 Erin L. Marcotte
Erin L. Marcotte Logan G. Spector
Logan G. Spector Daniela P. Mendes-de-Almeida
Daniela P. Mendes-de-Almeida Heather H. Nelson
Heather H. Nelson- 1Division of Epidemiology & Clinical Research, Department of Pediatrics, University of Minnesota, Minneapolis, MN, United States
- 2Masonic Cancer Center, University of Minnesota, Minneapolis, MN, United States
- 3Department of Hematology, Instituto Nacional de Infectologia Evandro Chagas, Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil
- 4Division of Molecular Carcinogenesis, Research Center, Instituto Nacional de Câncer (INCA), Rio de Janeiro, Brazil
- 5Division of Epidemiology and Community Health, School of Public Health, University of Minnesota, Minneapolis, MN, United States
Childhood leukemias are heterogeneous diseases with widely differing incident rates worldwide. As circulating tumors, childhood acute leukemias are uniquely accessible, and their natural history has been described in greater detail than for solid tumors. For several decades, it has been apparent that most cases of childhood acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) initiate in utero. Circumstantial evidence in support of this contention includes the young age of onset and high rate of concordance among identical twins. “Backtracking” of leukemic somatic mutations, particularly gene translocations, to cord blood and dried blood spots collected during the perinatal period has provided molecular proof of prenatal leukemogenesis. Detection of a patient's leukemia translocation in easily accessible birth samples, such as dried blood spots, is straightforward with the knowledge of their idiosyncratic breakpoints. However, to translate these findings into population-based screening and leukemia prevention requires novel methods able to detect translocations at all possible breakpoints when present in a low frequency of cells. Several studies have attempted to screen for leukemic translocations, mainly the common ETV6-RUNX1 translocation, in cord blood samples from healthy children. Most studies have reported finding translocations in healthy children, but estimates of prevalence have varied widely and greatly exceed the incidence of leukemia, leading to concerns that technical artifact or contamination produced an artificially inflated estimate of translocation prevalence at birth. New generation techniques that capture the presence of these translocations at birth have the potential to vastly increase our understanding of the epidemiology of acute leukemias. For instance, if leukemic translocations are present at birth in a far higher proportion of children than eventually develop acute leukemia, what are the exposures and somatic molecular events that lead to disease? And could children with translocations present at birth be targeted for prevention of disease? These questions must be answered before large-scale newborn screening for leukemia can occur as a public health initiative. Here, we review the literature regarding backtracking of acute leukemias and the prevalence of leukemic translocations at birth. We further suggest an agenda for epidemiologic research using new tools for population screening of leukemic translocations.
Introduction
Leukemia is the most common malignancy diagnosed in childhood. Acute lymphoblastic leukemia (ALL) comprises approximately 80% of all leukemia diagnoses among children age 0–19 years, and acute myeloid leukemia (AML) represents ~15–20%. ALL and AML demonstrate substantial differences in incidence patterns by age (1), race/ethnicity (2), and sex (3). Additionally, childhood forms of ALL and AML are distinct from those occurring in adulthood with respect to molecular (e.g., cytogenetic) features, demographic characteristics (e.g., incidence according to racial/ethnic group), risk factors, leukemogenic susceptibility associated with certain exposures, and prognosis. Advances in understanding of immunology and molecular/genetic features of the childhood acute leukemias along with laboratory improvements in immunophenotyping and cytogenetic characterization have led to the recognition of molecularly defined subtypes of ALL and AML, targeted therapeutics, and recognition of distinct prognostic groups (4). Notably, the past 30 years has also yielded insight into the natural history and etiology of acute leukemias, particularly with the discovery that many forms of childhood leukemia initiate in utero (5). This article reviews previous work identifying preleukemic clones present at birth, among both healthy children and those who later develop overt leukemia, and suggests a research agenda to capitalize on this knowledge.
Epidemiology of Childhood Leukemia
Acute Lymphoblastic Leukemia
ALL is the most common childhood cancer, with an overall incidence of 42 cases per million children. An early life peak in incidence is evident between 1 and 4 years of age, with rates nearing 100 cases per million (Figure 1). Males experience higher incidence rates compared with females at every age through 19 years (3), and both Hispanics and non-Hispanic whites have substantially higher risk of ALL than have African-American children (2). 5-year survival is high overall, approximately 87% for cases in the United States (4), but can be very low with unfavorable cytogenetics such as KMT2A rearrangements, BCR/ABL translocation, and intrachromosomal amplification of chromosome 21 (iAMP21) (6). ALL mortality does not completely describe the public health burden of the disease, as survivors later have markedly higher risks of cognitive deficits, obesity, debilitating chronic conditions, congestive heart failure, second neoplasms, and early mortality (7).
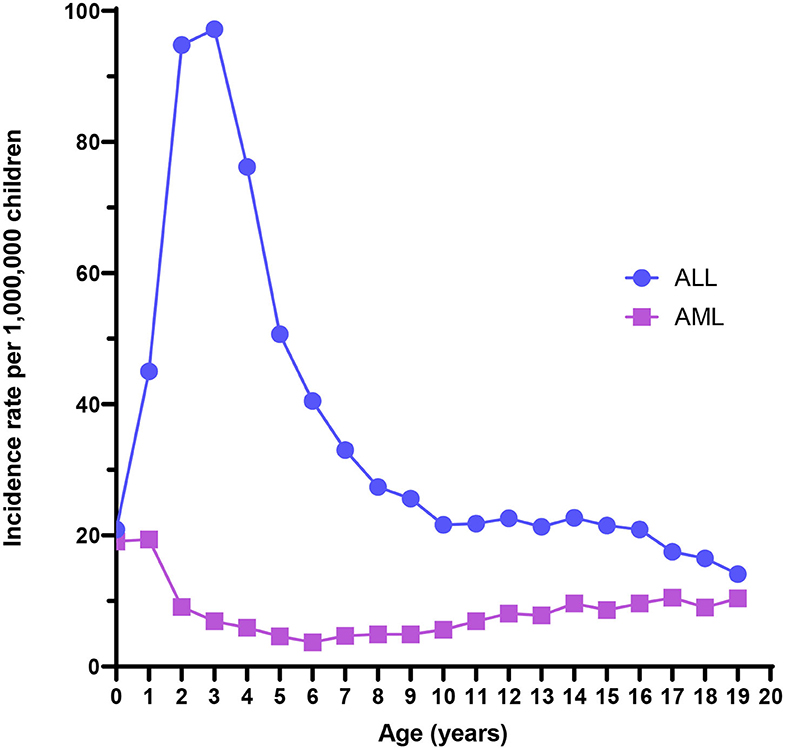
Figure 1. Acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) incidence rates from the US Surveillance, Epidemiology, and End Results program, 2000–2017.
Acquired chromosomal and genetic abnormalities are a hallmark of ALL, and numerous structural and ploidy aberrations have been discovered (Figure 2). The two most frequent molecular events that dominate the early life peak include hyperdiploidy and the t(12;21) translocation, which results in the ETV6/RUNX1 gene fusion. Together, these somatic events are present in over 70% of ALL diagnosed between the ages of 1 and 4 years (6). By contrast, infant leukemia is largely driven by rearrangements in the KMT2A gene; the t(4;11) translocation, which results in a KMT2A/AFF1 gene fusion, is the most common and is present in ~2% of childhood ALL overall and 32% of infant leukemia (6). Figure 2 shows the distribution of cytogenetic and molecular profiles among ALL cases by age at diagnosis.
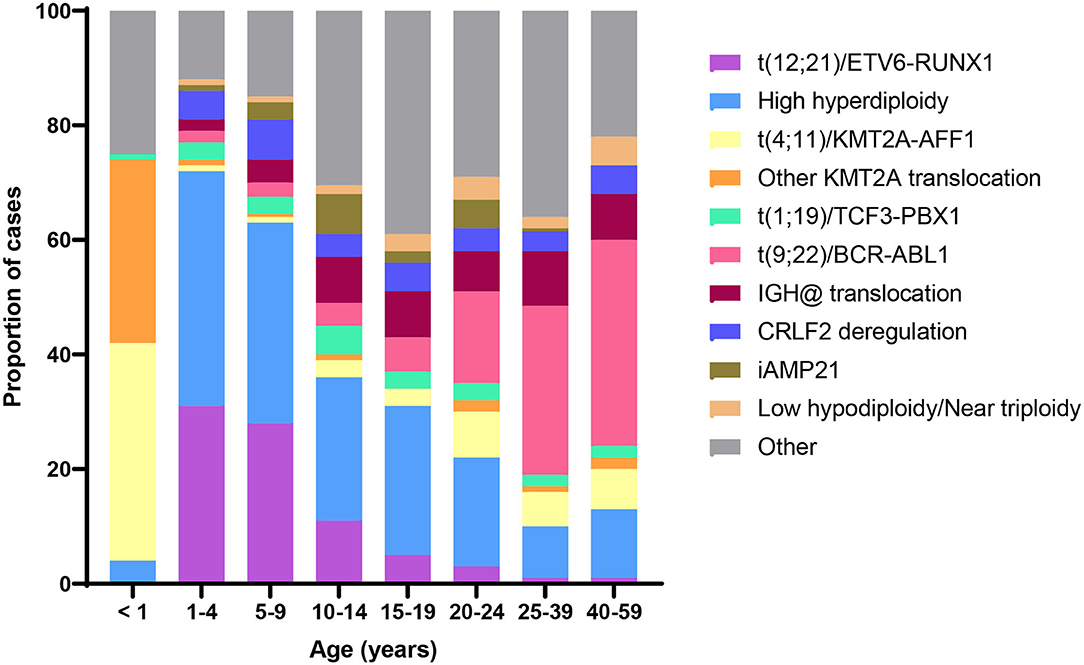
Figure 2. Distribution of B-cell acute lymphoblastic leukemia (ALL) cytogenetic subtypes by age at diagnosis. Data adapted from (6).
