 Marzieh Nejabat1
Marzieh Nejabat1 Soroor Inaloo2Afsaneh Taghipour Sheshdeh3Shima Bahramjahan3Fatima Masoomi Sarvestani3Pegah Katibeh1Hamid Nemati4,5Seyed Mohammad Bagher Tabei6
Soroor Inaloo2Afsaneh Taghipour Sheshdeh3Shima Bahramjahan3Fatima Masoomi Sarvestani3Pegah Katibeh1Hamid Nemati4,5Seyed Mohammad Bagher Tabei6 Mohammad Ali Faghihi3,7*
Mohammad Ali Faghihi3,7*- 1Pediatric Neurology Ward, Shiraz University of Medical Sciences, Shiraz, Iran
- 2Neonatal Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 3Persian BayanGene Research and Training Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 4Epilepsy Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 5Shiraz Neuroscience Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 6Maternal-Fetal Medicine Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 7Express Gene Molecular Diagnostics Laboratory, Palmetto Bay, FL, United States
Purpose: Cerebral palsy (CP) is a heterogeneous permanent disorder impacting movement and posture. Investigations aimed at diagnosing this disorder are expensive and time-consuming and can eventually inconclusive. This study aimed to determine the diagnostic yield of next generation sequencing in patients with atypical CP (ACP).
Methods: Patient eligibility criteria included impaired motor function with onset at birth or within the first year of life, and one or more of the following conditions: severe intellectual disability, positive family history, brain imaging findings not typical for cerebral palsy, abnormal neurometabolic profile, intractable seizure, normal neuroimaging despite severe psychomotor disability, after pediatric neurologist assessment including neuroimaging and biochemical-metabolic study offered for genetic study.
Results: Exome sequencing was done for 66 patients which revealed pathogenic, likely pathogenic, and variants of unknown significance in 36.2, 9, and 43.9%, respectively. We also found 10 new mutations and were able to suggest specific and personalized treatments for nine patients. We also found three different mutations with different phenotypical spectrum in one gene that have not been reported for cerebral palsy.
Conclusion: An accurate history and physical examination and determination of patients with atypical cerebral palsy for doing exome sequencing result in improved genetic counseling and personalized management.
Introduction
Cerebral palsy is a group of chronic neurodevelopmental disorders that is the most common cause of childhood physical disability and shows heterogeneity in all of its aspects including etiology, presentation, functional severity, comorbidities, treatment options, and outcomes (1–4). Cerebral palsy definition derived from Swaiman's pediatric Neurology 2017 (6th Edition). Few cases are solely due to prematurity or severe hypoxic-Ischemia at birth (5, 6). Cerebral palsy is a non-progressive, but often changing, motor impairment syndrome secondary to brain lesions or anomalies in the early stages of its development (7). CP rates have remained the same for 50 years despite major advances in the obstetrics and neonatology. It is seen in around 2–2.5 for every 1,000 births. Although there have been small statistical fluctuations in the cerebral palsy rates amongst children born preterm, the rates of cerebral palsy at term remain stable (5). Along with motor disabilities, children with CP have disturbances of sensation, perception, learning, and behavior. CP imposes great demands on health, social, and educational services, as well as a large financial and emotional burden on families (1–4, 6). In this report we present our experience with a group of patients who were assessed at our institution with neurodevelopmental disorders and initial diagnosis of CP, but in whom the condition was not associated with known perinatal complications or with the brain lesions commonly related to CP. Atypical CP included: full term neonate without history of perinatal and postnatal insult; absence of brain MRI finding compatible with neonatal asphyxia; progressive neurological deterioration; severe or profound intellectual disability; severe hypotonia opposite spasticity; positive family history of one or more similarly affected relatives (8). The main goal of this work was to delineate the clinical manifestations, laboratory data and molecular findings of patients who are regarded as CP mimics or atypical CP, so that more targeted approaches to the diagnosis and management of this condition can be developed, and genetic counseling can further be provided to the families.
Materials and Methods
The study was approved by the Namazi Hospital, Shiraz University of Medical Science Ethics board. Each patient guardian provided informed consent for study participation and subsequent publication of established results. Exome sequencing was done for 66 patients and WES were not obtained for their parents.
