 Birce Sunman
Birce Sunman Dilber Ademhan Tural
Dilber Ademhan Tural Beste Ozsezen
Beste Ozsezen Nagehan Emiralioglu
Nagehan Emiralioglu Uğur Özçelik
Uğur Özçelik- Department of Pediatric Pulmonology, Hacettepe University Faculty of Medicine, Ankara, Turkey
Allergic bronchopulmonary aspergillosis (ABPA) is a complex pulmonary disorder characterized by a hypersensitivity reaction to Aspergillus fumigatus, and almost always seen in patients with cystic fibrosis (CF) and asthma. Fungal hyphae leads to an ongoing inflammation in the airways that may result in bronchiectasis, fibrosis, and eventually loss of lung function. Despite the fact that ABPA is thought to be more prevalent in CF than in asthma, the literature on ABPA in CF is more limited. The diagnosis is challenging and may be delayed because it is made based on a combination of clinical features, and radiologic and immunologic findings. With clinical deterioration of a patient with CF, ABPA is important to be kept in mind because clinical manifestations mimic pulmonary exacerbations of CF. Early diagnosis and appropriate treatment are important in preventing complications related to ABPA. Treatment modalities involve the use of anti-inflammatory agents to suppress the immune hyperreactivity and the use of antifungal agents to reduce fungal burden. Recently, in an effort to treat refractory patients or to reduce adverse effects of steroids, other treatment options such as monoclonal antibodies have started to be used. Intensive research of these new agents in the treatment of children is being conducted to address insufficient data.
Introduction
Aspergillus fumigatus (A. fumigatus) is the most common ubiquitous airborne fungus, which causes allergic bronchopulmonary aspergillosis (ABPA) (1). Aspergillus spores are found in high concentrations in nature, especially in fertile soil, decaying vegetation, swimming pool water, leaky basements, bedding, and dust from homes (2). Hypersensitivity reactions that occur because of A. fumigatus allergens are allergic asthma, hypersensitivity pneumonia, and ABPA (3), which is a localized hypersensitivity reaction to the lung that develops against aspergillus antigens in the colonized bronchial mucus.
The prevalence of ABPA changes according to the population (child/adult), geographic region, or diagnostic criteria that have been used. At the same time, ABPA is believed to be underdiagnosed, especially in developing countries, because its clinical features are much the same as cystic fibrosis (CF). In asthmatic patients the prevalence is reported to be about 1 to 2% and is more common in adults than in children (4, 5). The prevalence is higher in CF patients than in asthmatic patients and thought to be 8.9% (ranged from 3 to 25%) with a significantly higher occurrence among adults (6, 7).
In this review, immunopathogenesis, clinical features, diagnosis, and current treatment modalities have been tried to be summarized. Although this review is based on the studies and case reports with the pediatric age group, some studies in adults and asthmatics have also been mentioned due to limited number of publications in children. By the way the diagnosis and treatment in children are not much different from adults and the treatment in CF is similar to asthmatics (8).
Immunopathogenesis of ABPA
The pathogenesis of ABPA involves many immunologic reactions. These are Aspergillus-specific immunoglobulin (Ig)-E–mediated hypersensitivity, IgG-mediated immune complex hypersensitivity, and abnormal cell-mediated immune response (9). These hypersensitivity responses cause mucus impaction in the bronchi and bronchioles, as well as inflammatory cell infiltration in bronchial walls and peribronchial tissues. All of these reactions cause bronchiectasis and bronchocentric non-caseating granulomatosis (10, 11).
Aspergillus conidia, because of its small diameter (2–3 mm), can easily reach the pulmonary alveoli and deposit there. Once they reach the alveoli, spores germinate to produce fungal hyphae and continuously grow in the airways of patients with ABPA. A. fumigatus has several virulence factors to escape from the immune system including superoxide dismutases, catalases, mannitol, proteases, ribotoxin, phythiotic acid, phospholipases, gliotoxin, and hemolysin. Most of these proteins are known to be antigenic and are believed to be responsible for the immune response in ABPA. These virulence factors also damage the airway epithelium and cause a larger dose of antigenic factors to pass to the interstitial and vascular compartments. Antigenic cells with human leukocyte antigen (HLA)-DR5 or HLA-DR2 process these antigens together and present them to T lymphocytes in bronchoalveolar lymphoid tissue. In normal hosts, while the organism is eradicated with the T helper (Th)1 response, in patients with ABPA, an extreme Th2 response to the aspergillus antigen is seen, even if the Th1 response is not defective. Protease and antigen released by spores and hyphae cause activation of the innate immune system, and damage in the bronchial epithelium, which causes bronchiectasis and impaired mucociliary clearance. As a result, various chemokines including thymus and activation regulated chemokine (TARC), monocyte chemotactic protein 1, eotaxin, RANTES (regulated on activation, normal T-cell expressed, and secreted), interleukin (IL)-8, and macrophage inflammatory protein 1a are released in the airways. These cytokines activate the Th2 response and this causes the proliferation of CD4+ Th2 lymphocytes, which produce IL-4, IL-5, IL-9, IL-10, IL-13, and eosinophilic growth and survival, mast cell proliferation, IgG and IgE isotype switching occurs (9, 10).
Similarly in patients with CF, due to abnormal mucociliary clearence of secretions and defective innate immune responses, exposure to A. fumigatus spores results in accumulation and persistence of fungal spores within the smaller airways (12). Release of antigens, cytokins, and other virulence factors cause airway epithelial damage and antigenic factors are transmitted to the interstitial and vascular compartments (13). The immune response to ABPA in CF patients is also IL-4–mediated T helper cell (Th) type 2–predominant response, which is shown by CFTR mutant mouse expression profiling studies (14, 15).
Finally, there are some opinions about why some patients with CF develop ABPA. One of these is that the predominant CD4+ Th2 cell response can be related to genetic factors and this can explain why some patients with CF develop ABPA while others do not (16). Another conviction is that because patients with ABPA have an exaggerated response to IL-4 and produce a large amount of IgE, IgG, and IgA antibodies against A. fumigatus antigens, a gain of function polymorphism in the IL-4 receptor-α chain may be responsible for this situation (17). Lastly, some authors suggest that HLA-DRB1*1501 and HLA-DRB1*1503 confer the highest risk of developing ABPA, whereas HLA-DQ2 (HLA-DQB1*0201 in particular) provides relative protection against the development of ABPA (18–20).
Therefore, a combination of all these factors may determine the outcome of ABPA in patients with CF.
Clinical Features
ABPA symptoms are usually non-specific and resemble clinical findings in CF. One-third of patients with CF are asymptomatic and are diagnosed as having ABPA in routine follow-up (7). The most common clinical findings are chronic productive cough and wheezing. Other symptoms are pleuritic chest pain and blood-stained sputum. Expectoration of golden-brownish mucus plugs is a characteristic finding in ABPA and is found in half of all patients (5, 21, 22). The dark mucus plugs are due to the increased production of tenacious mucus in the respiratory tract and consist of inflammatory cells including eosinophils, desquamated epithelial cells, and mucin (23, 24). Hemoptysis can occur due to severe airway inflammation and bronchiectasis (3). Constitutional symptoms such as low-grade fever, myalgia, and weight loss are found in 26% of patients with CF (25, 26). Physical examination is usually not noticeable except for crackles, rhonchi unresponsive to bronchodilator treatment, absence of respiratory sounds distal to dark mucus plugs, and clubbing. In end-stage disease, cor pulmonale findings may be present (4). ABPA should be suspected in a patient with CF who develops wheezing or major reductions of forced expiratory volume in one second (FEV1) without evidence of a CF exacerbation, that do not respond to appropriate antibiotics, standard physiotherapy and which is not explained by another etiology (3).
Stages
The disease can be clinically divided into five stages as shown in Table 1 (2). Patients may be detected at any stage at the time of diagnosis and the transition from one stage to another may not be in order. It is also important to notify that ABPA serology is most likely to be positive in stage 1 and 3. Early diagnosis and treatment prevent the disease from progressing to stage 5.
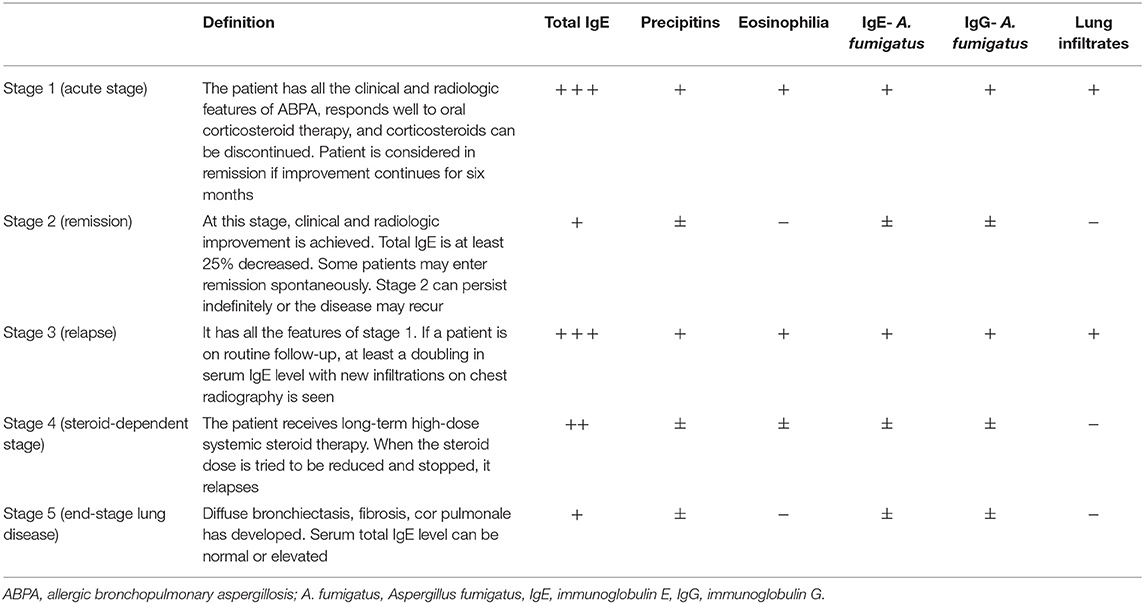
Table 1. Clinical and serological characteristics of ABPA stages.
Diagnosis
The diagnosis of ABPA in patients with CF is challenging and may be delayed because many of the diagnostic criteria crossover with the clinical manifestations of CF. There is no fully covered individual test that demonstrates the diagnosis of ABPA in patients with CF. The diagnosis is made through a combination of clinical characteristics, and radiologic and immunologic findings (27). There are different sets of diagnostic criteria for the diagnosis of ABPA in patients with CF. The diagnostic criteria are summarized in Table 2 according to historical date (28–31).
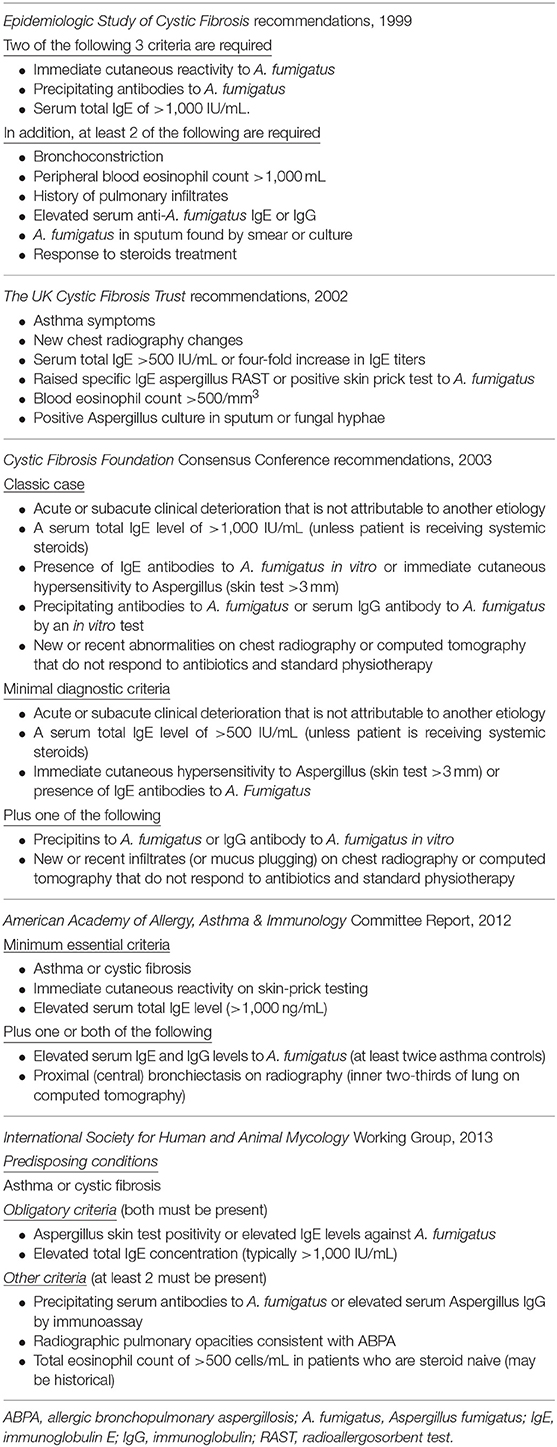
Table 2. History of acceptable criteria for diagnosing ABPA.
