 Omar I. Saadah1,2†
Omar I. Saadah1,2† Babajan Banaganapalli1,3†Naglaa M. Kamal4,5†Ahmed N. Sahly1,6Hadeel A. Alsufyani7
Babajan Banaganapalli1,3†Naglaa M. Kamal4,5†Ahmed N. Sahly1,6Hadeel A. Alsufyani7 Arif Mohammed8
Arif Mohammed8 Aftab Ahmad9
Aftab Ahmad9 Khalidah Khalid Nasser1,10Jumana Y. Al-Aama1,3*Noor Ahmad Shaik1,3*
Khalidah Khalid Nasser1,10Jumana Y. Al-Aama1,3*Noor Ahmad Shaik1,3* Ramu Elango1,3*
Ramu Elango1,3*- 1Princess Al-Jawhara Center of Excellence in Research of Hereditary Disorders, King Abdulaziz University, Jeddah, Saudi Arabia
- 2Pediatric Gastroenterology Unit, Department of Pediatrics, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 3Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 4Department of Pediatrics, Al-Hada Armed Forces Hospital, Taif, Saudi Arabia
- 5Pediatric Hepatology Unit, Department of Pediatrics, Faculty of Medicine, Cairo University, Cairo, Egypt
- 6Department of Neurosciences, King Faisal Specialist Hospital and Research Centre, Jeddah, Saudi Arabia
- 7Department of Physiology, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 8Department of Biology, College of Science, University of Jeddah, Jeddah, Saudi Arabia
- 9Department of Health Information Technology, Faculty of Applied Studies, King Abdulaziz University, Jeddah, Saudi Arabia
- 10Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Jeddah, Saudi Arabia
Background: Alström syndrome (AS) is a very rare childhood disorder characterized by cardiomyopathy, progressive hearing loss and blindness. Inherited genetic variants of ALMS1 gene are the known molecular cause of this disease. The objective of this study was to characterize the genetic basis and understand the genotype–phenotype relationship in Saudi AS patients.
Methods: Clinical phenotyping and whole-exome sequencing (WES) analysis were performed on six AS patients belonging to two unrelated consanguineous Saudi families. Sanger sequencing was performed to determine the mode of inheritance of ALMS1 variant in first-degree family relatives and also to ensure its rare prevalence in 100 healthy population controls.
Results: We identified that Alström patients from both the families were sharing a very rare ALMS1, 3′-splice site acceptor (c.11873−2 A>T) variant, which skips entire exon-19 and shortens the protein by 80 amino acids. This disease variant was inherited by AS patients in autosomal recessive mode and is not yet reported in any population-specific genetic databases. AS patients carrying this mutation showed heterogeneity in clinical presentations. Computational analysis of the mutant centroid structure of ALMS1 mRNA revealed that exon-19 skipping enlarges the hairpin loop and decreases the free energy, eventually affecting its folding pattern, stability, and function. Hence, we propose c.11873–2A as an AS causative potential founder mutation in Saudi Arabia because it is found in two families lacking a common lineage.
Conclusions: We conclude that WES analysis potentially helps in clinical phenotyping, early diagnosis, and better clinical management of Alström patients showing variable clinical expressivity.
Introduction
Alström syndrome (AS; OMIM 203800), first described in 1959, is an inherited ciliary disorder seen in early childhood. The affected children typically present a complex array of clinical characteristics including childhood obesity, early onset of blindness, neurosensory hearing loss, type 2 diabetes mellitus, progressive liver and kidney disease, hypertriglyceridemia, and cardiomyopathy (1). AS patients also present short stature, hypogonadotropic hypogonadism in affected males, and polycystic ovarian syndrome (PCOS) in affected females. In AS patients, clinical features may appear at different times throughout childhood followed by progressive multiorgan failure which reduces life expectancy where patients rarely live beyond their 5th decade of life (2). Although clinical assessment is considered as the first line of diagnosis, gradual multiorgan dysfunction and variable expressivity of the disease within the same family members make it more challenging (3). Clinical management of AS is also complicated as patients present a combination of endocrine, cardiac, renal, neural, and hepatic defects (4). At present, there is no specific therapy for AS; however, early diagnosis and symptom-targeted treatment can prevent or slow down the progression of the disease as well as improve the life span (5).
The genome-wide linkage and homozygosity mapping study in a large French kindred has demonstrated that molecular defects in the ALMS1 gene localized on Chr 2p13.1 are causal mutations (6). ALMS1 gene encompasses 23 exons and codes for a 461-kDa protein with 4,169 amino acids. ALMS1 protein shows ubiquitous expression and is located at centrosomes and basal bodies of ciliated cells, suggesting its probable role in the maintenance of cytoskeleton, regulation of cell division, intraciliary transport, cellular migration, and endosomal recycling (7). At present, ~330 disease-causing mutations (166 missense, 8 splice site, 156 indel) in the ALMS1 gene have been reported in mutation databases. Among the missense mutations, ~90% were observed in exons 8, 10, and 16, making them “mutational hotspot” regions of the ALMS1 gene.
AS is reported in all major ethnic populations like Caucasians, Asians, Africans, Americans, and Arabs (8–10). However, majority of the published studies on AS are conducted in patients belonging to non-Arab ethnicities. Owing to the distinct cultural traditions followed in Arab countries, we believe that studying AS patients from Saudi Arabia might present interesting clinical and genetic findings. Saudi Arabia is regarded as the epicenter of consanguinity with more than 50% marriages (close relatives among extended families). Consanguinity is known to enrich the prevalence of defective alleles in the population and is the predominant reason for high incidence of genetic diseases in certain countries such as Saudi Arabia and Pakistan (11, 12). So far, the number of studies reporting on sporadic AS cases is greater than on familial forms. Hence, familial cases have great potential to characterize the genetic basis of the disease by identifying novel mutations in ALMS1, deducing mode of inheritance, genotype–phenotype correlation, and distinct clinical phenotype presentations. In this study, we aim to use whole-exome sequencing (WES) technology on two unrelated Saudi families to simultaneously characterize the molecular basis of AS and identify casual genetic variants. We also employed various system biology approaches to explore the consequences of ALMS1 pathogenic mutation on the gene function.
