 Adrien Guillaume1,2*
Adrien Guillaume1,2* Benedetta Chiodini2†
Benedetta Chiodini2† Brigitte Adams2†
Brigitte Adams2† Karin Dahan3†Georges Deschênes4†
Karin Dahan3†Georges Deschênes4† Khalid Ismaili2†
Khalid Ismaili2†- 1Department of Neonatology, Hôpital Erasme, Université Libre de Bruxelles (ULB), Brussels, Belgium
- 2Department of Pediatric Nephrology, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles (ULB), Brussels, Belgium
- 3Department of Genetics, Institute Pathology and Genetic (IPG), Gosselies, Belgium
- 4Department of Pediatric Nephrology, Paris University Hospital Robert Debré, Paris, France
Introduction: Oxalate overproduction in Primary Hyperoxaluria type I (PH1) leads to progressive renal failure and systemic oxalate deposition. In severe infantile forms of PH1 (IPH1), end-stage renal disease (ESRD) occurs in the first years of life. Usually, the management of these infantile forms is challenging and consists in an intensive dialysis regimen followed by a liver-kidney transplantation (combined or sequential).
Methods: Medical records of all infants with IPH1 reaching ESRD within the first year of life, diagnosed and followed between 2005 and 2018 in two pediatric nephrology departments in Brussels and Paris, have been reviewed.
Results: Seven patients were included. They reached ESRD at a median age of 3.5 (2–7) months. Dialysis was started at a median age of 4 (2–10 months). Peritoneal dialysis (PD) was the initial treatment for 6 patients and hemodialysis (HD) for one patient. Liver transplantation (LT) was performed in all patients and kidney transplantation (KT) in six of them. A sequential strategy has been chosen in 5 patients, a combined in one. The kidney transplanted as part of the combined strategy was lost. Median age at LT and KT was 25 (10–41) months and 32.5 (26–75) months, respectively. No death occurred in the series. At the end of a median follow-up of 3 years, mean eGFR was 64 ± 29 ml/min/1.73 m2. All patients presented retinal and bone lesions and five patients presented bones fractures.
Conclusion: Despite encouraging survival figures, the morbidity in IPH1 patients remains extremely heavy and its management presents a huge challenge. Thanks to the newly developed RNA-interference drug, the future holds brighter prospects.
Introduction
Primary hyperoxaluria type I (PH1) is a rare autosomal recessive disease caused by an enzymatic defect of the alanine-glyoxylate amino-transferase, an hepatocyte peroxisomal enzyme coded by the AGXT gene (1, 2). In Europe, PH1 incidence is estimated at 1 per 120,000 living births (3). The infantile form of PH1 (IPH1) accounts for about 10% of PH1 cases in Europe and North America (4) and usually constitutes a life-threatening disease. It is symptomatic before the age of 1 year and quickly progresses to end-stage renal disease (ESRD) within the first 3 years of life in 80% of the patients (3, 5). In these infants, the renal clearance is exceeded by the liver production resulting in systemic oxalate accumulation (6). Oxalate depositions concern multiple organs such as the heart (7), retina (8), nervous system (9), skin (10), and bones (11, 12). The bone overload of oxalate can lead to spontaneous bone fractures but also to joint (13) and medullary (14) damages.
IPH1 has a non-specific clinical presentation, which frequently leads to a delay in diagnosis. Once suspected, the oxalate concentration in urine and blood should be measured and the disease should be confirmed by sequencing the AGXT gene.
Current management recommendations for IPH1 are mostly based on the 2012 guidelines of the European Hyperoxaluria Consortium (OxalEurope) (15). A conservative treatment should be started as soon as possible to slow the progression of the disease (15). Unfortunately, in IPH1 the kidney failure is often very severe at the time of diagnosis and an aggressive dialysis regimen is needed to provide maximal oxalate clearance. Nowadays the only curative option to stop the endogenous production of oxalate and restore the renal function is a double liver (LT) and kidney transplantation (KT). Depending on local practice, the two interventions can either be combined (simultaneous liver-kidney transplantation, CLKT) or sequential (liver first followed by a subsequent kidney transplantation, SLKT). The sequential procedure is usually preferred in infants for anatomical reasons and to prevent massive oxalate release from the systemic stock damaging the kidney graft (16).
