 Claudia L. Gaefke1†
Claudia L. Gaefke1† Jonathan Metts2†
Jonathan Metts2† Donya Imanirad1
Donya Imanirad1 Daime Nieves3Paola Terranova4Gianluca Dell'Orso4
Daime Nieves3Paola Terranova4Gianluca Dell'Orso4 Eleonora Gambineri5Maurizio Miano4Richard F. Lockey1
Eleonora Gambineri5Maurizio Miano4Richard F. Lockey1 Jolan Eszter Walter3,6*
Jolan Eszter Walter3,6* Emma Westermann-Clark1,3
Emma Westermann-Clark1,3- 1Department of Medicine, University of South Florida, Tampa, FL, United States
- 2Cancer and Blood Disorders Institute, Johns Hopkins All Children's Hospital, Saint Petersburg, FL, United States
- 3Department of Pediatrics, University of South Florida, Saint Petersburg, FL, United States
- 4Hematology Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 5NEUROFARBA, Meyer Children's Hospital, Florence, Italy
- 6Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States
Autoimmune Lymphoproliferative Syndrome (ALPS), commonly caused by mutations in the FAS gene, is a disease with variable penetrance. Subjects may be asymptomatic, or they may present with lymphadenopathy, splenomegaly, cytopenias, or malignancy. Prompt recognition of ALPS is needed for optimal management. We describe a multi-generational cohort presenting with clinical manifestations of ALPS, and a previously unreported heterozygous missense variant of uncertain significance in FAS (c.758G >T, p.G253V), located in exon 9. Knowledge of the underlying genetic defect permitted prompt targeted therapy to treat acute episodes of cytopenia. This cohort underscores the importance of genetic testing in subjects with clinical features of ALPS and should facilitate the reclassification of this variant as pathogenic.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is characterized by lymphadenopathy, splenomegaly, autoimmunity, especially autoimmune cytopenias, and an increased risk of lymphoma. Lymphoproliferation and autoimmunity result from failure of effector T-cells to undergo programmed cell death. The most common mutation associated with ALPS lies in the FAS gene, and is labeled as ALPS-FAS; other known mutations that cause ALPS include Fas ligand (FAS-L) and caspase 10 (CASP 10) (1, 2). Germline mutations in FAS, inherited in an autosomal dominant manner, comprise the largest group of ALPS cases, while somatic mutations in FAS are the second most common genetic cause of ALPS (2, 3). There is an expanding spectrum of ALPS-like disorders, including caspase 8 deficiency state (CEDS, resulting from a germline mutation in caspase 8 [CASP8]); RAS-associated autoimmune leukoproliferative disease (RALD), resulting from gain of function somatic mutation of the neuroblastoma RAS viral oncogene (NRAS) or proto-oncogene Kirsten Rat Sarcoma virus (KRAS); X-linked lymphoproliferative syndrome (XLP1) resulting from mutation or deletion of the SH2D1A gene; heterozygous loss of function mutation in nuclear factor kappa light chain enhancer of activated B cells (NFKB-1), heterozygous loss of function mutation of cytotoxic T-lymphocyte associated protein 4 (CTLA-4), and mutations of the FAS-associated protein with death domain (FADD), among others. Approximately one-third of subjects with clinical features of ALPS have unidentified genetic defects (1, 2, 4). Patients with unknown genetic defects but clinical features of ALPS are classified as Dianzani autoimmune lymphoproliferative disease (DALD) (2).
Children with confirmed genetic findings and clinical features of ALPS require monitoring for cytopenias, splenomegaly, and malignancy as they age. A sizeable proportion of subjects with ALPS-FAS (74%) require medical/surgical intervention during their lifetime, and intervention is often initiated due to autoimmune cytopenias, massive splenomegaly, or adenopathy (3).
The variable clinical phenotype of ALPS contributes to the complexity of management. Affected subjects can carry a pathogenic variant but be relatively asymptomatic, and biomarkers for development of lymphoproliferation and autoimmunity have not been fully established. It is challenging to determine when to initiate immunomodulating therapy with milder disease, such as intermittent thrombocytopenia. Targeted therapy, including T-cell modulation with sirolimus, can be beneficial, but timing and duration of therapy is unclear (5, 6). Splenectomy should be avoided because of the high risk of subsequent sepsis, increased risk of death, and failure to prevent recurrence of autoimmune cytopenia in many subjects (3, 7). Here we describe a family with a novel FAS mutation found across several generations and discuss challenges in long-term management.