Materials and Methods
Patients Recruitment and Clinical Assessment
This study is conducted upon the approval of ethics committees from Al-Hada Armed forces hospital, and King Abdulaziz University Hospital, Jeddah. Initially, AS patients were recruited from the pediatric hepatology and pediatric cardiology clinics, in Al-Hada Armed Forces Hospital, Saudi Arabia. Clinical examination of multiple organs among AS patients were conducted using combined investigations such as electrocardiogram, radiological tests (x-ray), ultrasonography of abdomen, audiometry, and visual evoked potential (13). The extensive family data of the patients were collected by careful interviews by genetic counselors. These families were subsequently referred to genetic medicine clinic for genetic testing, diagnosis, and counseling. Written informed consent was also obtained from all participants of both families.
DNA Isolation
Using a sterile EDTA tube, 2–3 ml of venous blood was collected from each participant and stored at −20°C. Total genomic DNA was isolated using a commercial kit as per the manufacturer's instructions (Wizard® Genomic DNA Purification Kit, Promega). DNA concentration and quality (260:280 ratios) were measured at 260 nm using a NanoDrop spectrophotometer. The integrity of genomic DNA was confirmed using agarose gel electrophoresis as part of qualitative assessment.
Whole-Exome Sequencing and Variants Filtering
One hundred microliters (55 ng/μl) of high-quality genomic DNA (260/280 ratio is 1.9) was used for WES. Library preparation was performed using “Sure Select QXT All human exon V6 (Agilent)” to prepare 60 Mb exome library followed by sequencing on the HiSeq 2000 Next Generation Sequencer platform. Sequencing reads were aligned against human genome reference assembly build 38 (GRCH38.p12) with the help of BLAST (version 0.6.4d). Base quality was recalibrated using the GATK tool (GATK; www.broadinstitute.org/gatk). The alignment of sequencing data revealed 100× coverage for up to 75% of the target region. SAM tools were used for base calling to identify single-nucleotide polymorphisms (SNPs) and indels (14). Variant prioritization and filtering steps were conducted as per the following criteria: a minimum Phred score of 40 (base quality), rare allele frequency (MAF < 0.01), functional variants (coding or regulatory regions), pathogenicity based on variant prediction tools (SIFT, Polyphen, and CADD), and allelic zygosity (homozygous or compound heterozygous in patients).
Sanger Sequencing Validation of ALMS1 Mutation
Potential ALMS1 candidate mutation in affected family members and population healthy controls was validated using the Sanger sequencing method. Primer sequences targeting an average amplicon size of 400–500 bp (forward primer: 5′-GTCTTTCTAACTTGGGATCAGAG-3′ and reverse primer: 5′-CCTCCAGGGTCTGGTCTTG-3′), were designed using the NCBI-primer BLAST online program, followed by PCR and bidirectional dideoxynucleotide sequencing reactions. The BioEdit program was used for sequence alignment and annotation of nucleotide sequence mismatches (http://www.mbio.ncsu.edu/). All ALMS1 mutations were annotated against the ENST00000613296.4 reference m-RNA sequence. The position of mutation was determined according to human genome variation society guidelines, which recommended to consider “A” of ATG codon as the 1st nucleotide in the mRNA.
Computational Biology Analysis of ALMS1 Mutation
Computational analysis of potential ALMS1 mutation was performed based on the results obtained from WES and Sanger sequencing. Our system biology approach was subjective to the nature of ALMS1 mutation and its effect on RNA or protein sequence. We have initially used Human Splicing Finder (HSF), a publicly available web server (http://www.umd.be/HSF/) to predict the impact of mutations on the strength of 5'ss, 3'ss, and branch splicing points. For predicting the impact of the mutation on ALMS1 RNA secondary structure, we used RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (29), which uses the loop-based energy model and the dynamic programming algorithm for prediction of the linear or circular single-stranded RNA structure (16). The minimum free energy (MFE) value differences were compared to measure mutation-induced stability differences between wild-type and mutant secondary structures of ALMS1 and RNA molecules.
Results
Identification of the ALMS1 Variant in AS Patients
In this study, we present the whole-exome sequencing results of six AS patients (three from each family) belonging to two unrelated families from Saudi Arabia. In both families, parents were first cousin-consanguineous marriages. Exome sequencing of each AS patient has yielded an average number of 6.9 billion bp read bases and 46 million bases, with 51.2% of GC content and 93.2% of Q30. The average read length was 148.96 base pairs, and the cumulative depth distribution value in the target region for >60% of bases was 50× or greater. Post quality control filtering, we found approximately 96.5% of known nucleotide variants, previously reported in public databases. From each exome, approximately 96,000 SNPs, including 12,153 synonymous variants, 12,000 missense variants, and 11,500 indels were identified.
Since ALMS1 pathogenic mutations are known to be clustered around hotspot regions, we have initially screened biallelic mutations located in exons 5, 8, 10, 11, 16, 17, and 18. Interestingly, the mutation search of all AS patients has not revealed the presence of either coding or regulatory region mutations in the hotspot regions of ALMS1 gene. However, interestingly, we found a novel ALSM1 mutation (c.11873–2A>T) at the intron 18–Exon-19 boundary, which is not reported in any of the public resource genetic databases like gnomAD,1000 genomes, ExAC, dbSNP-NCBI, and Saudi Human Genome Project (SGHP). Table 1 summarizes the genotype–phenotype correlations for all splice site mutations. The CADD score of this mutation is 24.5, which classifies it as likely pathogenic. Since all the six AS patients carried this mutation, we assumed that all the patients have inherited one copy of the defective allele from each parent. Then, the validation of exome sequencing derived c.11873–2A>T mutation in all the probands and other family members were carried out by Sanger sequencing, which confirmed the absence of this mutation in homozygous form among the remaining family members. None of the 100 healthy controls had this pathogenic mutation, whether hetero- or homozygous states.
