 Cécile Carl1,2,†
Cécile Carl1,2,† Lars Dinkelbach1,3*†
Lars Dinkelbach1,3*† Julia Mohr1Ruy Perez1Tobias Vera Lopez1
Julia Mohr1Ruy Perez1Tobias Vera Lopez1 Susanne Fricke-Otto1Tim Niehues1
Susanne Fricke-Otto1Tim Niehues1
- 1Centre for Child and Adolescent Health, HELIOS Klinikum Krefeld, Krefeld, Germany
- 2Medical Faculty, RWTH University Aachen, Aachen, Germany
- 3Department of Pediatrics III, University Children's Hospital Essen, University of Duisburg-Essen, Essen, Germany
We describe two female infants at the age of five and six months with urinary tract infections presenting with vomiting and reduced drinking behavior. On laboratory analysis, severe hyponatremia (106 mmol/L and 109 mmol/L) was seen with hyperkalemia and compensated metabolic acidosis. Endocrinological analyses revealed massively increased levels of aldosterone and renin, leading to the diagnosis of type III pseudohypoaldosteronism (PHA). A review of the current literature 2013–2023 revealed 26 type III PHA cases aged up to ten months with reduced drinking behavior, weight loss and/or failure to thrive being the most common clinical presentations. Given the severe presentation of PHA electrolyte measurements in infants with urinary tract infections and/or in infants with congenital anomalies of the kidney and urinary tract (CAKUT) are strongly recommended.
Background
Urinary tract infections (UTIs) in children are among the most common bacterial infections in childhood (1). Current guidelines suggest that oral treatment and thus outpatient care can be considered in infants older than 2 to 3 months of age (2). Severe complications are rare but include urosepsis and renal scarring (1). We present two cases of infants with UTI developing pseudohypoaldosteronism (PHA) and severe hyponatremia, a known but yet often unrecognized life threating complication of UTI in children less than 6 months of age (3). Further, a systematic review on the current literature is given.
Case report
Case 1
In September 2022, a five-month-old, previously healthy German female infant presented in our pediatric emergency room with a history of increased stool frequency and vomiting of four days. Physical examination revealed a pale child in reduced general condition, signs of moderate dehydration with a sunken fontanelle, mild tachycardia (129/min), a mildly prolonged peripheral capillary refill time of 2–3 s, dry lips, moist eyes without tears, dry nappies and cold extremities. The blood pressure on admission was 78/55 mmHg. Family history was unremarkable. The patient was admitted with the suspected diagnosis of gastroenteritis. Upon admission, blood gas analysis revealed severe hyponatremia (109 mmol/L), hypochloremia (81 mmol/L), hyperkalemia (8.5 mmol/L, venous serum 9.6 mmol/L) and compensated metabolic acidosis (pH 7.36; HCO3− 15.5 mmol/L, pCO2 22.2 mmHg, base excess −12.2 mmol/L). Repeated blood gas analyses as well as serum measurements confirmed these findings. Urine sodium was <20 mmol/L. Laboratory analyses revealed leukocyturia, erythrocyturia, nitrituria, an increased C-reactive protein (24.9 mg/L), leukocytosis (19.6 /nl) and thus the diagnosis of a urinary tract infection. Urine cultures were conducted which grew Enterobacter cloacae. Abdominal ultrasound showed a dilated right ureter, pyelectasis and a blurry differentiation between the renal medulla and cortex as an expression of inflammation.
Case 2
The second case is a six-month-old, German, female infant who presented in January 2023 in the emergency room with a history of vomiting and reduced drinking behavior of three weeks. Previous history included a respiratory infection at the age of five months, a COVID-19 infection at the age of four months as well as failure to thrive (5,380 g corresponding to the 91th Percentile at the age of 7 weeks, 6,300 g corresponding to the 12th percentile at admission). Family history revealed a benign paroxysmal postural vertigo of the mother but was otherwise unremarkable. The clinical picture on admission is one of mild dehydration, characterized as sunken eyes, mildly prolonged capillary refill time (centrally 3 s, peripheral 2–3 s), mild tachycardia (151/min) and a blood pressure of 105/52 mmHg. Laboratory findings upon admission showed severe hyponatremia (106 mmol/L), hyperkalemia (7.0 mmol/L), hypochloremia (81 mmol/L) and a compensated metabolic acidosis (pH 7.41, HCO3− 19.9 mmol/L, pCO2 27.8 mmHg, base excess −5.2 mmol/L), an elevated C-reactive protein (59.1 mg/L) as well as leukocyturia, erythrocyturia, proteinuria and nitrituria. Urine cultures grew Klebsiella oxytoca. Urine sodium was 23 mmol/L. Abdominal ultrasound led to the finding of II°–III° hydronephrosis and enlargement of the right kidney.
