 Ying Dai
Ying Dai Yan Wang
Yan Wang Youfei Fan
Youfei Fan Bo Han
Bo Han- Department of Pediatrics, Shandong Province Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, China
Dilated cardiomyopathy (DCM) in children is a severe myocardial disease characterized by enlargement of the left ventricle or both ventricles with impaired contractile function. DCM can cause adverse consequences such as heart failure, sudden death, thromboembolism, and arrhythmias. This article reviews the latest advances in genotype and phenotype research in pediatric DCM. With the development of gene sequencing technologies, considerable progress has been made in genetic research on DCM. Research has shown that DCM exhibits notable genetic heterogeneity, with over 100 DCM-related genes identified to date, primarily involving functions such as calcium handling, the cytoskeleton, and ion channels. As human genomic variations are linked to phenotypes, DCM phenotypes are influenced by numerous genetic variations across the entire genome. Children with DCM display high genetic heterogeneity and are characterized by early onset, rapid disease progression, and poor prognosis. The genetic architecture of pediatric DCM markedly differs from that of adult DCM, necessitating analyses through clinical phenotyping, familial cosegregation studies, and functional validation. Clarifying the genotype-phenotype relationship can improve diagnostic accuracy, enhance prognosis, and guide follow-up treatment for genotype-positive and phenotype-negative patients identified through genetic testing, providing new insights for precision medicine. Future research should further explore novel pathogenic genes and mutations and strengthen genotype-phenotype correlation analyses to facilitate precise diagnosis and treatment of DCM in children.
Introduction
Dilated cardiomyopathy (DCM) in children is a myocardial disease characterized by enlargement of the left ventricle or both ventricles, accompanied by systolic dysfunction (1). DCM is one of the most common causes of heart failure in children, accounting for approximately 30%–50% of cases (2). Pediatric DCM progresses rapidly and has a poor prognosis, with a 5-year survival rate of only 50%–60% and with most deaths resulting from progressive heart failure and its complications (3). The etiology of pediatric DCM is diverse and includes genetic, infectious, metabolic, toxic, and idiopathic causes. Approximately 30%–50% of pediatric patients with DCM have pathogenic gene mutations, primarily involving genes encoding sarcomere proteins, ion channels, Z-disc proteins, and nuclear envelope proteins (4). Currently, according to the Human Gene Mutation Database and Online Mendelian Inheritance in Man database,over 100 genes have been identified to be associated with monogenic hereditary DCM among the pathogenic genes in hereditary/familial DCM. These include TTN, LMNA, DSP, PLN, FLNC, RBM20, SCN5A, MYH7, MYBPC3, and others (5, 6). However, after in-depth data analysis, the associations of some of these genes with DCM were not supported. In recent years, with advancements in research on additional genes and genotype-phenotype correlations, phenotypic overlap and dynamic changes in DCM phenotypes have been discovered. Genetic heterogeneity makes it challenging to accurately classify DCM and guide clinical decision-making, thus posing challenges for traditional DCM diagnostic methods (7, 8). There are both similarities and significant differences between DCM and hypertrophic CM (HCM). There is notable phenotypic overlap between arrhythmogenic CM (ACM) and DCM, with specific genes such as LMNA, SCN5A, FLNC, RBM20, PLN, DSP, and DES potentially causing ACM (9). Clarifying genotype-phenotype relationships can enable rapid and precise interpretation of candidate variants in pediatric DCM and greater understanding of its specific variant spectrum, which is crucial for guiding clinical treatment strategies and developing new therapeutic approaches (10). Although understanding of the genotypes and phenotypes of pediatric DCM continues to deepen, several issues remain unresolved. This article reviews the latest advances in genotype and phenotype research in pediatric DCM and discusses future research directions with the aim of providing new insights for precision medicine in pediatric DCM.