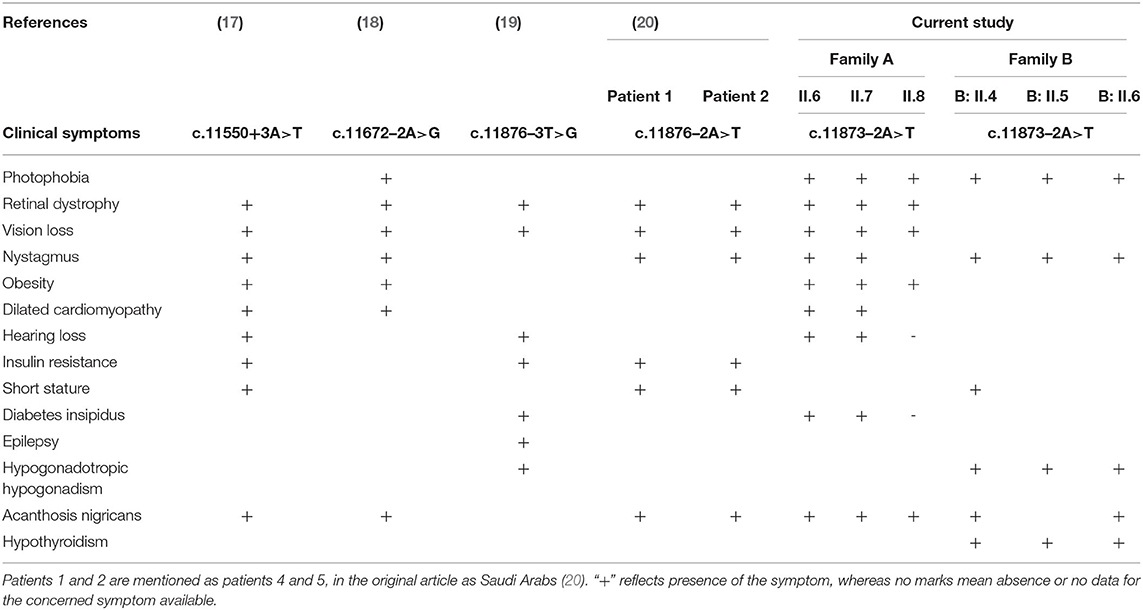
Table 1. Genotype–phenotype correlations for ALMS1 splice site mutations in Alström syndrome patients.
Inheritance Mode of ALMS1 Mutation
Family A
In this consanguineous family, both father (I.1) and mother (I.2) were heterozygous carriers of c.11873–2 A>T mutation (Figure 1). The index case (II.6, 17 years) and the two siblings (II.7, 15 years & II.8, 8 years) were homozygous to the T allele. Among other siblings, three sisters (II.3, II.4, II.5) and one brother (II.9) were homozygous to the wild-type allele “A.” The mode of inheritance of ALMS1 mutation confirms that both defective alleles were transmitted from parents to AS patients in an autosomal recessive pattern. All the three affected patients including the proband (IV.6, IV.7, IV.8) showed common clinical symptoms including severe photophobia, progressive visual loss, obesity, bilateral horizontal nystagmus, and flat feet. Diabetes mellitus, cardiomyopathy, and mental disability were observed in probands IV.6 and IV.7 only.
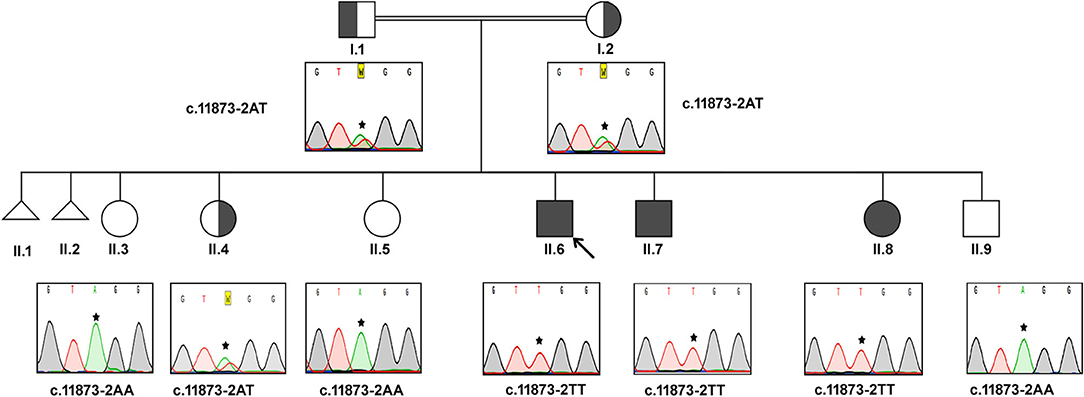
Figure 1. Pedigree and the DNA sequence analysis of AS Family A. Proband. (II.6) is indicated by the arrow. Each DNA chromatogram (reverse strand) shows the result from the respective Sanger sequencing confirming the status of the mutation. The proband is having the homozygous TT mutation at the splice site (c.11873–2). Both parents are heterozygous for the given mutation. All three affected siblings show homozygous mutations while the unaffected siblings were either heterozygous for the mutation or homozygous for the normal allele.
Proband II.6 is severely photophobic and experienced a progressive loss of visual acuity since the age of 9. His eyes showed bilateral horizontal nystagmus, and hearing loss since the age of 11. II.6 has also manifested hepatic dysfunction, sensorineural hearing loss (SNHL), speech impairment, and acanthosis nigricans. He reported mild continuous palpitations and was NYHA class III. He did not have recurring infections but had a history of balancing problems in gait. II.7 presented with progressive visual acuity loss since the age of 3. He was severely photophobic, like his elder brother (II.6). He had an acquired bilateral horizontal nystagmus. He only complained of tinnitus, not hearing loss. He has also complained of episodes of profuse epistaxis, sometimes during sleep. He manifested with acanthosis nigricans and was on oral hypoglycemic. His frequency of urination increased, and he had nocturia. He reported the sensation of pounding continuous palpitations. He was NYHA class II and reported general fatigue. Both II.6 and II.7 had a history of long standing cough with yellow sputum that is precipitated by moderate activity.
Patient II-8 presented with photophobia and visual loss since a very young age (<5). She did not complain of hearing loss or tinnitus. She manifested with acanthosis nigricans and had a history of persistent weight gain and increased appetite. She did not have urinary habit changes or nocturia. She had dental abnormalities and was diagnosed with hypertension. She had mood swings and numbness on her left hand and foot. Their clinical follow-up revealed that none of them have developed renal failure or recurrent infections or malignancies. This family reported thyroid pathology symptoms consistent with hypothyroidism (aversion to cold, dryness, and myxedema).
