 Reiner K. Mailer
Reiner K. Mailer- Institute of Clinical Chemistry and Laboratory Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
The transcription factor FOXP3 controls the immunosuppressive program in CD4+ T cells that is crucial for systemic immune regulation. Mutations of the single X-chromosomal FOXP3 gene in male individuals cause the inherited autoimmune disease immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome. Insufficient gene expression and impaired function of mutant FOXP3 protein prevent the generation of anti-inflammatory regulatory T (Treg) cells and fail to inhibit autoreactive T cell responses. Diversification of FOXP3 functional properties is achieved through alternative splicing that leads to isoforms lacking exon 2 (FOXP3Δ2), exon 7 (FOXP3Δ7), or both (FOXP3Δ2Δ7) specifically in human CD4+ T cells. Several IPEX mutations targeting these exons or promoting their alternative splicing revealed that those truncated isoforms cannot compensate for the loss of the full-length isoform (FOXP3fl). In this review, IPEX mutations that change the FOXP3 isoform profile and the resulting consequences for the CD4+ T-cell phenotype are discussed.
Introduction
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare condition affecting less than one in a million newborns. Because patients with IPEX syndrome may develop a wide range of symptoms with varying severity, it has been estimated that cases remain often undiagnosed and the prevalence of the disease is higher than reported. Clinical presentation of IPEX syndrome include systemic autoimmune manifestations (dermatitis/eczema, autoimmune hemolytic anemia, atopy), enteropathy (severe diarrhea, food allergies), endocrinopathy (type 1 diabetes mellitus, thyroiditis), and a failure to thrive. The triad of dermatitis, diarrhea and diabetes in male infants is applicable as disease indicator and if left untreated IPEX syndrome becomes a life-threatening condition early in life. The disease is commonly inherited from the maternal lineage and is disseminated in an X chromosome-linked recessive pattern. Because fetal immunity develops already during the second trimester of gestation, intrauterine immune dysregulation has been linked to fetal death and repeated miscarriages may point toward an IPEX mutation in one of the maternal X chromosomes [reviewed in Bacchetta et al. (1)]. IPEX syndrome is a monogenic disease caused by the mutation of the forkhead box P3 (FOXP3) gene, that encodes for the transcription factor FOXP3 (initially also named JM2 and Scurfin) (2, 3). Analysis of FOXP3 mutations from patients and patients' mothers promptly confirm IPEX syndrome diagnosis and identify IPEX mutation carrier, respectively.
IPEX Mutations Target FOXP3—The Indispensable Immune Regulator
Foxp3 gained intensive research interest since it was described in 2003 as the central lineage-defining marker for regulatory T (Treg) cells (4, 5) that maintains immunologic self-tolerance through the development of the immunosuppressive CD4+CD25+ T cell subset (6). Absence of Foxp3 in mice (7) and FOXP3 in humans (2, 8) cause fatal multiple organ autoimmunity termed scurfy and IPEX syndrome, respectively. In line with the pivotal role of the transcription factor retroviral transduction of murine Foxp3 (4, 5) and human FOXP3 (9) induces a Treg-cell phenotype and confers suppressive capacity on CD4+CD25− T cells. FOXP3 broadly regulates the gene expression profile (10), which is related to transcriptional and epigenetic changes triggered by T-cell antigen receptor (TCR) stimulation (11, 12). Moreover, TCR signaling is necessary to maintain Treg-cell functions (13, 14). Importantly, FOXP3 transcription is controlled by chromatin modifications at conserved non-coding sequences (CNS) (15). CNS3 located at FOXP3 intron 1 (i.e., between exon 1 and 2) acts as a genetic switch that initiates gene expression through interactions with c-rel (15, 16) and atypical NF-κB inhibitor (17). Accordingly, targeting CNS3 chromatin accessibility with a specific ribonucleoprotein (dCas9—catalytically inactive variant of clustered regularly inter-spaced short palindromic repeats-associated protein 9) has been shown to promote FOXP3 expression in Jurkat cells (18), whereas endogenous long non-coding RNA (FLICR—Foxp3 long intergenic non-coding RNA) induces a FOXP3low T-cell subset (19). Although high FOXP3 expression levels are commonly attributed to CD4+CD25high Treg cells, FOXP3 is also induced by TCR stimulation in naive CD4+CD25− T cells and persists for several days in activated CD4+ T cells (20). Notably, activation-induced FOXP3 expression seems to depend on the CNS3 element, because generation of transgenic mice with a bacterial artificial chromosome that encodes for a reporter protein inserted into the human FOXP3 gene (consisting of >50 kb each side of the start codon, including CNS1 and CNS2, but not CNS3) demonstrated that TCR stimulation alone (i.e., without transforming growth factor-β activation) does not induce transgenic FOXP3 expression in murine CD4+ T cells (21).