According to Cystic Fibrosis Foundation Consensus Conference recommendations (2003), which is the most common used definition for diagnosis (3):
Classic Case
1. Clinical deterioration, acute or subacute, which is unexplained by another etiology;
2. Serum total IgE concentration of >1,000 IU·mL−1 (unless the patient is receiving systemic corticosteroids);
3. Immediate cutaneous reactivity (skin prick test wheal >3 mm in diameter with surrounding erythema) to Aspergillus or in vitro presence of serum IgE antibody to A. fumigatus;
4. Precipitating antibodies to A. fumigatus or serum IgG antibody to A. fumigatus in an in vitro test;
5. New or recent abnormalities on chest radiography (infiltrates or mucus plugging) or chest computed tomography (CT) (bronchiectasis) that do not respond to appropriate antibiotics and standard physiotherapy.
Minimal Diagnostic Criteria
1. Clinical deterioration, acute or subacute, which is unexplained by another etiology;
2. Serum total IgE concentration of >500 IU·mL−1. If total IgE level is 200–500 and ABPA is suspected, repeat testing in 1–3 months;
3. Immediate cutaneous reactivity to Aspergillus (skin prick test wheal >3 mm in diameter with surrounding erythema while the patient is not being treated with systemic antihistamines) or in vitro presence of IgE antibody to A. fumigatus;
4. One of the following: (a) precipitins to A. fumigatus or in vitro demonstration of IgG antibody to A. fumigatus; or (b) new or recent abnormalities on chest radiography (infiltrates or mucus plugging) or chest CT (bronchiectasis) that do not respond to appropriate antibiotics and standard physiotherapy.
The suggestions of Cystic Fibrosis Foundation Consensus Conference for screening ABPA in CF (3):
1. Patients >6 years of age should be considered suspicious for ABPA.
2. Patients should be checked for serum total IgE concentration annually. If the serum total IgE concentration is >500 IU·mL−1, get immediate cutaneous reactivity to A. fumigatus or use an in vitro test for IgE antibody to A. fumigatus. If results are positive, consider diagnosis on the basis of minimal criteria;
3. If the serum total IgE concentration is 200–500 IU·mL−1, repeat the test in 1–3 months if there is a suspicion for ABPA.
The diagnostic algorithm created with the criteria suggested by the Cystic Fibrosis Foundation is showed in Figure 1.
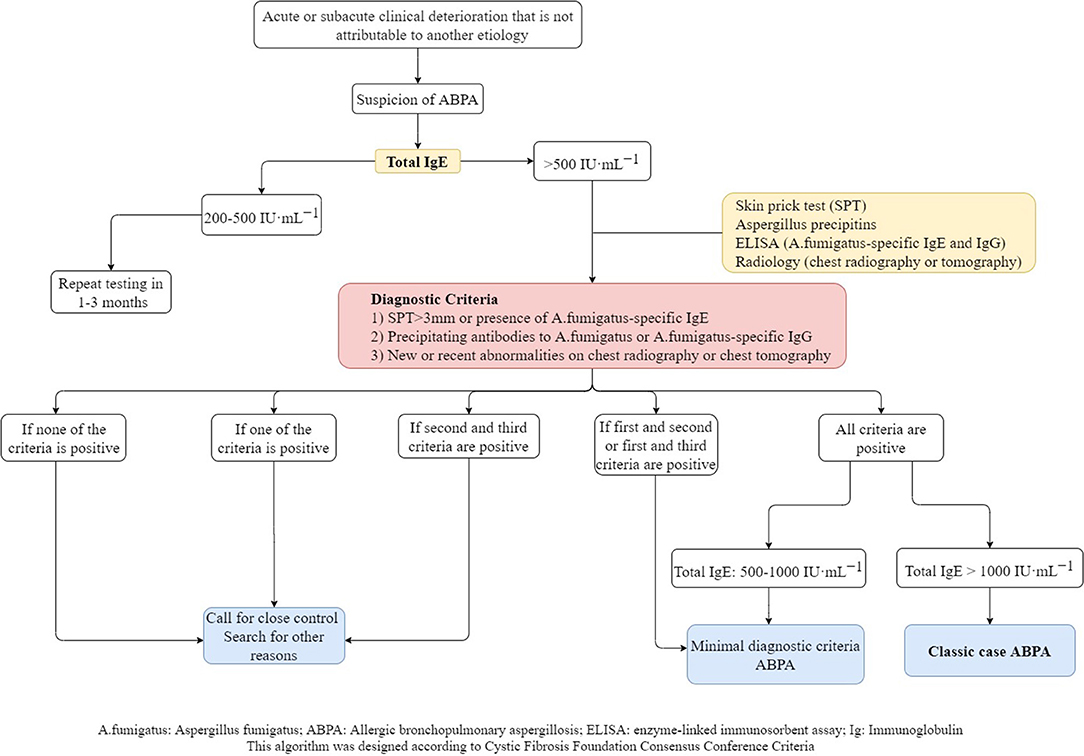
Figure 1. Diagnostic algorithm of ABPA in CF.
Although there are many diagnostic criteria for ABPA, Maleki et al. showed that there was no significant differences on the reported rate of ABPA prevelance between the Cystic Fibrosis Foundation and International Society for Human and Animal Mycology (ISHAM) diagnostic criteria (32).
Clinical Findings
Acute/subacute clinical worsening defined as cough, increased amount of sputum or changing in color of sputum, wheeze, dyspnea, the onset of new fever, weight loss, exercise-induced asthma and decrease in pulmonary function, that does not respond to appropriate treatment and is not explained with another etiology (3).
Serum Total IgE
This can be used for the detection of fungal sensitization (33). Values of total IgE as high as >500 IU·mL−1 (34) or even >1,000 IU·mL−1 (35) or >2-fold rise from baseline total IgE have been suggested as diagnostic markers unless the patient is receiving systemic corticosteroids. If the patient is using systemic steroids, retesting is recommended after the completion of steroid treatment (3). Irregular changes in IgE values with clinical symptoms can be a marker for exacerbations and responses to therapy. In patients with ABPA, despite total serum IgE levels often being in concordance with clinical activity and treatment, it is not sufficiently specific for the diagnosis (29, 36, 37).
Aspergillus Skin Test
Another investigation for the detection of sensitization to A. fumigatus is the Aspergillus skin test, which shows immediate cutaneous hypersensitivity to A. fumigatus (38). However, it shows heterogenity among different centers in terms of procedures, interpretation, and the use of different commercial fungal preparations (39). A skin prick test should be performed for Aspergillus skin testing; if the results are negative it should be confirmed by an intradermal test. Intradermal skin tests are usually preferred for the diagnosis of Aspergillus sensitization because they are more sensitive than the skin prick test (40).
Both type I (immediate) and type III (delayed) skin sensitivity with different Aspergillus antigens can be positive in patients with ABPA. Although type III responses are mainly suppressed by steroid treatment, there is little or no effect of steroids on the type I reactions. For the wheal of immediate skin sensitivity, the authorities have suggested a diameter of surrounding erythema >3 mm to be considered as a positive result (3). Aspergillus skin test is sensitive enough that the lack of a positive skin test reduces the likelihood of ABPA diagnosis; however, the specificity of the test is moderately low, which means it can be positive in patients with CF without ABPA (41). Therefore, a positive Aspergillus skin test must always be followed up with serologic and radiologic testing to confirm ABPA.
Serum Specific IgE to A. fumigatus
This is not an appropriate biomarker and lacks sufficient specificity for ABPA because nearly 35% to 50% of patients with CF demonstrate detectable sensitization to A. fumigatus despite not having ABPA. However, elevated specific IgE to A. fumigatus is a more sensitive marker than total IgE like the aspergillus skin test for ABPA in CF (38, 39, 42, 43). In 2013, ISHAM accepted 0.35 kUA·L−1 as a cut-off value for anti-Aspergillus sensitization and this value was changed and accepted as 0.10 kUA·L−1 by the US Food and Drug Administration (FDA) in 2008 and recommended by a 2015 consensus guideline (29, 39). The level of specific IgE to A. fumigatus can act as a marker of an exacerbation or remission like total IgE.
Precipitating Antibodies or Serum IgG Antibody to A. fumigatus
There are few methods to evaluate the A. fumigatus precipitating antibody or specific IgG to the crude antigen. These antibodies, have been found to be a sensitive sign for ABPA in patients with CF. Precipitating antibodies to A. fumigatus are usually in the IgG isotype, in particular of the IgG1, IgG2, and IgG4 subclasses, and their increased levels have been reported in patients with CF and ABPA (44, 45).
Traditionally, IgG antibodies against A. fumigatus were measured by immunoprecipitation and counterimmunoelectrophoresis (CIE) using double gel diffusion techniques and called Aspergillus precipitins (46–48). Because of poor sensitivity and subjective qualitative results of these methods, commercial ImmunoCAP systems using enzyme-linked immunosorbant assays (ELISA) have been developed and this method has increased the detection of cases with ABPA. Recently, this commercial system is the most widely used method for quantifying specific IgG. The results of few studies also suggest that A. fumigatus specific IgG measured by the ImmunoCAP system is more sensitive than Aspergillus precipitins measured by the double diffusion method (47, 48). The best cutoff value for A. fumigatus specific IgG using ImmunoCap system has been reported as 26.9 mgA/L with 88% sensitivity and 100% specificity. Although the sensitivity of A. fumigatus specific IgG detected by double gel diffusion technique has been reported low as 27%; the sensitivity of commercial ImmunoCAP systems was 89% in the diagnosis of ABPA (47). However, the cutoff values have been varied in patients with CF within different studies (47–50). The prevalence of serum IgG antibodies to A. fumigatus has been reported to increase with age in patients with CF, regardless of ABPA (51). Aspergillus precipitins may represent previous exposure, but high levels of precipitins may indicate increased probability of ABPA (31). A. fumigatus-specific IgG levels may suggest disease activity, which can be evidenced by radiographic changes and clinical exacerbations, whereas serum precipitins do not reflect disease activity in most cases (1).
Peripheral Blood Eosinophilia
Peripheral blood eosinophil counts of >1,000 cell/ μL were previously considered as a major criteria for the diagnosis of ABPA (52, 53). However, now it is known to be of limited value in diagnosing patients with CF and ABPA because high eosinophil counts may be present due to chronic Pseudomonas aeruginosa infection (3) and many other disorders other than ABPA (54, 55). In a recent study on children with CF reported that 75% of ABPA patients had eosinophil count >400 cells/μL and 40% of them having counts >1,000 cells/μL, while none of the patients in the A. fumigatus sensitized and non-sensitized groups had eosinophilia and these findings suggested that eosinophil count could be a specific biomarker for ABPA in children (56).
New Serologic Tests
Specific IgE Antibodies Against Recombinant A. fumigatus Allergens
Recombinant allergens are raw extracts from A. fumigatus which uses for immunological assays of ABPA. There are recognized 23 specific allergens of A. fumigatus but five of them (rAsp f1, f2, f3, f4, and f6) are commercially available (57). However, those allergens can cross-react with other fungal antigens. Due to rAsp f1 and f2 have minimal cross-reactivity with other fungal antigens, they are considered as the specific allergens for A. fumigatus (58). Specific IgE against a combination of rAsp f1 or f3 found to be the most sensitive (97%) and specific IgE against a combination of rAsp f4 or f6 had highest specificity (99%) for diagnosing ABPA in patients with asthma (57). Some authors suggested combining serum total IgE with specific IgE to recombinant A. fumigatus allergen (rAspf) to differentiate ABPA from sensitization (59). A recent study reported that IgE against rAsp f1 and f2 were found to be the most useful in differentiating ABPA from A. fumigatus sensitization in patients with asthma (60).