Family B
The Sanger sequencing findings, after the exome sequencing identified the mutation, of this consanguineous AS family are as follows; the proband (II.4) and her two brothers (II.5 and II.6) were homozygous to T minor allele, whereas their healthy siblings were homozygous to A major allele (II.1, II.2, and II.3) and both parents (I.1 and I.2) were heterozygous (AT) for ALMS1, c.11873–2A>T mutation. These results confirm that the AS patients in this family have inherited one copy of defective T allele from each parent, confirming the autosomal recessive mode of inheritance (Figure 2). The probands II.4 (12 years), II.5 (9 years), and II.7 (4 years) showed initial symptoms of vision dysfunction (severe progressive retinal dystrophy and cataract) and dilated cardiomyopathy. The common eye manifestations are photophobia, nystagmus, and squint easily identified by parents early between 3 and 36 months. All the three patients had visual manifestations before the first 36 months of their life. The proband (11.4; 12 years) had reported photophobia since her first year. She had bilateral horizontal nystagmus, progressive loss of visual acuity, and bilateral sensorineural hearing loss. She used to sleep during the day and mostly avoided daytime because of her photophobia. She was obese (>95 percentile) with polyphagia and acanthosis nigricans. At the age of 5, she was diagnosed with autism spectrum disorder. She was short for her age and had microcephaly but did not have any extra or fused digits. She was also diagnosed with hypothyroidism. She did not have any allergies. Probands II.5 and II.6 presented SNHL and endocrine-related disorders like impaired glucose homeostasis, hypothyroidism, hypogonadism evidenced by small testes, and precocious puberty. Acanthosis nigricans and sparse hair were also observed in both of them. The remaining three sisters (II.1, II.2, and II.3) were healthy and did not present any symptoms of the disease. Both parents were apparently healthy and mention no significant health complications related to Alström Syndrome (I.1 and I.2).
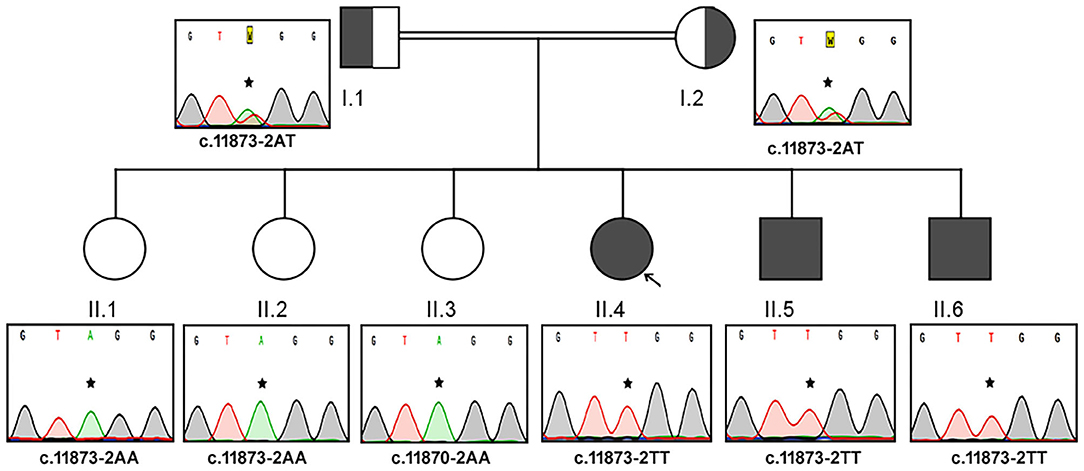
Figure 2. Pedigree and the DNA sequence analysis of AS Family B. Each DNA chromatogram (reverse strand) shows the result from the Sanger sequencing confirming the status of the mutation. Both parents (I.1 and I.2) are heterozygotes, three AS patients are homozygous for TT, while the three unaffected siblings are homozygotes for AA genotype for the given genetic location.
Computational Analysis of ALMS1 Mutation
Splice Site Prediction
The HSF analysis of ALMS1 variant (c.11873–2A>T) in the splice site (tgtttcctgtAGga >> tgtttcctgtTGga) predicted the consensus values (CV) of 87.32 and 59.45 for the wild-type active sites and mutant active sites, respectively. The CV variation score of −31.92% between wild-type and mutant alleles suggests that the loss of wild-type splice site acceptor (AG>TG) would result in the skipping of exon 19 and instead accept the splice site acceptor sequence (AG) of exon 20 of ALMS1.
RNA Secondary Structure Analysis
The ALMS1, RNA secondary structure is characterized by helix, loops, stem, and dangling ends. Although both wild-type and mutant RNAs showed structural similarity, their centroid structures revealed differences in their minimum free energy (MFE) values. In the native state, A nucleotide at c.11873–2, located in the hairpin loop, confers stability (144 K.Cal/Mol of free energy) to the secondary structure of ALMS1, RNA compared to the variant state (c.11873–2 A>T), where both sizes of the hairpin loop is seen enlarged and free energy is decreased (−175.8 K.Cal/Mol of free energy). Hence, it is assumed that the lower stability of mRNA with c.11873–2 A>T is likely to affect the mRNA folding pattern and tertiary structure formation (Figure 3).