New therapeutic options based on RNA interference are forthcoming. They seek to selectively silence the glycolate oxidase or the lactate deshydrogenase A enzymes in hepatocytes, thus reducing oxalate production.
Phase 3 clinical trials are now underway (17, 18), and preliminary results showed a normalization of urine oxalate excretion without significant adverse effect in most of the PH1 treated patients. This could be an important turning point as such new treatments will most probably transform the management and prognosis of the disease.
This retrospective study aims to highlight the numerous complications occurring in IPH1 affected patients and their outcome after transplantation. It illustrates the challenges and difficulties we are still facing in the management of infants affected by this condition.
Methods
Ethics
This study protocol, involving human patient data, was reviewed and approved by the Ethics Committee of the Queen Fabiola Children's University Hospital in Brussels (N° CEH 43/19).
We reviewed the clinical data of all infants affected by IPH1 followed at Hôpital Universitaire des Enfants - Reine Fabiola (HUDERF) in Brussels and at Hôpital Robert Debré in Paris between January 2005 and December 2018. All patients had genetically proven PH1, reached ESRD during the first year of life and were enrolled in a liver-kidney transplantation program.
The data recorded included: patient and family history, clinical, laboratory, and genetic findings as well as additional examinations such as, renal ultrasound (US), standard bone radiographies, and ophthalmic examinations. Frequency, mode and duration of dialysis (pre and post liver transplantation) have also been recorded, as well as interval between kidney and liver transplantation. Follow-up data included patient and grafts outcomes, including post-transplantation surgical and oxalosis related complications. Glomerular filtration rates have been estimated according to the Schwartz formula (19).
Oxalate depositions were assessed in the retina by an eye fundus examination. Bone involvement was defined by the presence of one or several of the following features on standard radiographies: cystic bone changes, deformations, fractures, dense metaphyseal bands, cortical thickening in long bones, and dense rims around the ossification nucleus in the calcaneum and carpal/tarsal bones (13).
Numerical results have been presented as proportions for categorical variables, means and confidence interval for normally distributed continuous variables or medians with range in case of skewed distribution.
Plasma oxalate levels (POx) of all patients were measured before and after LT. Normal distribution of the two samples was confirmed using Shapiro-Wilk test. The means were compared using Welch-Shattertwhaite T-test for two independent samples with unequal variance. A p-value <0.05 was considered statistically significant. Statistical analysis was performed using XLSTAT.
Results
Patient Characteristics
Seven children were included in the study, four boys and three girls. Four patients had consanguineous parents. None had known antenatal anomalies. Patients characteristics are reported in Table 1.

Table 1. Characteristics of included patients.
At time of first presentation, five children had clinical features of ESRD (oligo-anuria, vomiting, and asthenia) and all of them presented with failure to thrive (see Figure 1). They all showed poorly differentiated and hyperechogenic kidneys at first ultrasound examination compatible with nephrocalcinosis.
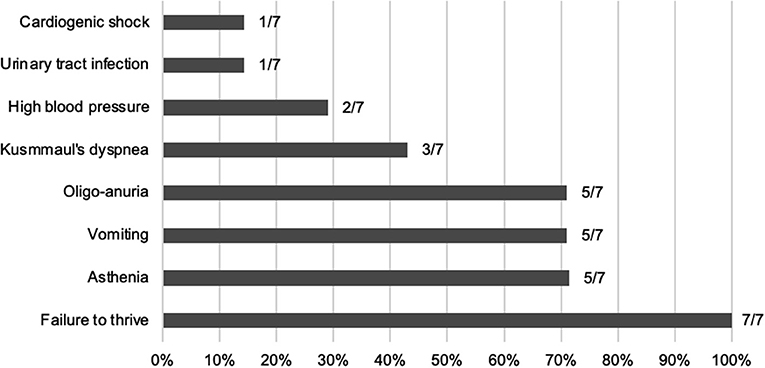
Figure 1. Frequency of the clinical features at diagnosis.
In all patients, PH1 was suspected at first presentation and rapidly confirmed by AGXT sequencing (see Table 1) except patient 1, for whom PH1 was diagnosed at the age of 14 months. At the end of the study, all patients were alive with a median age of 6.4 (3.4–12.3) years.