Treatment
In the light of life-threatening electrolyte deviations, both patients were admitted to the intensive care unit and initially treated according to the suspicion diagnosis adrenal insufficiency (4), receiving a hydrocortisone loading dose of 100 mg/m² followed by a parenteral hydrocortisone supplementation of 100 mg/m² over 24 h according to national guidelines (4). In Case 1, this was accompanied by fludrocortisone after the third day of treatment. Case 2 developed hypertension, most likely secondary to cortisol supplementation. Blood pressures spontaneously normalized (below the 95. Percentile for age) after day 8. Case 2 also had a short period of bradycardia (minimally 46 /min) on day 6, the electrocardiogram showed ventricular extrasystoles, echocardiography remained unremarkable.
Hydrocortisone and fludrocortisone were given for 6 days in Case 1. Hydrocortisone was given orally after day 5 and stopped after three additionally days in reduced dosage. Parenteral electrolyte supplementation to correct the decreased sodium levels were initiated directly after admission and slowly titrated under frequent controls (initially every two hours). Case 1 received 15.3 mmol/kg/d sodium parenterally while Case 2 required 8.6 mmol/kg/d sodium on day 1. In the following days, parenteral supplementation with 1–3 mmol/kg/d were required. In both cases, sodium levels normalized within three days after admission. A cerebral MRI in Case 1 on day 7 remained unremarkable, excluding central pontine myelinolysis (5). Potassium levels normalized within hours in both cases.
In both cases, an empiric parenteral antibiotic therapy with ampicillin and ceftazidim was initiated. Urine culture grew Enterobacter cloacae (Case 1) and Klebsiella oxytoca (Case 2) which led to the antibiogram-based therapy with piperacillin/tazobactam and ceftazidim. Antibiotics were consecutively given orally (sulfamethoxazole/trimethoprim and cefpodoxim) and stopped after 15 days (Case 1) and 9 days (Case 2) of treatment.
Hormone analyses, revealed massively increased aldosterone (>100 ng/dl) and renin (>500 µIU/ml) levels, indicating a reduced sensitivity against aldosterone and excluding adrenal insufficiency as the cause of the electrolyte deviations. ACTH values were within the normal range and cortisol levels were slightly increased in both cases (case 1: 21.2 µg/dl (measured at 3 a.m.) and case 2: 30.8 µg/dl (measured at 16 p.m.)), in line with a stress response to the patients’ severe infections. In combination with urine tract infections and structural abnormalities of the kidneys and ureters in both cases, the diagnosis of PHA type 3 was made. Hydrocortisone and fludrocortisone supplementation was reduced and stopped after 5 days in Case 1 and 8 days in Case 2, without recurrence of electrolyte deviations. Further hormone analyses (Division of Pediatric Endocrinology and Diabetes, Christian-Albrechts-University Kiel, Head: Prof. Dr. P.-M. Holterhus) showed increased levels of the steroid precursors 11-desoxycortisone and corticosterone, confirming the diagnosis.
Both infants could be discharged 10 days (Case 1) respectively 13 days (Case 2) after admission. Figure 1 describes the clinical course during the hospitalisation with laboratory chemistry of the electrolytes and the initiated therapy of case 1 and 2.
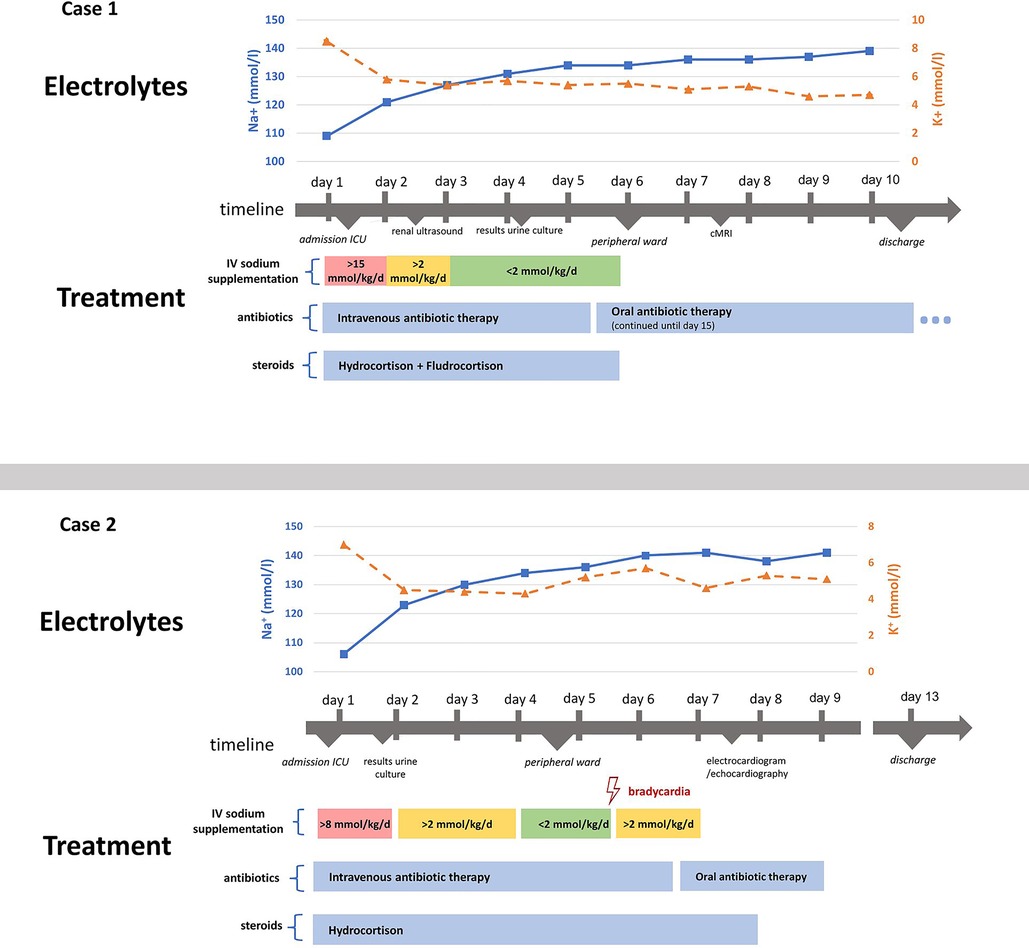
