 Won Kyung Kwon1†
Won Kyung Kwon1† Jee Ah Kim1†Jong-Ho Park2†Doo Ri Kim3†Su Eun Park4†Yae Jean Kim3†Keon Hee Yoo3†Ja-Hyun Jang1*†
Jee Ah Kim1†Jong-Ho Park2†Doo Ri Kim3†Su Eun Park4†Yae Jean Kim3†Keon Hee Yoo3†Ja-Hyun Jang1*† Eun Suk Kang1*†
Eun Suk Kang1*†- 1Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea
- 2Clinical Genomics Center, Samsung Medical Center, Seoul, South Korea
- 3Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea
- 4Department of Pediatrics, School of Medicine, Pusan National University, Children's Hospital, Yangsan, South Korea
X-linked lymphoproliferative disease type 1 (XLP1), an X-linked recessive genetic disorder, is associated with primary immunodeficiency. Patients with XLP1 are susceptible to Epstein–Barr virus (EBV) infection. SH2D1A gene is known as the causative gene. We found a novel hemizygous variant of SH2D1A, c.162_201+31delinsTACAAGGACATATACA, from a 5-year-old male patient who had been diagnosed with EBV infection and Hodgkin's lymphoma. In targeted next-generation sequencing (NGS), complex variants at exon 2 were not consistently identified with two software programs. They showed a soft-clipped read pattern. The variant had a 71-bp deletion and a 16-bp insertion across exon 2 as confirmed by direct sequencing. As the variant was located within the exon–intron boundary, two aberrant transcripts were shown by RNA study. Although NGS method has a limitation in detecting large deletion/duplication variants, proper bioinformatics pipeline and careful review of data might enable the detection of complex variants.
Introduction
X-linked lymphoproliferative disease type 1 (XLP1) is an inherited immunodeficiency characterized by susceptibility to Epstein–Barr virus (EBV) infection (1). SH2D1A gene, located on chromosome Xq25, is known as the causative gene. SH2D1A encodes the cytoplasmic protein SAP (signaling lymphocyte activation molecule-associated protein), which has an important role in regulating T cells, NK cells, NKT cells (2, 3), and possibly B cells (4). The incidence of XLP1 in the United States was reported to be one in one million male individuals (5). About 120 different variants of SH2D1A were found in Human Gene Mutation Database (HGMD). Typical manifestations of XLP1 include EBV-associated hemophagocytic lymphohistiocytosis (HLH) and lymphomas (6). Up to 35% of patients have no evidence of previous EBV infection (7, 8). It has been reported that dysgammaglobulinemia (51.8 vs. 37.2%) and lymphoma (25.0 vs. 19.6%) have higher rates in EBV-positive XLP1 patients than in EBV-negative patients (8). On the other hand, EBV-negative XLP1 patients experience progression to HLH more frequently (51.0 vs. 21.4%) (8). Therefore, XLP1 is an immune dysregulation disorder not specifically associated with EBV infection (9). Due to various clinical manifestations, the clinical diagnosis for XLP1 is often difficult. Molecular approaches are helpful for its confirmatory diagnosis. Here we report a 5-year-old male patient with XLP1 caused by a novel pathogenic variant, c.162_201+31delinsTACAAGGACATATACA, in SH2D1A. This complex variant was suspected to have a 71-bp deletion and a 16-bp insertion based on next-generation sequencing (NGS). Such deletion and insertion were eventually confirmed by direct sequencing. It was found that the variant affected the splicing and yielded two aberrant transcripts by RNA study.