Indeed these patients suffer various neurodevelopmental disorders, some of them presented with atypical cerebral palsy phenotype. The most important complaint of our patients and the reason for their referral was physical disability and delayed motor development and in most of them it has been accompanied by significant degrees of cognitive disability.
This represents a descriptive-analytical cross-sectional retrospective study of patients diagnosed with CP without history of perinatal injury and asphyxia (especially result NICU or prolonged neonatal ward admission by delivery chart review), brain MRI compatible with HIE (7), assessed by pediatric neurologist in the pediatrics clinics of the Shiraz University of Medical Science from 2016 to 2020 years.
The population study is children and adolescents (ages 6 months to 18 years) with delayed motor development from the birth or early infancy assessed by a pediatrician and then referred to a pediatric neurologist.
History and physical examination, brain MRI imaging and laboratory tests were recommended, which included metabolic tests (serum amino acids, urine organic acids, urine and serum acyl carnitines) and in certain cases, such as autism, other metabolic tests such as creatine and purine pyrimidine panel. The patients had clinical and brain imaging (Red flags) findings that led us to perform genetic testing.
For the studied patients, 5 cc of peripheral blood was collected in EDTA tube. After DNA extraction, whole exome sequencing was conducted using Illumina HiSeq 4,000 sequencing platform. Various bioinformatics tools and databases such as ANNOVAR, GATK, and BWA aligner were used for the bioinformatics analysis of the WES results.
Pathogenic and likely pathogenic variants were defined according to the standards and guidelines recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology for the interpretation of genetic variants (9).
Inclusion Criteria
Non progressive disorder of the development of movement and posture leading to the limitation of activity with onset at birth or within the first year of life.
1. Normal MRI findings despite motor disabilities, atypical white matter lesions or other structural findings that are not typical of CP.
2. Severe symptoms in the absence of a history of perinatal injury.
3. A pattern of disease inheritance, or consanguinity.
4. Isolated muscular hypotonia.
5. Rigidity (as opposed to spasticity) on physician examination.
Exclusion Criteria
1. Gestational age of 36-week gestation or less.
2. Perinatal complications: asphyxia, respiratory distress syndrome requiring mechanical ventilation, meningitis/encephalitis, non-physiological jaundice.
3. Presence of acquired and/or progressive lesions on brain MRI, such as ischemic lesions, hemorrhage, calcification.
4. Patient with major dysmorphic features and Patients with multiple congenital anomalies.
5. Positive neonatal metabolic screening tests that was confirmed by more accurate tests.
Patients do not have to cover all inclusion criteria at the same time but all of our patients met all the exclusion criteria at the same time.
Results
A total of 66 affected individual with atypical CP were examined. The general characteristics for all 66 propends are listed in Table 1. Clinical findings, including details of neurologic exam and seizure and intellectual disability and developmental status can be found in Table 2. It should be noted that intellectual disability was the most frequent sign after motor symptoms. Their MRI findings were as follows: normal findings were the most frequent (36.2%), brain atrophy (24.2%), White matter lesion (23%), Neuronal migration defects (7.5%), Vermian and cerebellar hypoplasia (3%), Basal ganglia lesion (1.5%), Corpus callosum agenesis (1.5%), Molar tooth sign (3%).

Table 1. Patients general characteristics.
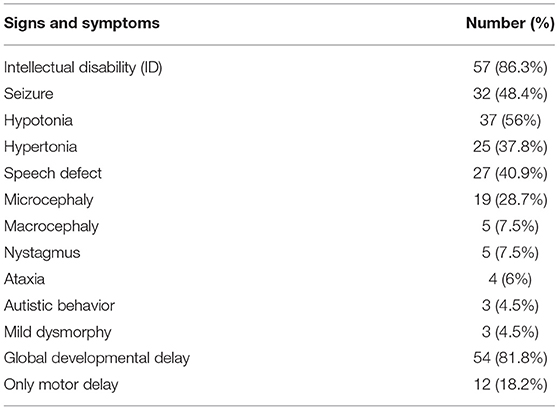
Table 2. Patients clinical finding and developmental status.