Figure 1. Illustration of both cases’ clinical course and treatment. Depicts the clinical course and treatment of both cases. Intravenous sodium supplementation is presented as mmol/kg/d, meaning the sodium amount in mmol given per kilogram body weight per day, summarizing different intravenous fluids given, including normal saline or balanced electrolyte solutions.
Due to the sonographic findings, a voiding cystourethrography was conducted in Case 1 and revealed an ectopic megaureter and vesicoureteral reflux leading to III° hydronephrosis. The patient is currently receiving antibiotic prophylaxis with cefaclor and may undergo surgical correction in the future. Patient 2 is currently being followed as an outpatient in our pediatric nephrology department. In the age of 16 months, she was diagnosed with developmental delay and muscular hypotonia and is currently undergoing further neuropediatric and genetic diagnostics.
Discussion
We present two cases of PHA type 3 secondary to urinary tract infections as complications of urinary tract malformations. These presented with very severe hyponatremia, hyperkalemia and massively increased aldosterone and renin levels. Correction of hyponatremia in both cases proved uncomplicated.
Pathomechanism
Aldosterone causes the retention of sodium via epithelial sodium channels (ENaC) as well as the elimination of potassium via renal outer medullary K+ channels (ROMK), finally leading to hyponatremia and hyperkalemia. In PHA type 3, there is a resistance of aldosterone to renal mineralcorticoid receptors, leading to an electrolyte shift with hyponatremia, hyperkalemia and metabolic acidosis. It is closely associated with infections and/or congenital anomalies of the kidney and urinary tract (CAKUT) and mainly affects infants less than six months of age with a tendency of an increased risk in male infants (3). Recently, a case series described the occurrence of PHA type 3 in infants (mostly preterm) with high-output stomata, however the underlying mechanism and link to UTI or CAKUT-associated PAH type 3 remains unclear (6). Here, the combination of inflammation of the kidneys with its age-dependent immaturity appear to result in renal unresponsiveness for mineralocorticoids (7–10). The specific underlying mechanisms are not yet fully understood. One theory postulates that bacterial endotoxins damage the aldosterone receptor (10, 11). However, the presence of PHA type 3 with CAKUT without the presence of UTI, even representing a minority of cases, has repeatedly been reported (3, 10, 12). Another theory postulates that a rise in proinflammatory cytokines, either due to bacterial infection or increased intrarenal pressure, leads to an inhibition of renal mineralocorticoid receptors (10). However, neither of these mechanisms have been studied in detail. Additionally, it remains unclear why some infants suffer from those severe electrolyte alterations while most infants with urinary tract infections and/or CAKUT do not (7).
Additional yet unknown genetic alterations might contribute to the susceptibility of some infants. Tuoheti et al. conducted genetic testing in two cases with PHA and identified several heterozygote variants with unknown clinical significance (10). Future studies should systematically address potentially underlying genetic alterations which might help to identify infants at risk as well as to clarify potentially underlying pathomechanisms.