Case Description
Clinical Manifestations and Laboratory Findings
A 5-year-old Korean male patient who had recurrent fever (up to 39°C) with high EBV titer was transferred from an outside hospital for further evaluation of primary immunodeficiency (PID). At 3 years old, he had been hospitalized for a total of three times due to multiple episodes of mycoplasma pneumonia. At that time, his immunoglobulin G was nearly absent (10.4 mg/dl, reference range: 480–900 mg/dL for 3–5 years old). However, he had normal B cell counts (1,082 × 109/L, reference range: 200–2,100 × 109/L). His NK cell count (67 × 109/L, reference range: 100–1,000 × 109/L) was decreased, although his T cell count was normal. With a presumptive diagnosis of common variable immunodeficiency, Intravenous immunoglobulin (IVIG) treatment was started. At that time, he had visited the previous hospital due to recurrent fever and diagnosis of pneumonia and bronchiectasis. He was treated with cefotaxime for 3 days. His C-reactive protein (CRP) decreased from 5.31 to 2.2 mg/dl. He was found to have an EBV titer of 35,100 copies/ml. Regarding his family history, his uncle (older brother of his mother) died from meningitis at 3 years old (Figure 1). When admitted, his body temperature was 39.0°C, and his pulse rate was 124/min. However, his breathing sound was normal. Palpable lymph node enlargement and organomegaly (except swelling in both parotid gland sites) were not observed. He had mild leukocytosis (19,450 × 109/L) with neutrophilia (13,830 × 109/L) and monocytosis (1,670 × 109/L). He showed a markedly increased CRP level (14.16 mg/dl). Due to continuous administration of IVIG before he was transferred, his immunoglobulin levels were within the normal range. Tests for EBV were performed, including serologic tests of EBV and quantitation of EBV. The serologic tests for anti-VCA, IgG, and anti-EBNA antibodies were positive. The EBV DNA titer measured with a Real-Q EBV Quantification Kit (BioSewoom, Seoul, Korea) was 530 IU/ml. Immunophenotyping of lymphocyte subsets and naïve/memory B and T cell subsets was performed. The results are shown in Table 1. His CD16+56+3- NK cell fraction was markedly decreased both in proportion and count (1.0%, 33 × 109/L). Although the CD19 + B cell proportion and count were within the reference ranges as before, both switched/non-switched (CD10-CD27+IgD+/-) and differentiated (IgD-CD27-) memory B cells were rarely detected (2.3%, 30 × 109/L) with increased IgD + CD27- naïve B cell fraction. T cells also showed a skewed distribution of CD45RA+CCR7+ naïve T cells (73.2%, 441 × 109/L) with decreased CD45RO + memory T cells. Radiologic studies revealed low-density lesions in the thymus, spleen, liver, and T12 level of spine bone. Multiple cervical and mediastinal lymph node enlargement with swollen left parotid gland which needed to be distinguished between inflammatory sialadenitis and EBV-related lymphoma were observed (Table 1). The pathology of lymph nodes based on video-assisted thoracoscopic surgery revealed classic Hodgkin's lymphoma, nodular sclerosis type with positive EBV by in situ hybridization. With a diagnosis of Hodgkin's lymphoma, a combination chemotherapy of COPP-ABV including cyclophosphamide, vincristine sulfate (Oncovin), prednisone, procarbazine hydrochloride, doxorubicin hydrochloride (Adriamycin), bleomycin, and vinblastine sulfate was started at admission day 16. After five cycles of chemotherapy, the patient underwent thoracoscopic lobectomy to remove persistent pulmonary nodules suspected to be a fungal infection. The pathology findings were consistent with inflammatory nodules composed of histiocyte aggregates and some lymphocytes. He developed a headache and left side motor weakness. The brain MRI showed multifocal brain lesions suspicious of lymphoproliferative disorder or encephalitis associated with EBV infection. After four cycles of rituximab, the abnormal hypermetabolic lesions due to EBV-related LPD resolved, with EBV DNA undetectable in the cerebrospinal fluid. Currently, the patient is awaiting haploidentical peripheral blood stem cell transplantation from his father.
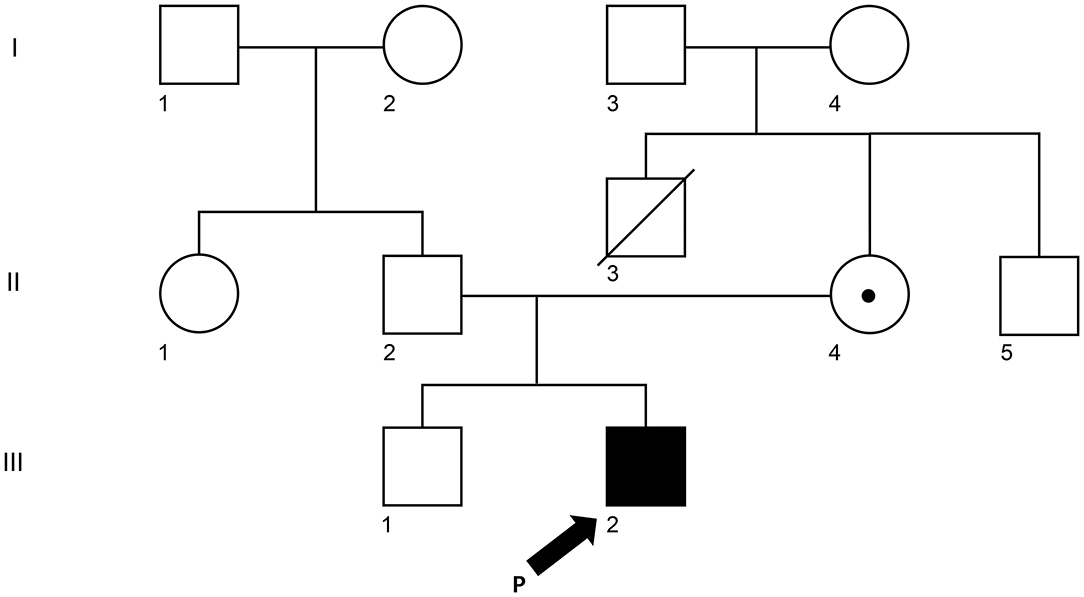
Figure 1. Pedigree of the XLP1 case child. The arrow (III-2) is the proband. II-3 (his mother) and III-1 (his brother) were tested for the same variant. The white square represents a male individual who was unaffected, the white circle was for an unaffected female individual, and the dot represents the mother who is a carrier.