Different inheritance models of candidate ACP variants have been shown in Table 3. Furthermore, types of pathogenicity of genetic variants in candidate ACP genes have been displayed in Table 4. Missense variants were the most frequent (56%) type. Number of patients with each clinical feature, MRI and metabolic finding evaluated; associated causal genes in Table 5. Detailed results of exome sequencing in our atypical CP patients; associated diseases, causal genes and their location and Inheritance pattern has been described in Table 6.

Table 3. Different inheritance models of candidate ACP variants.

Table 4. Types of pathogenicity of genetic variants in candidate ACP genes.

Table 5. Clinical feature, MRI and metabolic finding evaluated and associated causal genes.
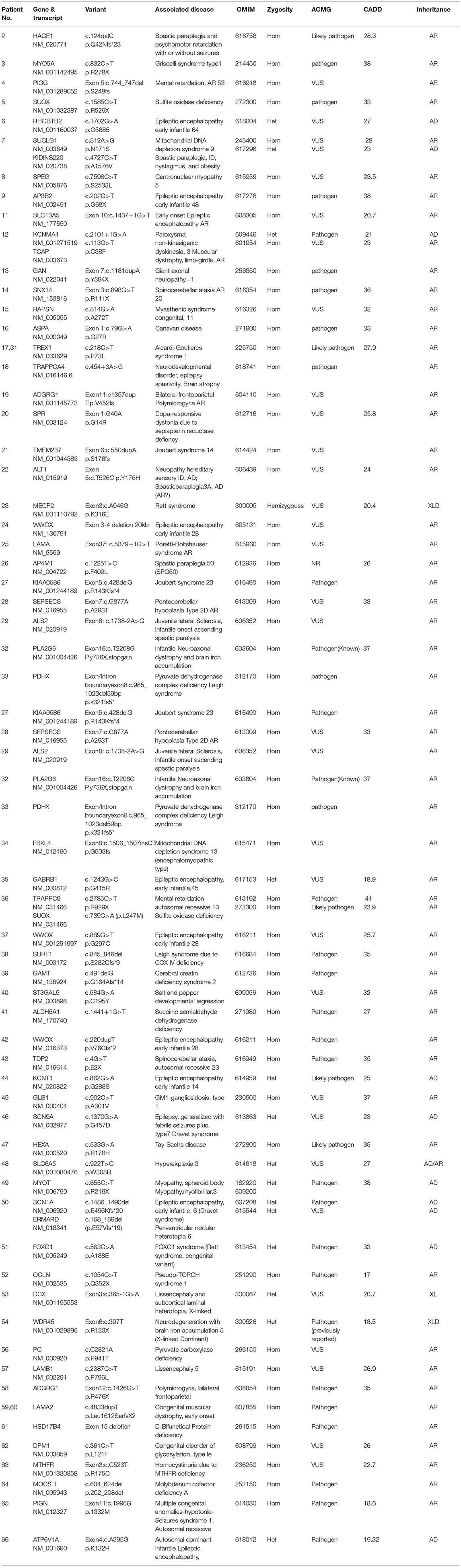
Table 6. Exome sequencing results in selected atypical CP patients.
In this study, 30 patients had proband and parental Sanger confirmation for mutations and 32 patients didn't have. The following genes were confirmed in our patients by Sanger sequencing: HACE1, SPEG, SLC13A5, TRAPPC4, FBXL4, TDP2, GAMT, LAMB1, OCLN, WWOX (37), TREX1 (31, 17), SURF1, WDR45, LAMA2 (59, 60), MTHFR, MOCS1, DOM1, SEPSECS, GABRB1, KCNT1, AP4M1, PDHX, FOXG1, ATP6V1A, SPR, PIGG, ATL1, LAMA.
For others families, Sanger confirmation of the identified variant was not carried out but genotype-phenotype correlation was confirmed.