Moreover, in the absence of antigenic stimulation, FOXP3 expression is promoted in CD4+CD25− T cells through STAT5-signaling cytokines IL-2, IL-7, and IL-15 (22) and short term exposure with these cytokines has been reported to reveal higher portions of “latent” Treg cells among human peripheral blood mononuclear cells (23). Although addition of cytokines increased the number of Treg cells for autologous transfer trials, this treatment may increase plasticity of Treg cells and reduce susceptibility of reactive T cells toward Treg-mediated suppression (24–26). In contrast to peripheral blood, cytokine treatment does not induce FOXP3 expression in CD4+ T cells obtained from lymph nodes (23); thus consequences for T-cell responses in disease settings remain unknown. Flow cytometry analysis of CD4+ T cells from peripheral blood demonstrated that FOXP3 expression varies up to 40-fold between individual cells. Strength and duration of FOXP3 induction may therefore explain discrepancies in the suppressive capacity of T cells with ectopically expressed or activation- and cytokine-induced FOXP3 (20, 22, 23, 27–30). In vivo, CD4+ T cells with activation-induced FOXP3 expression should accumulate at active sites of inflammation. Indeed CD4+CD25−FOXP3+ and CD4+CD45RA−FOXP3low populations increase in inflamed tissue of patients with autoimmune diseases and this has been attributed to self-reactive non-Treg cell responses (31–34). The notion that CD4+ T-cell subsets in different tissues express distinct patterns of CD25 and other Treg-cell markers in both mice and men (35–38), adds even more complexity to the task to safely distinguish FOXP3+ Treg from FOXP3+ non-Treg cells. One important target for FOXP3-mediated transcriptional repression is the promoter of the IL-7 receptor (CD127). Accordingly, the most suppressive human Treg-cell population has been identified to express a CD4+CD25+CD127low phenotype (39, 40). However, the corresponding CD4+CD25−CD127+ conventional T (Tcon) cells rapidly decrease CD127 upon IL-7 signaling and TCR stimulation (41), whereas CD127 expression has been reported to increase in activated Treg cells in mice (42).
Taken together, FOXP3 induction in human CD4+ T cells prevents unambiguous identification of immunosuppressive and immunoreactive T-cell subsets that challenges assessment of T-cell responses in autoimmunity [reviewed in Scheinecker et al. (43) and Buckner (44)]. So far it remains elusive, why murine Tcon cells do not induce Foxp3 upon stimulation likewise to human Tcon cells, because both species have the CNS3 as well as a Treg-associated gene expression profile similar to stimulated Tcon cells in common (45, 46). In human Tcon cells, activation-induced FOXP3 intrinsically restricts T-cell proliferation and differentiation (47–49) and protects stimulated Tcon cells from premature restimulation-induced cell death (50). Further studies are required to investigate susceptibility, spatio-temporal expression and isoform-specific functions of activation-induced FOXP3 in stimulated Tcon cells.
FOXP3 Isoform Expression
Alternative splicing of human FOXP3 leads to isoforms lacking exon 2 (FOXP3Δ2), exon 7 (FOXP3Δ7), or both exon 2 and 7 (FOXP3Δ2Δ7) (51). Several studies revealed that FOXP3 splice variants confer partly overlapping but also isoform-specific functions (Table 1). FOXP3Δ2 induces immunosuppressive phenotype and function cooperatively with FOXP3fl in human and murine CD4+ T cells (27, 52). Although FOXP3Δ2 lacks the LXXLL interaction motif for binding retinoid orphan receptor-γt (the transcription factor ROR-γt that promotes the differentiation of pro-inflammatory Th17 cells) (53), FOXP3Δ2 has not been associated with Th17-related disease (54–56). Moreover, exclusion of exon 2 in activation-induced FOXP3 through splice-shifting antisense oligonucleotides significantly decreases expression of IL-17A, but not IL-2, and IFN-γ, demonstrating that FOXP3Δ2 facilitates restraint on T cell responses and effectively prevents rather than promotes Th17-cell differentiation (55). Consistently with immunosuppressive function of FOXP3Δ2, decreasing ratios of FOXP3fl/FOXP3Δ2 has been interpreted as functional gain in long-term activated Treg cells (57), whereas a functionally exhausted Treg-cell status has been proposed for this phenotype in autoimmunity (58, 59). A recent study by the latter group showed that FOXP3 isoforms containing exon 2 increase stronger than FOXP3 isoforms lacking exon 2 in peripheral CD4+ T cells of patients suffering from a moderate stage of chronic obstructive pulmonary disease (60). Moreover, expression of the proliferation marker Ki-67 increases in cell populations stained with FOXP3 exon 2-specific antibodies compared to those stained with antibodies that detect all FOXP3 isoforms. This is in line with our results that TCR stimulation has an impact on the FOXP3fl/FOXP3Δ2 ratio (61). Detection of FOXP3 exon 2 in relation to total FOXP3 by specific antibodies revealed that alternative splicing shifted toward increased inclusion of exon 2 upon antigenic stimulation. Moreover, this activation-induced FOXP3 isoform pattern has been observed in immunostimulatory settings, such as coronary artery disease (61), inflammatory bowel disease (54) and myelodysplastic syndrome (62). The latter report also describes a prevalence for FOXP3fl expression during T cell development in the thymus. Consistently, we recently found a higher expression of FOXP3fl compared to FOXP3Δ2 in thymocytes and that FOXP3fl precedes alternative splicing of exon 2 in TCR-stimulated naive T cells (manuscript in preparation). The development of IPEX caused by FOXP3 exon 2 mutations that abrogate expression of FOXP3fl but otherwise enable unchanged FOXP3Δ2 expression, highlights the importance of FOXP3fl for T cell homeostasis (see below).
