 Megan F. Baxter
Megan F. Baxter Michele Hansen3,4
Michele Hansen3,4 Gareth Baynam
Gareth Baynam- 1Emergency Department, Perth Children’s Hospital, Perth, WA, Australia
- 2School of Medicine and Dentistry, Griffith University, Gold Coast, QLD, Australia
- 3Telethon Kids Institute, University of Western Australia, Perth, WA, Australia
- 4Western Australian Register of Developmental Anomalies, King Edward Memorial Hospital, Perth, WA, Australia
- 5Rare Care Centre, Perth Children’s Hospital, Perth, WA, Australia
- 6Undiagnosed Diseases Program, WA, Genetic Services of WA, Perth, WA, Australia
The diagnostic odyssey for people living with rare diseases (PLWRD) is often prolonged for myriad reasons including an initial failure to consider rare disease and challenges to systemically and systematically identifying and tracking undiagnosed diseases across the diagnostic journey. This often results in isolation, uncertainty, a delay to targeted treatments and increase in risk of complications with significant consequences for patient and family wellbeing. This article aims to highlight key time points to consider a rare disease diagnosis along with elements to consider in the potential operational classification for undiagnosed rare diseases during the diagnostic odyssey. We discuss the need to create a coding framework that traverses all stages of the diagnostic odyssey for PLWRD along with the potential benefits this will have to PLWRD and the wider community.
1. Introduction
Rare diseases (RD) are conditions with a specific pattern of clinical signs, symptoms, and findings that affect fewer than 1 in 2,000 persons living in any World Health Organisation-defined region of the world (1). There are more than 7,000 rare diseases, largely genetic (>80%) in origin, with more than half affecting children (2, 3). Whilst rare diseases individually represent a small population burden, collectively they represent a much larger healthcare burden with between 3.5% and 5.9% of the world's population living with a rare disease, equating to an estimated 262.9–446.2 million people globally (3). The direct and indirect costs of only a subset of rare diseases in the USA alone is approximately $1 trillion p.a. and two thirds of national paediatric inpatient expenditure (105 Billion p.a. RD vs. 70 billion p.a. for common diseases) (4, 5). Early and accurate rare disease diagnosis is critical to ensure that there is informed decision making, targeted treatments, reduced complication risks and improved patient wellbeing (6) and healthcare sustainability. However, people living with a rare disease often share a long arduous diagnostic process with a mean time to a diagnosis between 5 and 7 years (7, 8). Every rare disease starts out undiagnosed, and currently globally the majority of people living with a rare disease (PLWRD) are undiagnosed. Many, likely the majority, are not referred for a specialist assessment nor get access to (genomic) diagnostics. Even for those referred and assessed in specialist centres, the typical diagnostic rate is approximately 50% (9). The individual burden and population magnitude of impact of undiagnosed rare disease necessitates measures to improve rare diseases diagnosis, however the fundamental issue of defining a systematic approach to surfacing potential undiagnosed rare diseases and the related coding approach for undiagnosed rare diseases that can be applied to different data sets at various points in the diagnostic odyssey remains to be comprehensively addressed.
If you don't look for something, you will not find it. Colloquially, rare diseases are often referred to as “zebras” with advice given to clinicians to not consider rare diseases first; “When you hear hoofbeats behind you, don’t expect to see a zebra” (10). Focusing on individually common diseases first, or exclusively, prolongs the diagnostic process. Similarly, there is a systemic bias that hinders tracking the undiagnosed due to the dearth of specific and systematically unified processes and codes for a suspected or likely undiagnosed rare disease. If you can't measure it, you can't improve it. ICD-10 limitations for rare diseases coding are well documented (11, 12). When developing ICD-11 the WHO established Topic Advisory Groups, including a Rare Disease Group, ensuring the ICD-11 coding system had a greater focus on rare disease. By utilising the Orphanet database, and a poly-hierarchal classification structure the ICD-11 includes a unique identifier for classification of over 5,500 rare diseases (11) It also has a dedicated code MG48—“unknown and unspecified causes of morbidity”, similar to the Orphanet code ORPHA: 616874—“rare disease without a determined diagnosis after full investigation”. The IDC-11 code is only applicable after extensive specialist assessments are completed and neither codes alone can be used to track across the journey prior to such an assessment (11, 13). This means that often for numerous years, pending specialist assessments, if these in fact occur, undiagnosed individuals are invisible within systems with no comprehensive or consistent identification, flags for consideration, or operational definitions of “suspected” or “likely” undiagnosed rare disease. Accordingly, there is an inability to identify and track the hundreds of millions of people living with undiagnosed rare diseases. This prohibits health, and other system planning for this enormous group of people, which in turn threatens the value and sustainability of healthcare. Herein, we apply an Australian lens whilst noting that much of the below is generalizable internationally.