Genetic architecture of pediatric DCM
When comparing the genetic composition of DCM in children and adults, although the guidelines recommend genetic testing, there are significant differences in actual clinical practice (Figure 1). In DCM, the presence of variants of uncertain significance (VUS) alone [odds ratio [OR] 4.0, 95% confidence interval [CI] 1.9–8.3] and in combination with pathogenic variants (OR 5.2, 95% CI: 1.7–15.9) is associated with major adverse cardiac events (11). Studies have shown that children with cardiomyopathy have a higher incidence of neuromuscular diseases, congenital metabolic defects, mitochondrial diseases, and malformation syndromes (12). However, due to the lack of large-scale clinical studies, understanding of the genetic composition of pediatric DCM is limited, and the existence of child-specific DCM pathogenic genes cannot be ruled out. Pediatric DCM exhibits high genetic heterogeneity with a higher number of rare variants, most of which are VUS. This necessitates analyses using clinical phenotyping, familial cosegregation studies, and functional validation (13). A Finnish single-center study conducted a 20-year follow-up of 66 children with DCM and found that 39% of the cases had at least one disease-related gene mutation. These diseases included metabolic disorders, sarcomere-related diseases, and various other syndromes. The predominant mutations were missense variants (91.5%) and loss-of-function mutations (5.4%). This Finnish single-center study reported that NRAP is a new cause of severe DCM and other atypical clinical phenotypes in children. PPA2 deficiency involving sudden death may lead to DCM, and junctophilin-2 variants may cause recessive DCM with childhood onset (12).
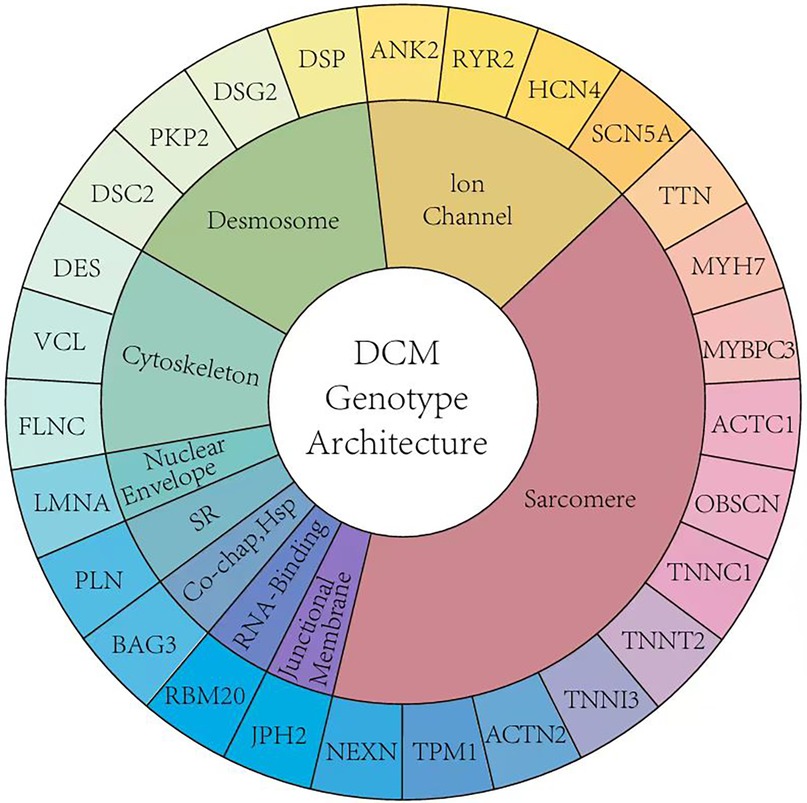
Figure 1. The genetic architecture of pediatric DCM.