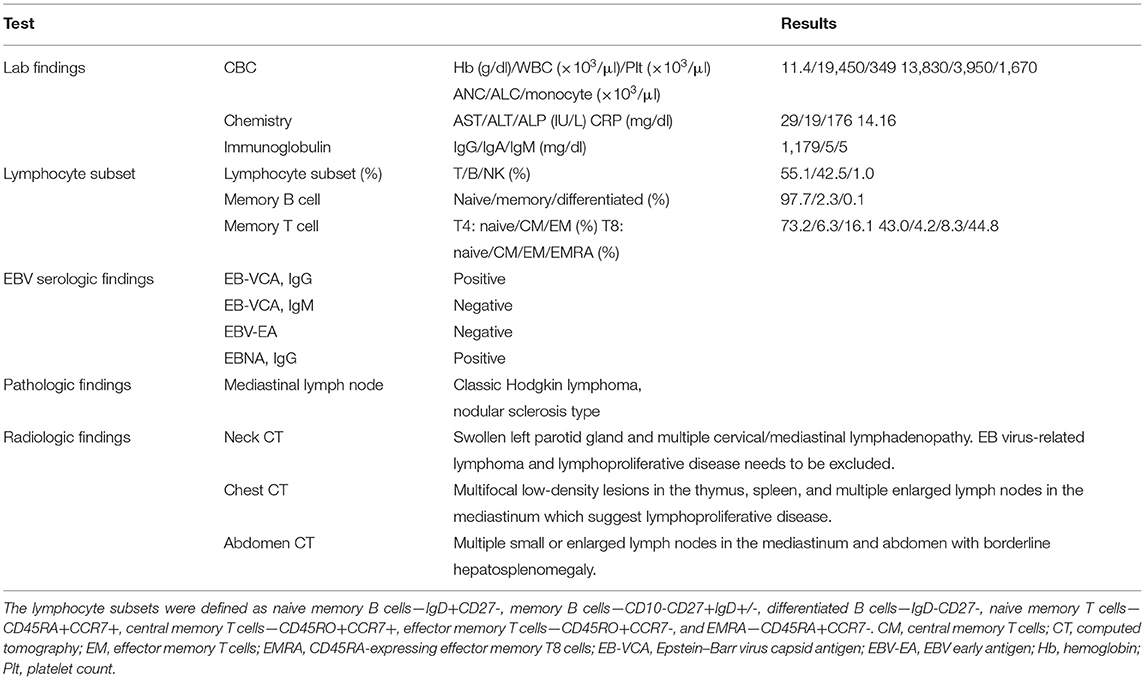
Table 1. Clinical characteristics and laboratory, radiologic, and pathologic findings of the patient at initial diagnosis.
Genetic Diagnosis
NGS for targeted panel of B cell and humoral immune deficiency, including 74 related genes including SH2D1A, was performed. Target enrichment with customized probes (IDT, Coralville, IA, USA) and subsequent massively parallel sequencing with NextSeq550 (Illumina, San Diego, CA, USA) were performed. The sequence reads were aligned to the reference genome, hg19, with decoy sequence using the BWA-mem algorithm implemented in BWA 0.7.17 (10). Duplicate reads were marked using Picard 2.19 (https://broadinstitute.github.io/picard/). Recalibration of base quality scores was performed using previously known sites (from dbSNP138, Mills and 1000G gold standard INDELs b37 sites, and 1000G phase1 INDELs b37) after removing duplicate reads using GenomeAnalysisTK-4.1.2.0 (GATK) (10). Variants were called using HaplotypeCaller in GATK, VarDict, and Strelka2 and annotated with ANNOVAR. All variants were interpreted based on the 2015 American College of Medical Genetics (ACMG) and Genomics and the Association for Molecular Pathology (AMP) guideline (11) and, additionally, the ClinGen Sequence Variant Interpretation Workgroup's recommendation (12).