Thymus and Activation-Regulated Chemokine (TARC)
TARC is produced as a result of the antifungal immune response. The serum levels of TARC are found to be increased in patients with CF and ABPA (61). TARC was found to be a more sensitive and specific marker of ABPA when it was compared with other serum markers. TARC levels are increased even before the development of clinical features of ABPA and before total IgE increment (59). TARC stays elevated for a prolonged period of time; therefore, the changes in TARC levels can be a sign of exacerbations and remissions of ABPA (59, 61). However, it has not been added to classic case definitions.
Cellular Allergen Stimulation Test (CAST)
CAST measures cysteinyl-leukotrienes, which are produced in vitro by allergen-stimulated basophils. CAST is used for the diagnosis of allergic and pseudoallergic reactions. CAST has a high sensitivity (100%) but a low specificity (74%) for ABPA. The combination of a positive CAST, serum total IgE >500 IU·mL−1, and positive IgE antibodies against rAsp f4 and f6 was found only in those with ABPA, giving rise to 100% specificity (62).
Further studies with a larger number of patients are required to investigate CAST and TARC before they become routine investigations for ABPA.
Basophil Activation Test (BAT)
BAT is an in vitro flow cytometry-based cellular assay that measures the activation of basophils with Ig-E mediated mechanism and using stimulation with A. fumigatus extract and CD63, CD193, and CD203c as activation surface markers which they found to have high diagnostic accuracy for ABPA. This test should be performed within the four hour of blood collection to increase viability and functionality of basophils; because basophil reactivity decreases over time (63). Different studies suggest that BAT is a useful, reliable diagnostic tool especially for ABPA in patients with CF (64, 65) and it is useful in distinguishing ABPA from Aspergillus colonization and sensitization in patients with CF (66, 67) but not in patients with asthma (68). However, BAT needs a flow cytometer and the requirement of performing this test immediately after the collection of blood sample limit its usability.
Culture of Sputum for A. fumigatus
Sputum culture is a supportive marker for ABPA (43, 69), whereas others have considered it as only a minor criterion because A. fumigatus hyphae in sputum smears or A. fumigatus in sputum cultures may not be detected in ABPA or can also be seen in other pulmonary diseases (70, 71). In ABPA, rates of culture positivity were reported as 40% to 60% in different studies (72). Also, A. fumigatus culture-negative patients with ABPA were found to have A. fumigatus DNA in their sputum (73). Aspergillus polymerase chain reaction (PCR) is more sensitive than culture in ABPA diagnosis and it may be used to monitor the efficacy of antifungal therapy (72). Both real-time Aspergillus PCR and galactomannan in respiratory samples have also been used for enhanced recognition of ABPA in CF with traditional immunological tests (serum total IgE, A. fumigatus-specific IgE and IgG) (74).
Pulmonary Function Tests
In mild or early stages of ABPA, partially reversible airflow obstruction is a common pulmonary function test finding. Prolonged airflow obstruction and decreased lung volumes in total lung capacity (TLC), vital capacity (VC), and FEV1 suggest interstitial changes in progressive disease (75, 76). The diffusing capacity of lung for carbon monoxide (DLCO) may be decreased during an exacerbation and it remains low at the end stage of ABPA (5). Despite pulmonary function tests not being diagnostic for ABPA, they are useful during follow-up to monitor improvement.
Bronchoscopy
Bronchoscopic evaluation and histology are not necessary for the diagnosis of ABPA and bronchoscopy may be performed in patients with ABPA when the diagnosis is unclear. Bronchoalveolar lavage (BAL) shows elevated levels of IgA, IgG, IgM, and IgE, as well as elevated eosinophil counts. However, the sensitivity of staining BAL washes or sputum samples for Aspergillus is poor. Detecting Aspergillus species in BAL is not specific for active disease of ABPA because it may reflect colonization (75, 77).
Interpretation of diagnostic findings of ABPA are summarized in Table 3.
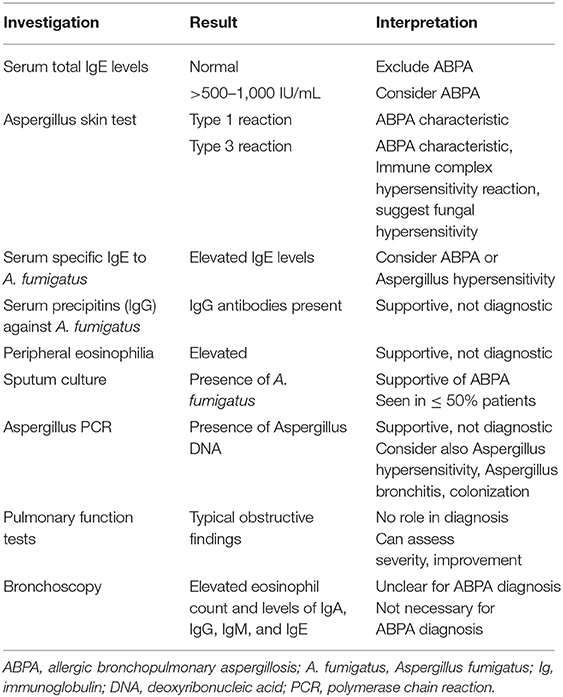
Table 3. Interpretation of diagnostic findings of ABPA.
Radiologic Manifestations of ABPA
Both chest radiography and chest CT are useful for the diagnosis of ABPA. Radiologic findings are summarized in Table 4.
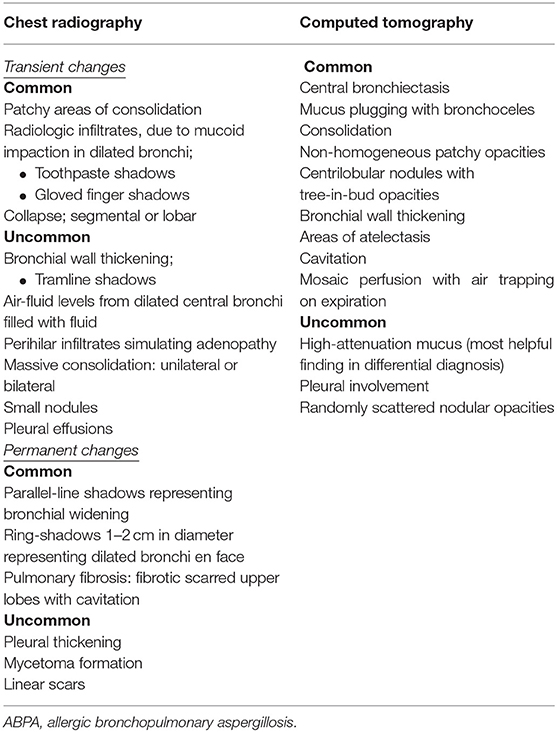
Table 4. Radiologic findings in ABPA.
Chest radiography has 50% sensitivity for the diagnosis of ABPA. It can be normal in the early stages but temporary or permanent parenchymal opacities can also be seen. Parenchymal infiltrate and bronchiectasis are mostly in the upper lobes; however, all lobes may be affected (40). Central bronchiectasis is one of the hallmarks of ABPA, and bronchiectasis affecting more than three lobes is highly suggestive of diagnosis (78, 79). A massive homogeneous shadow without fissure displacement usually located in the upper and middle lobes that frequently shifts from one side to another is the most common abnormality seen on a chest radiography with ABPA. The shadow may be patchy, triangular, oblong, or lobar (6). “Ring sign” indicating bronchial inflammation with or without plugs can be a sign of bronchial wall thickening or bronchiectasis (80). “Tramline” shadows and “finger-in-glove” opacities are temporary findings that indicate bronchial wall edema and thickening, whereas once the mucus plug is expectorated, it can remain as permanent parallel line shadows (5).
High-resolution CT (HRCT) of the lungs is more sensitive for detecting bronchiectasis distribution and other abnormalities that cannot be detected in chest radiography (40). The findings of ABPA on chest HRCT include centrilobular nodules, central bronchiectasis often with mucoid impaction “tree-in-bud” pattern, mosaic attenuation, fibrosis, and cavitation (81). Bronchiectasis seen in ABPA is typically central (bronchiectasis that involves two-thirds of the central part of the lung parenchyma) but peripheral bronchiectasis is not infrequent. Central or cystic varicose bronchiectasis, infiltrations that completely resolve with corticosteroid treatment, and mucus plugs are common in CF and ABPA, high-attenuation mucus (HAM) has been reported in 28% of patients with ABPA on HRCT. Mucoid impaction causes “toothpaste” shadows or “gloved-finger” shadows (75, 81, 82). HAM is the name given to mucus that appears denser than skeletal muscles (radiodensity of >70 Hounsfield units). Mucus plugs may cause segmental, lobar or total atelectasis. HAM and bronchiectasis are indicators of serious disease and recurrent exacerbations. Also, the presence of bronchiectasis makes it difficult to enter remission (83). In the later stages, pneumothorax might be seen in patients who develop pulmonary fibrosis (84). Pleural thickening is seen in ABPA and in advanced CF (85, 86). Radiographic findings in ABPA are demonstrated in Figure 2.
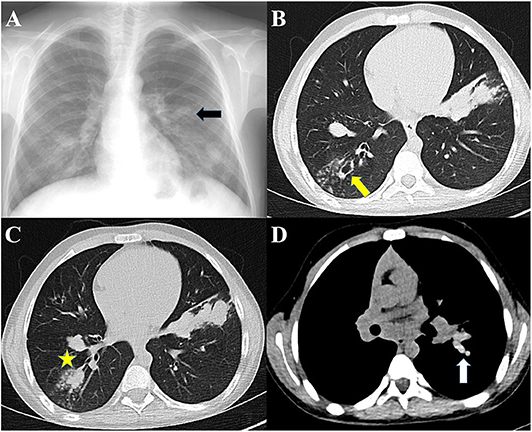
Figure 2. Radiographs of patients with ABPA. (A) Chest radiograph showing finger-in-glove sign, (B) HRCT showing central bronchiectasis, (C) HRCT showing mucus plugging in dilated bronchi, (D) HRCT showing high-attenuation mucus.
Chest HRCT findings in ABPA may correlate with the immunologic severity. Agarwal et al. reported that the presence of HAM plugs was more consistent with serologic severity and recurrent relapses (75, 83). ABPA can be classified radiologically based on clinical and HRCT findings (83) (Table 5).

Table 5. Radiologic classification of ABPA based on clinical and HRCT findings.
Magnetic resonance imaging (MRI) has not been traditionally used for the evaluation of lung parenchyma due to the low proton density of the tissue and high susceptibility of artifacts. Recently with the major technological advancements; MRI has been used in the diagnosis of respiratory pathologies including ABPA. In the diagnosis of ABPA; MRI demonstrated high specificity and positive predictive value; but less sensitivity and negative predictive value compared with the HRCT scan in children (87). The match of HAM on MRI seems to be inverted mucus impaction, which is characterized by a high signal intensity on T1-weighted images and low signal intensity on T2-weighted images (87, 88). However; MRI is not being used as a routine clinical practice of patients with ABPA and there is a need for new clinical investigations to use MRI in the diagnosis of ABPA.
The differences and similarities of clinical features and diagnostic criteria of ABPA in patients with CF or asthma are summarized in Table 6.
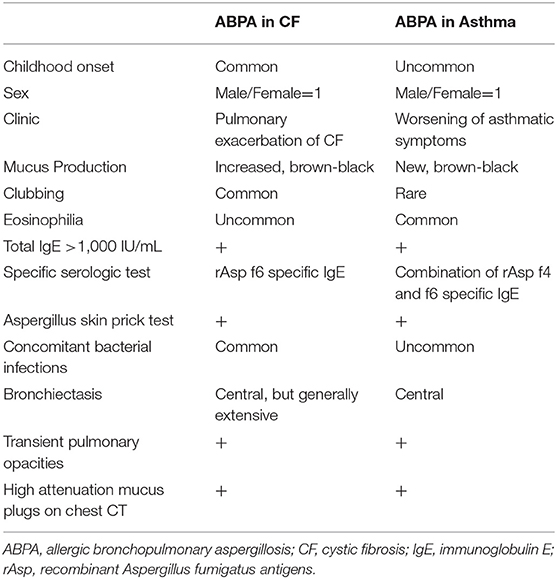
Table 6. Similarities and differences of ABPA in patients with cystic fibrosis or asthma.