Herein we:
1. Highlight key time points and flags for considering a rare disease diagnosis, and
2. Provide thematic suggestions of some elements or characteristics that could help inform the definition of codes along the diagnostic process.
We do not endeavour to create a definitive list of codes or their elements, but seek to both raise awareness of the need and deliver an initial framework for considering an approach to surfacing and monitoring undiagnosed rare diseases.
2. Benefits of shortened diagnostic odyssey
There are numerous benefits to shortening the diagnostic odyssey. From a patient perspective there are psychological, social and functional implications (14). Earlier diagnosis allows for better informed decision making with the patients able to access more accurate information with regards to likely progression and potential disease symptoms along with more accurate life expectancy information. Given these diseases are largely genetic, patients and their families are also able to access better information to support further family planning. This includes correct information from a genetic counselling perspective with regards to the risk of existing siblings and further children being affected and the option to access carrier screening or preimplantation genetic testing if desired (7). Patients and families are also better able to access appropriate targeted support and advocacy groups (15). Overall, shortening the diagnostic odyssey can significantly improve patients quality of life (14).
With regards to investigations, a specific diagnosis allows for the minimization of unnecessary, often painful, tests and assessments with the focus shifting to monitoring disease (16).
From a treatment perspective a single diagnosis allows patients to make more informed decisions with regards to treatment. Earlier disease identification can result in an increased range of treatments available. It also ensures that specific targeted treatments can be offered when available rather than just symptomatic management and can prevent unnecessary, ineffective treatments from being commenced. Finally, it allows for consideration of inclusion in appropriate clinical trials and research groups for which patients are often unlikely to meet inclusion criteria without a formal diagnosis (17).
All of these benefits of earlier disease diagnosis also result in a reduced healthcare burden allowing for streamlining of healthcare services, having a significant fiscal benefit (18, 19).
3. Elements that could flag the consideration of a rare disease
The following elements can be considered in the diagnosis of rare diseases (see Table 1):
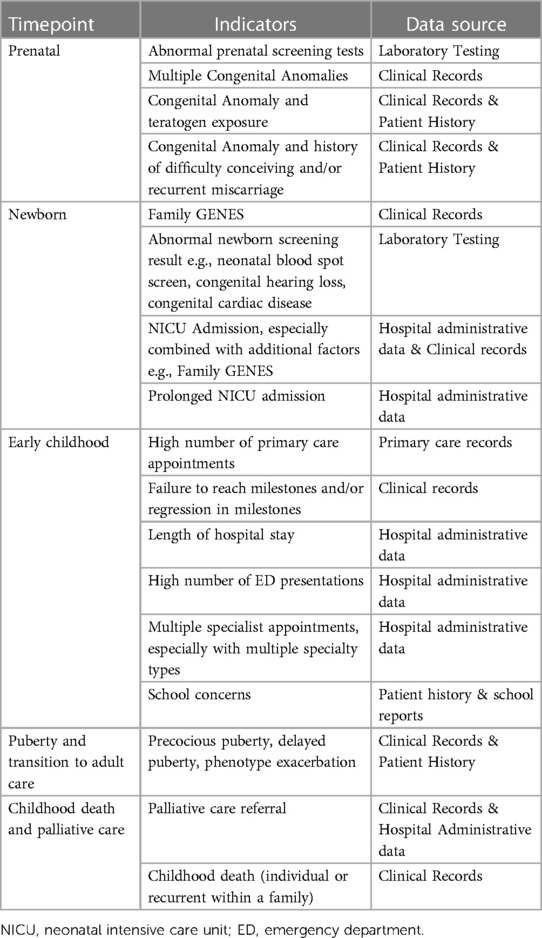
Table 1. A framework of potential flags for consideration of an undiagnosed rare disease over time.
3.1. Clinical phenotypic factors
The mnemonic “Family GENES” has been proposed in order to identify risk factors for rare disease (20); Family—history with multiple generations or siblings affected with a similar phenotype
G—Groups of congenital abnormalities
E—Extremes or exceptional presentations of common disease
N—Neurodevelopmental delay or regression
E—Extremes or exceptional pathology
S—Surprising lab values
This mnemonic broadly highlights clinical red flags to consider, but does not reflect key timepoints at which to consider them, with one of the main barriers to rare disease diagnosis being the initial consideration of the possibility of a rare disease. This approach can be applied “manually” through care pathways or converted to algorithmic approaches that can be applied automatically from electronic health records. However, it may not be readily applicable to existing e.g., administrative data sets which may lack these data elements.
3.2. Administrative data elements
The often-complex nature and high burden of rare diseases generates flags that could be identified in administrative data, these include:
• Presentation to multiple specialty types/multisystem disorder.
• Multiple investigations, and multiple types of investigations.
• Frequent inpatient and outpatient admissions.
• Multiple medications, especially in children.