Three missense variants in COPA, NLRC4, and DOCK8 genes, deletion/duplication, and missense variants in SH2D1A were identified. According to the 2015 ACMG-AMP guideline, the missense variants in COPA, NLRC4, and DOCK8 genes were classified as variants of uncertain significance. Variants of the COPA gene (NM_004371.3:c.3502A > G) had one moderate and support pathogenic evidences [low allele frequency (0.00119%) and a low rate of benign missense variant in this gene] but one support benign evidence [in silico analysis (REVEL 0.1)]. The variant of the NLRC4 gene showed no evidence. Only a heterozygous variant was detected in the DOCK8 gene with an autosomal recessive inheritance pattern and a low allele frequency (0.00080%). For SH2D1A, several variants including deletion/duplication and missense changes located at exon 2 were called. Called by only GATK with low depths (5-11X) but not with other callers (Vardict and Strelka2), it was assumed as a false positive calling at first. A manual review of BAM file using the Integrated Genome Browser was performed to exclude whether exon deletion was present. Visual inspection revealed soft-clipped reads which suggested a large deletion/insertion event at the 3′ of exon 2, including the flanking intron (Figure 2A). Targeted direct sequencing revealed that the variant had a deletion of 71 bp and an insertion of 16 bp across exon 2 with a flanking intron [NM_002351.4 (SH2D1A):c.162_201+31delinsTACAAGGACATATACA] (Figure 2B). Since deletion/insertion involved the splice donor site, RNA study using peripheral blood leukocytes was performed to investigate the effect of such deletion/insertion on splicing. Reverse transcription-PCR and complementary DNA sequencing were performed across exons 1–3 of SH2D1A using primers designed by the authors. Gel electrophoresis showed two aberrant mRNA transcripts. One transcript corresponded to exon 2 skipping (r.138_201del, p.Tyr47_Glu67del). The other lacked exon 2 and 55 bp of exon 3 by creating a new splice acceptor site (r.138_256del, p.Tyr47Ilefs*17) (Figure 3). Finally, the variant could be classified as likely a pathogenic variant (PVS1, PM2). By targeted direct sequencing, it was confirmed that his mother was a heterozygous carrier, while his unaffected brother did not have this variant.
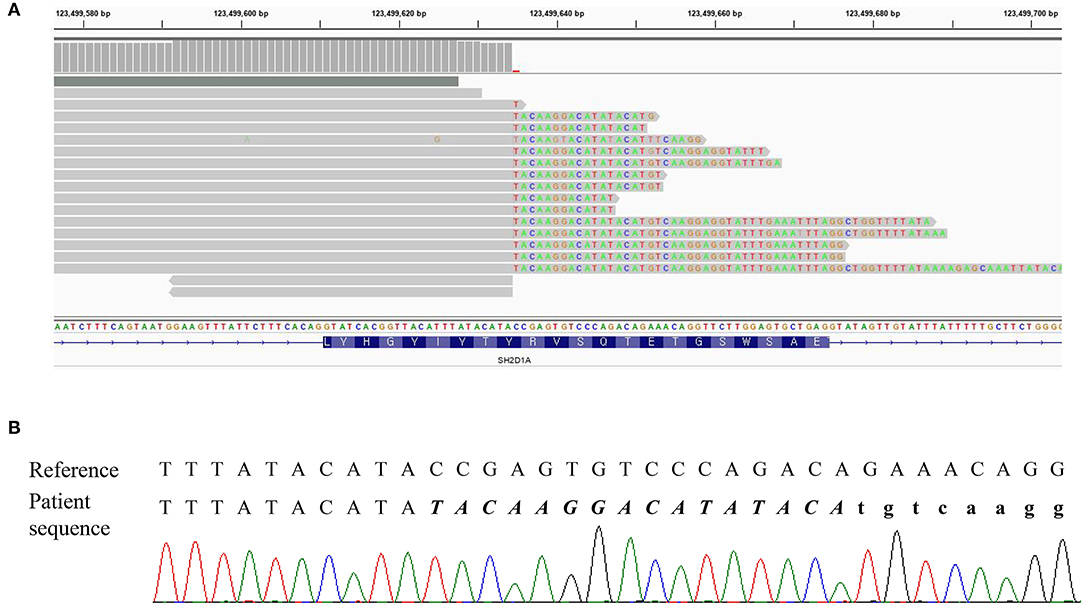
Figure 2. DNA result of the patient. (A) IGV snapshot of a next-generation sequencing panel showing soft clipped reads. (B) Sequencing chromatogram showing a hemizygous variant of c.162_201+31delinsTACAAGGACATATACA. The inserted sequences were written in italic letters, and the intron sequences were written in small letters.