Dialysis
All children started dialysis at a median age of 4 (2–10 months). Daily nocturnal peritoneal dialysis (PD) was the initial modality for six patients and hemodialysis (HD) for one. HD followed PD in four patients and was combined with PD in two children. In all patients, PD was ended after liver transplantation (LT) (see Table 2).
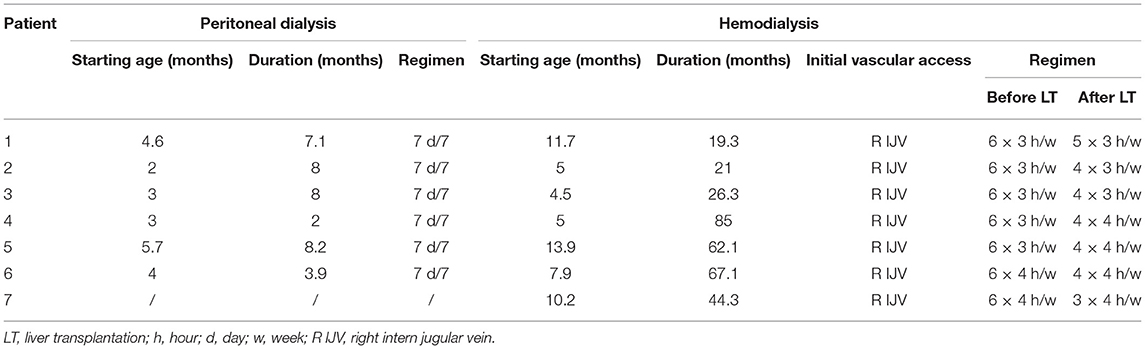
Table 2. Renal replacement therapy.
Up to LT, HD was performed six times a week for 3–4 h. The number of HD sessions after LT was adapted to plasma oxalate levels and progressively reduced in all patients.
In six patients, dialysis was stopped after successful kidney transplantation (KT). At the end of follow-up, one patient was still on a waiting list for a kidney transplant.
Transplantation
Liver Transplantation
All patients received a splitted liver graft from cadaveric donor, and one of them needed a second LT. Six transplantations were sequential and one was combined. The median age at first LT was 25 (10–41) months. The survival graft rate was of 88% after a mean follow-up of 55 ± 40 months (see Table 3).
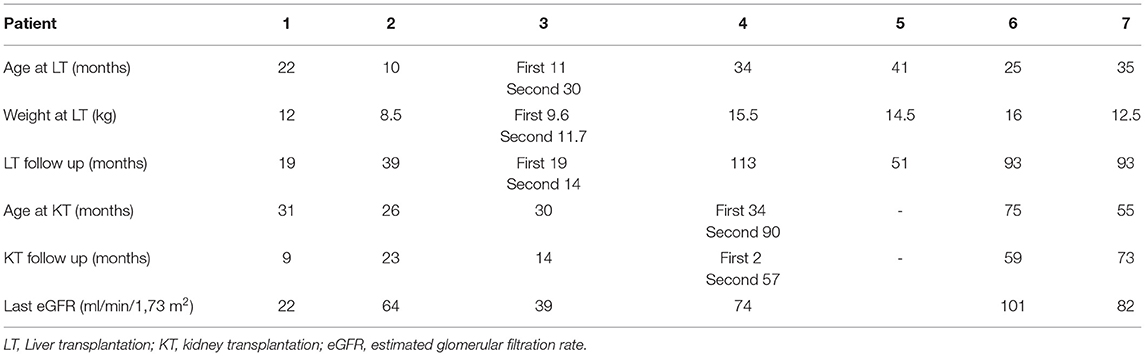
Table 3. Liver and Kidney transplantations.
Post-LT complications were as followed: five acute rejections, four intra-abdominal infections, one biliary leak, one biliary stenosis, and one vascular stenosis. A surgical revision was needed in five patients. In patient 3, liver graft loss occurred 19 months post-transplantation secondary to biliary and infectious complications. He received a second liver graft at the time of KT at the age of 30 months.
Kidney Transplantation
Six patients received a kidney graft, one of them needed a second KT (patient 4) and one is still on the waiting list (patient 5).