The mutation found in patients 44 and 66 (ATP6V1A, KCNT1) was confirmed by Sanger sequencing but was not segregated possibly due to gonadal mosaism. In the study of exome sequencing, 4 patients with cerebral palsy in this investigation (No. 1, 10, 30, 55) were found to have no mutations despite adequate coverage and re-analysis. In other words, they were exome negative. The diagnostic yield for our patients was 93.9%. Four patients (No. 7, 12, 36, and 50) had two pathogenic variants and did not seek further genetic testing of other family members. In four patients, the identified mutation was the same. However, these patients were not related to each other but were of the same ethnic background, TREX1 (No.31, 17) and LAMA2 (No.59, 60). In exome sequencing of three patients, pathogenic mutations were found in a same specific gene (WWOX gene) but in different locations; WWOX was the most common disease-causing gene in this study. The most common genetic causes of atypical cerebral palsy in our study were neurometabolic (16 patients) and epileptic encephalopathies (14 patients). After these two groups of diseases, the most common disorder was neuromuscular diseases with 6 cases (9%) identified to be due to this condition. After that, 3 cases of spastic paraplegia were found in patients (No. 2, 22, and 26) with responsible genes including (HACE1, ATL1, AP4M1), respectively, and two cases of cerebellar ataxia in patients (No. 14 and 43), whose responsible genes were SNX14, TDP2, respectively. Although in this study we excluded primary microcephaly and dysmorphies and obvious syndromes from the beginning, we found 4 syndromes, three of which were Joubert spectrum syndrome (No. 21, 25, and 27) due to pathogenic variants in TMEM237, LAMA, and KIAA0586, respectively. Furthermore, one patient with Griscelli syndrome (No. 3) had disease-causing variants in MYO5A gene. Novel and Private mutations were found in ten patients (No. 20, 21, 22, 19, 4, 25, 30, 29, 34, and 24).
Discussion
In our study, the most common clinical sign was intellectual disability with a prevalence of 86.3% and then seizures with a prevalence of 48.4%. Many similar studies examined only the patient's motor symptoms and did not report any degree of intellectual disability or seizures (10, 11). Brain MRI results in our atypical CP patients showed 36% normal findings, 29.2% cerebral atrophy, and 13% white matter lesions in the studied population. The correlation between the results of the metabolic test and the genetic test in our study was 7% (three out of 43 cases), which was lower than the world reports about 20% (10). In Iran, especially in Fars province, Mass Spectrometry (MS/MS) metabolic screening test has been performed since 2018, which screens 44 of the most common causes of metabolic diseases. Individuals confirmed to be affected with these conditions were excluded from this study. Exome sequencing was performed in 66 patients with atypical cerebral palsy. In four patients, the test was negative (exome negative) and positive findings were found in 62 patients and in 62 different genes that indicates significant heterogeneity of the underlying genetic causes of CP. Other studies have confirmed this severe heterogeneity (2, 3, 5, 8, 10).
The most common inheritance pattern was autosomal recessive (75.7%) that was observed in 88% of consanguineous parents in our study. However, in similar studies, autosomal recessive inheritance was 9 and 10% of the patients' parents were relatives (8). These results show the relationship between autosomal recessive inheritance and parental kinship marriage.
The most common type of pathogenic variants found in this study was VUS (43.9%). Although in other studies, only pathogenic or likely pathogenic variants have been reported (12). Most of detected VUS mutations have been confirmed by Sangar sequencing in probands and their parents and good phenotype-genotype correlation has exist and these mutations have had a very low frequency in community, with these three conditions can even be advised to give prenatal diagnosis after careful genetic counseling. The most genetic variant found in patients with cerebral palsy in our study was missense (56%), but in a similar study, a missense mutation (67%) was reported (8). Our patients, despite having VUS and missense mutations, had a good phenotypic-genotypic correlation, and some of them were not referred for further studies (e.g., Sanger confirmation and family segregation and other complementary genetic studies).
In our study, three patients (patients 24, 37, and 42) all had developmental delay, microcephaly, and recurrent seizures, and their parents were first cousin, and all three had a sibling who had died. Patients 24 and 42 had severe motor impairment with a gross motor function classification system (GMFCS) of 5 but patient 37 had a GMFCS of 2. Patients 24 and 37 also had spasticity but patient 42 had hypotonia. Case 24 has a new and private homozygous deletion of exons 3 and 4, which is 20kb long, but the patient's parents did not undergo Sanger sequencing in order to confirm this variant. Patient 37 had a homozygous mutation c.889G> T (p.G297C) which has been confirmed in patients and parents. Patient number 42 has homozygous mutation duplication c.220dupT (p.V76Cfs * 2) but unfortunately, the patient's parents did not undergo Sanger sequencing for confirmation. Thus, mutations in the WWOX gene have led to developmental delay, microcephaly, recurrent seizures, and motor dysfunction, but mutations in different parts of the gene have resulted in varying severity and type of motor dysfunction (spasticity and hypotonia).