Treatment
The principles in the treatment of ABPA include the use of anti-inflammatory agents to suppress the immune hyperreactivity and the use of antifungal agents to attenuate immune hyperresponsiveness by reducing the fungal burden in the airways (89). There are several goals of treatment such as treating the acute stage of ABPA, controlling symptoms of CF, preventing or treating pulmonary exacerbations of ABPA, and reducing progression to end-stage disease. Inadequate and delayed treatment can lead to complications such as pulmonary fibrosis, bronchiectasis, and loss of lung function (90). At the same time, treatment options should also have minimal or no adverse reactions.
Treatment of ABPA in CF is not much different from ABPA in asthma, and involves the use of corticosteroids, antifungal agents, and monoclonal antibodies, mainly prednisolone, itraconazole, and omalizumab, respectively. The doses, side effects and important comments of administration of the drugs used in the treatment of ABPA are summarized in the Table 7.
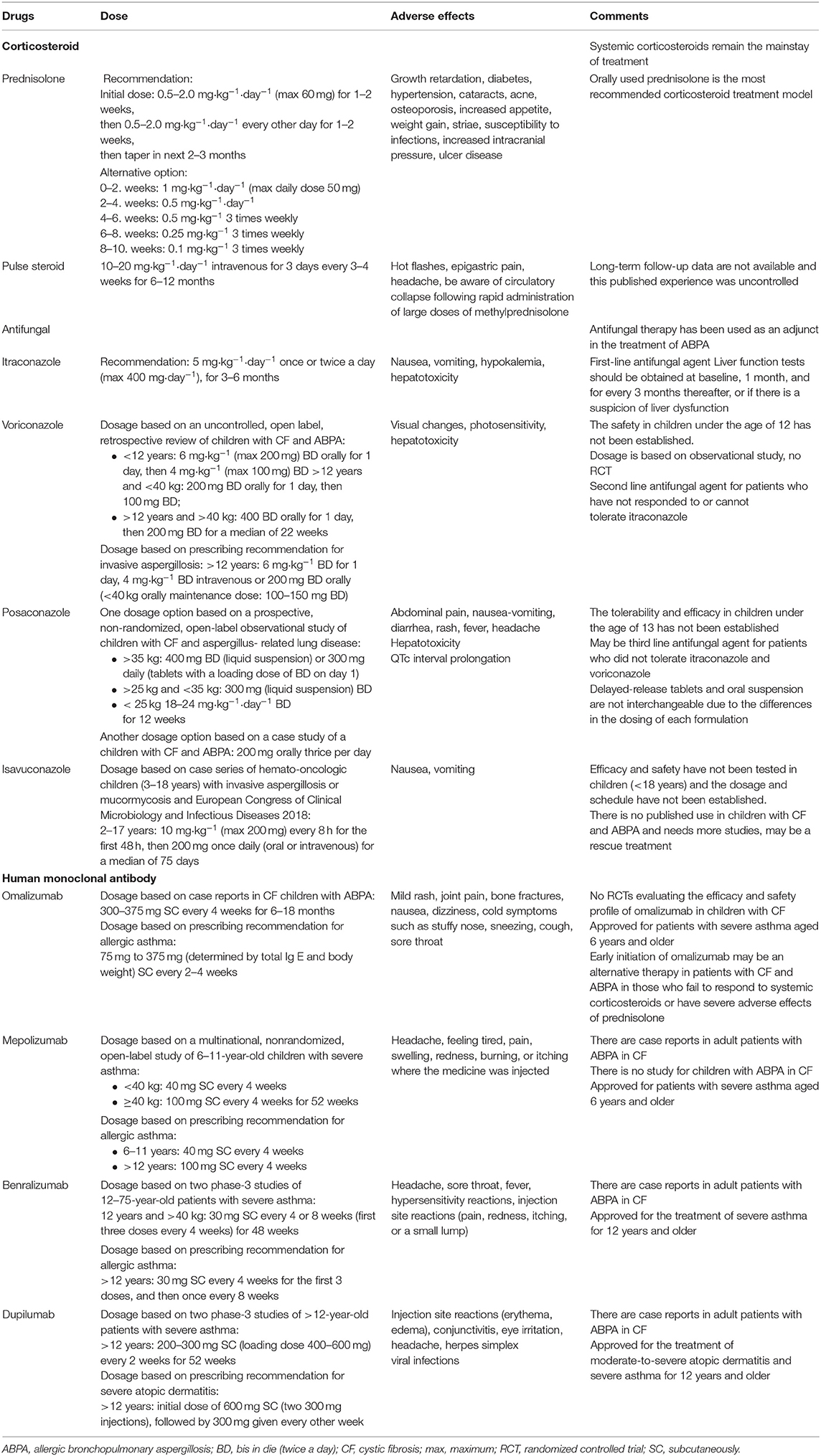
Table 7. Summary of drugs for children with ABPA in CF.
Corticosteroids
Oral Corticosteroids
Systemic corticosteroids are currently the most effective agents in the treatment of ABPA. Use of corticosteroids is based on clinical experience because randomized trials do not exist and are unlikely to be performed due to ethical concerns. Prednisolone is the most widely used corticosteroid, and the dosage and duration of treatment have been investigated in several trials.
The Cystic Fibrosis Foundation Consensus Conference recommends 0.5–2.0 mg·kg−1·day−1 prednisone equivalent (maximum 60 mg·day−1) for 1–2 weeks, then 0.5–2.0 mg·kg−1·day−1 prednisone equivalent every other day for 1–2 weeks, then tapering on the basis of clinical and immunologic improvement. An attempt should be made to begin to taper off corticosteroids in 2–3 months (3).
In patients with asthma with ABPA, Agarwal et al. compared two steroid regimens in a randomized controlled trial (RCT). The medium-dose regimen (0.5 mg·kg−1·day−1 for 1–2 weeks, then on alternate days for 6–8 weeks, taper by 5–10 mg every 2 weeks, and discontinue after 3–5 months) was equally efficacious as a high-dose regimen (0.75 mg·kg−1·day−1 for 6 weeks, 0.5 mg·kg−1·day−1 for 6 weeks, taper by 5 mg every 6 weeks to complete a total duration of 6–12 months) in terms of the improvement in lung function, time to first exacerbation, the number of subjects with exacerbation at 1 year, and the occurrence of corticosteroid-dependent ABPA at 2 years (91).
A third dose regimen of prednisolone different from the other two above (0.5 mg·kg−1·day−1 for 4 weeks, 0.25 mg·kg−1·day−1 for 4 weeks, 0.125 mg·kg−1·day−1 for 4 weeks, then taper by 5 mg every 2 weeks and discontinue after 4 months) was associated with 100% early composite response at 6 weeks (clinical, immunologic, and radiologic improvement) in three studies of patients with asthma with ABPA (92–94). Therefore, the third regimen may offer the right balance between early treatment response and toxicity.
The main goal in these studies was to investigate the treatment protocol with fewer adverse effects while providing similar efficacy. In order to reduce the adverse effects of steroid therapy, the addition of an antifungal agent to the treatment was investigated. Very recently, a study was performed to see the effectiveness of combining short-term prednisone (2 mg·kg−1·day−1 for 3 days, taper every 5 days to 1, 0.5, and 0.25 mg·kg−1·day−1 and discontinued after 18 days in total) and long-term itraconazole (10 mg·kg−1·day−1 for capsules and 5 mg·kg−1·day−1 for suspension for at least 12 months) in treatment of patients with CF and ABPA. It was shown that a combination of itraconazole with short-term prednisone improved long-term pulmonary outcome in patients with ABPA without undesired glucocorticoid adverse effects (95). In a recent survey, although the majority of consultants were found to use both corticosteroids and itraconazole to treat a first diagnosis of ABPA, only one-third was reported to use prednisolone alone (96).
Pulse Steroid Therapy
The role of intravenous (iv) corticosteroids in ABPA, especially “pulse” steroid therapy is still being investigated. Pulse steroid therapy consists of iv methylprednisolone infused daily for three consecutive days every month. Intravenous pulse steroid therapy in ABPA has been used in patients who have adverse effects with daily corticosteroids or do not respond to standard doses of oral steroid therapy, usually associated with prolonged use of steroids. However, there are no controlled trials comparing oral steroids with iv steroids. In several reports, pulse methylprednisolone was successfully used in oral steroid-dependent patients with CF and ABPA (10–20 mg·kg−1·day−1 for three consecutive days every month) (97, 98). In another report of an 11-year-old child with CF who was unresponsive to oral steroids, the use of iv pulse methylprednisolone made an improvement in clinical stabilization and better control of ABPA (20 mg·kg−1 for 3 days followed by 10 mg·kg−1 for 3 days) (99). In most of the studies, iv pulse steroid therapy was well tolerated and patients were able to stop the pulse therapy after 6–12 months with disease control (100).
Inhaled Corticosteroids (ICS)
ICS are known to have significantly fewer adverse effects compared with oral corticosteroids. Several case reports and small case series suggest some benefit of ICS in the management of ABPA without CF, but a study in 32 patients of ABPA found no benefit of using low doses of ICS (400 μg of beclomethasone per day) compared with placebo (101). In another study conducted retrospectively, 21 adult patients with asthma and serologic ABPA who refused conventional treatment received higher doses of ICS (1,600 μg of budesonide per day). The authors found that ICS were ineffective in controlling the immunologic activity because the total IgE levels continued to increase (102). As a result, ICS alone have no role as first-line therapy in ABPA.
Antifungal Therapy
Antifungal azoles are the most frequently combined agents with steroids. It is frequently used in steroid-resistant cases or for the purpose of reducing steroid dose and duration (103). Antifungal agents are thought to decrease the fungal burden in the respiratory tract, hence reducing the antigenic stimulus responsible for the inflammation, improving symptoms, and possibly slowing progression (104). Thus, antifungal drugs can act as steroid-sparing agents.
Itraconazole
The most widely used azole in the management of ABPA is itraconazole. Although azoles are considered to be fungostatic drugs, itraconazole seems to be efficacious in the treatment because fungal burden in ABPA is thought to be lower than in other invasive disorders of fungi (27). Studies on treatment for ABPA in CF are outdated and contain few patients. Recently, two RCTs evaluated the role of itraconazole, but only adult patients with asthma with ABPA were included. In a study involving 55 patients who were using oral corticosteroids regularly, the subjects were randomized to receive itraconazole and placebo for 16 weeks. It was shown that the rate of response to therapy was significantly higher in the itraconazole group than in the placebo group (105). The other study included 29 patients with ABPA randomized to receive itraconazole or placebo for 16 weeks. In this study, itraconazole was found to be effective in normalizing eosinophilic airway inflammation, reducing systemic immune activation, and reducing severe exacerbations (106).
In an RCT, 131 adult patients with asthma with ABPA were randomized to receive either oral itraconazole or prednisolone. All subjects treated with prednisolone showed a composite response after 6 weeks of treatment, whereas 12% of subjects in the itraconazole group did not exhibit a composite response. All subjects who failed to respond to itraconazole were treated with prednisolone, and showed a composite response after 6 weeks of treatment. This study suggests that oral corticosteroids are more effective than itraconazole (100 vs. 88%) in the treatment of acute-stage ABPA (93). The Cystic Fibrosis Foundation Consensus Conference recommends that the initial dose should be 5 mg·kg−1·day−1, which may be given once or twice daily (maximum 200 mg·dose−1). The daily dosage should not exceed 400 mg·day−1 unless low serum itraconazole levels are obtained. The duration of therapy should be short (3–6 months) because of the emerging risk of azole-resistant Aspergillus species. In addition, itraconazole cannot be recommended for initial therapy in patients with CF and ABPA. However, it should be added to therapy if there is a slow or poor response to corticosteroids, for relapse of ABPA, in corticosteroid toxicity, and corticosteroid-dependent ABPA. For patients receiving itraconazole, liver function tests should be obtained before therapy. Routine liver function testing after 1 month and every 3–6 months thereafter should be considered. There are several medications that are known to interact with itraconazole. Therefore, determining serum concentrations of other drugs and/or itraconazole may be required. Itraconazole concentrations should also be determined when there is a lack of clinical response or if there is concern about adequate drug absorption or patient compliance. Besides, itraconazole is associated with gastrointestinal symptoms, congestive heart failure, and rash (3).