Definition of ALPS
For this study, ALPS was defined as proposed by Oliveira et al. (2). These diagnostic criteria divide symptoms and laboratory values into required and accessory criteria. Required criteria include chronic (>6 months), non-malignant, non-infectious lymphadenopathy or splenomegaly or both, and elevated CD3+ T-cell receptor (TCR)αβCD4-CD8- double negative cells (DNTCs) (≥1.5% of total lymphocytes or 2.5% of CD3+ lymphocytes) in the setting of normal or elevated lymphocyte counts. Accessory criteria are divided into primary accessory [(1) defective lymphocyte apoptosis in two separate assays; (2) somatic or germline pathogenic mutation in FAS, FASLG, or CASP10] and secondary accessory criteria which include useful biomarkers (elevated soluble FAS-ligand [sFASL], interleukin-10 (IL-10), vitamin B12, interleukin-18 (IL-18) levels, elevated immunoglobulin G (IgG) levels), typical immuno-histological findings, autoimmune cytopenias (which must be accompanied by elevated IgG), or family history of non-malignant/non-infectious lymphoproliferation with or without autoimmunity. A definitive diagnosis is based on the presence of both required criteria plus one primary accessory criterion; a probable diagnosis is based on the presence of both required criteria plus one secondary accessory criterion (2).
Case Presentation
The proband (V-1) (Figure 1) is a 7-month-old female who presented to clinic with greater than 6 months of mild anterior and posterior cervical and axillary lymphadenopathy, with the largest lymph node measuring 1 cm in diameter in the neck. She also had a spleen of 7.5 cm in length, slightly larger than normal for age, with the upper limit being 7 cm in length for a 12-month old (8). She had a history of asthma triggered by viral respiratory tract infections and no history of recurrent or severe bacterial sinopulmonary infections. She was normal developmentally for her age. Complete blood count (CBC) with differential was normal at presentation, but at 2 years of age she experienced an episode of autoimmune hemolytic anemia, with hemoglobin nadir of 5.3 g/dL and mild thrombocytopenia (platelets 108,000). Immune evaluation was notable for normal immunoglobulin levels and normal diphtheria and tetanus antibody titers; however, pneumococcal titers were only protective for 50% of a 10-serotype panel, and patient had a low Haemophilus influenzae type B (Hib) titer (Table 1).
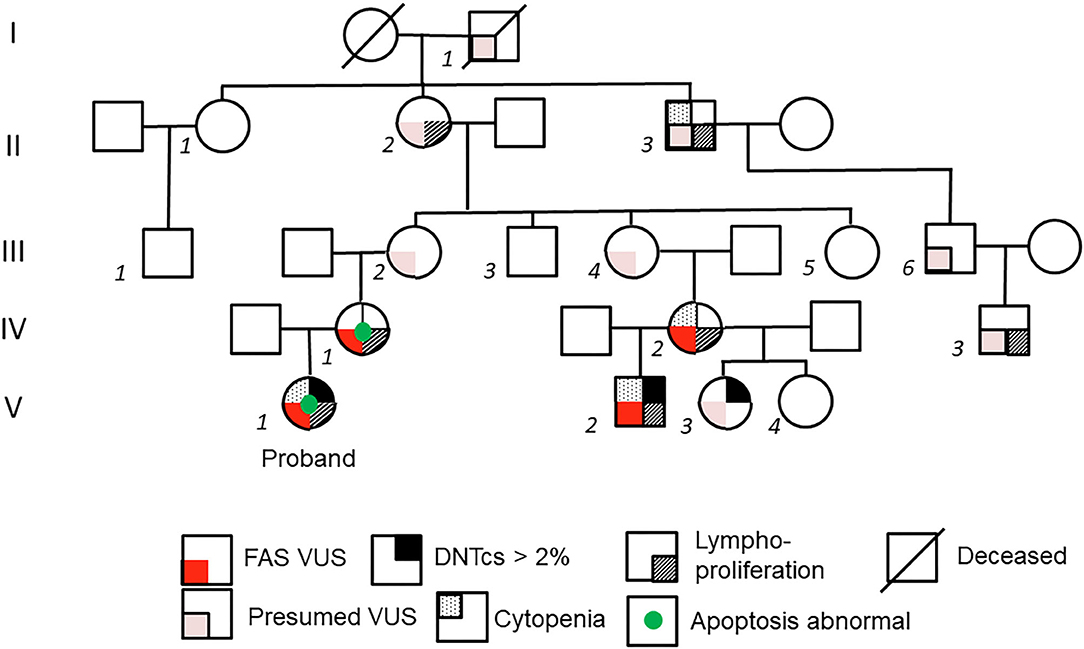
Figure 1. Pedigree of kindred. On left axis, generations are labeled with Roman numerals I-V. Individual members of the family are labeled with italicized Arabic numerals. Family members related only by marriage are not numbered. Detailed family history was obtained from subject II-2, the maternal great-grandmother of the proband V-1. Subject II-2 postulates that the FAS variant stems from her father's side of the family (I-1) because his sister (not depicted) died of leukemia at age 17.