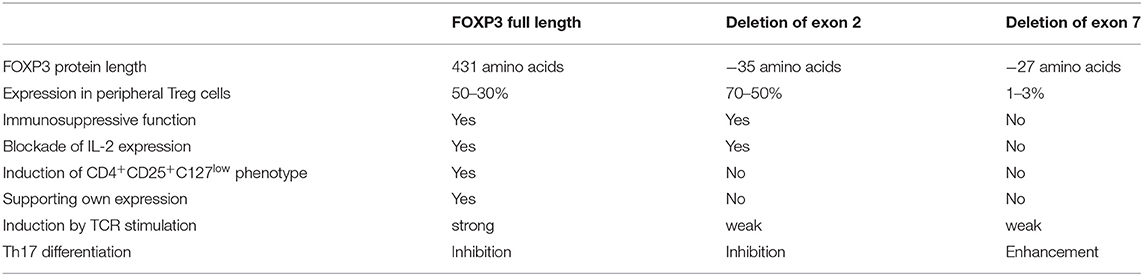
Table 1. Characteristics of FOXP3 isoforms.
Additional splicing of FOXP3 exon 7 generates the isoform FOXP3Δ2Δ7 in human CD4+ T cells. FOXP3Δ2Δ7 does not contribute to the suppressive phenotype and function of Treg cells in mice and men (52, 63). Exclusion of exon 7 is significantly decreased in activated Treg cells (55). However, IL-1β strongly induces alternative splicing of FOXP3 exon 7 in Treg cells and expression of FOXP3 lacking exon 7 is elevated in colon biopsies obtained from Crohn's disease patients. Moreover, antisense oligonucleotide-mediated splicing of FOXP3 exon 7 in naive CD4+ T cells promotes the differentiation of pro-inflammatory Th17 cells (55). Mostly described for inflamed tissues, phenotypically mixed CD4+FOXP3+IL-17A+/ROR-γt+ T cells have been identified [reviewed in Du et al. (64)]. However, the extent of FOXP3 exon 7 splicing and the threshold that sufficiently drives Treg-cell plasticity in these cells is currently unknown. FOXP3 exon 7 is part of the protein's leucine zipper domain regulating the composition and stability of the multi-protein transcriptional repressor complex (65, 66); nevertheless about one third of FOXP3 target genes are similarly expressed in stimulated CD4+ T cells in the absence of FOXP3 exon 7 (67) and FOXP3Δ2Δ7 inhibits FOXP3fl in a dominant negative manner (52). Thus, both complementary and competing functions are carried out by FOXP3 isoforms with alternative splicing of exon 7. IPEX mutations within FOXP3 exon 7 and particularly intronic mutations that mediate excessive splicing of FOXP3 exon 7 demonstrate that endogenous FOXP3Δ2Δ7 or aberrant FOXP3Δ7/FOXP3Δ2Δ7 expression cannot compensate for the loss of FOXP3 isoforms with exon 7 (see below).
Taken together, the analysis of FOXP3 isoforms can elucidate impaired tolerance induction and exaggerated T-cell responses in autoimmunity and chronic diseases. In healthy conditions, FOXP3 isoforms are co-expressed in CD4+ T cells but may differ in their respective ratio depending on TCR stimulation and cytokine activation. The assessment of isoform-specific functions is challenging because modulation of human CD4+ T cells (e.g., via transfection or transduction) induces FOXP3 expression. However, IPEX mutations that target alternative splicing or that cause nonsense-mediated decay of frameshifted mRNA facilitate the analysis of naive CD4+ T cells with limited FOXP3 isoform variation. Thus, FOXP3 sequencing in IPEX patients and female relatives together with flow cytometric analysis of CD4+CD25+CD127− T cells in pediatric case reports contribute to our understanding of FOXP3 functions and Treg-cell development.
Differential Effect of IPEX Mutations That Target the FOXP3 Isoform Ratio
Since the monogenic primary immunodeficiency IPEX syndrome was defined more than 170 distinct FOXP3 mutations have been reported (68). These mutations likely represent severe cases with various clinical symptoms, whereas milder forms may go undetected. However, it has become clear that identical FOXP3 mutations can cause different disease manifestation in IPEX patients (69). This reflects the stochastic nature of autoreactive responses [reviewed in Richards et al. (70)], compensatory mechanisms to suppress autoreactive T-cell responses [e.g., FOXP3-independent type 1 regulatory T cells (71)] and the balance of Treg- and Tcon-cell functions affected by the IPEX mutation (47, 48). Depending on disease severity, combinatorial therapy with immunosuppressive drugs (e.g., rapamycin, glucocorticoids, cyclosporine, tacrolimus) can alleviate autoimmunity in IPEX patients, whereas the only curative approach for IPEX syndrome is allogeneic hematopoietic stem cell transplantation. An analysis of currently known IPEX cases with FOXP3 mutations that abrogate the correct expression of FOXP3 exon 2 (five frameshift mutations) and FOXP3 exon 7 (four deletion/missense mutations, seven splicing mutations) and the impact on clinical parameters and the corresponding T cell phenotype is shown (Table 2).