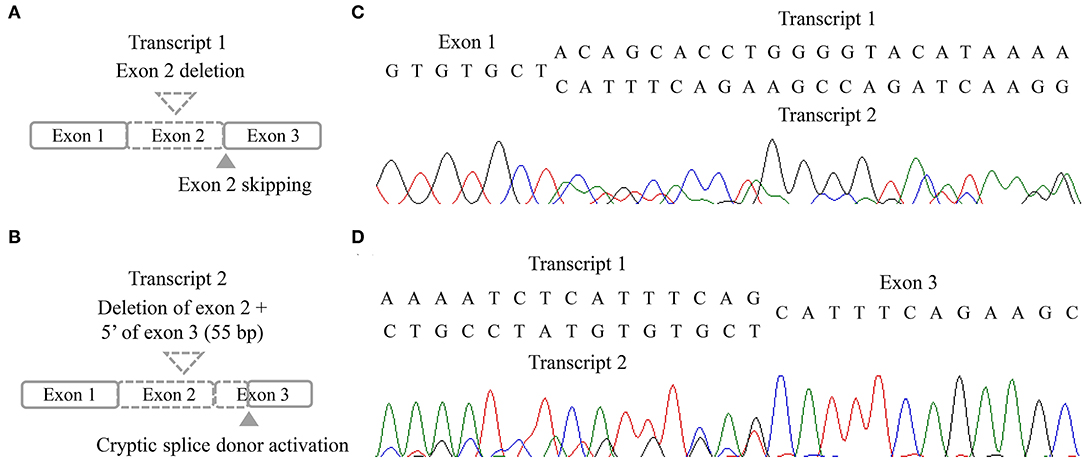
Figure 3. RNA results of the patient. (A,B) Schematic illustration of two aberrant transcripts. (C,D) Sequencing chromatogram in forward direction (C) and reverse direction (D).
Discussion
XLP1 is a rare disease caused by SH2D1A gene which encodes for SAP protein with 128 amino acids in length (2). XLP1 patients are known to be associated with fatal infectious mononucleosis, lymphoma, and dysgammaglobulinemia (13). We report a case of a 5-year-old male patient diagnosed as XLP1 based on clinical manifestation and genetic test. He presented with multiple episodes of infection, markedly reduced IgG level, multiple lymph node enlargements, and lymphoproliferative lesions with elevated EBV DNA titer comparable with typical XLP1. The patient was found to have a disease-causing variant in SH2D1A. However, the variant was not accurately called by the NGS method initially due to “soft clipping” which masked reads that did not align. We suspected abnormality in that region by visual inspection of BAM file and identified the deletion/insertion variant, NM_002351.4:c.162_201+31delinsTACAAGGACATATACA. The RNA study revealed two aberrant transcripts. There are approximately 120 pathogenic/likely pathogenic variants of SH2D1A gene in HGMD, of which gross deletion/duplication variants, including 31 unique variants, account for more than 20% (14). Since the introduction of NGS method for clinical practice, the genetic diagnosis of single-gene disorders, including PID, has become more feasible, and diverse causative variants have been discovered. Considering that SH2D1A has high rates of large deletion/duplication variants, caution is needed for NGS data interpretation of SH2D1A. Although the NGS method has a limitation in detecting large structural variants, review of coverage, especially for X-linked disorders for male patients, use of multiple variant callers, and visual inspection of BAM file could raise the diagnostic yield. In addition, in case of including splicing sites, more detailed studies, such as mRNA studies, are needed.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Samsung Medical Center IRB. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
DK, SP, YJK, and KY cared for the patient and collected the clinical data of the patient. J-HP performed the genetic test. WK, JK, J-HJ, and ESK analyzed the gene test results and drafted the manuscript. All authors read and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Purtilo DT, Cassel CK, Yang JP, Harper R. X-linked recessive progressive combined variable immunodeficiency (Duncan's disease). Lancet. (1975) 1:935–40. doi: 10.1016/s0140-6736(75)92004-8
2. Morra M, Simarro-Grande M, Martin M, Chen AS, Lanyi A, Silander O, et al. Characterization of SH2D1A missense mutations identified in X-linked lymphoproliferative disease patients. J Biol Chem. (2001) 276:36809–16. doi: 10.1074/jbc.M101305200