Newer Azoles
Few studies have evaluated the newer azoles (voriconazole, posaconazole and isavuconazole) for their efficacy in ABPA. In the largest study, Chishimba et al. retrospectively analyzed the efficacy and safety of voriconazole and posaconazole in 20 adult patients with asthma with ABPA. Overall, clinical improvement with voriconazole or posaconazole therapy was seen in about 70–75%, so both drugs were found to be alternative treatments to itraconazole (107). Monotherapy of voriconazole vs. prednisolone in patients with asthma with ABPA was evaluated in an RCT (92). Fifty subjects were randomized to receive either prednisolone or voriconazole. In this study, voriconazole monotherapy had similar efficacy to prednisolone. According to this study, voriconazole appeared to be as effective as corticosteroids in acute-stage ABPA. Fewer studies have been conducted on voriconazole therapy in patients with CF and ABPA. Glackin et al. reported that serum total IgE level was decreased with voriconazole therapy in patients with CF (108). In the other study, an uncontrolled, retrospective study in 21 patients with ABPA in CF showed an improvement in lung function with voriconazole therapy (109). Unique adverse reactions among patients receiving voriconazole include transient vision changes, visual hallucinations, and photosensitivity. However, voriconazole is a reasonable alternative to itraconazole because it is better tolerated in some patients and is well-absorbed.
In a retrospective study that compared posaconazole with other azoles in the treatment of ABPA in 32 adult patients with CF, it was found that there was a significant reduction in specific IgE to Aspergillus with posaconazole compared with itraconazole and voriconazole (110). Recently, Patel et al. reported a prospective single-center, non-randomized, open-label observational study over a 53-months period evaluating the safety, tolerability, and efficacy of posaconazole in pediatric patients with CF. A total of 23 episodes of Aspergillus-related lung disease were treated. Posaconazole was well-tolerated in children with CF and an improvement in FEV1 and serologic parameters in response to posaconazole was noted in this study (111). It is also associated with gastrointestinal symptoms depending on the formulation, and there are sparse data supporting its use for ABPA.
Isavuconazole is also a new azole that is approved for primary therapy of invasive aspergillosis. However, the use of isavuconazole in ABPA is less well-studied. The first report of the use of isavuconazole for the treatment of ABPA presented an adult patient with asthma who was successfully treated with isavuconazole after unsuccessful treatment with corticosteroids, itraconazole, and voriconazole (112). The patient tolerated isavuconazole well, had marked symptomatic improvement, and demonstrated a normal FEV1/FVC ratio for the first time in 7 years after being diagnosed as having ABPA. Treatment with isavuconazole is generally safe and well-tolerated but there are no studies on isavuconazole treatment of ABPA either in the pediatric population or patients with CF. However, a non-randomized open-label multicenter study on isavuconazole treatment of invasive aspergillosis or invasive mucormycosis in pediatric subjects is underway and planned to be completed in 2 years (ClinicalTrials.gov; NCT03816176).
Nebulized Amphotericin B (NAB)
NAB is an option for maintaining remission in recurrent exacerbations. In one RCT (21 adults with asthma), non-liposomal NAB without concomitant azole therapy was found to be efficacious in maintaining remission in those with recurrent exacerbations. On the other hand, NAB had poor efficacy in inducing a response in patients with acute-stage ABPA or during an exacerbation of ABPA (113).
In a case report of a pediatric patient with end-stage CF lung disease with progressing symptoms, very poor lung function and severe bronchiectasis, treatment with NAB resulted in improvement of cough, dyspnea, hypoxia, 6-min walk test, reduction in oral corticosteroid dosages, and pulmonary function with no adverse events (114).
NAB has the potential to precipitate or worsen bronchospasm, especially the deoxycholate preparation; therefore, the first dose should be administered with caution and short-acting bronchodilator may be administered 15–30 min prior to NAB (113).
Monoclonal Antibodies
Corticosteroids can control the symptoms of most patients and their combination with antifungal drugs increases treatment success. However, some patients become steroid-dependent or experience adverse effects. In an effort to treat refractory patients or to reduce adverse effects, many physicians have tried monoclonal antibody treatment. As a monoclonal antibody, although omalizumab is the most preferred, mepolizumab and benralizumab are the most investigated.
Omalizumab
Omalizumab is a humanized monoclonal antibody against IgE recommended for the treatment of uncontrolled allergic asthma and chronic spontaneous urticaria. It is considered for use in the treatment of other allergic disorders such as ABPA because its mechanism of action is via IgE antagonism. Although omalizumab seems to facilitate ABPA control in asthma, evidence for use in patients with CF is currently limited to data from case reports. In addition, an RCT evaluating the safety and efficacy of omalizumab for the treatment of ABPA in patients with CF aged 12 years and older was designed, but this study was terminated prematurely due to adverse events (115). Another RCT was reported in 2015 which evaluated the clinical and immunologic effects of omalizumab in asthmatic ABPA patients. Thirteen patients with chronic ABPA were randomized to a 4-months treatment with omalizumab or a placebo followed by a 3-months washout period. The ABPA exacerbations were significantly less frequent during the active treatment phase compared with the placebo period (116). Van der Ent et al. reported the first case of a single dose of omalizumab treatment in a 12- year-old girl with CF and ABPA who showed a rapid and good improvement of clinical signs and lung functions (117). In a case series of Emiralioglu et al. six patients with CF and ABPA who had received omalizumab were reported. Omalizumab (300 mg dosage) was administered subcutaneously every 4 weeks to the patients who were treated previously with oral prednisolone and itraconazole. Decreased IgE levels, improvement in respiratory symptoms, and a steroid-sparing effect was shown with omalizumab treatment (118). A retrospective multicenter observational French study retrieved 32 patients with ABPA and CF (11 children and 21 adults) who had received omalizumab for more than 3 months. Among them, 14 patients were able to discontinue steroid treatment or reduce their daily dose during follow-up (119). Very recently, a retrospective study of 27 adult CF patients receiving omalizumab for asthma or ABPA was conducted by Koutsokera et al. to evaluate the efficacy and safety of treatment. Omalizumab was found to be effective in the improvement of respiratory functions in adult CF patients with difficult-to-control asthma or ABPA with no significant adverse effects during the study period (120).
Furthermore, a standard dose has not been established for omalizumab in patients with CF and ABPA. Different studies used various doses (ranging from 225 mg to 750 mg) and frequency of treatment (ranging from once per week to once monthly) according to the weight and serum IgE level. However, the most commonly used regimen was 375 mg every 2 weeks (121). The duration of treatment is also controversial. As an example, Wong et al. have reported 2 patients with CF and steroid-dependent ABPA who were successfully able to be weaned off steroid therapy with the treatment of omalizumab monthly for 2 years (122).
In contrast to the above case reports, two studies on patients with CF with ABPA found no benefit with omalizumab treatment. In one of them, Brinkman et al., reported a 15-year-old patient who could not be weaned from steroid therapy for over 12 months with omalizumab treatment (123). Ashkenazi et al. also presented nine patients with CF and ABPA who were treated with 300–375 mg omalizumab every month but did not respond to treatment (124). However, in both studies, they administered the drug every month with a lower dose compared with other cases.
Omalizumab may be a promising treatment option also for CF patients with chronic bacterial infections since use of corticosteroids is of concern due to compromised immunity for these patients. In a recent retrospective cross-sectional study, no significant adverse events or worsening of infection due to omalizumab treatment were observed in patients with ABPA and chronic bacterial airway infection. Consequently, treatment with omalizumab was found to be effective and safe in patients with ABPA, regardless of concurrent chronic respiratory tract infections since it does not exhibit immonusuppressive effects (125).
Mepolizumab
Mepolizumab is an anti-IL-5 monoclonal antibody used for severe refractory eosinophilic asthma. Altman et al. were first to demonstrate mepolizumab as an additional and effective treatment option for severe ABPA resistant to corticosteroids, antifungal therapy, and omalizumab in a 58-year-old asthmatic woman (126). In this case, clinical improvement was achieved with the addition of 100 mg mepolizumab every 4 weeks to high-dose omalizumab treatment.
In another case, mepolizumab was shown to be effective as a monotherapy in a 64-year-old woman treated for severe bronchial asthma with ABPA exacerbation (127). In this case, after a successful 3-years treatment period with systemic corticosteroids and itraconazole, deterioration occurred. With the addition of 100 mg mepolizumab every 4 weeks, dramatic improvements were observed in symptoms, lung function, peripheral eosinophil counts, and chest imaging.
Benralizumab
Benralizumab is a monoclonal antibody directed against the alpha chain of the IL-5 receptor. Recently, two adult cases of ABPA with asthma were reported to switch to benralizumab after treatment with mepolizumab (128). Benralizumab is thought to clear bronchial mucus plugs, prevent irreversible airway damage, and improve the prognosis of patients with ABPA.
Dupilumab
Dupilumab is a humanized monoclonal antibody. It is a dual inhibitor of IL-4 and IL-13 pathways. In a case series, the clinical course of three adult subjects with asthma and ABPA treated with dupilumab over 6 months was presented (129). In this study, dupilumab was found to facilitate disease control in ABPA, with reduced symptoms and oral corticosteroid use.
Treatment Practices
In acute ABPA, systemic corticosteroids are the first choice of treatment. Although any of corticosteroid regimens as detailed above can be used, high-dose steroids should not be preferred as the first choice in the management of ABPA due to the more frequent adverse effects. The steroid dose should be adjusted according to the clinical and immunologic characteristics of disease. Antifungal agents should be added to therapy if there is a slow or poor response to corticosteroids, for relapse of ABPA, in corticosteroid toxicity, and corticosteroid-dependent ABPA. Azole therapy is usually begun with itraconazole; newer azoles are reserved for those who fail therapy or experience adverse reactions with itraconazole or fail to achieve optimal serum levels of itraconazole, despite receiving the maximum dose (104). Nonetheless, a small number of patients may require chronic corticosteroid therapy (3).
After starting treatment for acute ABPA, monitoring is performed with clinical evaluation, serum total IgE levels, spirometry, and chest radiography. It is not helpful to measure Aspergillus-specific IgE and IgG during treatment because their levels are not correlated with the reduction in the serum total IgE or clinical or radiologic improvement. Serum total IgE concentrations should be measured every 6–8 weeks, especially in the first year (130). The clinical effectiveness of therapy is evaluated through serum total IgE levels. The goal of therapy is not to achieve normal IgE levels but to decrease its levels by 35–50% at 8 weeks, which leads to clinical and radiographic improvement. During the treatment, serum total IgE levels are measured to determine the new baseline IgE concentrations (5). The lowest value achieved after treatment is taken as the new baseline. An increasing level (>100% of the new baseline) of total IgE along with worsening respiratory symptoms and the appearance of consistent radiologic findings suggest an exacerbation of ABPA (131). Twenty to 35% of relapses are asymptomatic and are detected radiographically and serologically. The treatment of the first exacerbation is similar to the treatment of acute disease. Greater than or equal to two exacerbations within 6 months of stopping therapy or worsening of clinical and/or radiologic condition, along with immunologic worsening (rise in IgE levels) on tapering oral steroids/azoles is steroid-dependent ABPA. Pulse steroids may be considered in such patients. If they are currently taking itraconazole, newer azoles may be considered. Omalizumab has shown promise in such cases but its use in patients with ABPA and CF requires more definitive clinical trials. Furthermore, it is also very important to identify and exclude any potential environmental exposure source of A. fumigatus because it can initiate exacerbations.
Remission may be considered if the patient has remained asymptomatic with stable IgE levels (persisting at/below baseline or increase by <50%) for at least 6 months without the requirement of corticosteroid or antifungal therapy. In the remission period, monitoring may be performed every 3 months for a year and every 6 months thereafter with a clinical examination and serum total IgE levels. Chest radiography may be obtained if clinically indicated. Spirometry is performed in routine follow-ups and in response to changes in symptoms. Antifungal therapy is not used to prevent exacerbations given the potential toxicity and lack of proven benefit.