The etiology of ALL remains mostly obscure. Prenatal ionizing radiation is an accepted cause of ALL, but it is a rare exposure (8–11). No other environmental risk factors have emerged as definitively causal. For instance, meta-analyses of maternal alcohol use, paternal smoking, and moderate electromagnetic field exposure have reported odds ratios of 1.1, 1.11, and 1.25, respectively (12–16). Intrinsic risk factors have shown more consistent and stronger associations. Risk of ALL rises by about 8% for each 5-year increase in maternal age, while structural birth defects are associated with increases of about 50% (17, 18). ALL also independently rises as a linear function of birth weight (19–21). High-penetrance inherited syndromes underlie about 5% of cases, while common single nucleotide variants (SNVs) uncovered by genomewide association studies are estimated to account for 24% of the total variation in ALL risk (22). The highest magnitudes of association are seen with SNVs in ARID5B, IKZF1, CEBPE, GATA3, and CDK2NA; smaller associations are seen for SNVs in COMMD3/BMI1 and PIP4K2A. Recent data from studies, which have examined ALL by cytogenetic or molecular subtypes, have found heterogeneity in associations with birth weight and SNVs. For instance, SNVs in GATA3 and PIP4K2A have no association with ETV6/RUNX1 ALL, whereas SNVs in LHPP and ELK3 have modest effects on risk of ETV6/RUNX1. Polygenic risk scores have been developed for the ALL risk variants, and those in the top 1% of genetic risk have a 6.2-fold increased risk of developing ALL compared with those with the median level of genetic risk (23, 24). Additional susceptibility factors likely exist for ALL, but their discovery will require novel epidemiologic methods.
Acute Myeloid Leukemia
AML is a rare childhood malignancy that represents 15–20% of all leukemia diagnoses among children and has lower 5-year relative survival (67%) than childhood leukemia overall (85%) (25). The age-adjusted incidence rate of AML was 8.4 per million for children aged <19 years from 2000 to 2017 using data from the Surveillance, Epidemiology, and End Results program (Figure 1). The overall incidence rate is higher for males than females (3). Additionally, Asian/Pacific Islanders, American Indian/Alaska Natives, and Hispanics experience higher incidence rates than non-Hispanic whites or African-Americans. Finally, incidence is highest among infants compared with older age groups. The incidence has increased 0.7% per year from 2000 to 2017 (1). Few definitive risk factors for pediatric AML have emerged, and its etiology is largely unknown. Known risk factors for AML include race, exposure to ionizing radiation in utero, previous chemotherapy agents, bone marrow disorders such as myelodysplastic syndrome (MDS), and several genetic conditions, including Down syndrome (25, 26).
More than half of pediatric AML patients demonstrate an abnormal karyotype, with a subset exhibiting multiple abnormalities (27, 28). The most common include trisomy 8 and 21, monosomy 5 and 7, and various rearrangements such as t(8;21), t(15;17), and inv(16). Patients with a normal karyotype typically harbor recurrent mutations, frequently in NRAS, KRAS, NPM1, and FLT3 (29). Recent evidence shows distinct age-related patterns in AML cytogenetics and mutations (29). For example, in contrast to older children, the most common abnormality in infants with AML is 11q23 translocation involving the KMT2A gene. Figure 3 shows the distribution of cytogenetic and molecular profiles among AML cases by age at diagnosis.
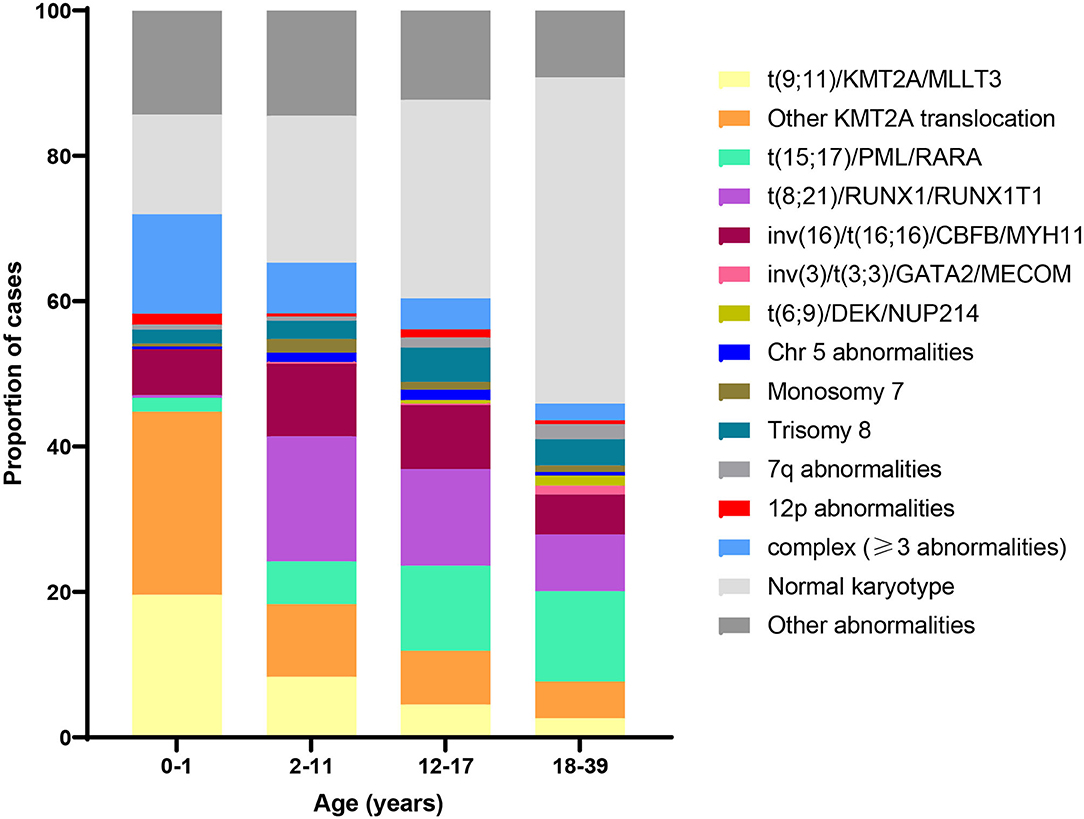
Figure 3. Distribution of acute myeloid leukemia (AML) cytogenetic subtypes by age at diagnosis. Data adapted from (30) and (29).
The diverse patterns of molecular characteristics observed in pediatric AML may result from specific exposome conditions that give rise to etiologically, clinically, and biologically distinct subsets of leukemia. The strongest evidence for this theory comes from studies of therapy-related AML, where different cytotoxic agents are associated with distinct molecular patterns (31, 32). Other studies suggest chemical exposure may be associated specifically with RAS mutation-positive AML (33, 34). Identifying the timing of molecular anomaly occurrence and the type of abnormalities may provide insight into AML etiology. Recognition that the 11q23 rearrangement is characteristic of exposure to topoisomerase II inhibitors in therapy-related AML has led to the investigation of maternal exposure to dietary and environmental inhibitors of topoisomerase II in relation to infant leukemia risk (35, 36). A prenatal origin also has been demonstrated for t(8;21) in patients with childhood AML (37), which may be related to prenatal exposures (38). Although most cases of AML are sporadic, studies of familial clusters have identified case subgroups associated with germline mutations (39). The 2016 edition of the World Health Organization classification of hematopoietic neoplasms created a separate category for myeloid neoplasms with a germline predisposition that includes CEBPA, RUNX1, ETV6, GATA2, DDX41, and ANKRD26 (40).
In utero Initiation
Twin Studies
Anecdotal evidence of a prenatal origin of childhood leukemia first came in the form of case reports of twins and triplets concordant for leukemia. There have been over 70 twin pairs with concordant leukemia reported in the literature, the first of which was published in 1882 (41). The first molecular evidence for a prenatal origin of childhood leukemia arose from twin studies of KMT2A-rearranged and ETV6/RUNX1 leukemia published in the 1990s (42–44). The first study published on this topic examined three pairs of identical infant twins with KMT2A-rearranged ALL and showed that the leukemia cells of each twin pair harbored KMT2A rearrangements that were identical within the pair but distinct from the other twin pairs as well as KMT2A rearrangements observed in eight infant ALL cases from singleton births. The first illustration of in utero origin of the ETV6/RUNX1 translocation in twins appeared in the literature a few years later. This study demonstrated that a pair of monozygotic twins, diagnosed with ALL at ages 3 years 6 months and 4 years 10 months, respectively, harbored an identical ETV6/RUNX1 fusion breakpoint. As the breakpoints for this translocation occur within a 14.7-kb region of ETV6 (intron 1) and a 166-kb region of RUNX1 (introns 1, 2, or 3), it would be nearly impossible for an identical breakpoint to occur by chance. The evidence provided by this study supports a model of leukemogenesis in which a single preleukemic clone arises in utero in one twin and is shared with the other twin via the shared placental blood. Since this time, several other publications have established identical translocations shared between identical twins.