Median age at first KT was 33 (26–75) months. The mean interval between LT and KT was 26 ± 17 months.
At the last visit, all the transplanted children had a functioning graft after a mean follow-up of 36 ± 29 months with a mean eGFR of 64 ± 29 ml/min/1.73 m2.
The only kidney transplantation loss occurred in patient 4 immediately after CLKT. Biopsies showed extended tubular necrosis and ischemic glomerular lesions. He eventually received a second kidney graft at the age of 90 months.
Patient 5 developed HLA hyperimmunization after LT and is still waiting for a compatible donor.
No graft nephrocalcinosis was observed on US at the end of the follow-up.
Plasma Oxalate Levels
POx levels were regularly measured in all patients before the dialysis session (see Figure 2). The overall POx mean levels before and after LT periods were of 120 ± 44 μmol/L and 79 ± 26 μmol/L, respectively. The difference was statistically significant (p <0.0001).
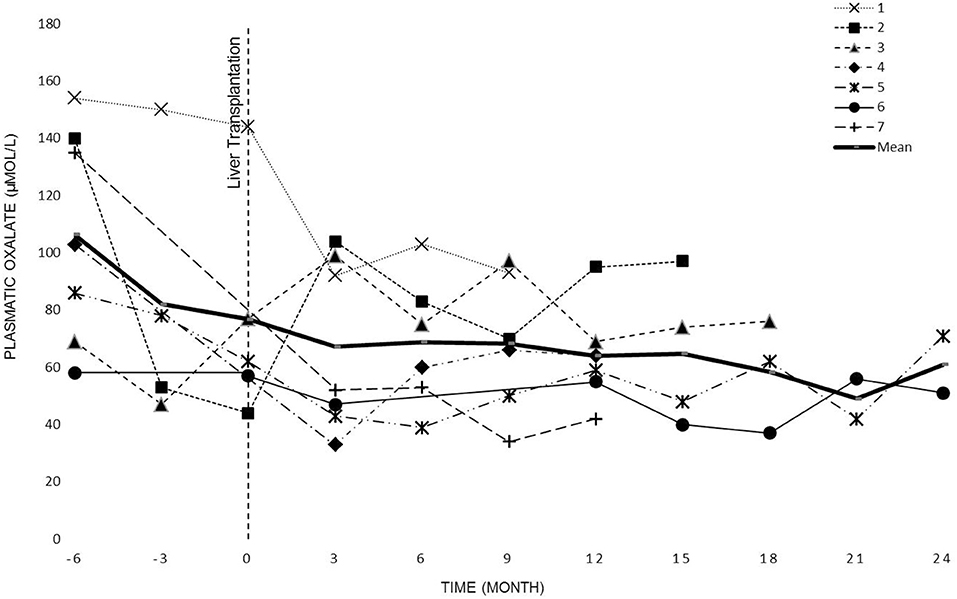
Figure 2. Patient's plasmatic oxalate evolution before KT.
The evolution of oxalate plasmatic levels before LT up to KT or 2 years after LT (data collection stops at KT) is shown in Figure 2.
Complications of Oxalate Depositions
From time of IPH1 diagnosis onwards, the presence of retinal and bone oxalate depositions was regularly assessed (see Table 4).

Table 4. Extra-renal complications of oxalosis.
Retinal oxalate depositions were observed in all patients before LT, they remained stable after LT and did not regress afterwards. Three patients developed a significant visual impairment (corrected visual acuity <5/10).
Radiographic signs of oxalate bone deposits were found in all patients. Four children presented bones fractures and two patients were particularly affected by several fractures before LT and in the first months after LT.
Discussion
IPH1 remains a rare and severe condition. In an historical European cohort of 38 IPH1 patients, Harambat et al. (20) observed a mortality rate of 26% 3 years after initiation of dialysis.
In our series no death was observed at the end of the follow-up. However, the intensive dialysis regimen, the complications of the double organ transplantation, and the systemic oxalate deposition led to a severe morbidity.
Because antenatal anomalies were not reported in this series, no specific management was undertaken at birth, and most of the patients presented severe chronic renal insufficiency, failure to thrive and nephrocalcinosis at the time of diagnosis.
