi: 10.1038/s41375-019-0600-z
141. Chen B, Lim S, Kannan A, Alford SC, Sunden F, Herschlag D, et al. High-throughput analysis and protein engineering using microcapillary arrays. Nat Chem Biol. (2016) 12:76–81. doi: 10.1038/nchembio.1978
142. Schultze-Florey RE, Tischer-Zimmermann S, Heuft HG, Priesner C, Lamottke B, Heim A, et al. Transfer of hexon- and penton-selected adenovirus-specific T cells for refractory adenovirus infection after haploidentical stem cell transplantation. Transpl Infect Dis. (2020) 22:e13201. doi: 10.1111/tid.13201
143. Meij P, Jedema I, Zandvliet ML, van der Heiden PL, van de Meent M, van Egmond HM, et al. Effective treatment of refractory CMV reactivation after allogeneic stem cell transplantation with in vitro-generated CMV pp65-specific CD8+ T-cell lines. J Immunother. (2012) 35:621–8. doi: 10.1097/CJI.0b013e31826e35f6
144. McDonald GB, Sandmaier BM, Mielcarek M, Sorror M, Pergam SA, Cheng GS, et al. Survival, nonrelapse mortality, and relapse-related mortality after allogeneic hematopoietic cell transplantation: comparing 2003-2007 versus 2013-2017 cohorts. Ann Intern Med. (2020) 172:229–39. doi: 10.7326/M19-2936
145. Willasch AM, Peters C, Sedlacek P, Dalle JH, Kitra-Roussou V, Yesilipek A, et al. Myeloablative conditioning for allo-HSCT in pediatric ALL: FTBI or chemotherapy?-A multicenter EBMT-PDWP study. Bone Marrow Transplant. (2020). doi: 10.1038/s41409-020-0854-0. [Epub ahead of print].
146. Locatelli F, Masetti R, Rondelli R, Zecca M, Fagioli F, Rovelli A, et al. Outcome of children with high-risk acute myeloid leukemia given autologous or allogeneic hematopoietic cell transplantation in the aieop AML-2002/01 study. Bone Marrow Transplant. (2015) 50:181–8. doi: 10.1038/bmt.2014.246
147. Brown P, Pieters R, Biondi A. How I treat infant leukemia. Blood. (2019) 133:205–14. doi: 10.1182/blood-2018-04-785980
148. Theunissen P, Mejstrikova E, Sedek L, van der Sluijs-Gelling AJ, Gaipa G, Bartels M, et al. Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood. (2017) 129:347–57. doi: 10.1182/blood-2016-07-726307
149. Wood BL. Principles of minimal residual disease detection for hematopoietic neoplasms by flow cytometry. Cytometry B Clin Cytom. (2016) 90:47–53. doi: 10.1002/cyto.b.21239
150. Pulsipher MA, Carlson C, Langholz B, Wall DA, Schultz KR, Bunin N, et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood. (2015) 125:3501–8. doi: 10.1182/blood-2014-12-615757
151. Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. (2018) 378:1189–99. doi: 10.1056/NEJMoa1716863
152. Patkar N, Shaikh AF, Kakirde C, Nathany S, Ramesh H, Bhanshe P, et al. A novel machine-learning-derived genetic score correlates with measurable residual disease and is highly predictive of outcome in acute myeloid leukemia with mutated NPM1. Blood Cancer J. (2019) 9:79. doi: 10.1038/s41408-019-0244-2
153. Walter C, Schneider M, Von Neuhoff N, Hanenberg H, Reinhardt D, Rasche M. Mutational landscape of pediatric acute myeloid leukemia: a report of the AML-BFM study group with a targeted NGS approach in 525 patients integrating de novo, relapsed and secondary AML. Blood. (2019) 134(Suppl. 1):1398. doi: 10.1182/blood-2019-129778
154. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
155. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
156. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
157. Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. (2017) 376:836–47. doi: 10.1056/NEJMoa1609783
158. von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. (2016) 34:4381–9. doi: 10.1200/JCO.2016.67.3301
159. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. (2018) 131:121–30. doi: 10.1182/blood-2017-07-793760
160. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. (2014) 123:2343–54. doi: 10.1182/blood-2013-09-529537
161. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. (2015) 29:1637–47. doi: 10.1038/leu.2015.52
162. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. (2010) 16:1245–56. doi: 10.1016/j.bbmt.2010.03.014
Keywords: minor histocompatibility antigen, T cell immunotherapy, hematopoietic stem cell transplantation, leukemia, pediatric, graft-vs.-leukemia, graft engineering, polymorphism
Citation: Summers C, Sheth VS and Bleakley M (2020) Minor Histocompatibility Antigen-Specific T Cells. Front. Pediatr. 8:284. doi: 10.3389/fped.2020.00284
Received: 15 April 2020; Accepted: 06 May 2020;
Published: 03 June 2020.
Edited by:
Patrick J. Hanley, Children's National Hospital, United StatesReviewed by:
Daniele Zama, Sant'Orsola-Malpighi Polyclinic, ItalyMarieke Griffioen, Leiden University Medical Center, Netherlands
Copyright © 2020 Summers, Sheth and Bleakley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie Bleakley, bWJsZWFrbGVAZnJlZGh1dGNoLm9yZw==