Backtracking Studies
The observations from twin studies led to a series of “backtracking” studies providing further support for an in utero origin for preleukemic clones. In these studies, leukemia cells collected at the time of diagnosis were evaluated for cytogenetic and molecular abnormalities, including the exact patient-specific sequence of any gene translocations specific to the leukemia. Using this information, researchers examined each patient's DNA from dried blood spots (DBS) collected at birth for the purposes of newborn screening. Backtracking studies have revealed that, for each of the molecular events investigated, at least a portion of children diagnosed with leukemia harbored preleukemic cells at birth. The proportion of children whose unique leukemic event was detected in their newborn sample varies by subtype. Studies of hyperdiploid ALL have generally shown that a high proportion (83% to 100%) of patients harbored preleukemic cells at birth (45–48), although these studies were limited in the number of children screened (Table 1). By contrast, previous studies have reported a wide range of estimates for prevalence of other translocation events for ETV6/RUNX1 (0–100%) (47–49, 54, 55, 62), KMT2A-rearranged (0% to 100%) (45, 47, 48, 53–57), TCF3/PBX1 (13% to 100%) (47, 59), and BCR/ABL1 (0% to 100%) (48, 60).
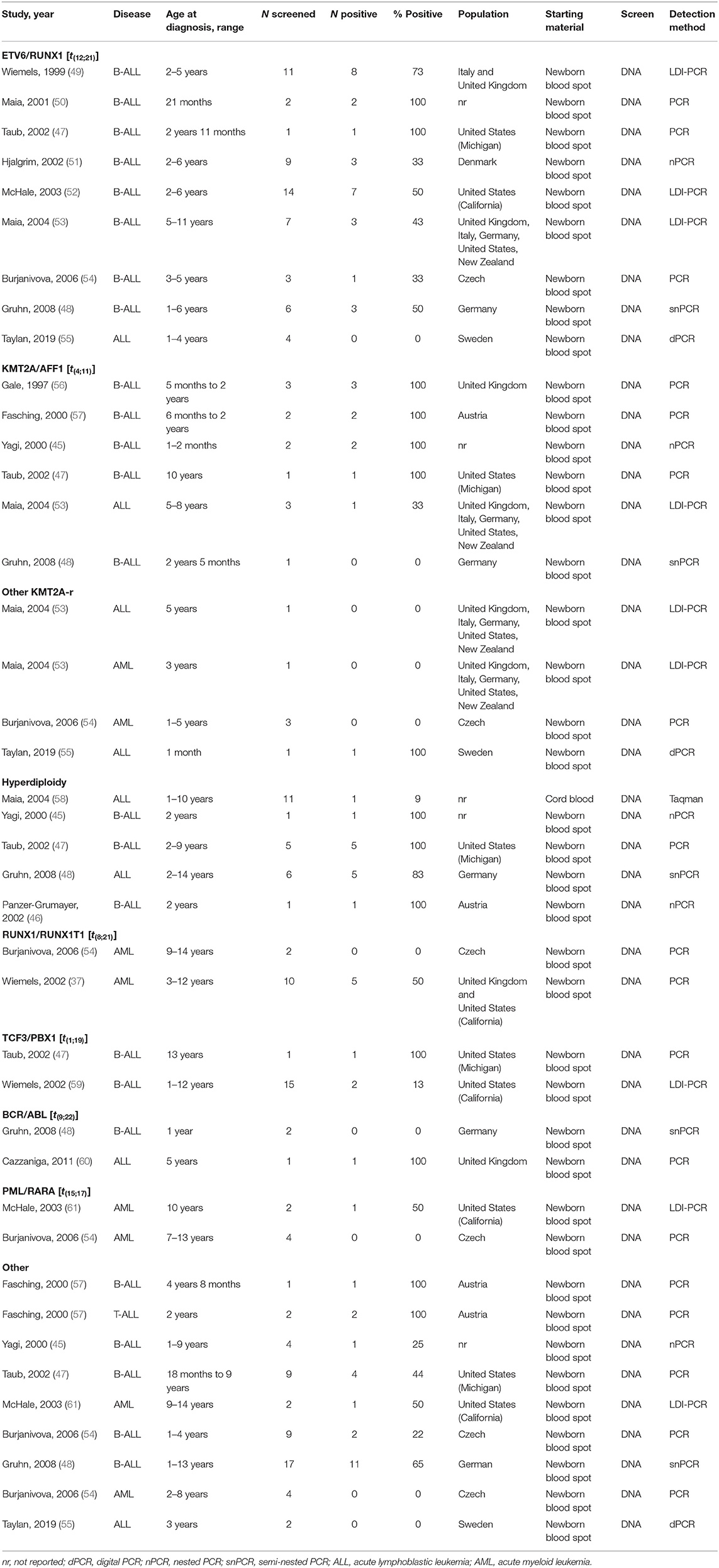
Table 1. Backtracking studies of childhood leukemia.
Previous Screening Studies of Healthy Newborns
There is substantial evidence that at least some of the common leukemia translocations are not necessarily deterministic for future development of leukemia and are present at varying prevalence in umbilical cord blood (UCB) of healthy newborns (Table 2) (62–70, 73, 74). The ETV6/RUNX1 translocation is by far the most widely studied preleukemic event among an unselected pool of healthy newborns. To our knowledge, the TCF3/PBX1, KMT2A/AFF1, BCR/ABL, and RUNX1/RUNX1T1 translocations are the only other leukemia translocation events to be examined among a sample of unselected newborns (Table 2).
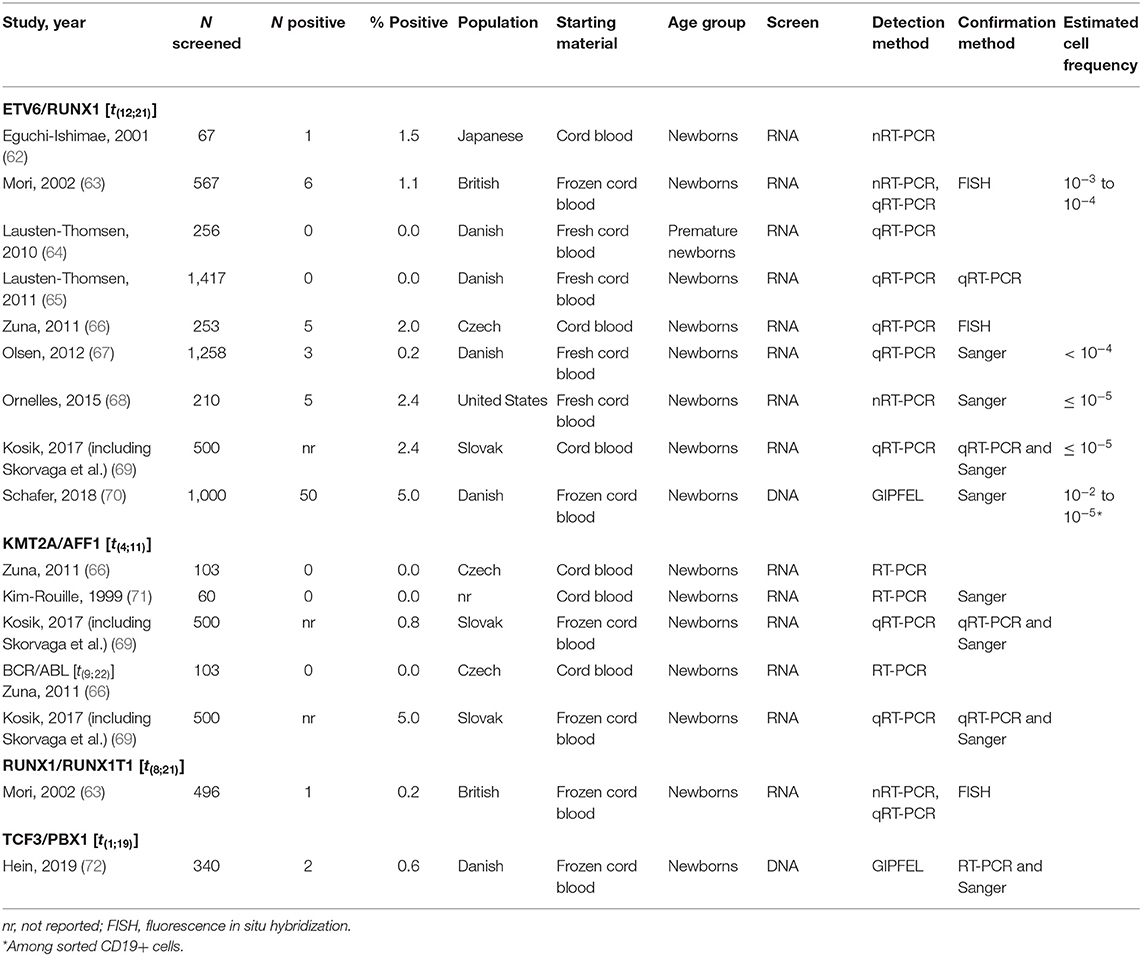
Table 2. Studies of healthy newborns that screened for leukemia translocations.