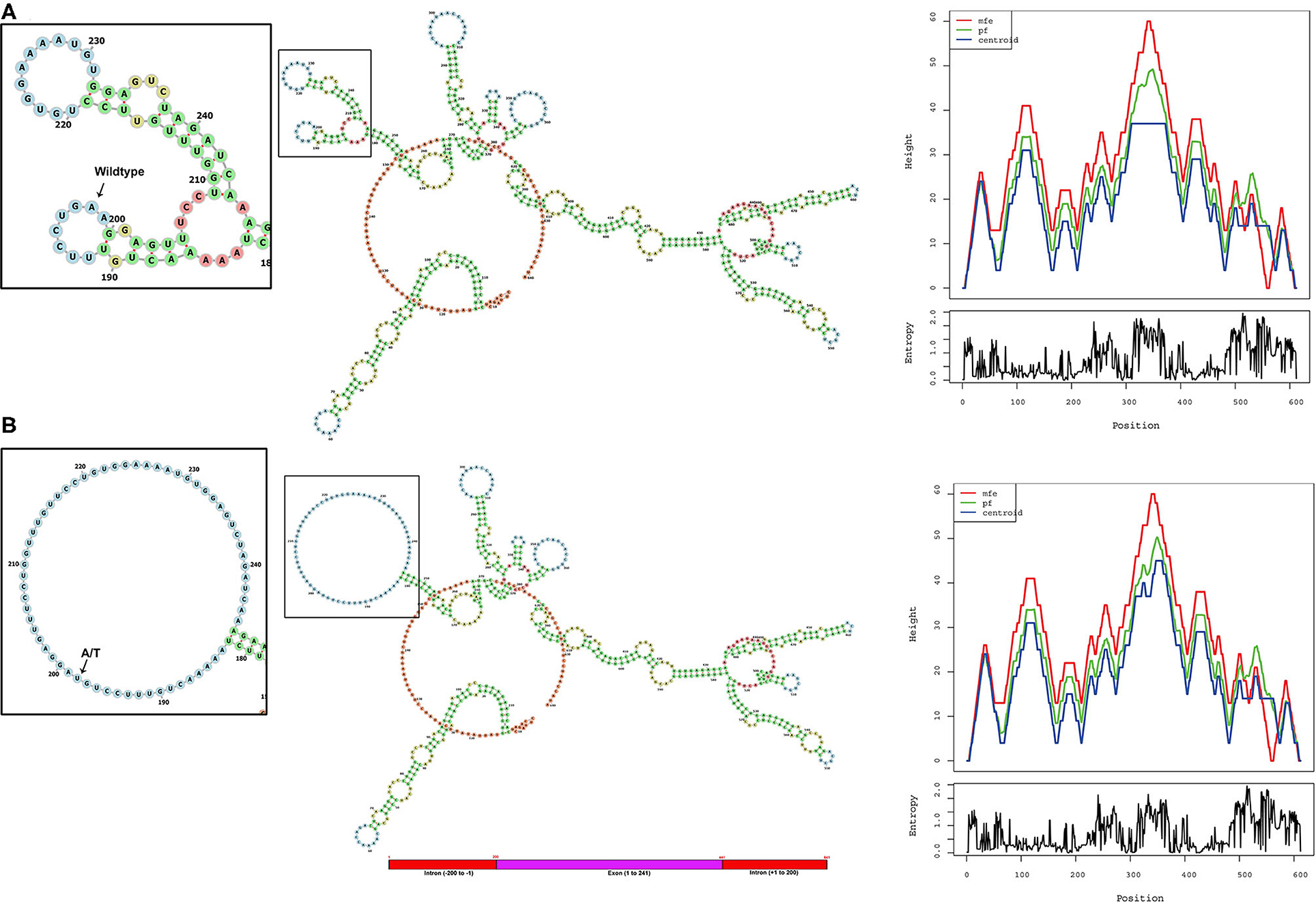
Figure 3. Computational analysis and the RNA secondary structure predictions. The secondary structure predictions of (A) native and (B) variant RNA structures of ALMS1 using RNAfold software. Inset shows the enlarged picture of the region containing the native or the variant part of the RNA. Base-pair probability is indicated in the key. Mountain plot shows the secondary structures in a height vs. position, where the helices are represented in slopes, loops in plateaus, and hairpin loops in the peaks.
Discussion
Homozygous or compound heterozygous mutations in the ALMS1 gene results in AS disorder. Hence, early identification of defective variants of ALMS1 in potential asymptomatic child and carriers will provide better clinical management and options for prenatal diagnosis, respectively. Whole-exome sequencing offers an efficient and cost-effective approach in identifying ALMS1 mutations with greater depth. Although whole-exome sequencing generates the unbiased exonic variant data from all coding genes, variant screening strategy first targets exons 8, 10, and 16 regions of the ALMS1 gene, which can solve the genetic basis in almost 90% of cases (1). However, the lack of ALMS1 exonic mutations in some AS patients suggests the possibility for the regulatory region mutations. The genetic and clinical data of Alström Syndrome of non-Arab patients is relatively well published compared to Arab patients, who inherently possess large blocks of homozygosity due to their traditional consanguineous marriage practices (8, 9, 18, 21–24). Many rare or novel mutations in different autosomal recessive diseases are widely reported in Saudi Arabian populations (25, 26).
There are few publications which reported the genetic basis of AS patients from Saudi Arabia. Table 2 summarizes the ALMS1 mutations and clinical phenotypes of Saudi AS patients. The first study conducted on four Saudi AS patients used genetic linkage mapping and Sanger sequencing and reported allelic heterogeneity of ALMS1 mutations, i.e., c.5534 C>G (S908X) and c.5981delCAGA(1992X) mutations in exon 8, c.8275C >T (R2720X) mutation in exon10, IVS18-2 A>T in exon 18 (20). Whole-exome sequencing of a 10-year-old Saudi girl presenting with diabetic ketoacidosis, hearing loss, and blindness found a homozygous frameshift deletion in exon 20 (c.12154_12166del) which creates premature termination codon (p.Arg4052Glyfs*2) in ALMS1 gene (27). Another whole-exome sequencing study on a 5-year-old Saudi girl, with photophobia, marked nystagmus, and retinal changes with short fingers tapering born to the consanguineous marriage, has reported a pathogenic mutation in ALMS1 gene (c. 8441C>A, p.S2814*) (31). By using whole-exome sequencing, earlier our group found causative rare biallelic mutations in the ALMS1 gene in exon 8 (T376S in exon 5, and S909*) and exon 10 (R2721*) among AS patients from two other unrelated Saudi families (28). These truncating mutations at residues S909 and R2721 possibly create an unstable protein due to loss of CC domain and ALMS motif on the C-terminal end which led to intracellular truncated protein degradation (31). Another retrospective study from Saudi Arabia has reported the identification of 10 ALMS1 different mutations (E3649*, Q2648*, p.E913Sfs*20, p.Ser2102*, p.Arg2928*, p.Ser2102*, p.Arg2722*, p.P3911QfsX16, p.Ser908*, IVS18-3A>T) in 19 Alström cases presenting different ophthalmic phenotypes from 13 Saudi families. Their finding underscored the point that visual phenotypes in children should raise suspicion for Alström syndrome (30).