Among all seven patients, seven different AGXT mutations were identified (Table 1). All except one have already been described in the literature as responsible for IPH1 (class 5) (21, 22). Patient 3 is compound heterozygote for two causative variants, previously reported without precise phenotypic description (22). Only Patient 7 is homozygous for a novel change, classifying the variant as likely pathogenic (class 4). This change is an insertion variant corresponding to the insertion of a G nucleotide between nucleotides c.349 and c.350, which is a frame-shift leading the introduction of an earlier termination codon (p.Glu117fs).
While waiting for liver-kidney transplantation, current guidelines recommend initiating intensive dialysis as soon as possible in order to target plasma oxalate levels below 30–45 μmol/L and avoid systemic oxalosis (3, 15). As HD provides a better oxalate clearance than PD (23), HD is recommended by the European Pediatric Working Group as the first-choice (24). However, treating small infants with chronic HD is challenging as it requires a functional vascular access and adapted equipment. Moreover, episodes of hypotension, and catheter infections or occlusions can occur (25, 26). Therefore, in our series PD was the initial dialysis modality in six out of seven patients and was combined or switched to HD as soon as possible. HD was performed six times a week, as short and frequent sessions proved to be more effective than a longer and less frequent dialysis regimen (27).
Previous studies (23, 28) have shown that even intensive dialysis regimens are insufficient to adequately remove the endogenous production of oxalate. Our experience confirms these findings. Despite intense dialysis we observed high oxalate levels in all patients (Figure 2).
It is commonly accepted that early liver-kidney transplantation is the best treatment option for IPH1 patients who have reached ESRD (15,16). Currently, there is no consensus on the best transplantation strategy between SLKT and CLKT, each strategy presenting advantages and drawbacks. In small children where anatomy may preclude a CKLT, SKLT offers the possibility of an earlier liver transplantation. However, several publications reported successful CKLT in IPH1 patients weighing 10–12 kg (29–32). In our cohort, six out of seven patients underwent a SLKT, with the aim of preserving the renal graft from new oxalate deposition, and to allow early LT even in small children. Their median weight at LT was 12 kg (the youngest was 10 months old and weighed 8.5 kg at LT). We did not observe any kidney graft loss after SLKT during the follow-up period. On the contrary, the only patient who received a CLKT experienced an immediate renal graft failure and needed a subsequent kidney transplantation.
As regard as the SKLT, the optimal interval between the two transplantations is unknown. Previous studies reported intervals from 2 to 17 months (33–41). In our patients, we were aiming to achieve POx levels as low as possible (ideally POx <20 μmol/L) (15), in order to protect the kidney graft from new oxalate deposition. However, the oxalate levels remained high with an overall mean of 79 μmol/L despite intense dialysis. Therefore, the mean interval between the two transplantations was extended to 26 months.
According to our experience, the decision about the timing of KT should probably not be based exclusively on the POx level, as this level can remain high for a too long period after LT.
Our data illustrates that even the combination of HD and PD and an early liver transplantation is not sufficient to protect against systemic oxalosis and extra-renal deposits. In our cohort, all children developed severe radiological signs of bone oxalate deposits during follow up. More than half of them presented pathological fractures, even months after LT. This is explained by the fact that both oxalate deposits and oxalate release from the bone compartment weaken the skeleton (11). We advise to thoroughly search patients for fractures and to handle the IPH1 patient with care during mobilization, both before and after LT. A good renal osteodystrophy prevention is mandatory in these patients.
The retina is also affected in IPH1. While at time of diagnosis most of our patients did not show any eye involvement, at time of LT they all showed significant oxalate retinopathy. Consistently with recent publications, oxalate retinopathy did not improve with transplantation and time (42).
Up to now, the management and the burden of IPH1 disease is extremely challenging. With the arrival of RNAi therapies, which seek to inhibit the endogenous oxalate production, a revolution in the management of IPH1 can be expected.
The avoidance of liver transplantation will probably be the most important improvement due to iRNA therapy.
Whilst it can be expected that dialysis will still be necessary for IPH1 patients with ESRD at diagnosis, the intensity of the dialysis regimen will probably be significantly lighter.
Although kidney transplantation will still be needed for PH1 patients having reached ESRD, graft survival could get closer to non-PH1 patients (20).