Translocation Prevalence Estimates and Variation
Estimates of the ETV6/RUNX1 translocation prevalence among all newborns at birth have varied widely (75), from 0.01% to 5% (65, 70). The sources of this variation may be due to the translocation detection methods and materials used (discussed below). Differences in populations tested may also lead to variation in estimated population frequency. Indeed, previous literature on the distribution of cytogenetic abnormalities among ALL patients of varying racial and ethnic groups has suggested that ETV6/RUNX1 leukemia is less common in Hispanics and East Asian populations compared with populations of European ancestry (76–78). Figure 4 shows estimates of cytogenetic abnormality frequency by country, from publications that reported proportions of at least four of the most common abnormalities (76–88). To our knowledge, two previous screening studies have focused on non-white populations. Eguchi-Ishimae et al. screened cord blood samples from 67 Japanese newborns and reported one (1.5%) as positive for the ETV6/RUNX1 translocation (62). Ornelles et al. conducted the only study with a majority African-American and Hispanic population. Of the 210 cord blood samples screened, 148 were from African-American newborn and 55 were from Hispanic newborns (68). Three (2.0%) samples from African-American children were positive for the ETV6/RUNX1 translocation, while two (3.6%) samples from Hispanic children were positive.
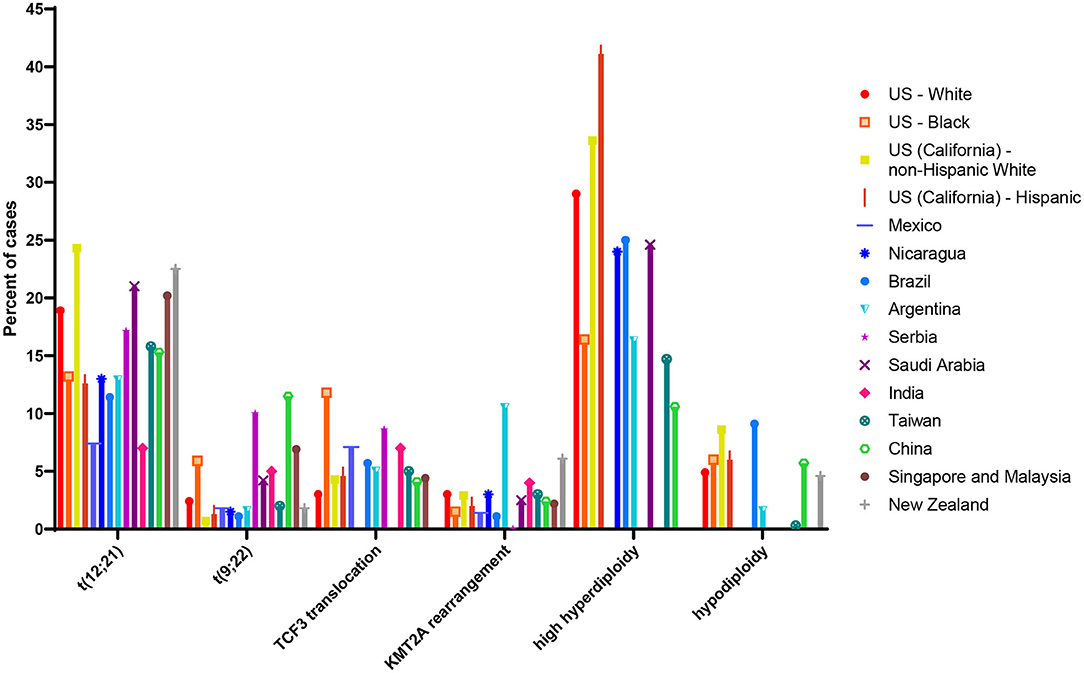
Figure 4. Proportion of common cytogenetic and molecular abnormalities among acute lymphoblastic leukemia (ALL) cases of different racial and ethnic groups or nations. Estimate of cytogenetic distribution among children diagnosed in the following countries: US white and black children and adolescents (age range not reported) taken from (79); US (California) Hispanic and non-Hispanic white children (age 0–14 years) taken from (76); Mexico (age 0–18 years) taken from (80); Nicaragua (age 0–16 years) taken from (81); Brazil (age 0–17 years) taken from (82); Argentina (age 0–16 years) taken from (83); Serbia (age 0–16 years) taken from (84); Saudi Arabia (age 0–14 years) taken from (85); India (age 0–20 years) taken from (86); Taiwan (age 0–18 years) taken from (77); China (age 0–18 years) taken from (87); Singapore and Malaysia (age 0–16 years) taken from (88); New Zealand (age 0–14 years) taken from (78). Taiwanese children (age 0–18 years) taken from (77); estimate of cytogenetic distribution among Chinese children (age 0–18 years) taken from (87).
It is notable that ALL-associated translocations have been reported more commonly than those associated with AML, in both backtracking (Table 1) and screening (Table 2) studies. There are several possible explanations for this phenomenon. The first is that the relative frequency of childhood ALL, and ETV6/RUNX1 ALL in particular, compared with childhood AML has led to more investigation of this leukemia subtype. In the published literature presented in Table 1, a total of 158 unique ALL patients have been examined in backtracking studies, of which 76 [48% (95% confidence interval, calculated by the Wilson score interval (89): 40–56%)] were positive for preleukemic clones at birth. By contrast, a total of only 28 AML patients have been examined in backtracking studies, of which seven [25% (95% confidence interval, calculated by the Wilson score interval (89): 13–43%)] were positive for preleukemic clones at birth. The apparent lower birth prevalence of preleukemic clones among children later diagnosed with AML is statistically indistinguishable from that of children later diagnosed with ALL and may simply be due to the small number of patients examined. Additionally, the presumption of an in utero origin is the strongest for cancers occurring near birth and diminishes with age. As AML is less skewed toward a younger age at onset than ALL (Figure 1), the observed detection of clones in birth samples may reflect true differences in frequency of in utero initiation. We examined whether the available data support the hypothesis that the age-specific incidence curves of ALL and AML indicate a likely lower frequency of in utero initiation of AML. We abstracted age at diagnosis and presence of preleukemic clones at birth for all 158 ALL cases reported in publications in Table 1, with the exception of 15 t(1;19)+ B-ALL cases in Wiemels et al. (59) because it was not possible to determine which of the patients presented in the publication were included in the Guthrie card analysis. Figure 5 shows number of ALL cases tested by age at diagnosis and proportion of those with detectable preleukemic clones at birth. We conducted a simple logistic regression to determine whether age at diagnosis (in months) was associated with odds of preleukemic clone presence at birth and observed no relationship (odds ratio = 0.99; 95% confidence interval: 0.98–1.01). Another explanation is that AML-associated translocations are more difficult to detect than the common ALL-associated translocations. Indeed, while the ETV6/RUNX1 translocation occurs in predictable regions of the genes involved—the breakpoints occur within a 14.7-kb region of ETV6 (intron 1) and a 166-kb region of RUNX1 (intron 1, 2, or 3) (90)—KMT2A rearrangements include more than 90 known translocation partner genes, 35 of which are known to occur recurrently (91). Additionally, the most common of these, KMT2A/AFF1 translocation, occurs more frequently among infant leukemia patients with ALL rather than AML (91, 92). The other common recurrent AML-associated translocations, including RUNX1/RUNX1T1, PML/RARA, and inv(16), have been investigated in few backtracking studies (Table 1); and only one of these, RUNX1/RUNX1T1, has been investigated in a screening study published in 2002 (Table 2). Thus, more research is needed on the in utero initiation of AML to determine its frequency, both in children who later develop AML and in the general population of healthy newborns.
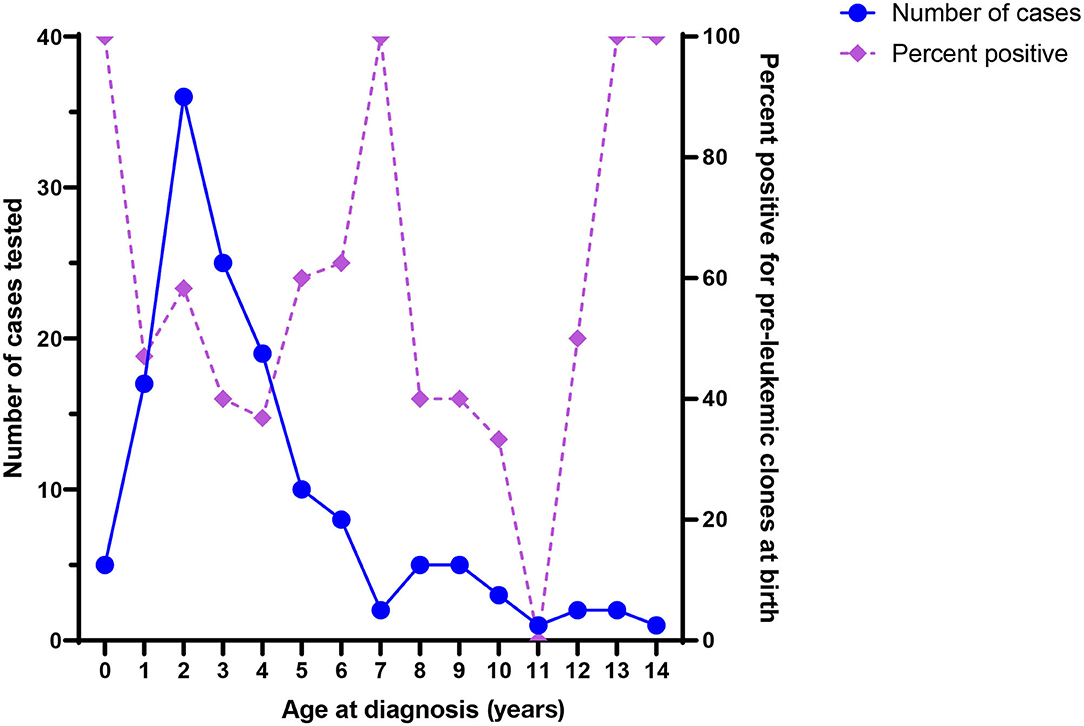
Figure 5. Number of acute lymphoblastic leukemia (ALL) cases tested in backtracking studies, by age at diagnosis (left axis), and the proportion of cases positive for preleukemic clones at birth, by age at diagnosis (right axis).