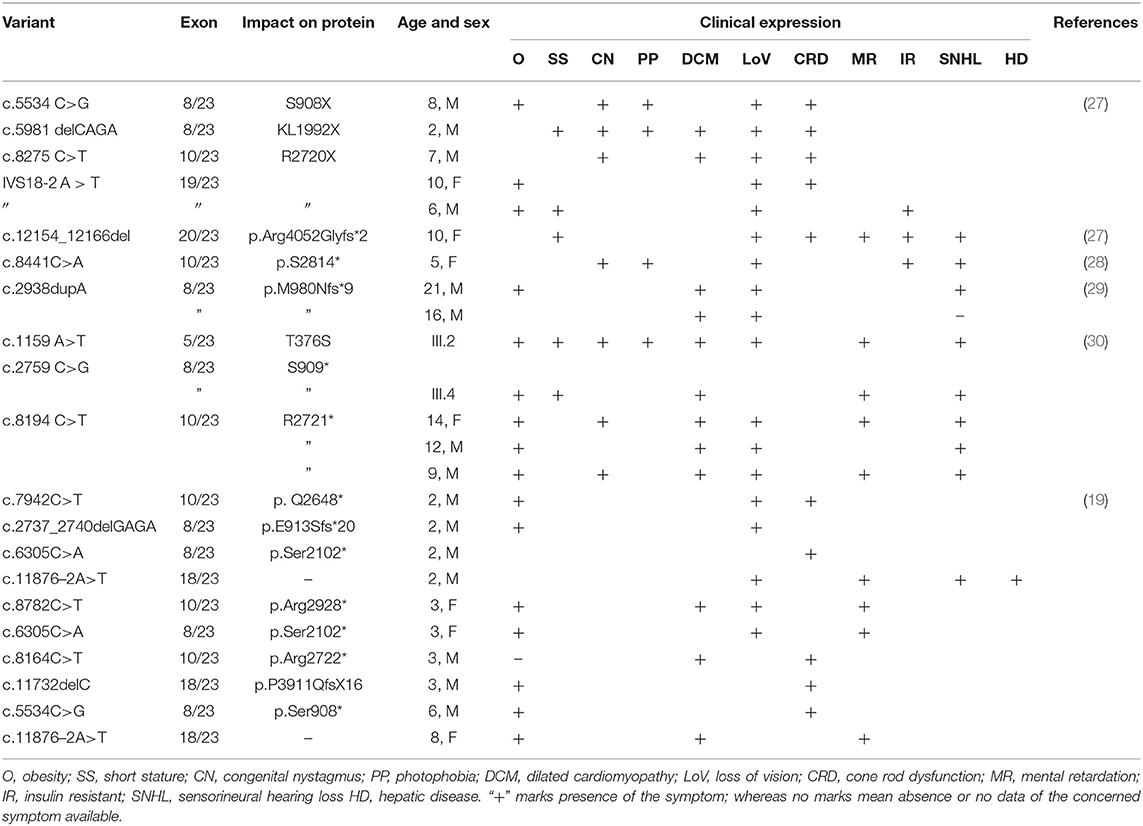
Table 2. Summary of genetic mutation vs. clinical features observed in Saudi Alström patients.
There are six ALMS1 splice site mutations (c.454–5T>G, c.7677+1G>T, c.10388–2A>G, c.11550+3A>T, c.11672–2A>G, c.11876–3T>G, c.11876–2A>T) reported in previous publications (7, 17–20, 30). In the current investigation, through whole-exome sequencing of AS patients from the consanguineous background, we observed a rare homozygous mutation (c.11873–2A>T) at the acceptor splice site of exon 19 of the ALMS1 gene. This mutation is similar to the previous publications which reported it in sporadic cases (19, 20, 30). From its autosomal recessive mode of inheritance pattern of ALMS1 mutation and subsequent computational functional analysis, we propose that this mutation is most likely disease causative in these families. Despite the lack of overlap between the lineages of the two studied families, we assume that c.11873–2A is possibly a founder mutation in Saudi Arabia. In eukaryotes, dinucleotides at the 5′ (GT) and 3′ (AG) ends of intronic sequences are recognized as exon boundaries, which are subsequently acted by the spliceosome complex to remove introns (32). The A>T transversion converts c.11873–2 the 3′ AG splice acceptor sequence to TG, due to which the RNA spliceosome complex skips the entire exon 19 (Chr2: 73,601,195–73,601,436) and accepts the next 3′ AG splice acceptor of exon 20, resulting in a shorter version of ALMS1 mRNA by 241 nucleotides.
In the absence of functional data, we tried to understand the putative functional role of the c.11873–2A variant on the secondary structural features of ALMS1 mRNA using RNA fold software, which predicted a free energy difference of −31 K.Cal/Mol between native and variant forms. Hence, it is assumed that the lower stability of mRNA with c.11873–2 A>T is likely to affect the mRNA folding pattern and tertiary structure formation. The change in the secondary structure of the splice site variant RNA may further alter its function which could primarily occur via the two possible mechanisms. Firstly, in the absence of the correct folding of the RNA, the physiological functioning of the RNA which is mediated through the potential interaction of RNA with the protein complexes like spliceosome complex (33) would be lost or change and thus affect proper RNA transport or mRNA splicing or both. Secondly, due to the improper folding of RNA, potential binding sites of micro-RNA (miRNA) could be lost or modified, leading to an altered cellular condition that may lead to the disease state (32, 34).
The mechanistic link between ALMS1 genotype vs. AS protein phenotype is not yet well-understood, partly because of limited functional data exploring the role of catalytic domains in protein function. Of the 4,166 aa long native ALMS1 protein, exon 19 encodes 80 amino acids (3,958–4,038 aa) lying in between serine-rich (3,857–3,873 aa) and ALMS1 motif regions (4,037–4,166 aa) at the c-terminal region of ALMS1 protein. It is assumed that exon 19 skipping by the c.11873–2A>T variant results in a short polypeptide (4086 aa), which could affect the loss of secondary (α-helices, and β-pleated sheets) and tertiary (3D) and quaternary (biomolecular complexes) structural features, in addition to biological functions (microtubule organization, intracellular transport, endosome recycling, and cell-cycle regulation) of ALMS1 protein. It remains plausible that the primary cilium or basal body dysfunction due to defective ALMS1 among homozygous carriers contributes to many aspects of the AS phenotype including obesity, retinal dystrophy, hearing loss, kidney dysfunction, neurological disturbances, and fibrosis (1).