Review of the literature
A systematic literature search of further case reports on transient pseudohypoaldosteronism was conducted in order to compare symptoms, diagnosis, therapy, and outcome of our two patients with previously published cases of pseudohypoaldosteronism type 3 and to update the comprehensive review by Delforge et al. (3). We searched PubMed for case reports that contained the term “pseudohypoaldosteronism” in their full text. To focus on the latest developments of the clinical picture, especially regarding therapy, the literature search was restricted to the period between 2013 and 2023. Only case reports published in English were considered. The last update of the literature search was 31.07.2023. In total, 100 papers were found and titles and abstracts were further screened for cases of type 3 pseudohypoaldosteronism, defined by transient pseudohypoaldosteronism secondary to UTI and/or hydronephrosis. In total, 17 case reports reporting on 26 cases of type 3 pseudohypoaldosteronism were found (8–10, 12–25). Table 1 depicts the clinical characteristics of the cases found in the literature search, together with our cases (N = 28).
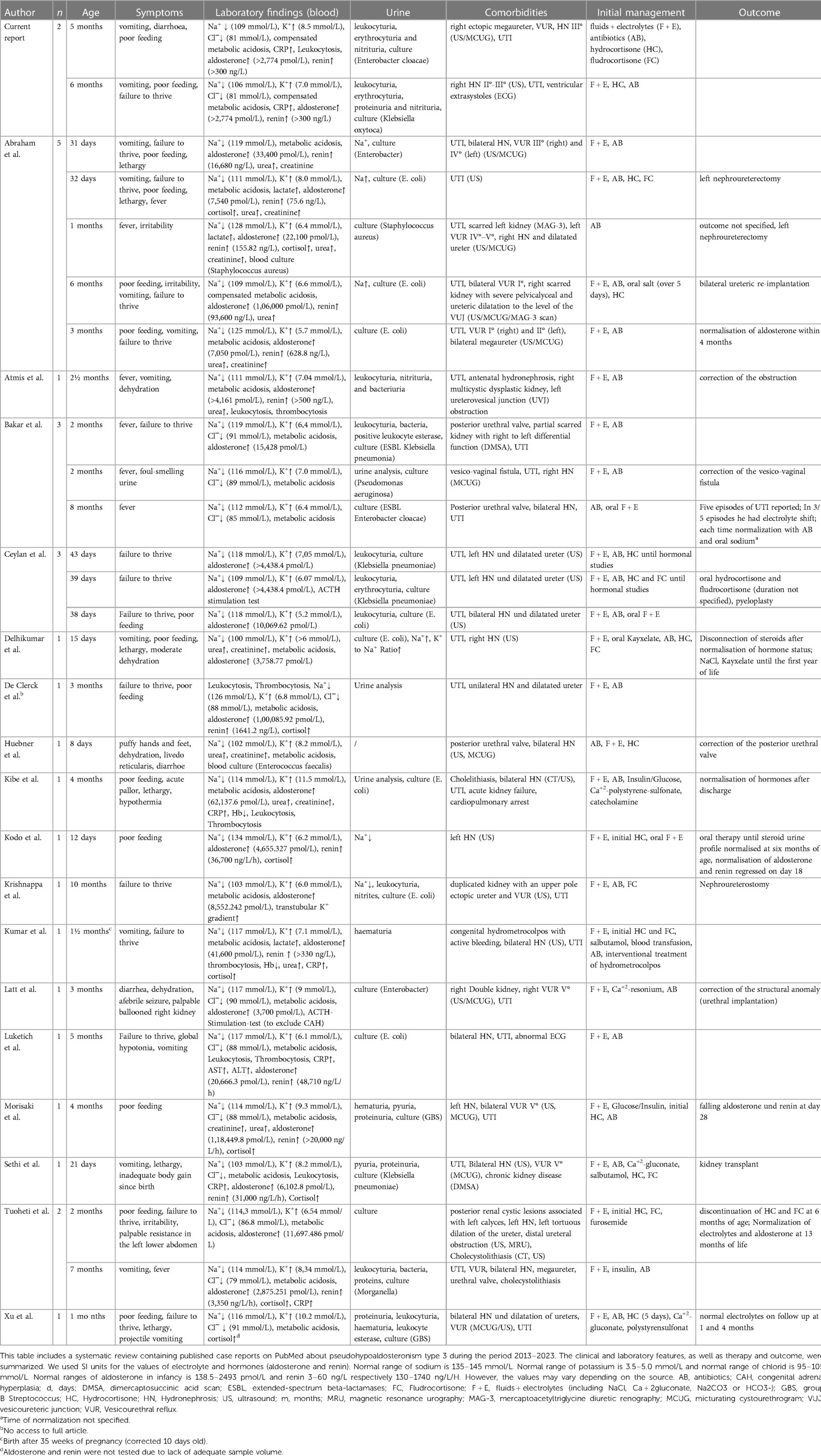
Table 1. Review of the literature.