We were able to recommend targeted and personalized treatment for 11 patients; KCNT1-related epilepsy (No. 44) quinidine has been used as an off-label anticonvulsant (13, 14), molybdenum cofactor deficiency type A (No. 64) with cyclic pyranopterin monophosphate (cPMP) (15),Succinic semialdehyde dehydrogenase deficiency (No. 41) with vigabatrin (16), cerebral creatin deficiency (No. 39) with creatin monophosphate (17), WWOX gene mutation (N0. 24-42-37) with lithium (18), Dopa-responsive dystonia (No. 20) with levodopa-carbidopa and other dopamine agonists (19), Congenital myasthenic syndromes RAPSYN deficiency (No. 15) with Pyridostigmine and 3,4 DAP (Diaminoprydine) (20), pyruvate dehydrogenase complex deficiency (No. 33) with Ketogenic Diet and Dichloroacetate (21), methylenetetrahydrofolate reductase deficiency (No. 63) with mefolinate (5-Methyltetrahydrofolate) (22).
Overall, Using a strict and accurate criteria for selecting atypical CP patients who are more likely to be genetic, we were able to identify the genetic cause of a significant proportion of the studied patients. This, in turn led to the reduction of psychological stress and guilt of parents. Furthermore, parents with better understanding of the cause of their child's disability are able to make proper decisions for future pregnancies and other family members can also have a better estimate of the risk of this condition in their offspring. Knowing the exact condition their child is affected with, parents can have a better understanding of its prognosis.
Whole exome sequencing (WES) was performed for all 66 patients with atypical cerebral palsy. The diagnostic yield of these genetic investigations was 93.9% for our patients. In various studies published by other researchers, this rate was lower. For example, in the United States, when examining all types of cerebral palsy (typical and atypical), this rate was 32.7% (10), but in another study, the diagnostic efficiency of WES genetic testing in atypical CP patients in the United states was 41% (10) but in another study in US it was 32.7% (10), in Japan this rate is 52.9% (12) and in the Greece 50% (11) and in a joint study of Canada and the United Kingdom, the diagnostic yield of exome sequencing is reported to be 65% (8).
The most important factors in the high diagnostic yield of genetic testing in our research are as follows:
• We set strict Exclusion and Inclusion Criteria to select specific patients with atypical cerebral palsy with a higher probability of being due to a genetic.
• Selection of severe phenotypes, CP patients in our study did not have the usual course of cerebral palsy (which usually improves with rehabilitation and occupational therapy) and often severe and resistant seizures (51.5%) and significant motor and mental disability (83% GMFCS 3 to 5 and 86.3 % had developmental delay), which sometimes showed a progressive pattern in their follow-up.
The financial constraints of patients and the limited assistance of the welfare department made us select CP patients with the highest probability of being inherited or genetic.
The results of our research contribute to the knowledge of the study of the genetics of cerebral palsy. Pathogenic variants located in a specific gene can lead to a wide range of clinical presentations; such as mutations in different locations within the WWOX gene, which in three of our patients caused a range of different symptoms of early epileptic encephalopathy type 28. WES is instrumental in enabling the recognition and definition of expanded phenotypes of single-gene disorders. However, it should be noted that it can be challenging to distinguish them from unidentified multi-locus variations. Multi-locus variation-pathogenic variants in two or more disease genes can potentially explain the underlying genetic basis for apparent phenotypic expansion but it is always possible that a pathogenic variant in a yet unknown disease-causing gene may be responsible for a second disease in these cases (8, 23).