In conclusion, this study confirmed the autosomal recessive inheritance of a very rare ALMS1 mutation among six AS patients from two unrelated consanguineous Saudi families. This mutation abolishes the 3′-AT splice site acceptor sequence of exon 19 due to which the entire exon 19 is skipped, shortening the mRNA by 241 nucleotides. Bioinformatics prediction tools demonstrated that this mutation destabilizes the secondary structural features of mature mRNA, hence disrupting the biological functions of ALMS1 protein. Future functional studies at the level of cDNA and protein could provide more insights about variant pathogenicity and also help to understand how this ALMS1 genotype contributes to AS clinical phenotypes. This study confirms the important utility of WES in detecting pathogenic variants occurring in either coding or regulatory regions among AS patients. Multidimensional molecular characterization of complex rare disorders is challenging owing to the variable clinical expressivity among AS patients. However, WES proved to be a valuable tool in helping clinicians in diagnosis, providing better clinical management and intervention for AS patents and their families.
Data Availability Statement
The datasets presented in this article are not readily available because(a) participants' refusal to store or distribute the genomic data in the public domain and (b) as per the local Institutional Ethics committee approval and national policy on genomic data sharing in the public domain outside the country. Requests to access the datasets should be directed to RE, OS, or NS.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of King Abdulaziz University, Saudi Arabia. Informed consent was obtained from each tested individual prior to genetic testing. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
OS, BB, NK, RE, and NS: conceptualization. NS and BB: data curation. RE: formal analysis. NA: funding acquisition and project administration. AS, HA, AM, NS, and BB: methodology. BB: software and visualization. OS, JA-A, NS, and RE: supervision. BB and KN: validation. OS, BB, NK, AA, JA-A, NS, and RE: writing of original draft and editing. All authors contributed to the article and approved the submitted version.
Funding
This project was funded by the National Plan for Science, Technology, and Innovation (MAARIFAH)—King Abdulaziz City for Science and Technology—the Kingdom of Saudi Arabia—award number 13-MED2225-03.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge with thanks the Science and Technology Unit, King Abdulaziz University, for technical support.
References
1. Hearn T. ALMS1 and Alström syndrome: a recessive form of metabolic, neurosensory and cardiac deficits. J Mol Med. (2019) 97:1–17. doi: 10.1007/s00109-018-1714-x
2. Kuburovic V, Marshall JD, Collin GB, Nykamp K, Kuburovic N, Milenkovic T, et al. Differences in the clinical spectrum of two adolescent male patients with Alstrom syndrome. Clin Dysmorphol. (2013) 22:7–12. doi: 10.1097/MCD.0b013e32835b9017
3. Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genomics. (2011) 12:225–35. doi: 10.2174/138920211795677912
4. Waldman M, Han JC, Reyes-Capo DP, Bryant J, Carson KA, Turkbey B, et al. Alstrom syndrome: renal findings in correlation with obesity, insulin resistance, dyslipidemia and cardiomyopathy in 38 patients prospectively evaluated at the NIH clinical center. Mol Genet Metab. (2018) 125:181–91. doi: 10.1016/j.ymgme.2018.07.010
5. Van Groenendael S, Giacovazzi L, Davison F, Holtkemper O, Huang Z, Wang Q, et al. High quality, patient centred and coordinated care for Alstrom syndrome: a model of care for an ultra-rare disease. Orphanet J Rare Dis. (2015) 10:149. doi: 10.1186/s13023-015-0366-y
6. Collin GB, Marshall JD, Cardon LR, Nishina PM. Homozygosity mapping at Alstrom syndrome to chromosome 2p. Hum Mol Genet. (1997) 6:213–9. doi: 10.1093/hmg/6.2.213
7. Marshall JD, Muller J, Collin GB, Milan G, Kingsmore SF, Dinwiddie D, et al. Alstrom Syndrome: mutation spectrum of ALMS1. Hum Mutat. (2015) 36:660–8. doi: 10.1002/humu.22796
8. Rethanavelu K, Fung JLF, Chau JFT, Pei SLC, Chung CCY, Mak CCY, et al. Phenotypic and mutational spectrum of 21 Chinese patients with Alström syndrome. Am J Med Genet A. (2020) 182:279–88. doi: 10.1002/ajmg.a.61412
9. Castro ÁB, Coronado BN, Costa RH, Chalita MR, Cella WP, Avila MP. Morphological and functional findings in Alström syndrome: a study of two families. Arq Bras Oftalmol. (2018) 81:524–8. doi: 10.5935/0004-2749.20180102
10. Piñeiro-Gallego T, Cortón M, Ayuso C, Baiget M, Valverde D. Molecular approach in the study of Alström syndrome: analysis of ten Spanish families. Mol Vis. (2012) 18:1794–802.