• Frequent emergency department presentations.
• High total medical treatment cost per child.
• Multiple surgeries.
• Death, especially in children.
• Frequent mental health presentations.
4. Timepoints at which to consider a rare disease diagnosis and related flags
4.1. Antenatal
Abnormal prenatal screening tests, such as biochemistry, imaging and molecular tests may prompt the consideration of diagnostic testing to detect a rare, and often genetic, disease cause. Individual morphological factors, such as intrauterine growth restriction (IUGR), may be indicators. Between 5% and 20% of IUGR is the result of an underlying genetic cause, in particular symmetrical IUGR, the result of early pregnancy growth restriction (21). Further, there may be a greater index of suspicion for a rare disease in the presence of multiple congenital anomalies, or with a history of difficulty conceiving or of recurrent miscarriage and a structural anomaly suggesting the possibility of a familial chromosomal rearrangement (22). Additionally, a history of teratogen exposure e.g., valproate should prompt the consideration of a rare disease (23).
4.2. Puerperal and Neonatal
The average hospital admission at time of birth is 6–48 h following a vaginal delivery and 3 days following a caesarean section (21). This time-period represents one of the key touchpoints where extensive assessments can be completed.
4.2.1. Dysmorphology assessment
In infants with congenital anomalies +/− facial dysmorphism clinical gestalt can lead to a key differential diagnosis and further clinical assessment and testing (24).
4.2.2. Routine screening
The Newborn Bloodspot Screening (NBS) test is completed between 48 and 72 h after birth (25).The test forms part of a screening process assessing for severe rare disease for which early identification significantly improves management. Formal confirmatory diagnostic testing should be considered for any positive results.
Newborn hearing screening (NHS) in Australia is performed by otoacoustic emission testing prior to hospital discharge (26). Eighty percent of prelingual deafness is genetic and rare and there are over 400 genetic syndromes that include hearing loss (27, 28). If an infant fails the otoacoustic emission testing, it is worth not only performing further audiology testing but considering other underlying conditions. Failure of NHS raises a high index of suspicion for a rare disease, especially if there are other red flags.
4.2.3. NICU admission
In Australia, 18% of infants require an admission to a Neonatal Intensive Care Unit (NICU) or Special Care Nursery (SCN) (21). Of those patients admitted to the NICU up to 13% have been estimated to have a congenital malformation which may indicate an underlying rare disease (28).
4.2.4. Symptoms
The following symptoms in a newborn should all be assessed to identify if further investigation is required: difficulty feeding, respiratory distress, and abnormal muscle tone. For example, difficulty feeding is often multifactorial in nature but can be due to craniofacial abnormalities, brainstem dysfunction and poor coordination along with aspiration and malabsorption (29).
4.3. Early childhood
4.3.1. Primary care
A Primary Care Provider (PCP) creates a common touch point for healthcare of PLWRD. PCPs are often the first to identify a potential underlying problem and document any changes over time. On average all children visit a General Practitioner (GP) 3.8 times per year (30). With 80% of children diagnosed with a rare disease visiting their GP at least once in the previous 12-month period (31). It is important to consider any underlying conditions during routine appointments including vaccinations and baby checks, and frequent GP visits alone may be an indicator of rare disease. It is also worth considering a rare disease in the setting of making multiple different specialist referrals in a short time-period.
A rare disease cause should be considered when there is a delay in reaching milestones in one or more domains, or loss or regression of skills. For example, when metabolic pathways are affected resulting in toxic metabolites as in Tay-Sachs Disease, or abnormal protein production preventing development as in Rett Syndrome, there is regression of skills (32). It is also important to consider in cases of extremes of growth or change in growth centiles, for example Noonan or Silver-Russell syndrome in short stature or Sotos syndrome with a larger stature (33–35). The Family GENES mnemonic provides a useful framework for Primary Care (20).
4.3.2. Hospital
Despite having significantly less granularity than clinical records, administrative data can often provide insights for further investigations. When admitted to hospital, patients with rare diseases have a significantly increased duration of hospitalisation, increased cost of care and larger number of procedures. Hence rare diseases should be considered in anyone who has a length of stay greater than one standard deviation from the mean within a given department (4, 36). This disparity is seen to a lesser extent in emergency departments however children with rare diseases are still over represented (4). Any child with 3 or more ED presentations in a one-year period requires consideration for an underlying disorder.
Outside of patients with rare disease, the average number of specialists seen is very low. In patients with a possible rare disease, the number of specialists can be significantly higher given the multiorgan involvement and prolonged diagnostic period. In a cohort study for patients diagnosed with a mitochondrial disease the average number of doctors seen was 8.19 (37). Hence in patients that have 2 or more active specialists with distinct organ pathology, or 3 or more distinct specialists in early childhood, rare disease should be considered. It is worth noting that multiple specialist referrals and investigations is often a significant rate limiting component to the diagnostic process (see Figure 1).