Finally, since the extra-renal complications are usually mild at diagnosis, we do expect an important decrease in their incidence and gravity with an early treatment by iRNA therapy.
Conclusion
Currently, the management of IPH1 remains particularly challenging. Hopefully this will change substantially in the near future.
In the meantime, and based on our experience we propose the following approach:
• Intensive HD regimen needs to be started as soon as possible, the combination of PD and HD may be needed to maximize oxalate clearance.
• The SLKT allows an earlier LT and thus an earlier correction of the metabolic defect and should be considered as first choice.
• Plasma oxalate level should not be the only factor to define the interval between LT and KT, as it can remain above saturation threshold for several months after LT.
• Efficient fracture screening and prevention is mandatory.
New therapeutics using RNA interference could be a real game changer for IPH1 children as it could stop the endogenous oxalate production, and thus, avoid the need for LT and intensive dialysis and also prevent the extra-renal complications of oxalosis.
Data Availability Statement
The datasets analyzed during the current study are not publicly available due to confidential reasons but are available from the corresponding author on reasonable request.
Ethics Statement
This study protocol, involving human patient data, was reviewed and approved by the Ethics Committee of the Queen Fabiola Children's University Hospital in Brussels (N° CEH 43/19).
Author Contributions
Material preparation, data collection, and analysis were performed by AG. The first draft of the manuscript was written by AG, BC, BA, KD, and KI. All authors contributed to the study conception and design, commented on previous versions of the manuscript, and read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Milliner D, Eickholt J, Bergstralh E, Wilson D, Smith L. Primary hyperoxaluria: results of long-term treatment with orthophosphate and pyridoxine. N Engl J Med. (1994) 331:1553–8. doi: 10.1056/NEJM199412083312304
3. Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. (2013) 369:649–58. doi: 10.1056/NEJMra1301564
4. Milliner DS, Harris PC, Cogal AG, Lieske JC. Primary hyperoxaluria type 1. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A, editors. GeneReviews(R). Seattle, WA: University of Washington, Seattle (2017).
5. Lieske JC, Monico CG, Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, et al. International registry for primary hyperoxaluria. Am J Nephrol. (2005) 25:290–6. doi: 10.1159/000086360
6. Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. (2009) 75:1264–71. doi: 10.1038/ki.2009.32
7. Mookadam F, Smith T, Jiamsripong P, Moustafa SE, Monico CG, Lieske JC, et al. Cardiac abnormalities in primary hyperoxaluria. Circ J. (2010) 74: 2403–9. doi: 10.1253/circj.CJ-10-0107
8. Punjabi OS, Riaz K, Mets MB. Crystalline retinopathy in primary hyperoxaluria. J AAPOS. (2011) 15:214–6. doi: 10.1016/j.jaapos.2010.12.015
9. Beck BB, Hoyer-Kuhn H, Göbel H, Habbig S, Hoppe B. Hyperoxaluria and systemic oxalosis: an update on current therapy and future directions. Expert Opin Investig Drugs. (2013) 22:117–29. doi: 10.1517/13543784.2013.741587
10. Bogle MA, Teller CF, Tschen JA, Smith CA, Wang A. Primary hyperoxaluria in a 27-year-old woman. J Am Acad Dermatol. (2003) 49:725–8. doi: 10.1067/S0190-9622(03)00119-1
11. Bacchetta J, Farlay D, Abelin-Genevois K, Lebourg L, Cochat P, Boivin G. Bone impairment in oxalosis: an ultrastructural bone analysis. Bone. (2015) 81:161–7. doi: 10.1016/j.bone.2015.07.010
12. Marangella M, Vitale C, Petrarulo M, Tricerri A, Cerelli E, Cadario A, et al. Bony content of oxalate in patients with primary hyperoxaluria or oxalosis-unrelated renal failure. Kidney Int. (1995) 48:182–7. doi: 10.1038/ki.1995.283
13. Bacchetta J, Boivin G, Cochat P. Bone impairment in primary hyperoxaluria: a review. Pediatr Nephrol. (2016) 31:1–6. doi: 10.1007/s00467-015-3048-z
14. Bakshi NA, Al-Zahrani H. Bone marrow oxalosis. Blood. (2012) 120:8. doi: 10.1182/blood-2011-12-400192
15. Cochat P, Hulton S-A, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. (2012) 27:1729–36. doi: 10.1093/ndt/gfs078
16. Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. (2012) 8:467–75. doi: 10.1038/nrneph.2012.113
17. Alnylam Pharmaceuticals. ILLUMINATE-A: A Phase 3 Randomized, Double-Blind, Placebo-Controlled Study With an Extended Dosing Period to Evaluate the Efficacy and Safety of Lumasiran in Children and Adults With Primary Hyperoxaluria Type 1. clinicaltrials.gov (2020). Available online at: https://clinicaltrials.gov/ct2/show/NCT03681184 (accessed July 27, 2020).