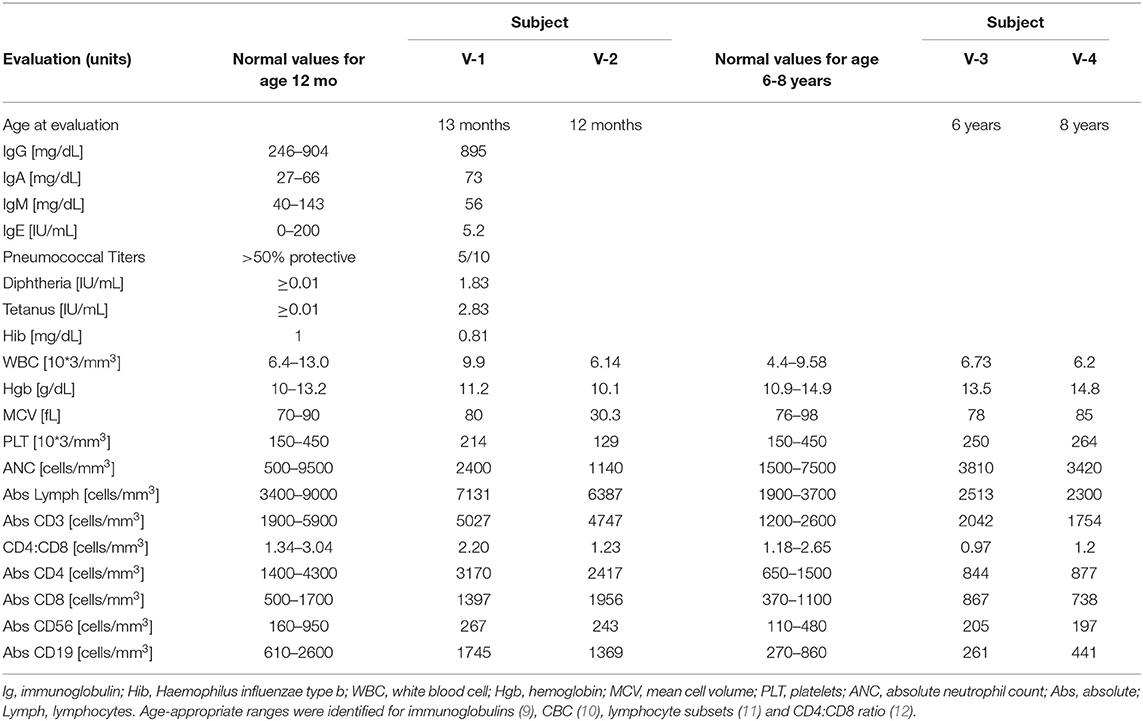
Table 1. Immunophenotypic evaluation of pediatric subjects.
Her mother (IV-1 in Figure 1) reported a personal history of splenomegaly, which had been diagnosed along with “kidney enlargement” during her pregnancy, and a family history of splenomegaly, lymphadenopathy, and hematologic malignancy in prior generation maternal relatives. The grandmother of subject IV-1, subject II-2, reported a personal history of neck lymphadenopathy requiring excision during her late teenage years. Subject II-2 suspected that she had inherited her tendency toward lymphadenopathy from her father (I-1 in Figure 1), whose sister had died of leukemia at age 17.