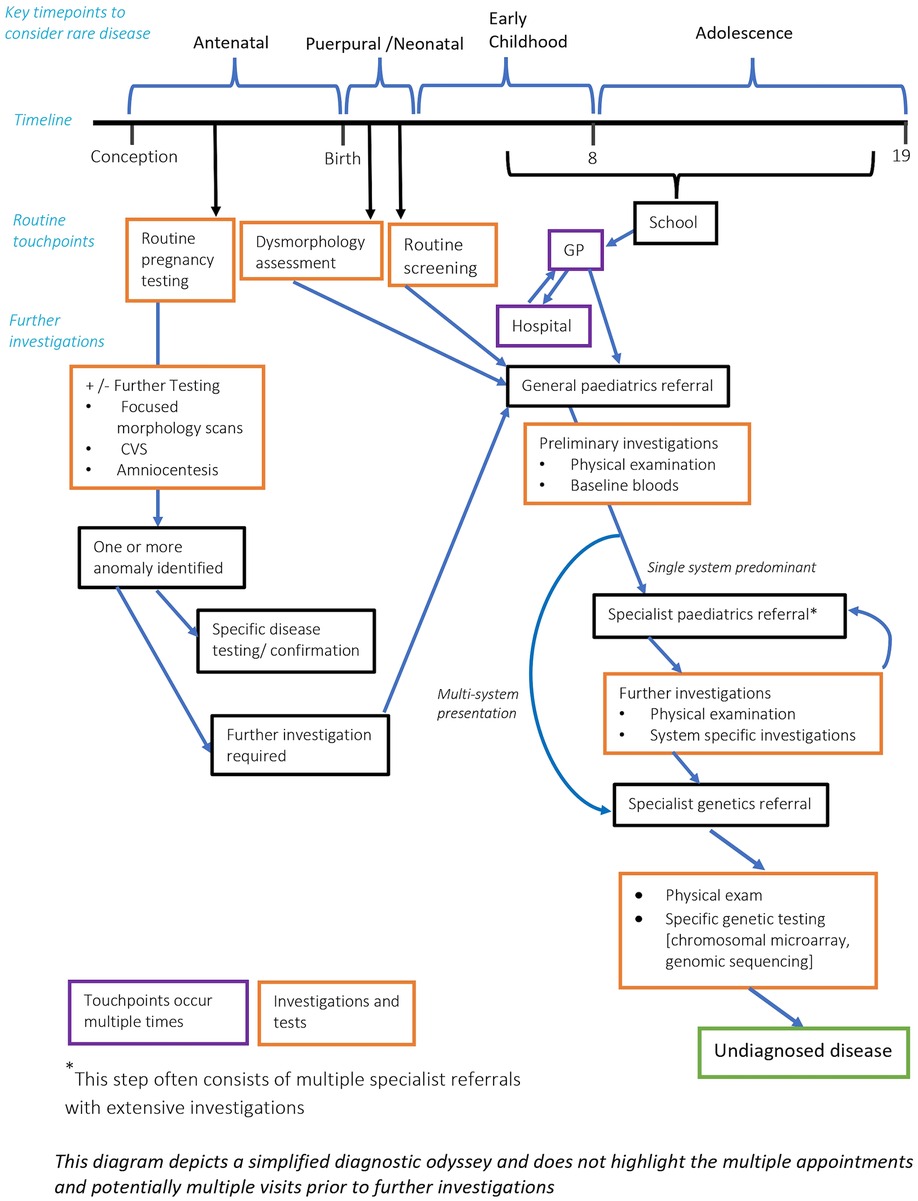
Figure 1. Timepoints to consider a rare disease diagnosis.
4.3.3. School
Given the extensive amount of time a child spends in the classroom, teacher's concerns should be noted. Failure to achieve milestones in developmental domains, consistently poor assessments, significantly small stature, hearing difficulties or vision issues, functional limitations, or strikingly abnormal behaviours should all prompt the need for further assessment.
4.4. Adolescence and transition to adult care
Whilst many rare diseases present in early childhood, they may also affect puberty. For example, patients with neurofibromatosis Type 1 have a significantly higher rate of precocious puberty than the general population. In cases of both peripheral and central precocious puberty an underlying genetic syndrome should be considered (38). This is also true in cases of delayed puberty and primary amenorrhoea (39). An individual's phenotype can also be exacerbated during puberty.
Transition to adult care provides another opportunity to revisit diagnosis. Children with complex undiagnosed diseases undergoing transition to adult services may not have had a diagnostic assessment for several years, and in that interval new diseases will have been identified and diagnostic techniques will have advanced (40). Similarly, those with a clinically diagnosed disorder, such as cerebral palsy, may not have had an assessment for an underlying rare genetic disease cause. Many children in transition clinics are likely to have an (undiagnosed) rare disease.