Another considerable point of management is that chronic respiratory tract infections are almost inevitable in ABPA patients with CF. These patients are especially vulnerable to Pseudomonas aeruginosa or nontuberculous mycobacteria because of the combined effects of structural deformities in the airways and compromised immunity caused by systemic and local administration of corticosteroids (132). In that case, monoclonal antibodies such as omalizumab may be a good choice since it can prevent the use or reduce the doses of systemic corticosteroids (125).
In summary, clinical improvement is generally achieved with proper diagnosis, follow-up, and treatment. At the same time, there is a considerable variation in treatment practices of patients with CF and ABPA, hence there is a pressing need for new guidance in both treatment and its duration.
Author Contributions
BS, DA, BO, NE, EY, and UÖ have made contributions to the design, editing, and writing of this manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sisodia J, Bajaj T. Allergic Bronchopulmonary Aspergillosis (ABPA). Treasure Island, FL: StatPearls (2020).
2. Bains SN, Judson MA. Allergic bronchopulmonary aspergillosis. Clin Chest Med. (2012) 33:265–81. doi: 10.1016/j.ccm.2012.02.003
3. Stevens DA, Moss RB, Kurup VP, Knutsen AP, Greenberger P, Judson MA, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis–state of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis. (2003) 37 (Suppl. 3):S225–64. doi: 10.1086/376525
4. Jat KR, Vaidya PC, Mathew JL, Jondhale S, Singh M. Childhood allergic bronchopulmonary aspergillosis. Lung India. (2018) 35:499–507. doi: 10.4103/lungindia.lungindia_216_18
5. Agarwal R. Allergic bronchopulmonary aspergillosis. Chest. (2009) 135:805–26. doi: 10.1378/chest.08-2586
6. Maturu VN, Agarwal R. Prevalence of Aspergillus sensitization and allergic bronchopulmonary aspergillosis in cystic fibrosis: systematic review and meta-analysis. Clin Exp Allergy. (2015) 45:1765–78. doi: 10.1111/cea.12595
7. Janahi IA, Rehman A, Al-Naimi AR. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ann Thorac Med. (2017) 12:74–82. doi: 10.4103/atm.ATM_231_16
8. Agarwal R, Sehgal IS, Dhooria S, Muthu V, Prasad KT, Bal A, et al. Allergic bronchopulmonary aspergillosis. Indian J Med Res. (2020) 151:529–49. doi: 10.4103/ijmr.IJMR_1187_19
9. Knutsen AP. Lymphocytes in allergic bronchopulmonary aspergillosis. Front Biosci. (2003) 8:d589–602. doi: 10.2741/994
10. Knutsen AP. Immunopathology and immunogenetics of allergic bronchopulmonary aspergillosis. J Allergy (Cairo). (2011) 2011:785983. doi: 10.1155/2011/785983
11. Greenberger PA. Allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol. (2002) 110:685–92. doi: 10.1067/mai.2002.130179
12. Chotirmall SH, McElvaney NG. Fungi in the cystic fibrosis lung: bystanders or pathogens? Int J Biochem Cell Biol. (2014) 52:161–73. doi: 10.1016/j.biocel.2014.03.001
13. Chotirmall SH, Al-Alawi M, Mirkovic B, Lavelle G, Logan PM, Greene CM, et al. Aspergillus-associated airway disease, inflammation, and the innate immune response. Biomed Res Int. (2013) 2013:723129. doi: 10.1155/2013/723129
14. Mueller C, Braag SA, Keeler A, Hodges C, Drumm M, Flotte TR. Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am J Respir Cell Mol Biol. (2011) 44:922–9. doi: 10.1165/rcmb.2010-0224OC
15. Skov M, Poulsen LK, Koch C. Increased antigen-specific Th-2 response in allergic bronchopulmonary aspergillosis (ABPA) in patients with cystic fibrosis. Pediatr Pulmonol. (1999) 27:74–9. doi: 10.1002/(sici)1099-0496(199902)27:2<74::aid-ppul2>3.0.co;2-l
16. Zeaske R, Bruns WT, Fink JN, Greenberger PA, Colby H, Liotta JL, et al. Immune responses to Aspergillus in cystic fibrosis. J Allergy Clin Immunol. (1988) 82:73–7. doi: 10.1016/0091-6749(88)90054-1
17. Hershey GK, Friedrich MF, Esswein LA, Thomas ML, Chatila TA. The association of atopy with a gain-of-function mutation in the alpha subunit of the interleukin-4 receptor. N Engl J Med. (1997) 337:1720–5. doi: 10.1056/NEJM199712113372403
18. Chauhan B, Santiago L, Hutcheson PS, Schwartz HJ, Spitznagel E, Castro M, et al. Evidence for the involvement of two different MHC class II regions in susceptibility or protection in allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol. (2000) 106:723–9. doi: 10.1067/mai.2000.109913
19. Chauhan B, Knutsen A, Hutcheson PS, Slavin RG, Bellone CJ. T cell subsets, epitope mapping, and HLA-restriction in patients with allergic bronchopulmonary aspergillosis. J Clin Invest. (1996) 97:2324–31. doi: 10.1172/JCI118675
20. Chauhan B, Santiago L, Kirschmann DA, Hauptfeld V, Knutsen AP, Hutcheson PS, et al. The association of HLA-DR alleles and T cell activation with allergic bronchopulmonary aspergillosis. J Immunol. (1997) 159:4072–6.
21. Mahdavinia M, Grammer LC. Management of allergic bronchopulmonary aspergillosis: a review and update. Ther Adv Respir Dis. (2012) 6:173–87. doi: 10.1177/1753465812443094
22. de Almeida MB, Bussamra MH, Rodrigues JC. Allergic bronchopulmonary aspergillosis in paediatric cystic fibrosis patients. Paediatr Respir Rev. (2006) 7:67–72. doi: 10.1016/j.prrv.2005.09.003
23. Kradin RL, Mark EJ. The pathology of pulmonary disorders due to Aspergillus spp. Arch Pathol Lab Med. (2008) 132:606–14. doi: 10.1043/1543-2165(2008)132[606:TPOPDD]2.0.CO;2
24. Kumar R. Mild, moderate, and severe forms of allergic bronchopulmonary aspergillosis: a clinical and serologic evaluation. Chest. (2003) 124:890–2. doi: 10.1378/chest.124.3.890
25. Prasad R, Garg R, Sanjay, Shukla AD. Allergic bronchopulmonary aspergillosis: a review of 42 patients from a tertiary care center in India. Lung India. (2009) 26:38–40. doi: 10.4103/0970-2113.48895
26. Zhang M, Gao J. Clinical analysis of 77 patients with allergic bronchopulmonary aspergillosis in peking union medical college hospital. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. (2017) 39:352–7. doi: 10.3881/j.issn.1000-503X.2017.03.009
27. Agarwal R, Sehgal IS, Dhooria S, Aggarwal AN. Developments in the diagnosis and treatment of allergic bronchopulmonary aspergillosis. Expert Rev Respir Med. (2016) 10:1317–34. doi: 10.1080/17476348.2016.1249853
28. Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A, et al. Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol. (2012) 129:280–91; quiz 292–283. doi: 10.1016/j.jaci.2011.12.970
29. Agarwal R, Chakrabarti A, Shah A, Gupta D, Meis JF, Guleria R, et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy. (2013) 43:850–73. doi: 10.1111/cea.12141
30. Geller DE, Kaplowitz H, Light MJ, Colin AA. Allergic bronchopulmonary aspergillosis in cystic fibrosis: reported prevalence, regional distribution, and patient characteristics. Scientific advisory group, investigators, and coordinators of the epidemiologic study of cystic fibrosis. Chest. (1999) 116:639–46. doi: 10.1378/chest.116.3.639
31. Thia LP, Balfour Lynn IM. Diagnosing allergic bronchopulmonary aspergillosis in children with cystic fibrosis. Paediatr Respir Rev. (2009) 10:37–42. doi: 10.1016/j.prrv.2009.01.001
32. Maleki M, Mortezaee V, Hassanzad M, Mahdaviani SA, Poorabdollah M, Mehrian P, et al. Prevalence of allergic bronchopulmonary aspergillosis in cystic fibrosis patients using two different diagnostic criteria. Eur Ann Allergy Clin Immunol. (2020) 52:104–11. doi: 10.23822/EurAnnACI.1764-1489.121
33. Mari A, Schneider P, Wally V, Breitenbach M, Simon-Nobbe B. Sensitization to fungi: epidemiology, comparative skin tests, and IgE reactivity of fungal extracts. Clin Exp Allergy. (2003) 33:1429–8. doi: 10.1046/j.1365-2222.2003.01783.x
34. Skov M, Koch C, Reimert CM, Poulsen LK. Diagnosis of allergic bronchopulmonary aspergillosis (ABPA) in cystic fibrosis. Allergy. (2000) 55:50–58. doi: 10.1034/j.1398-9995.2000.00342.x
35. Mastella G, Rainisio M, Harms HK, Hodson ME, Koch C, Navarro J, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. A european epidemiological study. epidemiologic registry of cystic fibrosis. Eur Respir J. (2000) 16:464–71. doi: 10.1034/j.1399-3003.2000.016003464.x
36. Hoehne JH, Reed CE, Dickie HA. Allergic bronchopulmonary aspergillosis is not rare. With a note on preparation of antigen for immunologic tests. Chest. (1973) 63:177–81. doi: 10.1378/chest.63.2.177
37. Rosenberg M, Patterson R, Roberts M, Wang J. The assessment of immunologic and clinical changes occurring during corticosteroid therapy for allergic bronchopulmonary aspergillosis. Am J Med. (1978) 64:599–606. doi: 10.1016/0002-9343(78)90579-x
38. Hutcheson PS, Knutsen AP, Rejent AJ, Slavin RG. A 12-year longitudinal study of Aspergillus sensitivity in patients with cystic fibrosis. Chest. (1996) 110:363–6. doi: 10.1378/chest.110.2.363
39. Carsin A, Romain T, Ranque S, Reynaud-Gaubert M, Dubus JC, Mege JL, et al. Aspergillus fumigatus in cystic fibrosis: An update on immune interactions and molecular diagnostics in allergic bronchopulmonary aspergillosis. Allergy. (2017) 72:1632–42. doi: 10.1111/all.13204
40. Bakare N, Rickerts V, Bargon J, Just-Nubling G. Prevalence of Aspergillus fumigatus and other fungal species in the sputum of adult patients with cystic fibrosis. Mycoses. (2003) 46:19–23. doi: 10.1046/j.1439-0507.2003.00830.x
41. Becker JW, Burke W, McDonald G, Greenberger PA, Henderson WR, Aitken ML. Prevalence of allergic bronchopulmonary aspergillosis and atopy in adult patients with cystic fibrosis. Chest. (1996) 109:1536–40. doi: 10.1378/chest.109.6.1536
42. Agarwal R, Gupta D, Aggarwal AN, Behera D, Jindal SK. Allergic bronchopulmonary aspergillosis: lessons from 126 patients attending a chest clinic in north India. Chest. (2006) 130:442–8. doi: 10.1378/chest.130.2.442