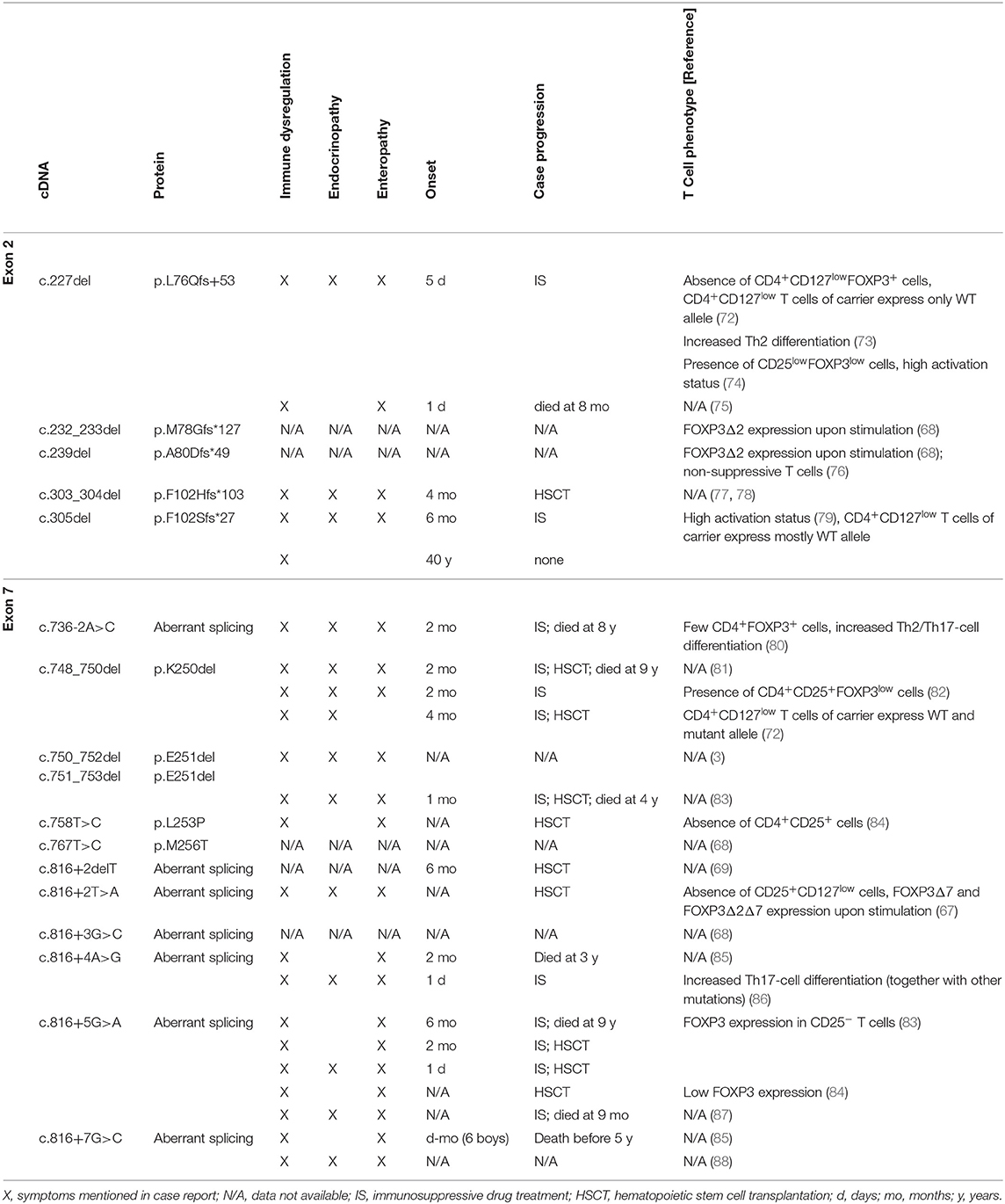
Table 2. Mutations that target FOXP3 isoform expression ratios cause IPEX.
Isoforms Fall Short to Preserve Immune Regulation in IPEX Patients
Frameshift mutations in FOXP3 exon 2 generate premature stop codons that target mRNAs for nonsense-mediated decay. The rapid mRNA degradation prevents translation into mutated protein, so that only FOXP3 transcripts lacking exon 2 yield considerable protein expression. Skipping mutations in exon 2 should ensue regular amino acid sequence and expression level of FOXP3Δ2. However, deletion of nucleotide 227 (c.227del) in FOXP3 exon 2 causes the absence of the CD4+CD127lowFOXP3+ T-cell population and only few CD4+CD25+FOXP3low T cells were found in the periphery (72, 74). Furthermore, due to random X-chromosome inactivation half of the Treg and Tcon cells lack FOXP3-mediated regulation in female mutation carriers. Although IPEX syndrome has not been reported, carriers display a bimodal FOXP3 expression within the CD4+CD25+CD127low T-cell population for FOXP3 mutations that do not affect Treg-cell development (e.g., c.1150G>A) (72). In contrast, only cells with activated wild-type allele but not IPEX mutations are present among the CD4+CD25+CD127low T-cell population of subjects carrying the FOXP3 exon 2 mutation c.227del and a similar trend has been reported for IPEX mutation c.305del (72, 79). Taken together, this suggests that FOXP3Δ2 expression alone is insufficient to promote the generation of Treg cells. Consistent with impaired T-cell homeostasis, T cells from IPEX patients with FOXP3 exon 2 mutations c.227del and c.305del have a highly activated/differentiated phenotype (73, 74, 79). Noteworthy, FOXP3Δ2 expression is induced in stimulated T cells from IPEX patients with FOXP3 exon 2 mutations c.232_233del, c.239del and c.305del, albeit individual expression levels differ and appear to be decreased compared to Treg cells from healthy controls (68, 79). The activation and proliferation status of Tcon cells may therefore explain variable FOXP3Δ2 expression patterns for IPEX patients with FOXP3 mutations c.227del, c.232_233del, c.239del, and c.305del. This notion is corroborated by the recent finding that T cells with enforced expression of FOXP3 c.239del via gene editing do not suppress T-cell proliferation (76). Thus, absence of FOXP3fl prevents phenotypical and functional Treg-cell development and FOXP3Δ2 expression alone in IPEX patients with FOXP3 exon 2 mutations does not characterize bona fide Treg cells.