18. Dicerna Pharmaceuticals Inc. A Placebo-Controlled, Single-Blind, Single-Center Phase 1 Study in Normal Healthy Volunteers and Open-Label Multi-Center Study in Patients With Primary Hyperoxaluria to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Ascending Doses of DCR-PHXC Solution for Injection (Subcutaneous Use). clinicaltrials.gov (2020). Available online at: https://clinicaltrials.gov/ct2/show/study/NCT03392896 (accessed July 27, 2020).
19. Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. (2009) 20:629–37. doi: 10.1681/ASN.2008030287
20. Harambat J, van Stralen KJ, Espinosa L, Groothoff JW, Hulton S-A, Cerkauskiene R, et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol. (2012) 7:458–65. doi: 10.2215/CJN.07430711
21. Harambat J, Fargue S, Acquaviva C, Gagnadoux M-F, Janssen F, Liutkus A, et al. Genotype–phenotype correlation in primary hyperoxaluria type 1: the p. Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. (2010) 77:443–9. doi: 10.1038/ki.2009.435
22. Mandrile G, van Woerden CS, Berchialla P, Beck BB, Acquaviva Bourdain C, Hulton S-A, et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. (2014) 86:1197–204. doi: 10.1038/ki.2014.222
23. Hoppe B, Graf D, Offner G, Latta K, Byrd DJ, Michalk D, et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol. (1996) 10:488–92. doi: 10.1007/s004670050145
24. Zurowska AM, Fischbach M, Watson AR, Edefonti A, Stefanidis CJ, Group EPDW. Clinical practice recommendations for the care of infants with stage 5 chronic kidney disease (CKD5). Pediatr Nephrol. (2013) 28:1739–48. doi: 10.1007/s00467-012-2300-z
25. Paul A, Fraser N, Manoharan S, Williams AR, Shenoy MU. The challenge of maintaining dialysis lines in the under twos. J Pediatr Urol. (2011) 7:48–51. doi: 10.1016/j.jpurol.2010.01.018
26. Symons JM. Renal replacement therapy for the critically ill infant. In: Ronco C, Bellomo R, Kellum JA, editors. Critical Care Nephrology. Philadelphia, PA: Elsevier. p. 1230–5. doi: 10.1016/B978-0-323-44942-7.00206-5
27. Illies F, Bonzel K-E, Wingen A-M, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int. (2006) 70:1642–8. doi: 10.1038/sj.ki.5001806
28. Bunchman T, Swartz R. Oxalate removal in type I hyperoxaluria or acquired oxalosis using HD and equilibration PD. Perit Dial Int. (1994) 14:81–4. doi: 10.1177/089686089401400117
29. Duclaux-Loras R, Bacchetta J, Berthiller J, Rivet C, Demède D, Javouhey E, et al. Pediatric combined liver–kidney transplantation: a single-center experience of 18 cases. Pediatr Nephrol. (2016) 31:1517–29. doi: 10.1007/s00467-016-3324-6
30. Herden U, Kemper M, Ganschow R, Klaassen I, Grabhorn E, Brinkert F, et al. Surgical aspects and outcome of combined liver and kidney transplantation in children. Transpl Int. (2011) 24:805–11. doi: 10.1111/j.1432-2277.2011.01278.x
31. Ranawaka R, Lloyd C, McKiernan PJ, Hulton SA, Sharif K, Milford DV. Combined liver and kidney transplantation in children: analysis of renal graft outcome. Pediatric Nephrol. (2016) 31:1539–1543. doi: 10.1007/s00467-016-3396-3
32. Szymczak M, Kaliciński P, Kowalewski G, Markiewicz-Kijewska M, Broniszczak D, Ismail H, et al. Combined liver-kidney transplantation in children: single-center experiences and long-term results. Transplant Proc. (2018) 50:2140–4. doi: 10.1016/j.transproceed.2018.04.061