In 20 of 28 cases electrolyte deviations rapidly resolved after sodium supplementation within hours up to six days, one case relied on sodium supplementation and polystyrene sulfonate up to the age of one year and in another case normalization of electrolytes and aldosterone was noted in follow up at 13 months. Both cases are questioning the diagnosis of transient PHA. In 6 cases, time of normalization of electrolytes was not specified.
Most patients were less than three months old (n = 16, 57% of cases), 7 cases (25%) were between 3 months and 6 months and 5 cases (18%) were older than 6 months up to 10 months. Most common presenting symptoms were general and unspecific in nature like reduced drinking behavior, weight loss and/or failure to thrive in 21 of cases (75%, multiple responses allowed) followed by vomiting (n = 13, 46%) or diarrhea in three cases (11%).
Our cases add specific facets to the spectrum of PHA type 3. First, our patients are relatively old as only five of the cases in the literature were older than four months. Second, we report two female cases, however, a strong tendency for PHA type 3 in male patients was previously reported (3). Third, our patients had severe hyponatremia below 110 mmol/L which highlight the potentially hazardous nature of PHA type 3.
Differential diagnosis and diagnostics
Hyponatremia, hyperkalemia and metabolic acidosis in infants always should rise the suspicion of adrenal insufficiency, e.g., due to congenital adrenal hyperplasia (CAH) with salt wasting syndrome or a form of primary hypoaldosteronism (e.g., Aldosterone-Synthase-deficiency). Even if 17-hydroxyprogesterone is covered in newborn screenings in multiple countries worldwide including Germany, patients with residual activity of 21-hydroxylase or other forms of adrenal enzyme deficiencies leading to salt wasting syndrome might be missed and thus an unremarkable newborn screening does not rule out CAH. PHA can be distinguished from adrenal insufficiency by an excess of aldosterone and renin with high ACTH levels as a result of missing negative feedback mechanisms due to resistance in mineralcorticoid receptors. Cortisol levels can be normal or elevated because of the stress-inducing underlying infection. An excess of 17-hydroxyprogesterone, the hallmark of CAH due to 21-hydroxylase deficiency, cannot be seen in PHA.
PHA type 3 occurs secondary to infections and/or CAKUT and is transient while PHA type 1 and 2 are autosomal dominant and recessively inherited. In PHA type 1, a further distinction is made between the renal (autosomal dominant) and the systemic (autosomal recessive) form (26, 27). In the generalized form, salt loss occurs in several organs, including the lung, colon, kidney, salivary- and sweat glands. Known genetic alterations lay in the genes for the subunits of ENaCs. Symptoms such as poor feeding and vomiting usually occur in the first weeks of life (26). Therapeutically, salt and fluid replacement with high sodium, ion exchange resins and low potassium diet is crucial (26). The renal form of PHA type 1 is the result of mutations in genes encoding for the mineral corticoid receptor and autosomal-dominantly inherited (26, 28). The clinical manifestation varies greatly but are, in general, milder. Vomiting, weight loss and dehydration occur in young infants. In later childhood, the children have no symptoms and show normal development. The prognosis of the renal form is good and therapy with sodium supplementation can usually be terminated in early childhood (26, 28). PHA type 2 [also known as Gordon's syndrome (29) or familial hyperkalemia and hypertension] is an autosomal-dominant inherited disease with the clinical hallmarks of hypertension, hyperkalemia and mild metabolic acidosis, but, in contrast to PHA type 1 and 3, without marked hyponatremia (30). Laboratory abnormalities can be detected as early as infancy, while hypertension may be detected two to four decades later (31). It responds well to treatment with thiazide diuretics (30). Underlying mutations vary but usually isoforms of the so-called with-no-lysine kinases (WNK) genes are affected (27, 31) which lead to alterations of the renal electrolyte exchange via NaCl cotransporter (NCC) and/or ROMK channels (27, 31). PHA type 1 or 2 should be considered in cases with unusual presentation e.g., congenital onset, absent trigger factors (UTI or CAKUT) or unsuccessful withdrawal from sodium supplementation as well as a suspicious family history and should be further evaluated via genetic testing.
In infants with severe hyponatremia and/or the suspected diagnosis of PHA an abdominal ultrasound is mandatory to screen for urinary tract malformations and to exclude adrenal hyperplasia. Urine analyses should be conducted to screen for possible urinary tract infections. Additionally, hypernatriuria might be seen as a result to the absent of aldosterone mediated resorption of sodium via renal ENaCs (32). In our cases, urine sodium was <20 mmol/L (Case 1, measured on day 2) or only slightly elevated (23 mmol/L, Case 2), however this might be the result of the severe hyponatremia and thus a general shortage of sodium in both patients.