Among living family members with suspected ALPS, a 17-month-old male second cousin (V-2) was also seen in the clinic for persistent lymphadenopathy. Subject V-2 had posterior cervical lymphadenopathy, splenomegaly, and history of thrombocytopenia. There was no history of recurrent or severe infections, bleeding, or other illnesses. His mother (IV-2) reported a personal history of recurrent lymphadenopathy, splenomegaly since childhood, and chronic anemia with no formal history or testing for ALPS. Subject IV-1 and IV-2 (mothers to proband and V-2, respectively) share a maternal grandmother (II-2), who reported neck lymphadenopathy in her teenage years, as mentioned previously. This maternal grandmother (II-2) had a sister (II-1) who was healthy, and a brother (II-3) who required splenectomy at age 15 and often had abnormal blood cell counts in childhood. A child of II-1, (male child III-1), was healthy. II-3 had a son, III-6, who was healthy, but his grandson IV-3 required splenectomy at 1 year of age due to splenomegaly. Subject II-2 also had four children (subjects III-2, III-3, III-4, and III-5) who did not show notable signs of ALPS. Based upon this information, further evaluation was pursued in available family members, which included two half-siblings of subject V-2: subjects V-3 and V-4. Subject V-3, a 6-year-old female, had a history of recurrent oral lesions resembling herpes simplex virus with no clinical evidence of ALPS. Subject, V-4, an 8-year-old female, had a history of recurrent ear infections and otherwise no clinical history suggestive of ALPS.
Laboratory Evaluation and Results
Given this clinical multi-generational history, laboratory evaluation of the kindred was undertaken to identify a possible shared mutation leading to ALPS.
The proband and her mother were evaluated for ALPS. As noted above, the proband had a relatively normal immune phenotype except for low recall response to certain vaccine antigens. Proband's mother (IV-1) had mild leukocytosis on CBC but normal absolute lymphocytes (3100 cells/μL), and full immune phenotyping was not performed. Both proband and mother had an abnormal apoptosis assay; proband (V-1) had FAS activity of 38% of control, and mother (IV-1) had FAS activity 41% of control (normal 68–93%). Flow cytometry in V-1 showed >2% TCR αβDNTcs (Cincinnati's Children's Hospital ALPS Flow Cytometry panel, Cincinnati, OH) which prompted further evaluation for accessory criteria. Secondary accessory criteria, including clinical and serological markers, were met as noted on Table 2. For confirmation, genetic sequencing was pursued using a 207-gene Primary Immunodeficiency Panel (Invitae, San Francisco, CA). Genetic sequencing confirmed a mutation in FAS, c.758G>T (p.G253V), exon 9, in the death domain. This mutation was identified in both the proband and her mother. This sequence change replaces glycine with valine at codon 253 of the FAS protein (p.Gly253Val). The glycine residue is moderately conserved and there is a moderate physicochemical difference between glycine and valine. This variant is not reported in the literature with FAS-related conditions and is not present in Genome Aggregation Databases (gnomAD June 2020, no allele frequency). The Genome Aggregation Database (gnomAD), also known as ExAC in its first release, does not include individuals with G253V amino acid substitution mutations. It does include 5 individuals with a nearby p.His285Arg mutation among 251,308 alleles sequenced.
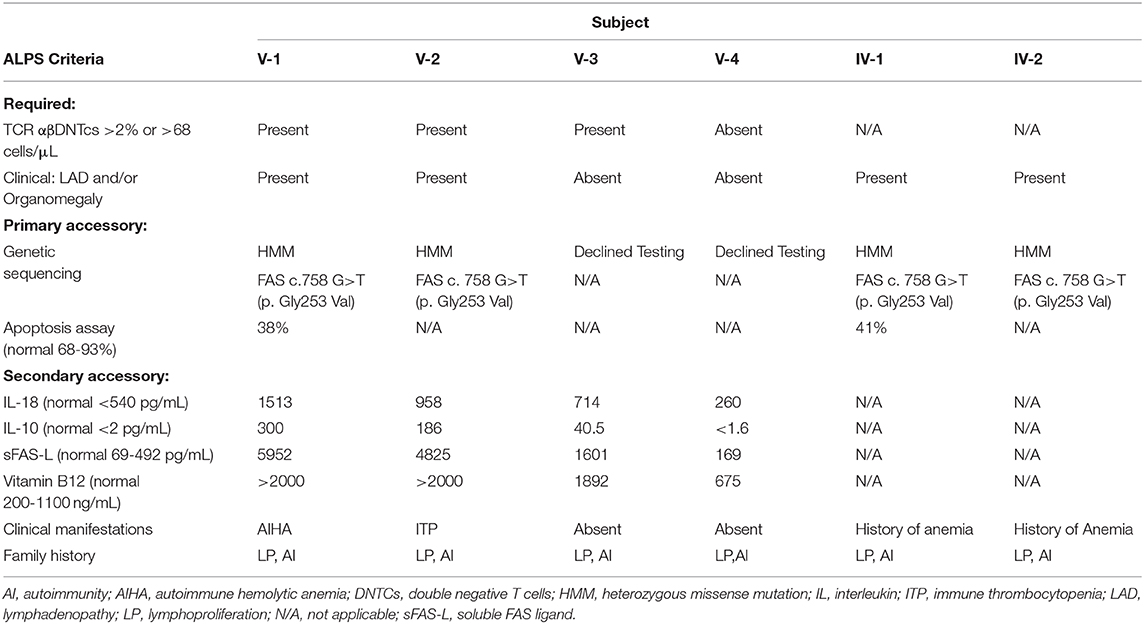
Table 2. Family members evaluated based on ALPS Criteria.