4.5. Childhood death and palliative care
Six in 10 deaths in children are due to rare diseases (41); rare diseases are the biggest killer of children in high income countries. Childhood death and palliative care are red flags for rare diseases. Any child without a confirmed diagnosis of a rare disease entering palliative care should be considered for diagnostic review, as should any child with a phenotype, such as cerebral palsy, which has a significant likelihood of an underlying rare disease. The possibility of a rare disease should be considered for any childhood death, especially unexplained and/or associated with other red flags for rare diseases.
A specific focus on considering rare disease at key timepoints is one of the ways the diagnostic odyssey will be better addressed.
5. The need for unified explanatory diagnosis classification during the diagnostic process
Currently there are significant multi-stakeholder challenges arising from the lack of systematic and comprehensive classification and coding along the patient journey:
5.1. Patients and their families
The diagnostic odyssey is often very discouraging to patients and their families, having a significant psychological burden (42, 43), and recurrently having to re-tell their history, especially in the absence of a diagnosis or descriptor that supports some level of validation and the opportunity for access to a tailored care pathway. A lack of provisional descriptor or a standardised code can also result in difficulties accessing the appropriate support groups and may impact access to financial support such as further access to disability services. For example, in Australia, National Disability Insurance Scheme (NDIS) often requires a diagnosis for access.
5.2. Healthcare workers
A single descriptive code that is not tailored to the nuances of rare disease is often selected for a patient's presentation. Given that many rare conditions are associated with numerous different symptoms, this can result in incorrect or incomplete coding with downstream consequences for inappropriate and fragmented treatment by the primary managing clinician. This also has implications for other healthcare workers as it does not convey that a currently unspecified disease is being considered, or should be considered.
5.3. Research
It is very difficult to identify all of the relevant patient data for rare diseases research because many of the presentations have been classified incorrectly in administrative data. From an epidemiological perspective, it is difficult to identify the burden of rare diseases; and from an individual disease perspective, it is difficult to obtain a complete cohort (44).
5.4. Hospital and government
As a result of difficulty assessing the burden of rare disease, funding cannot be allocated appropriately to patients or hospitals. This has significant implications with regards to staffing allocation, funding for investigations and health system planning and sustainability (44).
6. Functional classification at different stages of the diagnostic odyssey
As outlined above, there would be significant benefits to a universal classification approach that can be operationalized at various stages in the diagnostic odyssey for those with undiagnosed diseases. Currently within the hospital setting the coding process is laborious with clinical coders assigning codes from clinical notes including discharge summaries, results and patient notes. If specific criteria are met, as outlined in ICD 10 and now ICD ll, then a clinical code is assigned. This coding information is used to access activity-based funding and reimbursement and as such is a primary focus, however clinical coding is imperative to economic modeling, healthcare planning, along with research and education. It is important to note that if the specific criteria are not met or there is any uncertainty in the diagnosis then the coding is not applied. For example, it is possible to code notes citing “presumed”, “probable” or “definitive disease”. It is not possible to code “differential diagnosis”, “?” or “suspected” from within clinical notes (45). If the information is not present in the appropriate format within clinicians’ notes then no coding record of that part of the presentation or disease can be completed by clinical coders. Hence, the quality of documented information affects the quality of coded data (46).
From a rare disease perspective for a universal classification to be used during the diagnostic process it will require time to be developed and subsequently implemented in health systems. Whilst this is occurring, one interim approach is to adapt the use of existing e.g., ICD-10, or incoming e.g., ICD-11 coding, approaches combined with elements from the Orphanet nomenclature.
Both ICD-10 (R69) and ICD-11 (MG48) include a unique code to identify “unknown and unspecified causes of mortality and morbidity for use in undiagnosed diseases, not specified at the site or system involved”. A challenge to implementing and making inferences from these codes in isolation, is that they provide no information as to the nature or extent of diagnostic assessments that have been performed or the severity of the disease and affected organs. They do not allow for identification of a working diagnosis and the degree of diagnostic certainty associated with that.
The ICD-11 classification improves on ICD-10 by using multiple stem codes, or stem and extension codes, to define a patient's presentation. The stem codes provide information as to the clinical entity and can exist alone as a diagnosis, with the extension codes providing important additional descriptive information about a diagnosis (47). It is possible to use a stem on its own, multiple stems joined using a forward slash “/”, a stem and an extension joined using ampersand “&”, a stem and multiple extensions or multiple stems and multiple extensions joined using “/ and &”. For example, if a patient presented with moderate severity diabetic ketoacidosis with T1DM the following code could be used 5A22.0&XS0T/5A10, in which 5A22.0 is diabetic ketoacidosis without coma and XS0T is moderate severity and 5A10 is T1DM. Extension codes can provide information about anatomy and topography, laterality, aetiology, severity (e.g., stages of cancer diagnoses or mild, moderate, profound hearing loss), temporality, diagnosis method of confirmation (laboratory, genetics, imaging, etc.) and to a limited extent diagnostic certainty.