Methods and Material Used
Uncertainty around these prevalence estimates, in both cases and healthy controls, relates to the ability to detect a rare event in a vast excess of normal cells. Detection of rare translocations can be achieved using DNA if the exact translocation is known, as is the case for backtracking of known ALL cases (37, 41, 49, 52, 93). However, this approach is not feasible for screening unless the breakpoint region is small, which is not the case for ETV6/RUNX1; the breakpoints for this translocation occur within a 14.7-kb region of ETV6 (intron 1) and a 166-kb region of RUNX1 (intron 1, 2, or 3). Therefore, RNA approaches have typically been employed for the detection of the ETV6/RUNX1 translocation product in birth samples. The prior research has relied heavily on the use of nested reverse transcriptase PCR or quantitative RT-PCR to detect the low copy signal, and this is problematic, as RNA-based PCR methods are prone to contamination and false-positive results. To limit the contribution of false positives, most prior studies used at least one validation method (Table 2), but questions on the variation in prevalence between studies remain. RNA methods for detection of the ETV6/RUNX1 translocation product produce the same transcript for each breakpoint, which diminishes the ability to identify contaminants. Additionally, false-negative results are possible with low-quality input material.
Previous studies have relied on UCB or flow-sorted B cells from the cord blood. There are also differences in processing and storage of UCB in prior studies, some of which used fresh cord blood processed within 24 h of birth, while others used frozen UCB. Both of these methods are problematic for larger, population-based studies, as UCB is not routinely collected after birth and requires substantial resources for collection, processing, and storage. Thus, UCB-based methods are not scalable for very large population-based studies or implementation in newborn screening.
Development of highly sensitive DNA-based methods for translocation detection is a desirable step forward for this work. DNA is far more stable than RNA, and DNA-based translocation detection would facilitate identification of contamination since patient-specific amplification products can be compared with respect to size and exact sequence. Indeed, given these advantages, DNA-based methods have been utilized clinically in minimum residual disease monitoring (94). Recently, a DNA-based method for detection of translocation events at birth has been described, GIPFEL, and in among 1,000 unselected newborns has produced the highest estimate for birth prevalence of the ETV6/RUNX1 translocation (50/1,000; 5%) (70). While a DNA-based method has many advantages over RNA-based methods, the literature published using this method also required UCB processed within 24 h of birth as the input material to enable flow-sorting enrichment of CD19+ B cells (95). Currently, it is not feasible for use in population-wide screening; however, if the method is optimized to use whole blood with lower requirements for input material, it may 1 day be possible to deploy in newborn DBS.
Future Research Agenda
Advancing Novel Translocation Screening Methods
Utility of Newborn Dried Blood Spots
Newborn screening is a mandatory public health program in the United States and most high development index countries around the world (96). Under the US program, DBS are collected within 24–48 h of birth for >98% of the nearly 4 million infants born each year (97, 98). The DBS are sent to state laboratories to test infants for inborn errors of metabolism and other selected genetic and endocrine disorders. After screening is complete, residual DBS are stored for confirmation of positive results or re-testing as needed. Some states also retain residual DBS for research purposes, although the storage duration varies widely by state, from 30 days to indefinite, and policies regarding DBS retention and use in research continue to rapidly evolve (99).
DBS represent an ideal sample for preleukemic clone screening. They are easily stored, routinely collected for every infant, and, in some states, available for population-based research studies. Despite their many benefits, there are also barriers to using DBS for this purpose, such as variation in storage conditions among different states. For instance, while some states store residual DBS at −20°C, other states store DBS without temperature or humidity control (99). This represents a challenge for RNA-based methods, as RNA can degrade rapidly in DBS, particularly those stored in uncontrolled temperature conditions (100). Additionally, while UCB provides ample input material for many applications, DBS provide a limited quantity of cells. Each Guthrie card spot is ~6 to 10 mm in diameter and may be unevenly filled with blood (101). When a spot is completely filled to its borders, it contains approximately 50 μL of whole blood, and the amount of DNA or RNA that can be extracted will vary according to storage conditions and duration (102, 103). Despite these limitations, the widespread availability and ease of collection and storage of DBS make it an appealing target for new method development and eventual translational application. Additionally, recent studies have demonstrated that archived DBS are suitable for some RNA-based applications, including gene expression profiling (104–106).
Novel Detection Methods
As technology evolves, it may enable novel leukemia translocation detection methods. Detection of rare translocations can be achieved using DNA if the breakpoint region is small, by placing PCR primers in both translocation partner genes to create a translocation-specific PCR product. However, the breakpoints for most translocations, including the most common ETV6/RUNX1, occur within a large region of the genome (described in Translocation Prevalence Estimates and Variation section), and this has driven the use of RNA-based methods. Next-generation sequencing (NGS) is under-explored for the detection of translocations in newborn samples. Utilization of an NGS approach will require enrichment of target sequence. For instance, to detect ETV6/RUNX1 would require enrichment of RUNX1-specific cDNA using sequence specific oligos that are placed in exons 2, 3, and 4 in a reverse transcription assay (107). This procedure would create a pool of cDNA containing both wild-type RUNX1 and ETV6/RUNX1 fusion transcripts. The entire pool of cDNA could be used for library creation using Nextera XT transposase, which is optimal for use with low DNA inputs. However, an NGS approach has many challenges, including low quality or degradation of the input RNA, insufficient gene-specific cDNA, the need for high sequencing depths, and high cost, which limit feasibility for large-scale implementation. Additionally, this approach would require extensive bioinformatic resources.
Another possible innovative approach includes the application of digital PCR (dPCR), which has recently been utilized in a backtracking study of childhood ALL (55). dPCR is an attractive target for translocation screening given its sensitivity and specificity and the exceptional performance of dPCR for low copy number events. However, this approach requires careful design of primers and probes to enable translocation detection across multiple breakpoints and would require determination of a positive droplet threshold to determine true-positive samples. The innovative GIPFEL procedure, which utilizes DNA and inverse PCR, overcomes both the RNA quality/quantity problem and the “large breakpoint span” problem; it is an exciting development in the detection of translocations in the research setting.
Epidemiologic Studies Enabled by New Methods of Screening
The development of robust translocation detection methods utilizing DBS would open several opportunities for future epidemiologic and natural history studies of childhood leukemia. First, large studies of the prevalence of translocations in the general population of newborns could quickly determine whether the frequency of mutations varies in conjunction with known risk factors for overt ALL. Second, these methods would enable establishment of the first prospective epidemiologic studies of ALL risk by concentrating on children with translocations present at birth to determine the early life factors that promote or arrest their expansion. With serial sampling, researchers could also examine translocation persistence rather than overt leukemia as a study outcome. Should these studies suggest an intervention, they could motivate initiation of newborn screening and prevention of childhood leukemia. Success of these studies depends on a robust assay that captures translocation status in newborn DBS, even those not stored in ideal conditions.
Newborn Screening for Childhood Leukemia
Although there is evidence that some leukemic translocations, such as ETV6/RUNX1, occur at substantially higher rates in utero than the rate of overt leukemia (70), there may be translocations whose frequency of in utero initiation is near that of the specific leukemia subtype to which they give rise. For instance, although the studies examining the prevalence of KMT2A/AFF1 in healthy newborns were limited in sample size (Table 2), they suggest that this translocation occurs infrequently in the population of unselected newborns. If the incidence of the KMT2A rearrangements is close to that of infant leukemia, they may be nearly deterministic for infant leukemia development and newborn screening may be warranted. Indeed, infant leukemia is an attractive target for newborn screening given that it is a serious disease with onset in the 1st year of life for which there are available treatments. Despite the potential benefits of early detection and intervention for infant leukemia, a newborn screening program for infant leukemia presents several challenges. First, depending on the method of fusion detection, the addition of screening for preleukemic clones may be cost prohibitive on a population-wide basis. Additionally, a positive result would likely provoke a considerable amount of parental anxiety related to the newborn's well-being. Finally, infant leukemia patients experience high rates of glucocorticoid resistance (108), relapse, and emergence of chemoresistant cell populations (109), particularly among KMT2A-rearranged ALL. Therefore, the clinical utility of early intervention enabled by newborn screening is unclear for this population and requires additional research.