Laboratory evaluation for subjects V-2, V-3, and V-4 (second cousins of the proband) and their mother (IV-2) was performed. Subjects V-2, V-3, and V-4 did not undergo full immune phenotyping, but CBC and lymphocyte subsets, along with age-appropriate normal ranges, are reported in Table 1. ALPS panel testing was performed in V-2, V-3, and V-4. V-2 and V-3 had >2% TCR αβDNTcs and serological markers consistent with ALPS, while subject V-4 did not meet criteria for ALPS (Table 2). Subject IV-2 declined genetic sequencing for subject V-3 and V-4, but genetic sequencing in subject V-2 confirmed the same variant. Both mothers (subjects IV-1 and IV-2) did not wish to undergo immune phenotyping, serologic markers, or ALPS panel, but chose to pursue genetic testing; the same genetic variant was confirmed.
Evolution of Clinical Phenotype and Treatment
One year after initial presentation, the proband developed an acute episode of autoimmune hemolytic anemia, initially treated with corticosteroids and high dose intravenous immunoglobulin (IVIG). Since she had an established diagnosis of ALPS, targeted therapy with sirolimus was initiated at 2 mg/m2. She was able to discontinue corticosteroids and has not required further high dose IVIG or hospitalization (Figure 2). The family elected to discontinue sirolimus after 1 year of treatment, and she is currently being monitored for recurrence of cytopenias or progression of ALPS.
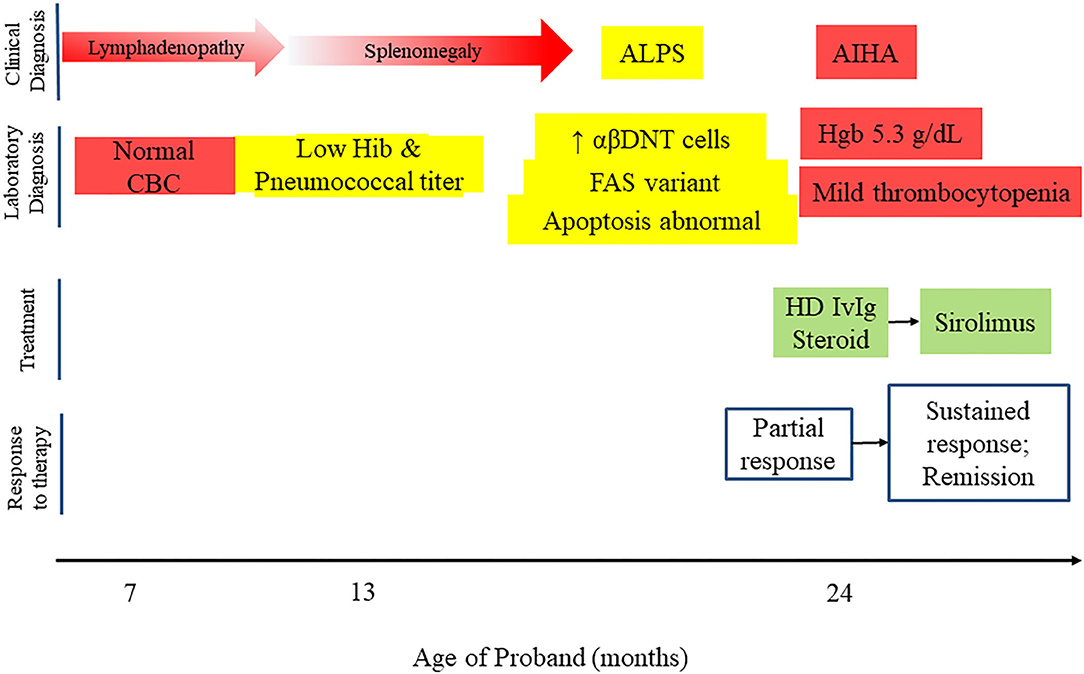
Figure 2. Timeline of proband's clinical course, including clinical and laboratory diagnosis, treatment and response to therapy.