ICD-11 incorporates specific codes for all the rare diseases currently included in the Orphanet nomenclature. For example, the code LCD2F.15 represents Noonan syndrome for which there is no specific code in ICD-10. For this ICD-11 code to be utilised a formal diagnosis must be reached. However, this code is under the parent code LCD2F; syndromes with multiple structural abnormalities, without predominant body system involvement—which could be used, either alone or in combination with other data, as part of a coding system to help flag a possible undiagnosed rare disease.
There are some limitations of the ICD-11 classification for coding undiagnosed diseases. For example, it does not allow for a diagnostic assertion beyond the extension code “diagnostic certainty” which provides only two options—“provisional diagnosis” (XY7Z) and “differential diagnosis” (XY75). The Orphanet Nomenclature advocates using a diagnostic assertion whenever possible in coding cases of rare (or suspected rare) disease. This means a case with suspected Noonan syndrome awaiting further confirmation would be coded using the Orpha code for Noonan Syndrome with the diagnostic assertion “Suspected rare disease” equivalent to the ICD11 extension code “provisional diagnosis”. If the diagnosis is confirmed (i.e., there is sufficient diagnostic and/or clinical evidence), the case would be coded to Noonan Syndrome with the diagnostic assertion “Confirmed rare disease”. Where a physician is unable to determine a clinical diagnosis because of the absence of suitable tests or non-contributory tests the diagnostic assertion is “Undetermined (unknown) diagnosis”. The latter has been assigned a specific code in the Orphanet Rare Disease Classification System—ORPHA: 616874. This code is not meant for use in coding patients along their diagnostic pathway and should only be used after all reasonable efforts to obtain a diagnosis according to the best diagnostic capabilities available have been performed. The addition of diagnostic assertion coding should help differentiate cases of likely undiagnosed rare disease requiring access or referral to diagnostic services from likely undiagnosed rare disease despite extensive specialist assessment.
With an updated more extensive clinical coding system it remains more imperative than ever that clear notes outlining clinicians' thinking including likely diagnosis along with diagnostic certainty and information regarding extension codes are completed to allow for improved clinical coding.
7. Conclusion
Reducing the diagnostic odyssey for rare diseases is critical to improving care for PLWRD and their families. This can be enabled through early consideration of rare disease along with an improved coding framework during the diagnostic process. Looking towards the future, existing coding systems could be adapted, whilst supporting and awaiting the development of new coding methods that ultimately will facilitate linking those with undiagnosed diseases to better care pathways and outcomes.
Author contributions
MB: Conceptualization, Project administration, Writing – original draft, Writing – review & editing, Methodology. MH: Writing – review & editing. DG: Writing – review & editing. TG: Writing – review & editing. GB: Conceptualization, Methodology, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
GB acknowledges that he is funded through the Angela Wright Bennett Foundation, The McCusker Charitable Foundation via Chanel 7 Telethon Trust, Mineral Resources Limited, the Stan Perron Charitable Foundation and the Perth Children's Hospital Foundation. This study received funding from Mineral Resources Limited. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rare Diseases: understanding this Public Health Priority (2005). Available at: https://www.eurordis.org/wp-content/uploads/2009/12/princeps_document-EN.pdf
2. Haendel M, Vasilevsky N, Unni D, Bologa C, Harris N, Rehm H, et al. How many rare diseases are there? Nat Rev Drug Discov. (2020) 19(2):77–8. doi: 10.1038/d41573-019-00180-y
3. Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. (2020) 28(2):165–73. doi: 10.1038/s41431-019-0508-0
4. Navarrete-Opazo AA, Singh M, Tisdale A, Cutillo CM, Garrison SR. Can you hear us now? The impact of health-care utilization by rare disease patients in the United States. Genet Med. (2021) 23(11):2194–201. doi: 10.1038/s41436-021-01241-7