43. Nelson LA, Callerame ML, Schwartz RH. Aspergillosis and atopy in cystic fibrosis. Am Rev Respir Dis. (1979) 120:863–73. doi: 10.1164/arrd.1979.120.4.863
44. Leung PS, Gershwin ME, Coppel R, Halpern G, Novey H, Castles JJ. Localization, molecular weight and immunoglobulin subclass response to Aspergillus fumigatus allergens in acute bronchopulmonary aspergillosis. Int Arch Allergy Appl Immunol. (1988) 85:416–21. doi: 10.1159/000234544
45. Skov M, Pressler T, Jensen HE, Hoiby N, Koch C. Specific IgG subclass antibody pattern to Aspergillus fumigatus in patients with cystic fibrosis with allergic bronchopulmonary aspergillosis (ABPA). Thorax. (1999) 54:44–50. doi: 10.1136/thx.54.1.44
46. Walter JE, Jones RD. Serologic tests in diagnosis of aspergillosis. Dis Chest. (1968) 53:729–35. doi: 10.1378/chest.53.6.729
47. Agarwal R, Dua D, Choudhary H, Aggarwal AN, Sehgal IS, Dhooria S, et al. Role of Aspergillus fumigatus-specific IgG in diagnosis and monitoring treatment response in allergic bronchopulmonary aspergillosis. Mycoses. (2017) 60:33–9. doi: 10.1111/myc.12541
48. Baxter CG, Denning DW, Jones AM, Todd A, Moore CB, Richardson MD. Performance of two Aspergillus IgG EIA assays compared with the precipitin test in chronic and allergic aspergillosis. Clin Microbiol Infect. (2013) 19:E197–204. doi: 10.1111/1469-0691.12133
49. Barton RC, Hobson RP, Denton M, Peckham D, Brownlee K, Conway S, et al. Serologic diagnosis of allergic bronchopulmonary aspergillosis in patients with cystic fibrosis through the detection of immunoglobulin G to Aspergillus fumigatus. Diagn Microbiol Infect Dis. (2008) 62:287–91. doi: 10.1016/j.diagmicrobio.2008.06.018
50. Harada K, Oguma T, Saito A, Fukutomi Y, Tanaka J, Tomomatsu K, et al. Concordance between Aspergillus-specific precipitating antibody and IgG in allergic bronchopulmonary aspergillosis. Allergol Int. (2018) 67S:S12–S17. doi: 10.1016/j.alit.2018.04.009
51. el-Dahr JM, Fink R, Selden R, Arruda LK, Platts-Mills TA, Heymann PW. Development of immune responses to Aspergillus at an early age in children with cystic fibrosis. Am J Respir Crit Care Med. (1994) 150:1513–8. doi: 10.1164/ajrccm.150.6.7952609
52. Patterson R, Fink JN, Pruzansky JJ, Reed C, Roberts M, Slavin R, et al. Serum immunoglobulin levels in pulmonary allergic aspergillosis and certain other lung diseases, with special reference to immunoglobulin E. Am J Med. (1973) 54:16–22. doi: 10.1016/0002-9343(73)90078-8
53. Wang JL, Patterson R, Rosenberg M, Roberts M, Cooper BJ. Serum IgE and IgG antibody activity against Aspergillus fumigatus as a diagnostic aid in allergic bronchopulmonary aspergillosis. Am Rev Respir Dis. (1978) 117:917–27. doi: 10.1164/arrd.1978.117.5.917
54. Laufer P, Fink JN, Bruns WT, Unger GF, Kalbfleisch JH, Greenberger PA, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis. J Allergy Clin Immunol. (1984) 73:44–8. doi: 10.1016/0091-6749(84)90482-2
55. Patterson K, Strek ME. Allergic bronchopulmonary aspergillosis. Proc Am Thorac Soc. (2010) 7:237–44. doi: 10.1513/pats.200908-086AL
56. Keown K, Abbott S, Kuzeljevic B, Rayment JH, Chilvers MA, Yang CL. An investigation into biomarkers for the diagnosis of ABPA and aspergillus disease in cystic fibrosis. Pediatr Pulmonol. (2019) 54:1787–93. doi: 10.1002/ppul.24465
57. Muthu V, Sehgal IS, Dhooria S, Aggarwal AN, Agarwal R. Utility of recombinant Aspergillus fumigatus antigens in the diagnosis of allergic bronchopulmonary aspergillosis: a systematic review and diagnostic test accuracy meta-analysis. Clin Exp Allergy. (2018) 48:1107–36. doi: 10.1111/cea.13216
58. Fukutomi Y, Tanimoto H, Yasueda H, Taniguchi M. Serological diagnosis of allergic bronchopulmonary mycosis: progress and challenges. Allergol Int. (2016) 65:30–6. doi: 10.1016/j.alit.2015.08.004
59. Latzin P, Hartl D, Regamey N, Frey U, Schoeni MH, Casaulta C. Comparison of serum markers for allergic bronchopulmonary aspergillosis in cystic fibrosis. Eur Respir J. (2008) 31:36–42. doi: 10.1183/09031936.00078107
60. Muthu V, Singh P, Choudhary H, Sehgal IS, Dhooria S, Prasad KT, et al. Diagnostic cutoffs and clinical utility of recombinant aspergillus fumigatus antigens in the diagnosis of allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol Pract. (2020) 8:579–87. doi: 10.1016/j.jaip.2019.08.041
61. Hartl D, Latzin P, Zissel G, Krane M, Krauss-Etschmann S, Griese M. Chemokines indicate allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Am J Respir Crit Care Med. (2006) 173:1370–6. doi: 10.1164/rccm.200508-1271OC
62. Ringer S, Hipler UC, Elsner P, Zintl F, Mainz J. Potential role of the cellular allergen stimulation test (CAST) in diagnosis of allergic bronchopulmonary aspergillosis (ABPA) in cystic fibrosis. Pediatr Pulmonol. (2007) 42:314–8. doi: 10.1002/ppul.20572
63. Hemmings O, Kwok M, McKendry R, Santos AF. Basophil activation test: old and new applications in allergy. Curr Allergy Asthma Rep. (2018) 18:77. doi: 10.1007/s11882-018-0831-5
64. Gernez Y, Waters J, Mirkovic B, Lavelle GM, Dunn CE, Davies ZA, et al. Blood basophil activation is a reliable biomarker of allergic bronchopulmonary aspergillosis in cystic fibrosis. Eur Respir J. (2016) 47:177–85. doi: 10.1183/13993003.01068-2015
65. Katelari A, Tzanoudaki M, Noni M, Kanariou M, Theodoridou M, Kanavakis E, et al. The role of basophil activation test in allergic bronchopulmonary aspergillosis and Aspergillus fumigatus sensitization in cystic fibrosis patients. J Cyst Fibros. (2016) 15:587–96. doi: 10.1016/j.jcf.2016.02.004
66. Gernez Y, Dunn CE, Everson C, Mitsunaga E, Gudiputi L, Krasinska K, et al. Blood basophils from cystic fibrosis patients with allergic bronchopulmonary aspergillosis are primed and hyper-responsive to stimulation by aspergillus allergens. J Cyst Fibros. (2012) 11:502–10. doi: 10.1016/j.jcf.2012.04.008
67. Mirkovic B, Lavelle GM, Azim AA, Helma K, Gargoum FS, Molloy K, et al. The basophil surface marker CD203c identifies Aspergillus species sensitization in patients with cystic fibrosis. J Allergy Clin Immunol. (2016) 137:436–43 e439. doi: 10.1016/j.jaci.2015.07.045
68. Prasad KT, Muthu V, Sehgal IS, Dhooria S, Singh P, Sachdeva MUS, et al. The utility of the basophil activation test in differentiating asthmatic subjects with and without allergic bronchopulmonary aspergillosis. Mycoses. (2020) 63:588–95. doi: 10.1111/myc.13081
69. Rosenberg M, Patterson R, Mintzer R, Cooper BJ, Roberts M, Harris KE. Clinical and immunologic criteria for the diagnosis of allergic bronchopulmonary aspergillosis. Ann Intern Med. (1977) 86:405–14. doi: 10.7326/0003-4819-86-4-405
70. Vlahakis NE, Aksamit TR. Diagnosis and treatment of allergic bronchopulmonary aspergillosis. Mayo Clin Proc. (2001) 76:930–8. doi: 10.4065/76.9.930
71. Chakrabarti A, Sethi S, Raman DS, Behera D. Eight-year study of allergic bronchopulmonary aspergillosis in an Indian teaching hospital. Mycoses. (2002) 45:295–9. doi: 10.1046/j.1439-0507.2002.00738.x
72. Hogan C, Denning DW. Allergic bronchopulmonary aspergillosis and related allergic syndromes. Semin Respir Crit Care Med. (2011) 32:682–92. doi: 10.1055/s-0031-1295716
73. Denning DW, Park S, Lass-Florl C, Fraczek MG, Kirwan M, Gore R, et al. High-frequency triazole resistance found In nonculturable Aspergillus fumigatus from lungs of patients with chronic fungal disease. Clin Infect Dis. (2011) 52:1123–9. doi: 10.1093/cid/cir179
74. Baxter CG, Dunn G, Jones AM, Webb K, Gore R, Richardson MD, et al. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol. (2013) 132:560–6 e510. doi: 10.1016/j.jaci.2013.04.007
75. Agarwal R, Gupta D, Aggarwal AN, Saxena AK, Chakrabarti A, Jindal SK. Clinical significance of hyperattenuating mucoid impaction in allergic bronchopulmonary aspergillosis: an analysis of 155 patients. Chest. (2007) 132:1183–90. doi: 10.1378/chest.07-0808
76. Nichols D, Dopico GA, Braun S, Imbeau S, Peters ME, Rankin J. Acute and chronic pulmonary function changes in allergic bronchopulmonary aspergillosis. Am J Med. (1979) 67:631–7. doi: 10.1016/0002-9343(79)90246-8
77. Riscili BP, Wood KL. Noninvasive pulmonary Aspergillus infections. Clin Chest Med. (2009) 30:315–35, vii. doi: 10.1016/j.ccm.2009.02.008
78. Lynch DA. Imaging of asthma and allergic bronchopulmonary mycosis. Radiol Clin North Am. (1998) 36:129–42. doi: 10.1016/s0033-8389(05)70010-5
79. Ward S, Heyneman L, Lee MJ, Leung AN, Hansell DM, Muller NL. Accuracy of CT in the diagnosis of allergic bronchopulmonary aspergillosis in asthmatic patients. AJR Am J Roentgenol. (1999) 173:937–42. doi: 10.2214/ajr.173.4.10511153
80. Neeld DA, Goodman LR, Gurney JW, Greenberger PA, Fink JN. Computerized tomography in the evaluation of allergic bronchopulmonary aspergillosis. Am Rev Respir Dis. (1990) 142:1200–5. doi: 10.1164/ajrccm/142.5.1200
81. Logan PM, Muller NL. High-attenuation mucous plugging in allergic bronchopulmonary aspergillosis. Can Assoc Radiol J. (1996) 47:374–7.
82. Goyal R, White CS, Templeton PA, Britt EJ, Rubin LJ. High attenuation mucous plugs in allergic bronchopulmonary aspergillosis: CT appearance. J Comput Assist Tomogr. (1992) 16:649–650.
83. Agarwal R, Khan A, Gupta D, Aggarwal AN, Saxena AK, Chakrabarti A. An alternate method of classifying allergic bronchopulmonary aspergillosis based on high-attenuation mucus. PLoS ONE. (2010) 5:e15346. doi: 10.1371/journal.pone.0015346
84. Ricketti AJ, Greenberger PA, Glassroth J. Spontaneous pneumothorax in allergic bronchopulmonary aspergillosis. Arch Intern Med. (1984) 144:151–2.