Another possible differential diagnosis in patients with hyperkalemia and metabolic acidosis is hyperkalemic renal tubular acidosis (RTA) type 4 which is characterized by reduced acid and potassium secretion (33). However, RTA type 4 is characterized by hyperchloremia, while our patients showed hypochloremia. RTA type 4 is usually secondary to kidney damage in patients with diabetes, interstitial nephritis (e.g., Lupus nephritis) or sickle cell nephropathy (33) and goes along with hypoaldosteronism or aldosterone resistance (33, 34). In the latter case, RTA type 4 can be seen as the clinical result of PHA. In children, patients with genetically proven PAH type 1 and type 2 and RTA type 4 have been described (34).
Other differential diagnoses of infants with severe hyponatremia include but are not limited to acute kidney or cardiac failure, gastroenteritis, especially in the light of concomitant vomiting and failure to thrive, as well as the syndrome of inappropriate antidiuretic hormone (SIADH) e.g., secondary to respiratory syncytial virus (RSV) bronchiolitis (35, 36).
Therapy
In patients with severe hyponatremia, hyperkalemia and metabolic acidosis and signs of dehydration, normal saline bolus should be administered for electrolyte and volume supplementation. If a urinary tract infection is suspected, empiric antibiotic therapy should promptly be initiated. Hormone analyses which confirm the diagnosis of PHA usually take some time, therefore we and others suggest treating all cases as acute adrenal insufficiency until proven otherwise, even if PHA is suspected (13). Thus, hormone supplementation with hydrocortisone and/or fludrocortisone according to common guidelines (4) should be initiated. In our literature review, most cases received hydrocortisone and/or fludrocortisone for initial treatment (see Table 1). If elevated aldosterone and renin confirm PHA, hydrocortisone and fludrocortisone can be discontinued. During therapy with hydrocortisone, attention should be paid to various side effects, for example hypertension or bradycardia, as described in case 2.
Complications
Hyponatremia can lead to cerebral edema and/or seizures and is associated with increased mortality and morbidity (35). Rapid correction of hyponatremia can lead central pontine myelinolysis associated with spastic quadriparesis (5). Luckily, despite the extreme hyponatremia in our patients, none of these complications were seen and cerebral MRI in Case 1 remained unremarkable.
Case 2 developed bradycardia (min. 46/min) and ventricular extrasystoles during a time when electrolytes already had been normalized. Bradycardia and hypertension can be seen with steroid replacement (37). In addition, elevated aldosterone levels, as excessively seen in our patient, might foster dysrhythmias (38). In addition, prior hyperkalemia might have promoted these dysrhythmias.
Conclusions
We report two cases of infants with PHA type 3 secondary to urinary tract infections who developed life-threatening hyponatremia (minimally 106 mmol/L). Clinicians should be aware of the possibility of severe electrolyte deviations in infants with symptoms as vomiting or food refusal and urinary tract infections. Electrolyte measurements in infants with urinary tract infections should be conducted with low threshold considering these unspecific symptoms. Hyponatremia then calls for an extensive diagnostic work-up, including hormone analyses and renal ultrasound as well as prompt treatment, including sodium supplementation under frequent electrolyte controls, supplementation with hydrocortisone as well as antibiotic treatment, if not already started. Further research is needed to recognize type III PHA earlier and better and to enlighten the underlying pathomechanisms as well as dynamics of electrolyte deviations to identify infants-at-risk and to guide recommendations for electrolyte monitoring in infants with urinary tract infections.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the patients' legal guardians for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceptualization: CC, LD, and TN. Conduction of the literature review: CC. Patient care: LD, JM, RP, TVL, and SFO. Writing – original draft: CC and LD. Writing – review and editing: TN, JM, RP, TVL, and SF. All authors contributed to the article and approved the submitted version.