6. Baynam G, Pachter N, McKenzie F, Townshend S, Slee J, Kiraly-Borri C, et al. The rare and undiagnosed diseases diagnostic service—application of massively parallel sequencing in a state-wide clinical service. Orphanet J Rare Dis. (2016) 11(1):77. doi: 10.1186/s13023-016-0462-7
7. Zurynski Y, Deverell M, Dalkeith T, Johnson S, Christodoulou J, Leonard H, et al. Australian children living with rare diseases: experiences of diagnosis and perceived consequences of diagnostic delays. Orphanet J Rare Dis. (2017) 12(1):68. doi: 10.1186/s13023-017-0622-4
8. Hendriksz C. Rare Disease Impact Report: Insights from patients and the medical community: Shire Genetics (2013). Available at: https://globalgenes.org/wp-content/uploads/2013/04/ShireReport-1.pdf
9. Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. (2018) 20(4):435–43. doi: 10.1038/gim.2017.119
10. Hart AM. When you hear hoofbeats, think horses—but be prepared for zebras. J Nurse Pract. (2019) 15(6):10. doi: 10.1016/j.nurpra.2019.03.025
11. Ayme S, Bellet B, Rath A. Rare diseases in ICD11: making rare diseases visible in health information systems through appropriate coding. Orphanet J Rare Dis. (2015) 10(35). doi: 10.1186/s13023-015-0251-8
12. Fung KW, Richesson R, Bodenreider O. Coverage of rare disease names in standard terminologies and implications for patients, providers, and research. AMIA Annu Symp Proc. (2014) 2014:564–72.25954361
13. Existing experiences and Guidelines about the coding of undiagnosed rare diseases patients: RD-CODE (2020). Available at: http://www.rd-code.eu/wp-content/uploads/2021/01/Existing-experiences-and-guidelines-about-coding-of-undiagnosed-RD-patients.pdf
14. Benito-Lozano J, Arias-Merino G, Gomez-Martinez M, Arconada-Lopez B, Ruiz-Garcia B, de la Paz M P, et al. Psychosocial impact at the time of a rare disease diagnosis. PLoS ONE. (2023) 18(7):e0288875. doi: 10.1371/journal.pone.0288875
15. Bauskis A, Strange C, Molster C, Fisher C. The diagnostic odyssey: insights from parents of children living with an undiagnosed condition. Orphanet J Rare Dis. (2022) 17(1):233. doi: 10.1186/s13023-022-02358-x
16. Carmichael N, Tsipis J, Windmueller G, Mandel L, Estrella E. Is it going to hurt?”: the impact of the diagnostic odyssey on children and their families. J Genet Couns. (2015) 24(2):325–35. doi: 10.1007/s10897-014-9773-9
17. Zozaya N, Villaseca J, Abdalla F, Ancochea A, Malaga I, Trapero-Bertran M, et al. Strategic discussion on funding and access to therapies targeting rare diseases in Spain: an expert consensus paper. Orphanet J Rare Dis. (2023) 18(1):41. doi: 10.1186/s13023-023-02635-3
18. Willmen T, Volkel L, Ronicke S, Hirsch MC, Kaufeld J, Rychlik RP, et al. Health economic benefits through the use of diagnostic support systems and expert knowledge. BMC Health Serv Res. (2021) 21(1):947. doi: 10.1186/s12913-021-06926-y
19. Yang G, Cintina I, Pariser A, Oehrlein E, Sullivan J, Kennedy A. The national economic burden of rare disease in the United States in 2019. Orphanet J Rare Dis. (2022) 17(1):163. doi: 10.1186/s13023-022-02299-5
20. Whelan AJ, Ball S, Best L, Best RG, Echiverri SC, Ganschow P, et al. Genetic red flags: clues to thinking genetically in primary care practice. Prim Care. (2004) 31(3):497–508, viii. doi: 10.1016/j.pop.2004.04.010
21. Mother and Baby. Camberra: Australian Institute of Health and Welfare (2023). Available at: https://www.aihw.gov.au/reports/mothers-babies/australias-mothers-babies/
22. Suzumori N, Sugiura-Ogasawara M. Genetic factors as a cause of miscarriage. Curr Med Chem. (2010) 17(29):3431–7. doi: 10.2174/092986710793176302
23. Gomes JDA, Olstad EW, Kowalski TW, Gervin K, Vianna FSL, Schuler-Faccini L, et al. Genetic susceptibility to drug teratogenicity: a systematic literature review. Front Genet. (2021) 12:645555. doi: 10.3389/fgene.2021.645555
25. Your newborn baby’s bloodspot screening test: Department of Health (2022). Available at: https://www.healthywa.wa.gov.au/Articles/U_Z/Your-newborn-babys-screening-test
26. Harvey Coates KG. Newborn hearing screening. Aust Prescr. (2003) 26:82–4. doi: 10.18773/austprescr.2003.062
27. Shearer AE, Hildebrand MS, Smith RJ. Hereditary hearing loss and deafness overview. GeneReviews. (2017) 33:458–63. doi: 10.1097/MOP.0000000000001033