Subject V-2, who met criteria for ALPS, was also found to have intermittent thrombocytopenia, with platelet counts between 84,000 and 200,000/μL. He is being monitored with plans to initiate sirolimus for platelet counts <50,000/μL or other persistent cytopenias.
Subject V-3 is being monitored for clinical manifestations of ALPS, as she meets required and secondary accessory criteria, but parents have declined genetic sequencing.
Subject V-4 did not manifest clinical or laboratory features of ALPS, and parents declined genetic sequencing.
Subjects IV-1 and IV-2, both of whom carry the VUS in FAS, are being monitored for further clinical manifestations of ALPS.
Discussion
Autoimmune lymphoproliferative syndrome is a disease with variable penetrance from asymptomatic to lymphadenopathy, splenomegaly, cytopenias, and malignancy. Though most patients develop lymphoproliferation at a median age of 11.5 months, seemingly unaffected family members with heterozygous FAS pathogenic variants are also predisposed to malignancy, most commonly non-Hodgkin lymphoma later in life, with a lifetime risk of up to 20% (13, 14). This underscores the importance of recognizing ALPS promptly to appropriately manage complications and to monitor asymptomatic kindred for prompt recognition of malignancy which may present later in life.
When the proband met criteria for ALPS, it was decided to pursue screening for willing kindred. By doing this, a second family member (subject V-2) was found to meet criteria for ALPS. A third family member (V-3) has not developed the required clinical manifestations of ALPS but does meet many laboratory benchmarks for ALPS diagnosis, including elevated DNTCs, IL-18, IL-10, sFAS-L, and vitamin B12. In total, four of the six family members agreed to genetic testing, and all four were found to share the same VUS in FAS.
The missense mutation identified by the genetic testing company (Invitae) that performed the 207-gene primary immunodeficiency panel was labeled as a “variant of uncertain significance (VUS)” because symptomatic individuals with this specific variant have not previously been reported in the literature or in population databases. The absence of the variant in population sequence databases increases the likelihood that it is pathogenic. A stop codon in the same residue G253V has been previously reported, with a FAS-induced apoptosis of 25% of control, slightly less than the apoptosis activity reported in this study (38–41%) (15). There have been multiple FAS mutations reported in the death domain of exon 9, and these mutations can show clinical variability among family members, including asymptomatic members with the mutation and diminution of signs and symptoms with increasing age (15, 16). The cohort described in this paper shows a similar pattern of clinical presentation, with the most affected subjects presenting during childhood. Taken together, the novel missense mutation, apoptosis assay/ALPS panel, and absence of the variant in population databases provide moderate to strong evidence that this variant should be reclassified as pathogenic, according to published guidelines for the interpretation of sequence variants (17). The clinical phenotype and cosegregation of the gene in multiple affected family members provide additional supporting evidence (17).