In line with a dominant-negative effect of FOXP3Δ2Δ7 (52), excessive splicing of FOXP3 exon 7 through IPEX mutations c.736-2A>C, c.816+2delT, c.816+2T>A, c.816+3G>C, c.816+4A>G, c.816+5G>A, and c.816+7G>C decreases FOXP3 expression and abrogates the development of CD4+CD25+CD127low T cells (67, 80, 84). Moreover, inducible expression of FOXP3 isoforms lacking exon 7 has been reported in TCR-stimulated CD4+CD25− T cells from IPEX patients with FOXP3 mutations c.816+2T>A and 816+5G>A (67, 83). Enhanced exclusion of FOXP3 exon 7 through splice-shifting antisense oligonucleotides negates FOXP3fl/FOXP3Δ2-mediated inhibition of IL-2 and promotes IL-17A expression (55). Consequently, differentiation of Th2 and Th17 cells increase in IPEX patients with mutations in intronic regions that regulate alternative splicing of FOXP3 exon 7 (80, 86). Only four reports analyzed the phenotype of T cells from IPEX patients with mutations within FOXP3 exon 7. The deletion mutations c.748_750del and c.750_752del/c.751_753del miss amino acid K250 and E251, respectively. Because reading frame and FOXP3 isoform expression ratio is maintained in these mutations, functional properties of this domain could be revealed (65). This is also reflected by the relatively mild T-cell phenotype with reduced FOXP3 expression in preserved CD4+CD25+ populations (82, 89), and bimodal FOXP3 expression among CD4+CD25+CD127low T cells in a mutation carrier (72). Similarly the missense mutation FOXP3 c.758T>C, that generates an amino acid exchange (p.L253P), has no effect on splice variants but prevents the CD4+CD25high T-cell phenotype (84). In summary, in-frame mutations of FOXP3 exon 7 cause dysfunctional FOXP3 isoforms, whereas mutations of intron 6 and 7 cause aberrant splicing that underlines the insufficient T-cell regulation by FOXP3 isoforms lacking exon 7.
Conclusions
Although activation and proliferation of T cells in IPEX patients may be affected by immunosuppressive drug treatment, important characteristics of FOXP3 isoforms can be deduced: FOXP3fl increases upon TCR stimulation and is indispensable to induce sustainable Treg-related phenotype and functions, whereas FOXP3Δ2Δ7 counteracts the action of FOXP3fl under pro-inflammatory conditions. FOXP3Δ2 does not promote its own expression but supports and may stabilize Treg-mediated immunosuppression. Data from IPEX patients with impaired isoform ratios indicate that modulated FOXP3 splicing could enhance T-cell responses via exon 7 exclusion (e.g., in tumor-specific T cells) or promote immunosuppression via exon 2 inclusion (e.g., in immunopathology). Essential for this strategy is the targeting of splice-shifting elements that has shown promising potential in other isoform-related diseases (90, 91). Alternatively, also gene editing that enhances FOXP3fl expression may provide a new approach to regulate human T-cell responses (76, 92).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by the German Heart Foundation (F/34/18) and the German Research Foundation (SFB 841, TP B08 and SFB 877, TP A11).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I like to thank Dr. Thomas Renné for critical reading of this manuscript.
References
1. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. (2018) 1417:5–22. doi: 10.1111/nyas.13011
2. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
3. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. (2000) 106:R75–81. doi: 10.1172/JCI11679
4. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
5. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
6. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. (1995) 155:1151–64.
7. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
8. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. (2001) 27:18–20. doi: 10.1038/83707
9. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. (2004) 16:1643–56. doi: 10.1093/intimm/dxh165
10. Sadlon TJ, Wilkinson BG, Pederson S, Brown CY, Bresatz S, Gargett T, et al. Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol. (2010) 185:1071–81. doi: 10.4049/jimmunol.1000082
11. Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. (2012) 151:153–66. doi: 10.1016/j.cell.2012.06.053
12. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. (2012) 37:785–99. doi: 10.1016/j.immuni.2012.09.010
13. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070–8. doi: 10.1038/ni.3004
14. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. (2014) 41:722–36. doi: 10.1016/j.immuni.2014.10.012
15. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. (2010) 463:808–12. doi: 10.1038/nature08750
16. Feng Y, van der Veeken J, Shugay M, Putintseva EV, Osmanbeyoglu HU, Dikiy S, et al. A mechanism for expansion of regulatory T-cell repertoire and its role in self-tolerance. Nature. (2015) 528:132–6. doi: 10.1038/nature16141
17. Schuster M, Glauben R, Plaza-Sirvent C, Schreiber L, Annemann M, Floess S, et al. IκB(NS) protein mediates regulatory T cell development via induction of the Foxp3 transcription factor. Immunity. (2012) 37:998–1008. doi: 10.1016/j.immuni.2012.08.023
18. Forstneric V, Oven I, Ogorevc J, Lainscek D, Praznik A, Lebar T, et al. CRISPRa-mediated FOXP3 gene upregulation in mammalian cells. Cell Biosci. (2019) 9:93. doi: 10.1186/s13578-019-0357-0
19. Zemmour D, Pratama A, Loughhead SM, Mathis D, Benoist C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci USA. (2017) 114:E3472–80. doi: 10.1073/pnas.1700946114
20. Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25− T cells. J Clin Invest. (2003) 112:1437–43. doi: 10.1172/JCI19441