28. Marouane A, Olde Keizer RACM, Frederix GWJ, Vissers LELM, de Boode WP, van Zelst-Stams WAG. Congenital anomalies and genetic disorders in neonates and infants: a single-center observational cohort study. Eur J Pediatr. (2022) 181(1):359–67. doi: 10.1007/s00431-021-04213-w
29. Schwemmle C, Arens C. Feeding, eating, and swallowing disorders in infants and children: an overview. HNO. (2018) 66(7):515–26. doi: 10.1007/s00106-017-0388-y
30. Britt H, Charles J, Harrison C, Bayram C. ‘The kids are alright’—changes in GP consultations with children 2000–15. Aust J Gen Pract. (2015) 44:877–9. doi: 10.1007/s12687-011-0043-3
31. Elliott E, Zurynski Y. Rare diseases are a ‘common’ problem for clinicians. Aust J Gen Pract. (2015) 44:630–3. doi: 10.1212/NXG.0000000000000230
32. Kruszka P, Regier D. Inborn errors of metabolism: from preconception to adulthood. Am Fam Physician. (2019) 99(1):25–32. doi: 10.1016/S2213-8587(15)00380-0
33. Zhou E, Hauser BR, Jee YH. Genetic evaluation in children with short stature. Curr Opin Pediatr. (2021) 33(4):458–63. doi: 10.1097/MOP.0000000000001033
34. Siklar Z, Berberoglu M. Syndromic disorders with short stature. J Clin Res Pediatr Endocrinol. (2014) 6(1):1–8. doi: 10.4274/Jcrpe.1149
35. Argente J, Tatton-Brown K, Lehwalder D, Pfaffle R. Genetics of growth disorders-which patients require genetic testing? Front Endocrinol (Lausanne). (2019) 10:602. doi: 10.3389/fendo.2019.00602
36. Dye DE, Brameld KJ, Maxwell S, Goldblatt J, Bower C, Leonard H, et al. The impact of single gene and chromosomal disorders on hospital admissions of children and adolescents: a population-based study. Public Health Genomics. (2011) 14(3):153–61. doi: 10.1159/000321767
37. Grier J, Hirano M, Karaa A, Shepard E, Thompson JLP. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol Genet. (2018) 4(2):e230. doi: 10.1212/NXG.0000000000000230
38. Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. (2016) 4(3):265–74. doi: 10.1016/S2213-8587(15)00380-0
39. Gohil A, Eugster EA. Delayed and precocious puberty: genetic underpinnings and treatments. Endocrinol Metab Clin North Am. (2020) 49(4):741–57. doi: 10.1016/j.ecl.2020.08.002
40. Van Lierde A, Menni F, Bedeschi MF, Natacci F, Guez S, Vizziello P, et al. Healthcare transition in patients with rare genetic disorders with and without developmental disability: neurofibromatosis 1 and williams–beuren syndrome. Am J Med Genet A. (2013) 161(7):1666–74. doi: 10.1002/ajmg.a.35982
41. Rare diseases—not as rare as you think: Sydney Child Health Network (2021). Available at: https://www.schn.health.nsw.gov.au/news/articles/2021/02/rare-diseases-not-as-rare-as-you-think
42. Anderson M, Elliott EJ, Zurynski YA. Australian families living with rare disease: experiences of diagnosis, health services use and needs for psychosocial support. Orphanet J Rare Dis. (2013) 8:22. doi: 10.1186/1750-1172-8-22
43. Rosenfeld LE, LeBlanc K, Nagy A, Ego BK, Network UD, McCray AT. Participation in a national diagnostic research study: assessing the patient experience. Orphanet J Rare Dis. (2023) 18(1):73. doi: 10.1186/s13023-023-02695-5
44. Walker CE, Mahede T, Davis G, Miller LJ, Girschik J, Brameld K, et al. The collective impact of rare diseases in Western Australia: an estimate using a population-based cohort. Genet Med. (2017) 19(5):546–52. doi: 10.1038/gim.2016.143
45. National Clinical Coding Standards ICD-10 5th Edition. NHS Digital. Accurate data for quality information (2022). Available at: http://systems.digital.nhs.uk/data/clinicalcoding
46. Alonso V, Santos JV, Pinto M, Ferreira J, Lema I, Lopes F, et al. Health records as the basis of clinical coding: is the quality adequate? A qualitative study of medical coders’ perceptions. Health Inf Manag J. (2020) 49(1):28–37. doi: 10.1177/1833358319826351
Keywords: rare disease, ICD-11, diagnostic odyssey, diagnostic coding, red flags, key timepoints
Citation: Baxter MF, Hansen M, Gration D, Groza T and Baynam G (2023) Surfacing undiagnosed disease: consideration, counting and coding. Front. Pediatr. 11:1283880. doi: 10.3389/fped.2023.1283880
Received: 1 September 2023; Accepted: 12 October 2023;
Published: 25 October 2023.
Edited by:
Arnold Munnich, Hôpital Necker-Enfants Malades, FranceReviewed by:
Zachary McPherson, Children’s Hospital at Westmead, AustraliaAmy Brower, American College of Medical Genetics and Genomics (ACMG), United States
© 2023 Baxter, Hansen, Gration, Groza and Baynam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan F. Baxter bWYuYmF4dGVyQGJpZ3BvbmQuY29t