3. Neves JF, Raga LT, Chiang SCC, Tesi B, Vieira JP, Cordeiro AI, et al. Fatal central nervous system lymphocytic vasculitis after treatment for burkitt lymphoma in a patient with a SH2D1A mutation. Pediatr Infect Dis J. (2019) 38:e29–31. doi: 10.1097/inf.0000000000002154
4. Ma CS, Pittaluga S, Avery DT, Hare NJ, Maric I, Klion AD, et al. Selective generation of functional somatically mutated IgM+CD27+, but not Ig isotype-switched, memory B cells in X-linked lymphoproliferative disease. J Clin Invest. (2006) 116:322–33. doi: 10.1172/jci25720
5. Sandlund JT, Shurtleff SA, Onciu M, Horwitz E, Leung W, Howard V, et al. Frequent mutations in SH2D1A (XLP) in males presenting with high-grade mature B-cell neoplasms. Pediatric Blood Cancer. (2013) 60:E85–7. doi: 10.1002/pbc.24525
6. Rezaei N, Mahmoudi E, Aghamohammadi A, Das R, Nichols KE. X-linked lymphoproliferative syndrome: a genetic condition typified by the triad of infection, immunodeficiency and lymphoma. Brit J Haematol. (2011) 152:13–30. doi: 10.1111/j.1365-2141.2010.08442.x
7. Sumegi J, Huang D, Lanyi A, Davis JD, Seemayer TA, Maeda A, et al. Correlation of mutations of the SH2D1A gene and epstein-barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood. (2000) 96:3118–25. doi: 10.1182/blood.V96.9.3118
8. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. (2011) 117:53–62. doi: 10.1182/blood-2010-06-284935
9. Panchal N, Booth C, Cannons JL, Schwartzberg PL. X-Linked lymphoproliferative disease type 1: a clinical and molecular perspective. Front Immunol. (2018) 9:666. doi: 10.3389/fimmu.2018.00666
10. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinform. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human Mutation. (2018) 39:1517–24. doi: 10.1002/humu.23626
13. Seemayer TA, Gross TG, Egeler RM, Pirruccello SJ, Davis JR, Kelly CM, et al. X-linked lymphoproliferative disease: twenty-five years after the discovery. Pediatric Res. (1995) 38:471–8. doi: 10.1203/00006450-199510000-00001
Keywords: XLP1, SH2D1A gene, mRNA studies, primary immunodeficiency, rare disease (RD)
Citation: Kwon WK, Kim JA, Park J-H, Kim DR, Park SE, Kim YJ, Yoo KH, Jang J-H and Kang ES (2022) Case Report: Novel Splicing Variant in SH2D1A in a Patient With X-Linked Lymphoproliferative Syndrome Type 1. Front. Pediatr. 10:812590. doi: 10.3389/fped.2022.812590
Received: 10 November 2021; Accepted: 18 January 2022;
Published: 23 March 2022.
Edited by:
Charlotte Jane Houldcroft, University of Cambridge, United KingdomReviewed by:
Claire Booth, University College London, United KingdomLamberto Torralba-Raga, Karolinska Institutet (KI), Sweden
Copyright © 2022 Kwon, Kim, Park, Kim, Park, Kim, Yoo, Jang and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ja-Hyun Jang, amFoeXVuLmphbmdAZ21haWwuY29t; Eun Suk Kang, ZXNrYW5nQHNra3UuZWR1
†ORCID: Won Kyung Kwon orcid.org/0000-0002-7676-5814
Jee Ah Kim orcid.org/0000-0002-7011-1537
Jong-Ho Park orcid.org/0000-0001-5883-8322
Doo Ri Kim orcid.org/0000-0001-5233-4043
Su Eun Park orcid.org/0000-0001-5860-821X
Yae Jean Kim orcid.org/0000-0002-8367-3424
Keon Hee Yoo orcid.org/0000-0002-5980-7912
Ja-Hyun Jang orcid.org/0000-0003-0516-4947
Eun Suk Kang orcid.org/0000-0001-6386-6520