Conclusion
More than 70% of infant leukemia cases and >40% of ALL cases diagnosed age 1–9 years contain cytogenetic profiles that have been found to occur in utero through backtracking and twin studies and have been positively identified in newborn blood spots. This includes KMT2A rearrangements, common in infant leukemia, which confer very poor survival, and both ETV6/RUNX1 and hyperdiploidy, which represent the majority of ALL diagnosed at ages 1 to 9 years.
Resolving the uncertainty regarding the prevalence of translocation at birth is an important public health issue with potential implications for newborn screening. However, previous studies among known leukemia cases require determination of the exact translocation breakpoint from diagnostic samples, while methods used in previous studies of healthy newborns are cumbersome and require the use of UCB. Developing a robust method using newborn DBS that can identify rare clones without false positives will have high value for future research efforts into the etiology of leukemia.
Author Contributions
ELM and LGS: article concept and design. ELM: drafting of the manuscript and statistical analysis. All authors: critical revisions of the manuscript for important intellectual content. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Children's Cancer Research Fund (Minneapolis, MN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Surveillance Epidemiology and End Results (SEER) Program, SEER*Stat Database: Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases Nov 2016 Sub (2000-2014) <Katrina/Rita Population Adjustment> - Linked To County Attributes - Total U.S., 1969-2015 Counties. Bethesda, MD: Naitonal Cancer Institute.
2. Chow EJ, Puumala SE, Mueller BA, Carozza SE, Fox EE, Horel S, et al. Childhood cancer in relation to parental race and ethnicity: a 5-state pooled analysis. Cancer. (2010) 116:3045–53. doi: 10.1002/cncr.25099
3. Williams LA, Richardson M, Marcotte EL, Poynter JN, Spector LG. Sex ratio among childhood cancers by single year of age. Pediatr Blood Cancer. (2019) 66:e27620. doi: 10.1002/pbc.27620
4. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. (2015) 125:3977–87. doi: 10.1182/blood-2015-02-580043
6. Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. (2012) 26:123–35. doi: 10.1016/j.blre.2012.01.001
7. Norsker FN, Pedersen C, Armstrong GT, Robison LL, McBride ML, Hawkins M, et al. Late effects in childhood cancer survivors: early studies, survivor cohorts, and significant contributions to the field of late effects. Pediatr Clin North Am. (2020) 67:1033–49. doi: 10.1016/j.pcl.2020.07.002
8. Axelson O, Fredrikson M, Akerblom G, Hardell L. Leukemia in childhood and adolescence and exposure to ionizing radiation in homes built from uranium-containing alum shale concrete. Epidemiology. (2002) 13:146–50. doi: 10.1097/00001648-200203000-00008
9. Hsu WL, Preston DL, Soda M, Sugiyama H, Funamoto S, Kodama K, et al. The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950-2001. Radiat Res. (2013) 179:361–82. doi: 10.1667/RR2892.1
10. Kendall GM, Little MP, Wakeford R, Bunch KJ, Miles JC, Vincent TJ, et al. A record-based case-control study of natural background radiation and the incidence of childhood leukaemia and other cancers in Great Britain during 1980-2006. Leukemia. (2013) 27:3–9. doi: 10.1038/leu.2012.151
11. Krille L, Dreger S, Schindel R, Albrecht T, Asmussen M, Barkhausen J, et al. Risk of cancer incidence before the age of 15 years after exposure to ionising radiation from computed tomography: results from a German cohort study. Radiat Environ Biophys. (2015) 54:1–12. doi: 10.1007/s00411-014-0580-3
12. Latino-Martel P, Chan DS, Druesne-Pecollo N, Barrandon E, Hercberg S, Norat T. Maternal alcohol consumption during pregnancy and risk of childhood leukemia: systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. (2010) 19:1238–60. doi: 10.1158/1055-9965.EPI-09-1110
13. Klimentopoulou A, Antonopoulos CN, Papadopoulou C, Kanavidis P, Tourvas AD, Polychronopoulou S, et al. Maternal smoking during pregnancy and risk for childhood leukemia: a nationwide case-control study in Greece and meta-analysis. Pediatr Blood Cancer. (2012) 58:344–51. doi: 10.1002/pbc.23347
14. Liu R, Zhang L, McHale CM, Hammond SK. Paternal smoking and risk of childhood acute lymphoblastic leukemia: systematic review and meta-analysis. J Oncol. (2011) 2011:854584. doi: 10.1155/2011/854584
15. Greenland S, Sheppard AR, Kaune WT, Poole C, Kelsh MA. A pooled analysis of magnetic fields, wire codes, childhood leukemia. Childhood Leukemia-EMF Study Group. Epidemiology. (2000) 11:624–34. doi: 10.1097/00001648-200011000-00003
16. Jaffa KC Pooled analysis of magnetic fields wire codes childhood leukemia. Epidemiology. (2001) 12:472–4. doi: 10.1097/00001648-200107000-00021
17. Botto LD, Flood T, Little J, Fluchel MN, Krikov S, Feldkamp ML, et al. Cancer risk in children and adolescents with birth defects: a population-based cohort study. PLoS ONE. (2013) 8:e69077. doi: 10.1371/journal.pone.0069077
18. Johnson KJ, Carozza SE, Chow EJ, Fox EE, Horel S, McLaughlin CC, et al. Parental age and risk of childhood cancer: a pooled analysis. Epidemiology. (2009) 20:475–83. doi: 10.1097/EDE.0b013e3181a5a332
19. Caughey RW, Michels KB. Birth weight and childhood leukemia: a meta-analysis and review of the current evidence. Int J Cancer. (2009) 124:2658–70. doi: 10.1002/ijc.24225
20. Hjalgrim LL, Westergaard T, Rostgaard K, Schmiegelow K, Melbye M, Hjalgrim H, et al. Birth weight as a risk factor for childhood leukemia: a meta-analysis of 18 epidemiologic studies. Am J Epidemiol. (2003) 158:724–35. doi: 10.1093/aje/kwg210
21. Koifman S, Pombo-de-Oliveira MS L. Brazilian Collaborative Study Group of Infant Acute. High birth weight as an important risk factor for infant leukemia. Br J Cancer. (2008) 98:664–7. doi: 10.1038/sj.bjc.6604202
22. Hsu LI, Briggs F, Shao X, Metayer C, Wiemels JL, Chokkalingam AP, et al. Pathway analysis of genome-wide association study in childhood leukemia among Hispanics. Cancer Epidemiol Biomarkers Prev. (2016) 25:815–22. doi: 10.1158/1055-9965.EPI-15-0528
23. Vijayakrishnan J, Kumar R, Henrion MY, Moorman AV, Rachakonda PS, Hosen I, et al. A genome-wide association study identifies risk loci for childhood acute lymphoblastic leukemia at 10q26.13 and 12q23.1. Leukemia. (2017) 31:573–9. doi: 10.1038/leu.2016.271
24. Enciso-Mora V, Hosking FJ, Sheridan E, Kinsey SE, Lightfoot T, Roman E, et al. Common genetic variation contributes significantly to the risk of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia. (2012) 26:2212–5. doi: 10.1038/leu.2012.89
25. Puumala SE, Ross JA, Aplenc R, Spector LG. Epidemiology of childhood acute myeloid leukemia. Pediatr Blood Cancer. (2013) 60:728–33. doi: 10.1002/pbc.24464
26. Smith MA, Ries LAG, Gurney JG, Ross JA. Leukemia. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL Jr., Bunin GR, editors. Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975-1995. Bethesda, MD: National Cancer Institute, SEER Program (1999). p. 17–34.
27. Manola KN Cytogenetics of pediatric acute myeloid leukemia. Eur J Haematol. (2009) 83:391–405. doi: 10.1111/j.1600-0609.2009.01308.x
28. Mrozek K, Heinonen K, Bloomfield CD. Clinical importance of cytogenetics in acute myeloid leukaemia. Best Pract Res Clin Haematol. (2001) 14:19–47. doi: 10.1053/beha.2000.0114
29. Bolouri H, Farrar JE, Triche T Jr.., Ries RELim EL, . The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. (2018) 24:103–12. doi: 10.1038/nm.4439
30. Creutzig U, Zimmermann M, Reinhardt D, Rasche M, von Neuhoff C, Alpermann T, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. (2016) 122:3821–30. doi: 10.1002/cncr.30220
31. Qian Z, Joslin JM, Tennant TR, Reshmi SC, Young DJ, Stoddart A, et al. Cytogenetic and genetic pathways in therapy-related acute myeloid leukemia. Chem Biol Interact. (2010) 184:50–7. doi: 10.1016/j.cbi.2009.11.025
32. Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. (2008) 22:240–8. doi: 10.1038/sj.leu.2405078
33. Taylor JA, Sandler DP, Bloomfield CD, Shore DL, Ball ED, Neubauer A, et al. ras oncogene activation and occupational exposures in acute myeloid leukemia. J Natl Cancer Inst. (1992) 84:1626–32. doi: 10.1093/jnci/84.21.1626
34. Barletta E, Gorini G, Vineis P, Miligi L, Davico L, Mugnai G, et al. Ras gene mutations in patients with acute myeloid leukaemia and exposure to chemical agents. Carcinogenesis. (2004) 25:749–55. doi: 10.1093/carcin/bgh057
35. Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, et al. Maternal diet and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the children's oncology group. Cancer Epidemiol Biomarkers Prev. (2005) 14:651–5. doi: 10.1158/1055-9965.EPI-04-0602
36. Alexander FE, Patheal SL, Biondi A, Brandalise S, Cabrera ME, Chan LC, et al. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Res. (2001) 61:2542–6.