Mutations in the sarcomere-related genes (TTN, MYH7, and MYBPC3) are common in DCM. Studies have found that approximately 10%–13% of patients aged 2–18 years have TTN variants. In DCM cohorts, mutations in genes such as BAG3, CRYAB, and DES can be observed, with some children showing evidence of skeletal muscle involvement (13, 14). These data indicate genotype-phenotype correlations, emphasizing the importance of genotype-directed therapy in pediatric DCM. However, Canadian literature suggests that the incidence of TTN-truncating variants (TTNtv) is higher in adult DCM patients than in pediatric DCM patients. Compared to adults, pediatric DCM patients have a lower variant burden of channelopathy genes (15).
In a North American multi-center study, genomic testing was conducted on 279 pediatric and adolescent patients with DCM (<18 years old) from 14 medical institutions in the United States and Canada. In this pediatric DCM cohort, no pathogenic/likely pathogenic gene had a frequency exceeding 4%. The frequencies of pathogenic/likely pathogenic variants of MYH7, MYBPC3, TNNT2, and RBM20 were similar to those in adult DCM. However, the frequencies of variants in TTN and LMNA were lower than those in adult patients with DCM (13).
A retrospective analysis was conducted on 299 pediatric patients with DCM (aged < 18 years) who received treatment at the Children's Hospital of Chicago in the United States between 2007 and 2016. The study found that the genetic architecture differed from that of adults; 37% of pediatric patients with DCM had pathogenic/likely pathogenic gene mutations in sarcomere-related genes. Mutations in LMNA, RBM20, and PLN, which are common in adult DCM, were found to have a lower frequency in pediatric DCM. Additionally, the study emphasized the age-dependent risk associated with TTNtv), which were associated with a later age of onset (average age at initial diagnosis: 9.7 years). TTNtv carriers had a poor prognosis, 60% of patients carrying TTNtv ultimately die or undergo heart transplantation (14).
Genotype insights in relation to pediatric DCM
Pediatric DCM is characterized by enlargement of the left or both ventricles and reduced contractile function (16). TTN is located on chromosome 2q31 and contains 364 exons. Extensive mRNA splicing can produce various titin isoforms, with N2B and N2BA being the main cardiac-related isoforms (17). TTN encodes titin, which is the largest protein in the muscles and is crucial for the structure and function of cardiomyocytes (18). TTN mutations have the highest detection rate in pediatric patients with DCM and are an important genetic factor in pediatric DCM (19). TTN-related DCM usually follows an autosomal-dominant inheritance pattern; however, complex inheritance patterns also exist (20), with TTNtv being the most common (21). TTNtv mutations occur predominantly in the A-band region, with smaller proportions occurring in the I-band, Z-disc, and M-line regions. Children with TTNtv mutations in the A-band or M-line regions have a poor prognosis (17). DCM caused by TTN mutations exhibits significant phenotypic variability. Even within the same family, there are considerable differences in disease severity and age of onset (22). In adult patients with DCM, TTNtv are not uniformly distributed but rather cluster in the A-band region (p = 3.4 × 10−4p = 3.4 × 10−4) (p = 3.5 × 10−3−3p = 3.5 × 10−3). However, this clustering is not observed in pediatric patients (15).
MYH7 encodes the myosin protein, which consists of 1,935 amino acids. Myosin is primarily expressed in myocardium and type 1 skeletal muscle fibers and is a important component of the human ventricular system. It plays a crucial role in supplying energy to cardiomyocytes and in maintaining intracellular and extracellular Ca2+ concentrations (23). MYH7 mutations can impair myocardial contractile function, resulting in DCM (24). In pediatric patients, pathogenic variants in the MYH7 gene are not evenly distributed but mainly cluster in the myosin head and neck regions (Kolmogorov–Smirnov goodness of fit test: p = 8.4 × 10−4, p = 8.4 × 10−4). In adults, variants are primarily concentrated in the myosin head and neck regions, but also appear in the tail region, similar to the reference population (15).