Our study had the following limitations: we didn't investigate these patients with other genetic tools such as Array CGH which evaluate deletion-duplications. Furthermore, we didn't evaluate intellectual disability patients as a first step with Array CGH and other molecular-cytogenetic study. In addition, we did not perform functional studies to confirm the pathogenicity of VUS mutations. We didn't perform exome sequencing for their parents simultaneously. In addition, although genotype-phenotype correlation was confirmed, half of our patients didn't do sanger confirmation for patients and their parents. Most of our patients had severe disabilities and significant sequelae due to delayed diagnosis. At this stage of disease, starting treatments cannot reverse the previous damage inflicted on the developing brain. Furthermore, most of the patients did not have further follow-up visits to evaluate the treatment effect due to the global health crisis caused by COVID-19 pandemic (24). In addition, patients in developing countries face various other challenges such as the high cost of genetic testing and lack of insurance. We, therefore, have to recommend these tests to a more selected group of patients in such settings. It should also be noted that in many instances the families refuse further genetic testing due to the unavailability of an effective targeted therapy in most of the cases.
Recommendation
Atypical cerebral palsy patients that require genetic studies including:
• Patients who had no risk factor for acquired cerebral palsy.
• Family history of same problems.
• Patients who have progressive symptoms and do not have any improvement in spite appropriate occupational therapy and physiotherapy.
• Patients who have normal brain MRI despite significant disability in various mental or motor areas.
• Patients with severe motor-mental disabilities.
• Patients with cerebral palsy who have severe and refractory seizures.
• Patients whose MRI shows abnormal lesions that are not usually seen on cerebral palsy.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Namazi Hospital, Shiraz University of Medical Science Ethics board. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
MF conceived and designed the study, collected, assembled, and interpreted NGS data. MN, SI, HN, and PK clinically evaluated the patients. MN drafted the manuscript. MF, AS, SB, FS, and ST performed the genetic studies. SI, HN, PK, AS, SB, FS, ST, and MF revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Swaiman KF, Ashwal S, Ferriero DM. Pediatric Neurology: Principles & Practice. Mosby, MO: Elsevier (2006).
2. Moreno-De-Luca A, Ledbetter DH, Martin CL. Genetic [corrected] insights into the causes and classification of [corrected] cerebral palsies. Lancet Neurol. (2012) 11:283–92. doi: 10.1016/S1474-4422(11)70287-3
3. Lee RW, Poretti A, Cohen JS, Levey E, Gwynn H, Johnston MV, et al. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med. (2014) 16:821–44. doi: 10.1007/s12017-014-8331-9
4. Maclennan AH, Lewis S, Moreno-De-Luca A, Fahey M, Leventer RJ, Mcintyre S, et al. Genetic or other causation should not change the clinical diagnosis of cerebral palsy. J Child Neurol. (2019) 34:472–6. doi: 10.1177/0883073819840449
5. Maclennan AH, Thompson SC, Gecz J. Cerebral palsy: causes, pathways, and the role of genetic variants. Am J Obstet Gynecol. (2015) 213:779–88. doi: 10.1016/j.ajog.2015.05.034
6. Fahey MC, Maclennan AH, Kretzschmar D, Gecz J, Kruer MC. The genetic basis of cerebral palsy. Dev Med Child Neurol. (2017) 59:462–9. doi: 10.1111/dmcn.13363
7. Himmelmann K, Horber V, De La Cruz J, Horridge K, Mejaski-Bosnjak V, Hollody K, et al. MRI classification system (MRICS) for children with cerebral palsy: development, reliability, and recommendations. Dev Med Child Neurol. (2017) 59:57–64. doi: 10.1111/dmcn.13166
8. Matthews AM, Blydt-Hansen I, Al-Jabri B, Andersen J, Tarailo-Graovac M, Price M, et al. Atypical cerebral palsy: genomics analysis enables precision medicine. Genet Med. (2019) 21:1621–8. doi: 10.1038/s41436-018-0376-y
9. Miller DT, Lee K, Gordon AS, Amendola LM, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2021) 23:1391–8. doi: 10.1038/s41436-021-01171-4
10. Millan F, Elloumi H, Teigen C, Scuffins J, Torene R, Mcknight D. Genetic testing of >1300 patients with cerebral palsy reveals an etiology in one-third of cases, underscoring the need for broad genetic testing and a significant recurrence risk for families. (P4.6-028). Neurology. (2019) 92:P4.6–028. Available online at: https://n.neurology.org/content/92/15_Supplement/P4.6-028
11. Zouvelou V, Yubero D, Apostolakopoulou L, Kokkinou E, Bilanakis M, Dalivigka Z, et al. The genetic etiology in cerebral palsy mimics: the results from a greek tertiary care center. Eur J Paediatr Neurol. (2019) 23:427–37. doi: 10.1016/j.ejpn.2019.02.001
12. Takezawa Y, Kikuchi A, Haginoya K, Niihori T, Numata-Uematsu Y, Inui T, et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann Clin Transl Neurol. (2018) 5:538–51. doi: 10.1002/acn3.551
13. Gertler T, Bearden D, Bhattacharjee A, Carvill G. KCNT1-Related Epilepsy. (2018). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK525917/ (accesssed June 15, 2021).