21. Tsuda M, Tone Y, Ogawa C, Nagaoka Y, Katsumata M, Necula A, et al. A bacterial artificial chromosome reporter system for expression of the human FOXP3 gene in mouse regulatory T-cells. Front Immunol. (2017) 8:279. doi: 10.3389/fimmu.2017.00279
22. Imamichi H, Sereti I, Lane HC. IL-15 acts as a potent inducer of CD4(+)CD25(hi) cells expressing FOXP3. Eur J Immunol. (2008) 38:1621–30. doi: 10.1002/eji.200737607
23. Tabares P, Berr S, Langenhorst D, Sawitzki B, Ten Berge I, Tony HP, et al. Short-term cytokine stimulation reveals regulatory T cells with down-regulated Foxp3 expression in human peripheral blood. Eur J Immunol. (2018) 48:366–79. doi: 10.1002/eji.201747244
24. Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1beta and IL-2 convert human Treg into T(H)17 cells. Clin Immunol. (2009) 131:298–307. doi: 10.1016/j.clim.2008.12.008
25. Heninger AK, Theil A, Wilhelm C, Petzold C, Huebel N, Kretschmer K, et al. IL-7 abrogates suppressive activity of human CD4+CD25+FOXP3+ regulatory T cells and allows expansion of alloreactive and autoreactive T cells. J Immunol. (2012) 189:5649–58. doi: 10.4049/jimmunol.1201286
26. Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abdeladhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. (2009) 182:6763–70. doi: 10.4049/jimmunol.0801792
27. Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, et al. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest. (2005) 115:3276–84. doi: 10.1172/JCI24685
28. Aarts-Riemens T, Emmelot ME, Verdonck LF, Mutis T. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(-) cells. Eur J Immunol. (2008) 38:1381–90. doi: 10.1002/eji.200737590
29. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. (2007) 19:345–54. doi: 10.1093/intimm/dxm014
30. Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated non-regulatory CD4+ T cells. Eur J Immunol. (2007) 37:129–38. doi: 10.1002/eji.200636435
31. Bonelli M, Savitskaya A, Steiner CW, Rath E, Smolen JS, Scheinecker C. Phenotypic and functional analysis of CD4+ CD25− Foxp3+ T cells in patients with systemic lupus erythematosus. J Immunol. (2009) 182:1689–95. doi: 10.4049/jimmunol.182.3.1689
32. Matsuki F, Saegusa J, Nishimura K, Miura Y, Kurosaka M, Kumagai S, et al. CD45RA-Foxp3(low) non-regulatory T cells in the CCR7−CD45RA−CD27+CD28+ effector memory subset are increased in synovial fluid from patients with rheumatoid arthritis. Cell Immunol. (2014) 290:96–101. doi: 10.1016/j.cellimm.2014.05.011
33. Mathian A, Parizot C, Dorgham K, Trad S, Arnaud L, Larsen M, et al. Activated and resting regulatory T cell exhaustion concurs with high levels of interleukin-22 expression in systemic sclerosis lesions. Ann Rheum Dis. (2012) 71:1227–34. doi: 10.1136/annrheumdis-2011-200709
34. Viisanen T, Gazali AM, Ihantola EL, Ekman I, Nanto-Salonen K, Veijola R, et al. FOXP3+ regulatory T cell compartment is altered in children with newly diagnosed type 1 diabetes but not in autoantibody-positive at-risk children. Front Immunol. (2019) 10:19. doi: 10.3389/fimmu.2019.00019
35. Mailer RKW, Gistera A, Polyzos KA, Ketelhuth DFJ, Hansson GK. Hypercholesterolemia induces differentiation of regulatory T cells in the Liver. Circ Res. (2017) 120:1740–53. doi: 10.1161/CIRCRESAHA.116.310054
36. Booth NJ, McQuaid AJ, Sobande T, Kissane S, Agius E, Jackson SE, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol. (2010) 184:4317–26. doi: 10.4049/jimmunol.0903781
37. Rodriguez-Perea AL, Arcia ED, Rueda CM, Velilla PA. Phenotypical characterization of regulatory T cells in humans and rodents. Clin Exp Immunol. (2016) 185:281–91. doi: 10.1111/cei.12804
38. Mohr A, Malhotra R, Mayer G, Gorochov G, Miyara M. Human FOXP3(+) T regulatory cell heterogeneity. Clin Transl Immunol. (2018) 7:e1005. doi: 10.1002/cti2.1005
39. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. (2006) 203:1701–11. doi: 10.1084/jem.20060772
40. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. (2006) 203:1693–700. doi: 10.1084/jem.20060468
41. Alves NL, van Leeuwen EM, Derks IA, van Lier AR. Differential regulation of human IL-7 receptor alpha expression by IL-7 and TCR signaling. J Immunol. (2008) 180:5201–10. doi: 10.4049/jimmunol.180.8.5201
42. Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S, et al. Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. Eur J Immunol. (2010) 40:2528–38. doi: 10.1002/eji.201040531
43. Scheinecker C, Goschl L, Bonelli M. Treg cells in health and autoimmune diseases. New insights from single cell analysis. J Autoimmun. (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376
44. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. (2010) 10:849–59. doi: 10.1038/nri2889
45. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. (2007) 445:931–5. doi: 10.1038/nature05478
46. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. (2007) 445:936–40. doi: 10.1038/nature05563
47. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. (2006) 116:1713–22. doi: 10.1172/JCI25112
48. McMurchy AN, Gillies J, Allan SE, Passerini L, Gambineri E, Roncarolo MG, et al. Point mutants of forkhead box P3 that cause immune dysregulation, polyendocrinopathy, enteropathy, X-linked have diverse abilities to reprogram T cells into regulatory T cells. J Allergy Clin Immunol. (2010) 126:1242–51. doi: 10.1016/j.jaci.2010.09.001
49. McMurchy AN, Gillies J, Gizzi MC, Riba M, Garcia-Manteiga JM, Cittaro D, et al. A novel function for FOXP3 in humans: intrinsic regulation of conventional T cells. Blood. (2013) 121:1265–75. doi: 10.1182/blood-2012-05-431023
50. Voss K, Lake C, Luthers CR, Lott NM, Dorjbal B, Arjunaraja S, et al. FOXP3 protects conventional human T cells from premature restimulation-induced cell death. Cell Mol Immunol. (2019). doi: 10.1038/s41423-019-0316-z. [Epub ahead of print].