37. Wiemels JL, Xiao Z, Buffler PA, Maia AT, Ma X, Dicks BM, et al. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood. (2002) 99:3801–5. doi: 10.1182/blood.V99.10.3801
38. Lafiura KM, Bielawski DM, Posecion NC Jr., Ostrea EM Jr., Matherly LH, Taub JW, et al Association between prenatal pesticide exposures and the generation of leukemia-associated T(8;21). Pediatr Blood Cancer. (2007) 49:624–8. doi: 10.1002/pbc.21283
39. West AH, Godley LA, Churpek JE. Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci. (2014) 1310:111–8. doi: 10.1111/nyas.12346
40. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
41. Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. (2003) 102:2321–33. doi: 10.1182/blood-2002-12-3817
42. Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc Natl Acad Sci U S A. (1998) 95:4584–8. doi: 10.1073/pnas.95.8.4584
43. Ford AM, Ridge SA, Cabrera ME, Mahmoud H, Steel CM, Chan LC, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature. (1993) 363:358–60. doi: 10.1038/363358a0
44. Gill Super HJ, Rothberg PG, Kobayashi H, Freeman AI, Diaz MO, Rowley JD. Clonal, nonconstitutional rearrangements of the MLL gene in infant twins with acute lymphoblastic leukemia: in utero chromosome rearrangement of 11q23. Blood. (1994) 83:641–4. doi: 10.1182/blood.V83.3.641.bloodjournal833641
45. Yagi T, Hibi S, Tabata Y, Kuriyama K, Teramura T, Hashida T, et al. Detection of clonotypic IGH and TCR rearrangements in the neonatal blood spots of infants and children with B-cell precursor acute lymphoblastic leukemia. Blood. (2000) 96:264–8. doi: 10.1182/blood.V96.1.264.013k08_264_268
46. Panzer-Grumayer ER, Fasching K, Panzer S, Hettinger K, Schmitt K, Stockler-Ipsiroglu S, et al. Nondisjunction of chromosomes leading to hyperdiploid childhood B-cell precursor acute lymphoblastic leukemia is an early event during leukemogenesis. Blood. (2002) 100:347–9. doi: 10.1182/blood-2002-01-0144
47. Taub JW, Konrad MA, Ge Y, Naber JM, Scott JS, Matherly LH, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood. (2002) 99:2992–6. doi: 10.1182/blood.V99.8.2992
48. Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Hafer R, et al. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. (2008) 22:1692–7. doi: 10.1038/leu.2008.152
49. Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. (1999) 354:1499–503. doi: 10.1016/S0140-6736(99)09403-9
50. Maia AT, Ford AM, Jalali GR, Harrison CJ, Taylor GM, Eden OB, et al. Molecular tracking of leukemogenesis in a triplet pregnancy. Blood. (2001) 98:478–82. doi: 10.1182/blood.V98.2.478
51. Hjalgrim LL, Madsen HO, Melbye M, Jorgensen P, Christiansen M, Andersen MT, et al. Presence of clone-specific markers at birth in children with acute lymphoblastic leukaemia. Br J Cancer. (2002) 87:994–9. doi: 10.1038/sj.bjc.6600601
52. McHale CM, Wiemels JL, Zhang L, Ma X, Buffler PA, Guo W, et al. Prenatal origin of TEL-AML1-positive acute lymphoblastic leukemia in children born in California. Genes Chromosomes Cancer. (2003) 37:36–43. doi: 10.1002/gcc.10199
53. Maia AT, Koechling J, Corbett R, Metzler M, Wiemels JL, Greaves M. Protracted postnatal natural histories in childhood leukemia. Genes Chromosomes Cancer. (2004) 39:335–40. doi: 10.1002/gcc.20003
54. Burjanivova T, Madzo J, Muzikova K, Meyer C, Schneider B, Votava F, et al. Prenatal origin of childhood AML occurs less frequently than in childhood ALL. BMC Cancer. (2006) 6:100. doi: 10.1186/1471-2407-6-100
55. Taylan F, Bang B, Ofverholm II, Tran AN, Heyman M, Barbany G, et al. Somatic structural alterations in childhood leukemia can be backtracked in neonatal dried blood spots by use of whole-genome sequencing and digital PCR. Clin Chem. (2019) 65:345–7. doi: 10.1373/clinchem.2018.293548
56. Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A. (1997) 94:13950–4. doi: 10.1073/pnas.94.25.13950
57. Fasching K, Panzer S, Haas OA, Marschalek R, Gadner H, Panzer-Grumayer ER. Presence of clone-specific antigen receptor gene rearrangements at birth indicates an in utero origin of diverse types of early childhood acute lymphoblastic leukemia. Blood. (2000) 95:2722–4. doi: 10.1182/blood.V95.8.2722.008k09_2722_2724
58. Maia AT, Tussiwand R, Cazzaniga G, Rebulla P, Colman S, Biondi A, et al. Identification of preleukemic precursors of hyperdiploid acute lymphoblastic leukemia in cord blood. Genes Chromosomes Cancer. (2004) 40:38–43. doi: 10.1002/gcc.20010
59. Wiemels JL, Leonard BC, Wang Y, Segal MR, Hunger SP, Smith MT, et al. Site-specific translocation and evidence of postnatal origin of the t(1;19) E2A-PBX1 fusion in childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. (2002) 99:15101–6. doi: 10.1073/pnas.222481199
60. Cazzaniga G, van Delft FW, Lo Nigro L, Ford AM, Score J, Iacobucci I, et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood. (2011) 118:5559–64. doi: 10.1182/blood-2011-07-366542
61. McHale CM, Wiemels JL, Zhang L, Ma X, Buffler PA, Feusner J, et al. Prenatal origin of childhood acute myeloid leukemias harboring chromosomal rearrangements t(15;17) and inv(16). Blood. (2003) 101:4640–1. doi: 10.1182/blood-2003-01-0313
62. Eguchi-Ishimae M, Eguchi M, Ishii E, Miyazaki S, Ueda K, Kamada N, et al. Breakage and fusion of the TEL (ETV6) gene in immature B lymphocytes induced by apoptogenic signals. Blood. (2001) 97:737–43. doi: 10.1182/blood.V97.3.737
63. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. (2002) 99:8242–7. doi: 10.1073/pnas.112218799
64. Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Lando A, Schmiegelow K. Increased risk of ALL among premature infants is not explained by increased prevalence of pre-leukemic cell clones. Blood Cells Mol Dis. (2010) 44:188–90. doi: 10.1016/j.bcmd.2009.12.007
65. Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Nersting J, Schmiegelow K. Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood. (2011) 117:186–9. doi: 10.1182/blood-2010-05-282764
66. Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K, et al. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. (2011) 117:368–9. doi: 10.1182/blood-2010-09-309070
67. Olsen M, Hjalgrim H, Melbye M, Madsen HO, Schmiegelow K. RT-PCR screening for ETV6-RUNX1-positive clones in cord blood from newborns in the Danish National Birth Cohort. J Pediatr Hematol Oncol. (2012) 34:301–3. doi: 10.1097/MPH.0b013e3182332268
68. Ornelles DA, Gooding LR, Garnett-Benson C. Neonatal infection with species C adenoviruses confirmed in viable cord blood lymphocytes. PLoS ONE. (2015) 10:e0119256. doi: 10.1371/journal.pone.0119256
69. Kosik P, Skorvaga M, Durdik M, Jakl L, Nikitina E, Markova E, et al. Low numbers of pre-leukemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget. (2017) 8:35824–34. doi: 10.18632/oncotarget.16211
70. Schafer D, Olsen M, Lahnemann D, Stanulla M, Slany R, Schmiegelow K, et al. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood. (2018) 131:821–6. doi: 10.1182/blood-2017-09-808402
71. Kim-Rouille MH, MacGregor A, Wiedemann LM, Greaves MF, Navarrete C. MLL-AF4 gene fusions in normal newborns. Blood. (1999) 93:1107–8. doi: 10.1182/blood.V93.3.1107
72. Hein D, Dreisig K, Metzler M, Izraeli S, Schmiegelow K, Borkhardt A, et al. The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in approximately 0.6% of healthy. Blood. (2019) 134:1355–8. doi: 10.1182/blood.2019002215
73. Skorvaga M, Nikitina E, Kubes M, Kosik P, Gajdosechova B, Leitnerova M, et al. Incidence of common preleukemic gene fusions in umbilical cord blood in Slovak population. PLoS ONE. (2014) 9:e91116. doi: 10.1371/journal.pone.0091116
74. Lausten-Thomsen U, Hjalgrim H, Marquart H, Lutterodt M, Petersen BL, Schmiegelow K. ETV6-RUNX1 transcript is not frequent in early human haematopoiesis. Eur J Haematol. (2008) 81:161–2. doi: 10.1111/j.1600-0609.2008.01091.x
75. Brown P TEL-AML1 in cord blood: 1% or 0.01%? Blood. (2011) 117:2–4. doi: 10.1182/blood-2010-09-304337
76. Aldrich MC, Zhang L, Wiemels JL, Ma X, Loh ML, Metayer C, et al. Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev. (2006) 15:578–81. doi: 10.1158/1055-9965.EPI-05-0833