14. Mesraoua B, Deleu D, Kullmann DM, Shetty AK, Boon P, Perucca E, et al. Novel therapies for epilepsy in the pipeline. Epilepsy Behav. (2019) 97:282–90. doi: 10.1016/j.yebeh.2019.04.042
15. Veldman A, Santamaria-Araujo JA, Sollazzo S, Pitt J, Gianello R, Yaplito-Lee J, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics. (2010) 125:e1249–54. doi: 10.1542/peds.2009-2192
16. Pearl PL, Wiwattanadittakul N, Roullet JB, Gibson KM. Succinic semialdehyde dehydrogenase deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews [(R)]. Seattle, WA: GeneReviews® [Internet] (1993).
17. Narayan V, Mahay SB, Verma IC, Puri RD. Case series of creatine deficiency syndrome due to guanidinoacetate methyltransferase deficiency. Ann Indian Acad Neurol. (2020) 23:347–51. doi: 10.4103/aian.AIAN_367_18
18. Cheng YY, Chou YT, Lai FJ, Jan MS, Chang TH, Jou IM, et al. Wwox deficiency leads to neurodevelopmental and degenerative neuropathies and glycogen synthase kinase 3beta-mediated epileptic seizure activity in mice. Acta Neuropathol Commun. (2020) 8:6. doi: 10.1186/s40478-020-0883-3
19. J F. Sepiapterin Reductase Deficiency. (2015). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK304122/ (accesssed June 15, 2021).
20. Jagtap SA, Abraham K, Sarada C, Nair MD. Congenital myasthenic syndromes: natural history and long-term prognosis. Ann Indian Acad Neurol. (2013) 16:338–41. doi: 10.4103/0972-2327.116918
21. Ferriero R, Manco G, Lamantea E, Nusco E, Ferrante MI, Sordino P, et al. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci Transl Med. (2013) 5:175ra131. doi: 10.1126/scitranslmed.3004986
22. Knowles L, Morris AA, Walter JH. Treatment with mefolinate (5-Methyltetrahydrofolate), but not folic acid or folinic acid, leads to measurable 5-methyltetrahydrofolate in cerebrospinal fluid in methylenetetrahydrofolate reductase deficiency. JIMD Rep. (2016) 29:103–7. doi: 10.1007/8904_2016_529
23. Karaca E, Posey JE, Coban Akdemir Z, Pehlivan D, Harel T, Jhangiani SN, et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med. (2018) 20:1528–37. doi: 10.1038/gim.2018.33
Keywords: atypical cerebral palsy, next-generation sequencing, motor disabilities, neuroimaging, developing brain
Citation: Nejabat M, Inaloo S, Sheshdeh AT, Bahramjahan S, Sarvestani FM, Katibeh P, Nemati H, Tabei SMB and Faghihi MA (2021) Genetic Testing in Various Neurodevelopmental Disorders Which Manifest as Cerebral Palsy: A Case Study From Iran. Front. Pediatr. 9:734946. doi: 10.3389/fped.2021.734946
Received: 01 July 2021; Accepted: 09 August 2021;
Published: 03 September 2021.
Edited by:
Wang-Tso Lee, National Taiwan University Hospital, TaiwanReviewed by:
Alastair MacLennan, University of Adelaide, AustraliaBrahim Tabarki Melaiki, University of Sousse, Tunisia
Copyright © 2021 Nejabat, Inaloo, Sheshdeh, Bahramjahan, Sarvestani, Katibeh, Nemati, Tabei and Faghihi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Ali Faghihi, bWZhZ2hpaGlAZXhwcmVzc2dlbmUudXM=