11. Monies D, Abouelhoda M, Alsayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. (2017) 136:921–39. doi: 10.1007/s00439-017-1821-8
12. Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. (2017) 22:1604–14. doi: 10.1038/mp.2016.109
13. Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum Mutat. (2007) 28:1114–23. doi: 10.1002/humu.20577
14. Al-Aama JY, Shaik NA, Banaganapalli B, Salama MA, Rashidi O, Sahly AN, et al. Whole exome sequencing of a consanguineous family identifies the possible modifying effect of a globally rare AK5 allelic variant in celiac disease development among Saudi patients. PLoS ONE. (2017) 12:e0176664. doi: 10.1371/journal.pone.0176664
15. Lorenz R, Bernhart SH, Siederdissen SHZ, Tafer H, Flamm C, Stadler PF, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. (2011) 6:26. doi: 10.1186/1748-7188-6-26
16. Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. (1981) 9:133–48. doi: 10.1093/nar/9.1.133
17. Weiss S, Cohen L, Ben-Yosef T, Ehrenberg M, Goldenberg-Cohen N. Late diagnosis of Alstrom syndrome in a Yemenite-Jewish child. Ophthal Genet. (2019) 40:7–11. doi: 10.1080/13816810.2018.1561900
18. Kilinç S, Yücel-Yilmaz D, Ardagil A, Apaydin S, Valverde D, Özgül RK, et al. Five novel ALMS1 gene mutations in six patients with Alström syndrome. J Pediatr Endocrinol Metab. (2018) 31:681–7. doi: 10.1515/jpem-2017-0418
19. Sanyoura M, Woudstra C, Halaby G, Baz P, Senée V, Guillausseau PJ, et al. A novel ALMS1 splice mutation in a non-obese juvenile-onset insulin-dependent syndromic diabetic patient. Eur J Hum Genet. (2014) 22:140–3. doi: 10.1038/ejhg.2013.87
20. Aldahmesh MA, Abu-Safieh L, Khan AO, Al-Hassnan ZN, Shaheen R, Rajab M, et al. Allelic heterogeneity in inbred populations: the Saudi experience with Alström syndrome as an illustrative example. Am J Med Genet A. (2009) 149a:662–5. doi: 10.1002/ajmg.a.32753
21. Tahani N, Maffei P, Dollfus H, Paisey R, Valverde D, Milan G, et al. Consensus clinical management guidelines for Alström syndrome. Orphan J Rare Dis. (2020) 15:253. doi: 10.1186/s13023-020-01468-8
22. Gatticchi L, Miertus J, Maltese PE, Bressan S, De Antoni L, Podracká L, et al. A very early diagnosis of Alstr?m syndrome by next generation sequencing. BMC Med Genet. (2020) 21:173. doi: 10.1186/s12881-020-01110-1
23. Das Bhowmik A, Gupta N, Dalal A, Kabra M. Whole exome sequencing identifies a homozygous nonsense variation in ALMS1 gene in a patient with syndromic obesity. Obes Res Clin Pract. (2017) 11:241–6. doi: 10.1016/j.orcp.2016.09.004
24. Ozantürk A, Marshall JD, Collin GB, Düzenli S, Marshall RP, Candan S, et al. The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey. J Hum Genet. (2015) 60:1–9. doi: 10.1038/jhg.2014.85
25. Alallasi SR, Kokandi AA, Banagnapali B, Shaik NA, Al-Shehri BA, Alrayes NM, et al. Exome analysis identifies a novel compound heterozygous alteration in TGM1 gene leading to lamellar ichthyosis in a child from Saudi Arabia: case presentation. Front Pediatr. (2019) 7:44. doi: 10.3389/fped.2019.00044
26. Gaboon NEA, Banaganapalli B, Nasser K, Razeeth M, Alsaedi MS, Rashidi OM, et al. Exome sequencing and metabolomic analysis of a chronic kidney disease and hearing loss patient family revealed RMND1 mutation induced sphingolipid metabolism defects. Saudi J Biol Sci. (2020) 27:324–34. doi: 10.1016/j.sjbs.2019.10.001
27. Bakar AA, Kamal NM, Alsaedi A, Turkistani R, Aldosari D. Alström syndrome: A novel mutation in Saudi girl with insulin-resistant diabetes. Medicine. (2017) 96:e6192. doi: 10.1097/MD.0000000000006192
28. Kamal NM, Sahly AN, Banaganapalli B, Rashidi OM, Shetty PJ, Al-Aama JY, et al. Whole exome sequencing identifies rare biallelic ALMS1 missense and stop gain mutations in familial Alström syndrome patients. Saudi J Biol Sci. (2020) 27:271–8. doi: 10.1016/j.sjbs.2019.09.006
29. Bdier AY, Al-Qahtani FA, Kumar Verma P, Alshoaibi NA, Mohammed Alrayes N, Shaik, et al. A novel homozygous ALMS1 protein truncation mutation (c.2938dupA) revealed variable clinical expression among Saudi Alström syndrome patients. Archives Med Sci. (2020) doi: 10.5114/aoms.2020.100635
30. Khan AO, Bifari IN, Bolz HJ. Ophthalmic features of children not yet diagnosed with alstrom syndrome. Ophthalmology. (2015) 122:1726–7.e1722. doi: 10.1016/j.ophtha.2015.03.001
31. Safieh LA, Al-Otaibi HM, Lewis RA, Kozak I. Novel mutations in two saudi patients with congenital retinal dystrophy. Middle East Afr J Ophthalmol. (2016) 23:139–41. doi: 10.4103/0974-9233.171779
32. Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. (2018) 59:253–68. doi: 10.1007/s13353-018-0444-7
33. Magnus M, Kappel K, Das R, Bujnicki JM. RNA 3D structure prediction guided by independent folding of homologous sequences. BMC Bioinform. (2019) 20:512. doi: 10.1186/s12859-019-3120-y
Keywords: Alström syndrome, ALMS1, splice site mutation, rare variant, Saudi Arabia
Citation: Saadah OI, Banaganapalli B, Kamal NM, Sahly AN, Alsufyani HA, Mohammed A, Ahmad A, Nasser KK, Al-Aama JY, Shaik NA and Elango R (2021) Identification of a Rare Exon 19 Skipping Mutation in ALMS1 Gene in Alström Syndrome Patients From Two Unrelated Saudi Families. Front. Pediatr. 9:652011. doi: 10.3389/fped.2021.652011
Received: 11 January 2021; Accepted: 17 February 2021;
Published: 26 April 2021.
Edited by:
Abjal Pasha Shaik, King Saud University, Saudi ArabiaReviewed by:
Syed Razvi, Community College of Qatar (CCQ), QatarMd Zubbair Malik, Jawaharlal Nehru University, India
Purna Ganji, Emory University, United States
Copyright © 2021 Saadah, Banaganapalli, Kamal, Sahly, Alsufyani, Mohammed, Ahmad, Nasser, Al-Aama, Shaik and Elango. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramu Elango, cmVsYW5nb0BrYXUuZWR1LnNh; Noor Ahmad Shaik, bnNoYWlrQGthdS5lZHUuc2E=; Jumana Y. Al-Aama, amFsYW1hQGthdS5lZHUuc2E=
†These authors have contributed equally to this work