Acknowledgments
We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
Conflict of interest
TN has received authorship fees from uptodate.com (Wellesley, Massachusetts, USA) and reimbursement of travel expenses during consultancy work for the European Medicines Agency (EMA), steering committees of the PENTA Paediatric European Network for Treatment of AIDS (Padua, Italy), the Juvenile Inflammatory Cohort (JIR) (Lausanne, Switzerland), and, until 2017, the FIND-ID Initiative (supported by the Plasma Protein Therapeutics Association [PPTA] [Brussels, Belgium]).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tullus K, Shaikh N. Urinary tract infections in children. Lancet. (2020) 395(10237):1659–68. doi: 10.1016/S0140-6736(20)30676-0
2. Okarska-Napierała M, Wasilewska A, Kuchar E. Urinary tract infection in children: diagnosis, treatment, imaging–comparison of current guidelines. J Pediatr Urol. (2017) 13(6):567–73. doi: 10.1016/j.jpurol.2017.07.018
3. Delforge X, Kongolo G, Cauliez A, Braun K, Haraux E, Buisson P. Transient pseudohypoaldosteronism: a potentially severe condition affecting infants with urinary tract malformation. J Pediatr Urol. (2019) 15(3):265.e1–e7. doi: 10.1016/j.jpurol.2019.03.002
4. Kamrath C. Handlungsempfehlung nach der leitlinie primäre nebenniereninsuffizienz im kindes-und jugendalter. Monatsschr Kinderheilkd. (2021) 169(8):748–50. doi: 10.1007/s00112-021-01196-8
6. Ou C-Y, Chen Y-J, Lin G-B, Chen M-F, Chia S-T. Case report: newborns with pseudohypoaldosteronism secondary to excessive gastrointestinal losses through high output stoma. Front Pediatr. (2021) 9:773246. doi: 10.3389/fped.2021.773246
7. Gil-Ruiz MA, Alcaraz AJ, Marañón RJ, Navarro N, Huidobro B, Luque A. Electrolyte disturbances in acute pyelonephritis. Pediatr Nephrol. (2012) 27:429–33. doi: 10.1007/s00467-011-2020-9
8. Delhikumar C, Narayanan P, Mahadevan S. Pseudohypoaldosteronism masquerading as congenital adrenal hyperplasia. Indian J Pediatr. (2012) 79:115–6. doi: 10.1007/s12098-011-0490-1
9. Morisaki A, Naruse Y, Shibata Y, Mori M, Hiramoto R. Transient Pseudohypoaldosteronism secondary to group B Streptococcus pyelonephritis. Cureus. (2021) 13(5):e15071. doi: 10.7759/cureus.15071
10. Tuoheti Y, Zheng Y, Lu Y, Li M, Jin Y. Transient pseudohypoaldosteronism in infancy mainly manifested as poor appetite and vomiting: two case reports and review of the literature. Front Pediatr. (2022) 10:895647. doi: 10.3389/fped.2022.895647
11. Peddle M, Joubert G, Lim R. Case 2: hyponatremia and hyperkalemia in a four-week-old boy. Paediatr Child Health. (2008) 13(5):387–90. doi: 10.1093/pch/13.5.387a
12. Kodo K, Goto S, Katsumi Y. Secondary pseudohypoaldosteronism associated with mild hydronephrosis in a newborn. Cureus. (2021) 13(2):e13462. doi: 10.7759/cureus.13462
13. Abraham MB, Larkins N, Choong CS, Shetty VB. Transient pseudohypoaldosteronism in infancy secondary to urinary tract infection. J Paediatr Child Health. (2017) 53(5):458–63. doi: 10.1111/jpc.13481
14. Abu Bakar K, Jalaludin MY, Zainal N, Woon SL, Mohd Zikre N, Samingan N, et al. Case report: severe hyponatremia in infants with urinary tract infection. Front Pediatr. (2021) 9:655010. doi: 10.3389/fped.2021.655010
15. Ceylan D, Bayramoğlu E, Polat E, Erdeve ŞS, Çetinkaya S, Aycan Z. A rare cause of salt-wasting in early infancy: transient pseudohypoaldosteronism. Turk Arch Pediatr. (2021) :75–7. doi: 10.14744/TurkPediatriArs.2020.38159
16. De Clerck M, Vande Walle J, Dhont E, Dehoorne J, Keenswijk W. An infant presenting with failure to thrive and hyperkalaemia owing to transient pseudohypoaldosteronism: case report. Paediatr Int Child Health. (2018) 38(4):277–80. doi: 10.1080/20469047.2017.1329889
17. Kibe T, Sobajima T, Yoshimura A, Uno Y, Wada N, Ueta I. Secondary pseudohypoaldosteronism causing cardiopulmonary arrest and cholelithiasis. Pediatr Int. (2014) 56(2):270–2. doi: 10.1111/ped.12267
18. Krishnappa V, Ross JH, Kenagy DN, Raina R. Secondary or transient pseudohypoaldosteronism associated with urinary tract anomaly and urinary infection: a case report. Urol Case Rep. (2016) 8:61–2. doi: 10.1016/j.eucr.2016.07.001
19. Kumar S, McDermott H, Kamupira S, Agwu JC. Rare case of pseudohypoaldosteronism in a neonate secondary to congenital hydrometrocolpos. BMJ Case Rep CP. (2020) 13(6):e234813. doi: 10.1136/bcr-2020-234813