Confirmation of ALPS with genetic sequencing facilitates clinical use of targeted therapies for complications of ALPS. Several potential therapies exist, including corticosteroids, IVIG, rituximab, mycophenolate mofetil (MMF), and mammalian target of rapamycin (mTOR) inhibitor therapy such as sirolimus (18). While the proband's autoimmune hemolytic anemia was initially treated with corticosteroids and intravenous immunoglobulin, due to the genetic diagnosis, she was successfully transitioned to sirolimus, minimizing steroid exposure. This is not surprising given published reports of durable complete response of autoimmunity, lymphadenopathy, and splenomegaly within 3 months of sirolimus initiation in ALPS subjects, as well as no detection of DNTCs in most subjects (19). Though rituximab is often used as a second-or third line option for pediatric subjects with autoimmune cytopenia, the risk of B-cell depletion and prolonged hypogammaglobulinemia is a concern, and long-term management with corticosteroids is not ideal. Splenectomy for autoimmune cytopenia is a last resort, given the risk of sepsis and failure to prevent recurrence of cytopenias in many subjects post-splenectomy (7). Mycophenolate mofetil (MMF) is generally well-tolerated and inactivates a key enzyme in purine synthesis required for lymphocyte proliferation (20, 21). However, MMF does not reduce DNTCs, which may account for suboptimal results such as a partial response or relapse in some ALPS subjects (13). Sirolimus shows good responses in ALPS subjects with autoimmune cytopenias, with possible side effects including hypercholesteremia, hypertension, and mucositis (22). Monitoring of clinical symptoms and ALPS biomarkers can be used to decide when and how treat patients with confirmed disease, as these biomarkers can increase as disease becomes active. DNTCs tend to decrease when patients are treated with sirolimus, and monitoring of DNTC percentage can therefore be useful. Additional serological biomarkers to monitor disease activity and treatment response include vitamin B12 levels, soluble FAS ligand, interleukin-10, and interleukin-18 levels. It is important to note that achieving the target plasma levels of sirolimus is not necessary to control the disease, as levels can be below 5 ng/mL and still be effective for disease control (23, 24). Additionally, in pediatric subjects who show decreased disease activity and eventually go into remission as they reach adolescence, sirolimus discontinuation can be considered, and the medication can be restarted if relapses occur.
We trust that this report will facilitate reclassification of this FAS variant as pathogenic and underscore the importance of genetic testing, which is useful for targeted therapy, family planning, and monitoring patients for malignancy.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Johns Hopkins Institutional Review Board IRB00103900. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
CG, JM, and JEW conceived the presented idea. CG, EW-C, JEW, and JM verified the methods used and reviewed the clinical information presented. CG, JM, DI, EW-C, and JEW assisted with data collection and manuscript review and editing. EG provided guidance regarding long-term management of subjects and serologic monitoring. EW-C oversaw the writing, data collection, and editing process. RFL provided critical review of the manuscript. MM, PT, and GD performed apoptosis assay on a research basis. JEW encouraged to describe this cohort's findings and supervised the findings of this work. All authors discussed the results and contributed and agreed to the final manuscript.
Funding
This research was partly funded by Johns Hopkins All Children's Hospital (JHACH) Institutional Grant entitled Feasibility study to assess the role of T and B cells in refractory cytopenias in children (JEW), the Jeffrey Modell Foundation, Jeffrey Modell Diagnostic and Research Center at JHACH and the Robert A. Good Endowment at the University of South Florida. The Jeffrey Modell Foundation funds supported genetic testing. The JHACH Institutional Grant supported a larger study on autoimmune cytopenia, in which the ALPS subjects were enrolled.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Alina Ramirez as liaison for the study who assisted with coordination of patient care. Maryssa Ellison and Sumai Gordon for research/sample coordination. Earle Trott for assistance with publication documents.
References
1. Geha R, Notarangelo L. Case Studies in Immunology: A Clinical Companion. 7th Edition edn. New York, NY: Garland Science (2016).
2. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. (2010) 116:e35–40. doi: 10.1182/blood-2010-04-280347
3. Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. (2011) 118:4798–807. doi: 10.1182/blood-2011-04-347641
4. Walter JE, Ayala IA, Milojevic D. Autoimmunity as a continuum in primary immunodeficiency. Curr Opin Pediatr. (2019) 31:851–62. doi: 10.1097/MOP.0000000000000833
5. Miano M, Rotulo GA, Palmisani E, Giaimo M, Fioredda F, Pierri F, et al. Sirolimus as a rescue therapy in children with immune thrombocytopenia refractory to mycophenolate mofetil. Am J Hematol. (2018) 93:E175–E7. doi: 10.1002/ajh.25119
6. Miano M, Scalzone M, Perri K, Palmisani E, Caviglia I, Micalizzi C, et al. Mycophenolate mofetil and Sirolimus as second or further line treatment in children with chronic refractory Primitive or Secondary Autoimmune Cytopenias: a single centre experience. Br J Haematol. (2015) 171:247–53. doi: 10.1111/bjh.13533
7. Price S SP, Kirk J, Davis J, Perkins K. Causes and Consequences of Splenectomy In ALPS-FAS. Blood. (2010) 116:3908. doi: 10.1182/blood.V116.21.3908.3908
8. Rosenberg HK, Markowitz RI, Kolberg H, Park C, Hubbard A, Bellah RD. Normal splenic size in infants and children: sonographic measurements. AJR Am J Roentgenol. (1991) 157:119–21. doi: 10.2214/ajr.157.1.2048509
9. Mayo Clinic Laboratories Pediatric Catalog Immunoglobulins Serum. Mayo Foundation for Medical Education Research. (2020) Available online at: pediatric.testcatalog.org/show/IMMG. (accessed September 24, 2020).