51. Mailer RKM. Alternative splicing of FOXP3-virtue and vice. Front Immunol. (2018) 9:530. doi: 10.3389/fimmu.2018.00530
52. Mailer RK, Falk K, Rotzschke O. Absence of leucine zipper in the natural FOXP3Delta2Delta7 isoform does not affect dimerization but abrogates suppressive capacity. PLoS ONE. (2009) 4:e6104. doi: 10.1371/journal.pone.0006104
53. Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol. (2008) 180:4785–92. doi: 10.4049/jimmunol.180.7.4785
54. Lord JD, Valliant-Saunders K, Hahn H, Thirlby RC, Ziegler SF. Paradoxically increased FOXP3+ T cells in IBD do not preferentially express the isoform of FOXP3 lacking exon 2. Dig Dis Sci. (2012) 57:2846–55. doi: 10.1007/s10620-012-2292-3
55. Mailer RK, Joly AL, Liu S, Elias S, Tegner J, Andersson J. IL-1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep. (2015) 5:14674. doi: 10.1038/srep14674
56. Chen L, Wu J, Pier E, Zhao Y, Shen Z. mTORC2-PKBα/Akt1 Serine 473 phosphorylation axis is essential for regulation of FOXP3 Stability by chemokine CCL3 in psoriasis. J Invest Dermatol. (2013) 133:418–28. doi: 10.1038/jid.2012.333
57. Joly AL, Seitz C, Liu S, Kuznetsov NV, Gertow K, Westerberg LS, et al. Alternative splicing of FOXP3 controls regulatory T cell effector functions and is associated with human atherosclerotic plaque stability. Circ Res. (2018) 122:1385–94. doi: 10.1161/CIRCRESAHA.117.312340
58. Sambucci M, Gargano F, De Rosa V, De Bardi M, Picozza M, Placido R, et al. FoxP3 isoforms and PD-1 expression by T regulatory cells in multiple sclerosis. Sci Rep. (2018) 8:3674. doi: 10.1038/s41598-018-21861-5
59. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. (2015) 16:1174–84. doi: 10.1038/ni.3269
60. Bruzzaniti S, Bocchino M, Santopaolo M, Cali G, Stanziola AA, D'Amato M, et al. An immunometabolic pathomechanism for chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. (2019) 116:15625–34. doi: 10.1073/pnas.1906303116
61. Lundberg AK, Jonasson L, Hansson GK, Mailer RKW. Activation-induced FOXP3 isoform profile in peripheral CD4+ T cells is associated with coronary artery disease. Atherosclerosis. (2017) 267:27–33. doi: 10.1016/j.atherosclerosis.2017.10.026
62. Dudina GA, Donetskova AD, Litvina MM, Mitin AN, Mitina TA, Polyakov SA. Regulatory T cells and profile of FOXP3 isoforms expression in peripheral blood of patients with myelodysplastic syndromes. Adv Hematol. (2018) 2018:8487403. doi: 10.1155/2018/8487403
63. Joly AL, Liu S, Dahlberg CI, Mailer RK, Westerberg LS, Andersson J. Foxp3 lacking exons 2 and 7 is unable to confer suppressive ability to regulatory T cells in vivo. J Autoimmun. (2015) 63:23–30. doi: 10.1016/j.jaut.2015.06.009
64. Du R, Zhao H, Yan F, Li H. IL-17+Foxp3+ T cells: an intermediate differentiation stage between Th17 cells and regulatory T cells. J Leukoc Biol. (2014) 96:39–48. doi: 10.1189/jlb.1RU0114-010RR
65. Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol. (2007) 19:825–35. doi: 10.1093/intimm/dxm043
66. Song X, Li B, Xiao Y, Chen C, Wang Q, Liu Y, et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep. (2012) 1:665–75. doi: 10.2210/pdb4i1l/pdb
67. Magg T, Wiebking V, Conca R, Krebs S, Arens S, Schmid I, et al. IPEX due to an exon 7 skipping FOXP3 mutation with autoimmune diabetes mellitus cured by selective TReg cell engraftment. Clin Immunol. (2018) 191:52–8. doi: 10.1016/j.clim.2018.03.008
68. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, De Boer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol. (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
69. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies. an international multicenter retrospective study. J Allergy Clin Immunol. (2018) 141:1036–49.e5. doi: 10.1016/j.jaci.2017.10.041
70. Richards DM, Kyewski B, Feuerer M. Re-examining the nature and function of self-reactive T cells. Trends Immunol. (2016) 37:114–25. doi: 10.1016/j.it.2015.12.005
71. Passerini L, Di Nunzio S, Gregori S, Gambineri E, Cecconi M, Seidel MG, et al. Functional type 1 regulatory T cells develop regardless of FOXP3 mutations in patients with IPEX syndrome. Eur J Immunol. (2011) 41:1120–31. doi: 10.1002/eji.201040909
72. Otsubo K, Kanegane H, Kamachi Y, Kobayashi I, Tsuge I, Imaizumi M, et al. Identification of FOXP3-negative regulatory T-like (CD4(+)CD25(+)CD127(low)) cells in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Clin Immunol. (2011) 141:111–20. doi: 10.1016/j.clim.2011.06.006