77. Liang DC, Shih LY, Yang CP, Hung IJ, Liu HC, Jaing TH, et al. Frequencies of ETV6-RUNX1 fusion and hyperdiploidy in pediatric acute lymphoblastic leukemia are lower in far east than west. Pediatr Blood Cancer. (2010) 55:430–3. doi: 10.1002/pbc.22628
78. Pettit T, Cole N, Leung W, Ballantine K, Macfarlane S. Analysis of common cytogenetic abnormalities in New Zealand pediatric ALL shows ethnically diverse carriage of ETV6-RUNX1, without a corresponding difference in survival. Pediatr Blood Cancer. (2017) 64:e26676. doi: 10.1002/pbc.26676
79. Pui CH, Sandlund JT, Pei D, Rivera GK, Howard SC, Ribeiro RC, et al. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA. (2003) 290:2001–7. doi: 10.1001/jama.290.15.2001
80. Bekker-Mendez VC, Miranda-Peralta E, Nunez-Enriquez JC, Olarte-Carrillo I, Guerra-Castillo FX, Pompa-Mera EN, et al. Prevalence of gene rearrangements in Mexican children with acute lymphoblastic leukemia: a population study-report from the Mexican Interinstitutional Group for the identification of the causes of childhood leukemia. Biomed Res Int. (2014) 2014:210560. doi: 10.1155/2014/210560
81. Ceppi F, Brown A, Betts DR, Niggli F, Popovic MB. Cytogenetic characterization of childhood acute lymphoblastic leukemia in Nicaragua. Pediatr Blood Cancer. (2009) 53:1238–41. doi: 10.1002/pbc.22169
82. Mesquita DR, Cordoba JC, Magalhaes IQ, Cordoba MS, Oliveira JR, Goncalves A, et al. Molecular and chromosomal mutations among children with B-lineage lymphoblastic leukemia in Brazil's Federal District. Genet Mol Res. (2009) 8:345–53. doi: 10.4238/vol8-1gmr582
83. Alonso CN, Gallego MS, Rossi JG, Medina A, Rubio PL, Bernasconi AR, et al. RT-PCR diagnosis of recurrent rearrangements in pediatric acute lymphoblastic leukemia in Argentina. Leuk Res. (2012) 36:704–8. doi: 10.1016/j.leukres.2011.12.003
84. Lazic J, Tosic N, Dokmanovic L, Krstovski N, Rodic P, Pavlovic S, et al. Clinical features of the most common fusion genes in childhood acute lymphoblastic leukemia. Med Oncol. (2010) 27:449–53. doi: 10.1007/s12032-009-9232-x
85. Al-Sudairy R, Al-Nasser A, Alsultan A, Al Ahmari A, Abosoudah I, Al-Hayek R, et al. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia in Saudi Arabia: a multi-institutional retrospective national collaborative study. Pediatr Blood Cancer. (2014) 61:74–80. doi: 10.1002/pbc.24584
86. Siraj AK, Kamat S, Gutierrez MI, Banavali S, Timpson G, Sazawal S, et al. Frequencies of the major subgroups of precursor B-cell acute lymphoblastic leukemia in Indian children differ from the West. Leukemia. (2003) 17:1192–3. doi: 10.1038/sj.leu.2402931
87. Chen B, Wang YY, Shen Y, Zhang WN, He HY, Zhu YM, et al. Newly diagnosed acute lymphoblastic leukemia in China (I): abnormal genetic patterns in 1346 childhood and adult cases and their comparison with the reports from Western countries. Leukemia. (2012) 26:1608–16. doi: 10.1038/leu.2012.26
88. Ariffin H, Chen SP, Kwok CS, Quah TC, Lin HP, Yeoh AE. Ethnic differences in the frequency of subtypes of childhood acute lymphoblastic leukemia: results of the Malaysia-Singapore Leukemia Study Group. J Pediatr Hematol Oncol. (2007) 29:27–31. doi: 10.1097/MPH.0b013e318030ac4c
89. Newcombe RG Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med. (1998) 17:857–72.
90. von Goessel H, Jacobs U, Semper S, Krumbholz M, Langer T, Keller T, et al. Cluster analysis of genomic ETV6-RUNX1 (TEL-AML1) fusion sites in childhood acute lymphoblastic leukemia. Leuk Res. (2009) 33:1082–8. doi: 10.1016/j.leukres.2008.11.001
91. Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. (2018) 32:273–84. doi: 10.1038/leu.2017.213
92. Britten O, Ragusa D, Tosi S, Kamel YM. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)-Current Treatment Options. Is there a role for CAR-T cell therapy? Cells. (2019) 8:1341 doi: 10.3390/cells8111341
93. Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. (2003) 3:639–49. doi: 10.1038/nrc1164
94. Hovorkova L, Zaliova M, Venn NC, Bleckmann K, Trkova M, Potuckova E, et al. Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood. (2017) 129:2771–81. doi: 10.1182/blood-2016-11-749978
95. Fueller E, Schaefer D, Fischer U, Krell PF, Stanulla M, Borkhardt A, et al. Genomic inverse PCR for exploration of ligated breakpoints (GIPFEL), a new method to detect translocations in leukemia. PLoS ONE. (2014) 9:e104419. doi: 10.1371/journal.pone.0104419
96. Howson CP, Cedergren B, Giugliani R, Huhtinen P, Padilla CD, Palubiak CS, et al. Universal newborn screening: a roadmap for action. Mol Genet Metab. (2018) 124:177–83. doi: 10.1016/j.ymgme.2018.04.009
97. Martin JA, Hamilton BE, Osterman MJK. Births in the United States, 2019. Atlanta, GA: NCHS Data Brief (2020). p. 1–8.
98. Ribeiro KB, Buffler PA, Metayer C. Socioeconomic status and childhood acute lymphocytic leukemia incidence in São Paulo, Brazil. Int J Cancer. (2008) 123:1907–12. doi: 10.1002/ijc.23738
99. Linabery AM, Slater ME, Spector LG, Olshan AF, Stork SK, Roesler MA, et al. Feasibility of neonatal dried blood spot retrieval amid evolving state policies (2009-2010): a Children's Oncology Group study. Paediatr Perinat Epidemiol. (2011) 25:549–58. doi: 10.1111/j.1365-3016.2011.01228.x
100. Wei C, Lu Q, Khoo SK, Lenski M, Fichorova RN, Leviton A, et al. Comparison of frozen and unfrozen blood spots for gene expression studies. J Pediatr. (2014) 164:189–91.e1. doi: 10.1016/j.jpeds.2013.09.025
101. Alsous MM, Hawwa AF, McElnay JC. Hematocrit, blood volume, and surface area of dried blood spots - a quantitative model. Drug Test Anal. (2020) 12:555–60. doi: 10.1002/dta.2776
102. McDade TW, Williams S, Snodgrass JJ. What a drop can do: dried blood spots as a minimally invasive method for integrating biomarkers into population-based research. Demography. (2007) 44:899–925. doi: 10.1353/dem.2007.0038
103. Pedersen L, Andersen-Ranberg K, Hollergaard M, Nybo M. Quantification of multiple elements in dried blood spot samples. Clin Biochem. (2017) 50:703–9. doi: 10.1016/j.clinbiochem.2017.01.010
104. Haak PT, Busik JV, Kort EJ, Tikhonenko M, Paneth N, Resau JH. Archived unfrozen neonatal blood spots are amenable to quantitative gene expression analysis. Neonatology. (2009) 95:210–6. doi: 10.1159/000155652
105. Ho NT, Busik JV, Resau JH, Paneth N, Khoo SK. Effect of storage time on gene expression data acquired from unfrozen archived newborn blood spots. Mol Genet Metab. (2016) 119:207–13. doi: 10.1016/j.ymgme.2016.08.001
106. Ho NT, Furge K, Fu W, Busik J, Khoo SK, Lu Q, et al. Gene expression in archived newborn blood spots distinguishes infants who will later develop cerebral palsy from matched controls. Pediatr Res. (2013) 73:450–6. doi: 10.1038/pr.2012.200
107. Pallisgaard N, Clausen N, Schroder H, Hokland P. Rapid and sensitive minimal residual disease detection in acute leukemia by quantitative real-time RT-PCR exemplified by t(12;21) TEL-AML1 fusion transcript. Genes Chromosomes Cancer. (1999) 26:355–65.
108. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. (2007) 370:240–50. doi: 10.1016/S0140-6736(07)61126-X
Keywords: leukemia, screening, newborns, translocation, epidemiology, childhood leukemia
Citation: Marcotte EL, Spector LG, Mendes-de-Almeida DP and Nelson HH (2021) The Prenatal Origin of Childhood Leukemia: Potential Applications for Epidemiology and Newborn Screening. Front. Pediatr. 9:639479. doi: 10.3389/fped.2021.639479
Received: 09 December 2020; Accepted: 22 February 2021;
Published: 23 April 2021.
Edited by:
Maria S. Pombo-de-Oliveira, National Cancer Institute (INCA), BrazilReviewed by:
Edward Anders Kolb, Alfred I. duPont Hospital for Children, United StatesMartina Pigazzi, University of Padua, Italy
Ute Fischer, University of Dusseldorf Medical School, Germany
Copyright © 2021 Marcotte, Spector, Mendes-de-Almeida and Nelson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erin L. Marcotte, bWFyY290dGVAdW1uLmVkdQ==