33. Rosenblatt GS, Jenkins RD, Barry JM. Treatment of primary hyperoxaluria type 1 with sequential liver and kidney transplants from the same living donor. Urology. (2006) 68:427.e7–8. doi: 10.1016/j.urology.2006.02.035
34. Büscher R, Büscher AK, Cetiner M, Treckmann JW, Paul A, Vester U, et al. Combined liver and kidney transplantation and kidney after liver transplantation in children: indication, postoperative outcome, and long-term results. Pediatr Transplant. (2015) 19:858–65. doi: 10.1111/petr.12595
35. Shapiro R, Weismann I, Mandel H, Eisenstein B, Ben-Ari Z, Bar-Nathan N, et al. Primary hyperoxaluria type 1: improved outcome with timely liver transplantation: a single-center report of 36 children. Transplantation. (2001) 72:428–32. doi: 10.1097/00007890-200108150-00012
36. Malla I, Lysy PA, Godefroid N, Smets F, Malaise J, Reding R, et al. Two-step transplantation for primary hyperoxaluria: cadaveric liver followed by living donor related kidney transplantation. Pediatr Transplant. (2009) 13:782–4. doi: 10.1111/j.1399-3046.2008.01049.x
37. Becker T, Nyibata M, Lueck R, Bektas H, Demirci G, Lehner F, et al. Results of combined and sequential liver-kidney transplantation. Liver Transplant. (2003) 9:1067–78. doi: 10.1053/jlts.2003.50210
38. Nakamura M, Fuchinoue S, Nakajima I, Kitajima K, Tojimbara T, Takasaki K, et al. Three cases of sequential liver–kidney transplantation fromliving-related donors. Nephrol Dial Transplant. (2001) 16:166–8. doi: 10.1093/ndt/16.1.166
39. Sato S, Fuchinoue S, Kimikawa M, Tojimbara T, Nakajima I, Teraoka S, et al. Sequential liver-kidney transplantation from a living-related donor in primary hyperoxaluria type 1 (oxalosis). Transplant Proc. (2003) 35:373–4. doi: 10.1016/S0041-1345(02)03911-8
40. Motoyoshil Y, Hattori M, Chikamoto H, Nakakura H, Furue T, Miyakawa S, et al. Sequential combined liver-kidney transplantation for a one-year-old boy with infantile primary hyperoxaluria type 1. Nihon Jinzo Gakkai shi. (2006) 48:22–28.
41. Ozer A, Aktas H, Bulum B, Emiroglu R. The experience of combined and sequential liver and kidney transplantation from a single living donor in patients with primary hyperoxaluria type 1. Pediatr Transplant. (2019) 23:e13406. doi: 10.1111/petr.13406
Keywords: kidney transplanation, dialysis (ESKD), hyperoxaluria type 1, infant, liver transplatation
Citation: Guillaume A, Chiodini B, Adams B, Dahan K, Deschênes G and Ismaili K (2021) The Struggling Odyssey of Infantile Primary Hyperoxaluria. Front. Pediatr. 9:615183. doi: 10.3389/fped.2021.615183
Received: 08 October 2020; Accepted: 22 March 2021;
Published: 20 April 2021.
Edited by:
Michael L. Moritz, University of Pittsburgh, United StatesReviewed by:
Dagmara Borzych-Duzalka, Medical University of Gdansk, PolandRainer Büscher, Essen University Hospital, Germany
Copyright © 2021 Guillaume, Chiodini, Adams, Dahan, Deschênes and Ismaili. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrien Guillaume, QWRyaWVuLkd1aWxsYXVtZUBlcmFzbWUudWxiLmFjLmJl
†ORCID: Benedetta Chiodini orcid.org/0000-0002-4550-5702
Brigitte Adams orcid.org/0000-0001-9693-2205
Karin Dahan orcid.org/0000-0003-3036-7981
Georges Deschêsnes orcid.org/0000-0002-5158-2915
Khalid Ismaili orcid.org/0000-0001-6791-9634