73. Kobayashi I, Shiari R, Yamada M, Kawamura N, Okano M, Yara A, et al. Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J Med Genet. (2001) 38:874–6. doi: 10.1136/jmg.38.12.874
74. Fuchizawa T, Adachi Y, Ito Y, Higashiyama H, Kanegane H, Futatani T, et al. Developmental changes of FOXP3-expressing CD4+CD25+ regulatory T cells and their impairment in patients with FOXP3 gene mutations. Clin Immunol. (2007) 125:237–46. doi: 10.1016/j.clim.2007.08.004
75. Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. (2009) 32:111–6. doi: 10.2337/dc08-1188
76. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene editing to induce FOXP3 expression in human CD4(+) T cells leads to a stable regulatory phenotype and function. Sci Transl Med. (2020) 12:eaay6422. doi: 10.1126/scitranslmed.aay6422
77. Moudgil A, Perriello P, Loechelt B, Przygodzki R, Fitzerald W, Kamani N. Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy. Pediatr Nephrol. (2007) 22:1799–802. doi: 10.1007/s00467-007-0532-0
78. Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. (2007) 109:383–5. doi: 10.1182/blood-2006-05-025072
79. Frith K, Joly AL, Ma CS, Tangye SG, Lohse Z, Seitz C, et al. The FOXP3Δ2 isoform supports Treg cell development and protects against severe IPEX syndrome. J Allergy Clin Immunol. (2019) 144:317–20.e8. doi: 10.1016/j.jaci.2019.03.003
80. Chen CA, Chung WC, Chiou YY, Yang YJ, Lin YC, Ochs HD, et al. Quantitative analysis of tissue inflammation and responses to treatment in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, and review of literature. J Microbiol Immunol Infect. (2016) 49:775–82. doi: 10.1016/j.jmii.2015.10.015
81. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. (2002) 39:537–45. doi: 10.1136/jmg.39.8.537
82. Hashimura Y, Nozu K, Kanegane H, Miyawaki T, Hayakawa A, Yoshikawa N, et al. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Nephrol. (2009) 24:1181–6. doi: 10.1007/s00467-009-1119-8
83. Baris S, Schulze I, Ozen A, Karakoc Aydiner E, Altuncu E, Karasu GT, et al. Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol. (2014) 34:601–6. doi: 10.1007/s10875-014-0059-7
84. Nademi Z, Slatter M, Gambineri E, Mannurita SC, Barge D, Hodges S, et al. Single centre experience of haematopoietic SCT for patients with immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Bone Marrow Transplant. (2014) 49:310–2. doi: 10.1038/bmt.2013.181
85. Harbuz R, Lespinasse J, Boulet S, Francannet C, Creveaux I, Benkhelifa M, et al. Identification of new FOXP3 mutations and prenatal diagnosis of IPEX syndrome. Prenat Diagn. (2010) 30:1072–8. doi: 10.1002/pd.2613
86. Passerini L, Olek S, Di Nunzio S, Barzaghi F, Hambleton S, Abinun M, et al. Forkhead box protein 3 (FOXP3) mutations lead to increased TH17 cell numbers and regulatory T-cell instability. J Allergy Clin Immunol. (2011) 128:1376–9.e1. doi: 10.1016/j.jaci.2011.09.010
87. Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. (2008) 122:1105–12.e1. doi: 10.1016/j.jaci.2008.09.027
88. Tsuda M, Torgerson TR, Selmi C, Gambineri E, Carneiro-Sampaio M, Mannurita SC, et al. The spectrum of autoantibodies in IPEX syndrome is broad and includes anti-mitochondrial autoantibodies. J Autoimmun. (2010) 35:265–8. doi: 10.1016/j.jaut.2010.06.017
89. Moes N, Rieux-Laucat F, Begue B, Verdier J, Neven B, Patey N, et al. Reduced expression of FOXP3 and regulatory T-cell function in severe forms of early-onset autoimmune enteropathy. Gastroenterology. (2010) 139:770–8. doi: 10.1053/j.gastro.2010.06.006
90. Tsoumpra MK, Fukumoto S, Matsumoto T, Takeda S, Wood MJA, Aoki Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine. (2019) 45:630–45. doi: 10.1016/j.ebiom.2019.06.036
91. van der Wal E, Bergsma AJ, Pijnenburg JM, van der Ploeg AT, Pijnappel W. Antisense oligonucleotides promote exon inclusion and correct the common c-32-13T>G GAA splicing variant in pompe disease. Mol Ther Nucleic Acids. (2017) 7:90–100. doi: 10.1016/j.omtn.2017.03.001
Keywords: Foxp3, isoform, alternative splicing, IPEX, CD4+ T cell
Citation: Mailer RK (2020) IPEX as a Consequence of Alternatively Spliced FOXP3. Front. Pediatr. 8:594375. doi: 10.3389/fped.2020.594375
Received: 13 August 2020; Accepted: 21 September 2020;
Published: 21 October 2020.
Edited by:
Talal A. Chatila, Boston Children's Hospital and Harvard Medical School, United StatesReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilOskar A. Haas, St. Anna Children's Cancer Research Institute (CCRI), Austria
Copyright © 2020 Mailer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reiner K. Mailer, ci5tYWlsZXJAdWtlLmRl