85. Hansell DM, Strickland B. High-resolution computed tomography in pulmonary cystic fibrosis. Br J Radiol. (1989) 62:1–5. doi: 10.1259/0007-1285-62-733-1
86. Currie DC, Goldman JM, Cole PJ, Strickland B. Comparison of narrow section computed tomography and plain chest radiography in chronic allergic bronchopulmonary aspergillosis. Clin Radiol. (1987) 38:593–6. doi: 10.1016/s0009-9260(87)80333-1
87. Sodhi KS, Gupta P, Shrivastav A, Saxena AK, Mathew JL, Singh M, et al. Evaluation of 3 T lung magnetic resonance imaging in children with allergic bronchopulmonary aspergillosis: pilot study. Eur J Radiol. (2019) 111:88–92. doi: 10.1016/j.ejrad.2018.12.021
88. Dournes G, Berger P, Refait J, Macey J, Bui S, Delhaes L, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis: MR imaging of airway mucus contrasts as a tool for diagnosis. Radiology. (2017) 285:261–9. doi: 10.1148/radiol.2017162350
89. Patterson KC, Strek ME. Diagnosis and treatment of pulmonary aspergillosis syndromes. Chest. (2014) 146:1358–68. doi: 10.1378/chest.14-0917
90. Patel AR, Patel AR, Singh S, Singh S, Khawaja I. Treating allergic bronchopulmonary aspergillosis: a review. Cureus. (2019) 11:e4538. doi: 10.7759/cureus.4538
91. Agarwal R, Aggarwal AN, Dhooria S, Singh Sehgal I, Garg M, Saikia B, et al. A randomised trial of glucocorticoids in acute-stage allergic bronchopulmonary aspergillosis complicating asthma. Eur Respir J. (2016) 47:490–8. doi: 10.1183/13993003.01475-2015
92. Agarwal R, Dhooria S, Sehgal IS, Aggarwal AN, Garg M, Saikia B, Chakrabarti A. A randomised trial of voriconazole and prednisolone monotherapy in acute-stage allergic bronchopulmonary aspergillosis complicating asthma. Eur Respir J. (2018) 52:1801159. doi: 10.1183/13993003.01159-2018
93. Agarwal R, Dhooria S, Singh Sehgal I, Aggarwal AN, Garg M, Saikia B, et al. A randomized trial of itraconazole vs prednisolone in acute-stage allergic bronchopulmonary aspergillosis complicating asthma. Chest. (2018) 153:656–64. doi: 10.1016/j.chest.2018.01.005
94. Dodamani MH, Muthu V, Thakur R, Pal A, Sehgal IS, Dhooria S, et al. A randomised trial of vitamin D in acute-stage allergic bronchopulmonary aspergillosis complicating asthma. Mycoses. (2019) 62:320–7. doi: 10.1111/myc.12879
95. Gothe F, Schmautz A, Häusler K, Tran NB, Kappler M, Griese M. Treating Allergic bronchopulmonary aspergillosis with short-term prednisone and itraconazole in cystic fibrosis. J Allergy Clin Immunol Pract. (2020) 8:2608–14.e3. doi: 10.1016/j.jaip.2020.02.031
96. Boyle M, Moore JE, Whitehouse JL, Bilton D, Downey DG. The diagnosis and management of respiratory tract fungal infection in cystic fibrosis: a UK survey of current practice. Med Mycol. (2019) 57:155–60. doi: 10.1093/mmy/myy014
97. Cohen-Cymberknoh M, Blau H, Shoseyov D, Mei-Zahav M, Efrati O, Armoni S, et al. Intravenous monthly pulse methylprednisolone treatment for ABPA in patients with cystic fibrosis. J Cyst Fibros. (2009) 8:253–7. doi: 10.1016/j.jcf.2009.04.008
98. Thomson JM, Wesley A, Byrnes CA, Nixon GM. Pulse intravenous methylprednisolone for resistant allergic bronchopulmonary aspergillosis in cystic fibrosis. Pediatr Pulmonol. (2006) 41:164–70. doi: 10.1002/ppul.20333
99. Thomas MF. Life-threatening allergic bronchopulomnary aspergillosis treated with methylprednisolone and anti-IgE monoclonal antibody. J R Soc Med. (2009) 102 (Suppl. 1):49–53. doi: 10.1258/jrsm.2009.s19011
100. Singh Sehgal I, Agarwal R. Pulse methylprednisolone in allergic bronchopulmonary aspergillosis exacerbations. Eur Respir Rev. (2014) 23:149–52. doi: 10.1183/09059180.00004813
101. Inhaled beclomethasone dipropionate in allergic bronchopulmonary aspergillosis. Report to the research committee of the british thoracic association. Br J Dis Chest. (1979) 73:349–56. doi: 10.1016/s0007-0971(79)80172-2
102. Agarwal R, Khan A, Aggarwal AN, Saikia B, Gupta D, Chakrabarti A. Role of inhaled corticosteroids in the management of serological allergic bronchopulmonary aspergillosis (ABPA). Intern Med. (2011) 50:855–60. doi: 10.2169/internalmedicine.50.4665
103. Denning DW, Van Wye JE, Lewiston NJ, Stevens DA. Adjunctive therapy of allergic bronchopulmonary aspergillosis with itraconazole. Chest. (1991) 100:813–9. doi: 10.1378/chest.100.3.813
104. Agarwal R. What is the current place of azoles in allergic bronchopulmonary aspergillosis and severe asthma with fungal sensitization. Expert Rev Respir Med. (2012) 6:363–71. doi: 10.1586/ers.12.35
105. Stevens DA, Schwartz HJ, Lee JY, Moskovitz BL, Jerome DC, Catanzaro A, et al. A randomized trial of itraconazole in allergic bronchopulmonary aspergillosis. N Engl J Med. (2000) 342:756–62. doi: 10.1056/NEJM200003163421102
106. Wark PA, Hensley MJ, Saltos N, Boyle MJ, Toneguzzi RC, Epid GD, et al. Anti-inflammatory effect of itraconazole in stable allergic bronchopulmonary aspergillosis: a randomized controlled trial. J Allergy Clin Immunol. (2003) 111:952–7. doi: 10.1067/mai.2003.1388
107. Chishimba L, Niven RM, Cooley J, Denning DW. Voriconazole and posaconazole improve asthma severity in allergic bronchopulmonary aspergillosis and severe asthma with fungal sensitization. J Asthma. (2012) 49:423–33. doi: 10.3109/02770903.2012.662568
108. Glackin L, Leen G, Elnazir B, Greally P. Voriconazole in the treatment of allergic bronchopulmonary aspergillosis in cystic fibrosis. Ir Med J. (2009) 102:29. Available online at: http://archive.imj.ie//ViewArticleDetails.aspx?ContentID=3804
109. Hilliard T, Edwards S, Buchdahl R, Francis J, Rosenthal M, Balfour-Lynn I, et al. Voriconazole therapy in children with cystic fibrosis. J Cyst Fibros. (2005) 4:215–20. doi: 10.1016/j.jcf.2005.05.019
110. Periselneris J, Nwankwo L, Schelenz S, Shah A, Armstrong-James D. Posaconazole for the treatment of allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. J Antimicrob Chemother. (2019) 74:1701–3. doi: 10.1093/jac/dkz075
111. Patel D, Popple S, Claydon A, Modha DE, Gaillard EA. Posaconazole therapy in children with cystic fibrosis and Aspergillus-related lung disease. Med Mycol. (2020) 58:11–21. doi: 10.1093/mmy/myz015
112. Jacobs SE, Saez-Lacy D, Wynkoop W, Walsh TJ. Successful treatment of allergic bronchopulmonary aspergillosis with isavuconazole: case report and review of the literature. Open Forum Infect Dis. (2017) 4:ofx040. doi: 10.1093/ofid/ofx040
113. Ram B, Aggarwal AN, Dhooria S, Sehgal IS, Garg M, Behera D, et al. A pilot randomized trial of nebulized amphotericin in patients with allergic bronchopulmonary aspergillosis. J Asthma. (2016) 53:517–24. doi: 10.3109/02770903.2015.1127935
114. Hayes D, Jr, Murphy BS, Lynch JE, Feola DJ. Aerosolized amphotericin for the treatment of allergic bronchopulmonary aspergillosis. Pediatr Pulmonol. (2010) 45:1145–8. doi: 10.1002/ppul.21300
115. Jat KR, Walia DK, Khairwa A. Anti-IgE therapy for allergic bronchopulmonary aspergillosis in people with cystic fibrosis. Cochrane Database Syst Rev. (2015) CD010288. doi: 10.1002/14651858.CD010288.pub3
116. Voskamp AL, Gillman A, Symons K, Sandrini A, Rolland JM, O'Hehir RE, et al. Clinical efficacy and immunologic effects of omalizumab in allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol Pract. (2015) 3:192–9. doi: 10.1016/j.jaip.2014.12.008
117. van der Ent CK, Hoekstra H, Rijkers GT. Successful treatment of allergic bronchopulmonary aspergillosis with recombinant anti-IgE antibody. Thorax. (2007) 62:276–7. doi: 10.1136/thx.2004.035519
118. Emiralioglu N, Dogru D, Tugcu GD, Yalcin E, Kiper N, Ozcelik U. Omalizumab treatment for allergic bronchopulmonary aspergillosis in cystic fibrosis. Ann Pharmacother. (2016) 50:188–93. doi: 10.1177/1060028015624204
119. Nove-Josserand R, Grard S, Auzou L, Reix P, Murris-Espin M, Bremont F, et al. Case series of omalizumab for allergic bronchopulmonary aspergillosis in cystic fibrosis patients. Pediatr Pulmonol. (2017) 52:190–7. doi: 10.1002/ppul.23612
120. Koutsokera A, Corriveau S, Sykes J, Coriati A, Cortes D, Vadas P, et al. Omalizumab for asthma and allergic bronchopulmonary aspergillosis in adults with cystic fibrosis. J Cyst Fibros. (2020) 19:119–24. doi: 10.1016/j.jcf.2019.07.011
121. Li JX, Fan LC, Li MH, Cao WJ, Xu JF. Beneficial effects of Omalizumab therapy in allergic bronchopulmonary aspergillosis: a synthesis review of published literature. Respir Med. (2017) 122:33–42. doi: 10.1016/j.rmed.2016.11.019
122. Wong R, Wong M, Robinson PD, Fitzgerald DA. Omalizumab in the management of steroid dependent allergic bronchopulmonary aspergillosis (ABPA) complicating cystic fibrosis. Paediatr Respir Rev. (2013) 14:22–4. doi: 10.1016/j.prrv.2012.11.004
123. Brinkmann F, Schwerk N, Hansen G, Ballmann M. Steroid dependency despite omalizumab treatment of ABPA in cystic fibrosis. Allergy. (2010) 65:134–5. doi: 10.1111/j.1398-9995.2009.02147.x
124. Ashkenazi M, Sity S, Sarouk I, Bar Aluma BE, Dagan A, Bezalel Y, et al. Omalizumab in allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. J Asthma Allergy. (2018) 11:101–7. doi: 10.2147/JAA.S156049
125. Tomomatsu K, Oguma T, Baba T, Toyoshima M, Komase Y, Taniguchi M, et al. Effectiveness and safety of omalizumab in patients with allergic bronchopulmonary aspergillosis complicated by chronic bacterial infection in the airways. Int Arch Allergy Immunol. (2020) 181:499–506. doi: 10.1159/000507216
126. Altman MC, Lenington J, Bronson S, Ayars AG. Combination omalizumab and mepolizumab therapy for refractory allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol Pract. (2017) 5:1137–9. doi: 10.1016/j.jaip.2017.01.013
127. Terashima T, Shinozaki T, Iwami E, Nakajima T, Matsuzaki T. A case of allergic bronchopulmonary aspergillosis successfully treated with mepolizumab. BMC Pulm Med. (2018) 18:53. doi: 10.1186/s12890-018-0617-5
128. Tomomatsu K, Sugino Y, Okada N, Tanaka J, Oguma T, Asano K. Rapid clearance of mepolizumab-resistant bronchial mucus plugs in allergic bronchopulmonary aspergillosis with benralizumab treatment. Allergol Int. (2020) 1. doi: 10.1016/j.alit.2020.03.003
129. Ramonell RP, Lee FE, Swenson C, Kuruvilla M. Dupilumab treatment for allergic bronchopulmonary aspergillosis: a case series. J Allergy Clin Immunol Pract. (2020) 8:742–43. doi: 10.1016/j.jaip.2019.11.031
130. Ricketti AJ, Greenberger PA, Patterson R. Serum IgE as an important aid in management of allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol. (1984) 74:68–71. doi: 10.1016/0091-6749(84)90089-7
131. Agarwal R, Gupta D, Aggarwal AN, Saxena AK, Saikia B, Chakrabarti A, et al. Clinical significance of decline in serum IgE levels in allergic bronchopulmonary aspergillosis. Respir Med. (2010) 104:204–10. doi: 10.1016/j.rmed.2009.09.005
Keywords: ABPA, allergic bronchopulmonary aspergillosis, aspergillus, cystic fibrosis, children
Citation: Sunman B, Ademhan Tural D, Ozsezen B, Emiralioglu N, Yalcin E and Özçelik U (2020) Current Approach in the Diagnosis and Management of Allergic Bronchopulmonary Aspergillosis in Children With Cystic Fibrosis. Front. Pediatr. 8:582964. doi: 10.3389/fped.2020.582964
Received: 13 July 2020; Accepted: 16 September 2020;
Published: 20 October 2020.
Edited by:
Bülent Taner Karadag, Marmara University, TurkeyReviewed by:
Ritesh Agarwal, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaRichard B. Moss, Stanford University, United States
Koichiro Asano, Tokai University, Japan
Copyright © 2020 Sunman, Ademhan Tural, Ozsezen, Emiralioglu, Yalcin and Özçelik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uğur Özçelik, b3pjZWxpa3VAZ21haWwuY29t