10. Complete Blood Count Normal Pediatric Values. Mayo Medical Laboratories. (2020). Available online at: a1.mayomedicallaboratories.com/webjc/attachments/110/30a2131-complete-blood-count-normal-pediatric-values.pdf (accessed September 24, 2020).
11. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. (2003) 112:973–80. doi: 10.1016/j.jaci.2003.07.003
12. Tosato F, Bucciol G, Pantano G, Putti MC, Sanzari MC, Basso G, et al. Lymphocytes subsets reference values in childhood. Cytometry A. (2015) 87:81–5. doi: 10.1002/cyto.a.22520
13. Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr. (2012) 24:1–8. doi: 10.1097/MOP.0b013e32834ea739
14. Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rosen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. (2001) 98:194–200. doi: 10.1182/blood.v98.1.194
15. Rieux-Laucat F, Blachere S, Danielan S, De Villartay JP, Oleastro M, Solary E, et al. Lymphoproliferative syndrome with autoimmunity: A possible genetic basis for dominant expression of the clinical manifestations. Blood. (1999) 94:2575–82.
16. Infante AJ, Britton HA, DeNapoli T, Middelton LA, Lenardo MJ, Jackson CE, et al. The clinical spectrum in a large kindred with autoimmune lymphoproliferative syndrome caused by a Fas mutation that impairs lymphocyte apoptosis. J Pediatr. (1998) 133:629–33. doi: 10.1016/s0022-3476(98)70102-7
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
18. George LA, Teachey DT. Optimal management of autoimmune lymphoproliferative syndrome in children. Paediatr Drugs. (2016) 18:261–72. doi: 10.1007/s40272-016-0175-3
19. Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. (2016) 127:17–28. doi: 10.1182/blood-2015-07-657981
20. Rao VK, Dugan F, Dale JK, Davis J, Tretler J, Hurley JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. (2005) 129:534–8. doi: 10.1111/j.1365-2141.2005.05496.x
21. Izeradjene K, Quemeneur L, Michallet MC, Bonnefoy-Berard N, Revillard JP. Mycophenolate mofetil interferes with interferon gamma production in T-cell activation models. Transplant Proc. (2001) 33:2110–1. doi: 10.1016/s0041-1345(01)01965-0
22. Hartford CM, Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther. (2007) 82:381–8. doi: 10.1038/sj.clpt.6100317
23. Klemann C, Esquivel M, Magerus-Chatinet A, Lorenz MR, Fuchs I, Neveux N, et al. Evolution of disease activity and biomarkers on and off rapamycin in 28 patients with autoimmune lymphoproliferative syndrome. Haematologica. (2017) 102:e52–e56. doi: 10.3324/haematol.2016.153411
Keywords: ALPS (autoimmune lymphoproliferative syndrome), novel mutation, Fas, cytopenia, lymphoproliferation
Citation: Gaefke CL, Metts J, Imanirad D, Nieves D, Terranova P, Dell'Orso G, Gambineri E, Miano M, Lockey RF, Walter JE and Westermann-Clark E (2021) Case Report: A Novel Pathogenic Missense Mutation in FAS: A Multi-Generational Case Series of Autoimmune Lymphoproliferative Syndrome. Front. Pediatr. 9:624116. doi: 10.3389/fped.2021.624116
Received: 30 October 2020; Accepted: 25 January 2021;
Published: 18 March 2021.
Edited by:
Ivan K. Chinn, Baylor College of Medicine, United StatesReviewed by:
Oskar A. Haas, St. Anna Children's Cancer Research Institute (CCRI), AustriaNicholas L. Rider, Baylor College of Medicine, United States
Copyright © 2021 Gaefke, Metts, Imanirad, Nieves, Terranova, Dell'Orso, Gambineri, Miano, Lockey, Walter and Westermann-Clark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jolan Eszter Walter, am9sYW53YWx0ZXJAdXNmLmVkdQ==
†These authors share first authorship