20. Latt TN, Rahman SI, Nor NSM. Transient pseudohypoaldosteronism in an infant: a case report. J ASEAN Feder Endocr Soc. (2018) 33(1):45. doi: 10.15605/jafes.033.01.07
21. Luketich SK, Deskins SJ, Downey S, Lynch J, Lynch JD. A five-month-old boy with hypotonia, electrolyte derangements, and failure to thrive. Cureus. (2023) 15(1):e34226. doi: 10.7759/cureus.34226
22. Xu M, Di Blasi C, Dickerson J, Jack R, Rutledge JC. A 5-week-old boy with failure to thrive, marked hyperkalemia, and hyponatremia. Clin Chem. (2016) 62(11):1439–43. doi: 10.1373/clinchem.2015.252320
23. Atmis B, Turan I, Melek E, Bayazit AK. An infant with hyponatremia, hyperkalemia, and metabolic acidosis associated with urinary tract infection: answers. Pediatr Nephrol. (2019) 34:1739–41. doi: 10.1007/s00467-019-04254-2
24. Sethi SK, Wazir S, Bansal S, Khokhar S, Wadhwani N, Raina R. Secondary pseudohypoaldosteronism masquerading congenital adrenal hyperplasia in a neonate. Kidney Int Rep. (2018) 3(3):752–4. doi: 10.1016/j.ekir.2018.01.004
25. Huebner K, Davis TK, Jackson T, Dawson J. Severe hyponatremia in a 1-week-old male infant. Clin Pediatr (Phila). (2015) 54(4):396–400. doi: 10.1177/0009922814553437
26. Amin N, Alvi N, Barth J, Field H, Finlay E, Tyerman K, et al. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. (2013) 2013(1). doi: 10.1530/EDM-13-0010
27. Furgeson SB, Linas S. Mechanisms of type I and type II pseudohypoaldosteronism. J Am Soc Nephrol. (2010) 21(11):1842–5. doi: 10.1681/ASN.2010050457
28. Geller DS, Zhang J, Zennaro M-C, Vallo-Boado A, Rodriguez-Soriano J, Furu L, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol. (2006) 17(5):1429–36. doi: 10.1681/ASN.2005111188
29. Gordon RD, Geddesj RA, Pawseynl CG, O'Halloran MW. Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction. Australas Ann Med. (1970) 19(4):287–94. doi: 10.1111/imj.1970.19.4.287
30. Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. (2002) 87(7):3248–54. doi: 10.1210/jcem.87.7.8449
31. Pathare G, Hoenderop JG, Bindels RJ, San-Cristobal P. A molecular update on pseudohypoaldosteronism type II. Am J Physiol Renal Physiol. (2013) 305(11):F1513–F20. doi: 10.1152/ajprenal.00440.2013
32. Bogdanović R, Stajić N, Putnik J, Paripović A. Transient type 1 pseudo-hypoaldosteronism: report on an eight-patient series and literature review. Pediatr Nephrol. (2009) 24(11):2167–75. doi: 10.1007/s00467-009-1285-8
33. Palmer BF, Kelepouris E, Clegg DJ. Renal tubular acidosis and management strategies: a narrative review. Adv Ther. (2021) 38:949–68. doi: 10.1007/s12325-020-01587-5
34. Santos F, Ordóñez FA, Claramunt-Taberner D, Gil-Peña H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. (2015) 30:2099–107. doi: 10.1007/s00467-015-3083-9
35. Storey C, Dauger S, Deschenes G, Heneau A, Baud O, Carel JC, et al. Hyponatremia in children under 100 days old: incidence and etiologies. Eur J Pediatr. (2019) 178:1353–61. doi: 10.1007/s00431-019-03406-8
36. Szabo FK, Lomenick JP. Syndrome of inappropriate antidiuretic hormone secretion in an infant with respiratory syncytial virus bronchiolitis. Clin Pediatr (Phila). (2008) 47(8):840–2. doi: 10.1177/0009922808315222
37. van der Gugten A, Bierings M, Frenkel J. Glucocorticoid-associated bradycardia. J Pediatr Hematol Oncol. (2008) 30(2):172–5. doi: 10.1097/MPH.0b013e31815dcfeb
Keywords: pseudohypoaldosteronism, hyponatremia, hyperkalemia, urinary tract infections, hydronephrosis, congenital anomalies of the kidney and urinary tract
Citation: Carl C, Dinkelbach L, Mohr J, Perez R, Vera Lopez T, Fricke-Otto S and Niehues T (2024) Case report: Life threatening hyponatremia in infants with urinary tract infections: two cases of type III pseudohypoaldosteronism and review of the literature. Front. Pediatr. 11:1233205. doi: 10.3389/fped.2023.1233205
Received: 1 June 2023; Accepted: 11 December 2023;
Published: 5 January 2024.
Edited by:
Maria G. Vogiatzi, University of Pennsylvania, United StatesReviewed by:
Paul B. Kaplowitz, Children's National Hospital, United StatesAysun Karabay Bayazit, Cukurova University, Türkiye
© 2024 Carl, Dinkelbach, Mohr, Perez, Vera Lopez, Fricke-Otto and Niehues. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Dinkelbach bGFycy5kaW5rZWxiYWNoQHVrLWVzc2VuLmRl
†These authors share first authorship