MYBPC3 encodes myosin-binding protein C and is the second most common pathogenic gene in DCM after MYH7. It plays a crucial role in maintaining myocardial sarcomere structure and regulating myocardial contraction (25). Mutations in MYBPC3 can lead to the abnormal binding of protein C, resulting in myocardial contraction and relaxation dysfunction (26). TNNT2 and TNNI3 encode troponins T and I, respectively. These genes encode key proteins involved in myocardial contractions. Mutations in these genes typically lead to impaired myocardial contractile function, thereby causing DCM (27, 28). TPM1 encodes the cardiac-specific α-chain of tropomyosin. Mutations in this gene affect the assembly and function of myofilaments, leading to decreased myocardial contractility (29). Variants in MYBPC3 cluster mainly in the C5, C7, and C10 regions, with no significant differences between children and adults (15).
LMNA is located on chromosome 1q21.1–21.3, and contains 12 exons. It encodes type A nuclear lamins, primarily lamins A/C. Vertebrate nuclear lamins include A and B forms. Human lamin A/C produces three type A isoforms through alternative splicing (30, 31). Mutations in LMNA can lead to various cardiac diseases, including DCM, arrhythmias, and conduction system diseases (32). LMNA-related DCM typically has an earlier age of onset, faster disease progression, and poorer prognosis. DCM caused by LMNA mutations is often accompanied by arrhythmias and conduction system abnormalities, which increase the risk of sudden death in patients (33). Currently, the treatment for LMNA-related DCM includes standard heart failure therapy and implantable cardioverter-defibrillator (ICD) implantation to prevent sudden death. LMNA is the only gene in the current guidelines with a Class I recommendation for ICD implantation (34).
FLNC encodes the actin-binding protein filamin C, which plays important structural and signaling roles in muscle cells (35). Mutations in FLNC can lead to abnormal myofilament structures and muscle cell dysfunction, resulting in cardiomyopathy (36). Studies have shown that FLNC mutations are significantly associated with pediatric DCM. Compared to the average clinical course of DCM, FLNC-related DCM is more malignant and characterized by a high risk of ventricular arrhythmias, myocardial fibrosis, and sudden cardiac death. The average age of onset is 39.7 ± 14.5 years, and FLNC-related DCM is more commonly found in adults (37). However, a pediatric study discovered biallelic FLNC variants in a family with previously unreported pediatric DCM. Biallelic FLNC variants can lead to congenital DCM (38). Understanding the role of FLNC may provide new insights for the development of targeted therapeutic strategies (39).
SCN5A encodes the α-subunit of the cardiac sodium channel, which plays a crucial role in determining the action potential of cardiomyocytes (40). Mutations in the SCN5A gene have been confirmed to be associated with DCM. These mutations can lead to abnormal sodium channel function, thereby affecting myocardial contraction (41). Pediatric patients with DCM carrying SCN5A mutations may present with symptoms such as ventricular enlargement, reduced contractile function, and arrhythmias (42, 43). Certain SCN5A mutations may also be associated with more severe disease progression and poorer prognosis, emphasizing the importance of early genetic testing (44).
DSP encode desmoplakin, a protein that plays a crucial role in cell-cell junctions. DSP-related DCM typically follows an autosomal dominant inheritance pattern, although autosomal recessive inheritance has also been reported (45, 46). Cardiomyopathy caused by DSP mutations is usually associated with arrhythmogenic right ventricular cardiomyopathy (ARVC) or DCM. DCM caused by DSP mutations may be accompanied by skin and hair abnormalities, such as curly hair and palmoplantar keratoderma (47). Although DSP mutation-related DCM can occur at any age, cases of childhood-onset have been increasingly reported (48).
RBM20 is located on chromosome 10 and contains 14 exons. It encodes a protein of 1,227 amino acids that is primarily expressed in striated muscle, with the highest expression in cardiac muscle and almost no expression in non-muscle tissues. DCM-related RBM20 gene mutations often occur in the arginine/serine-rich (RS) region, leading to loss of function in the expressed protein. Mutations in other regions occur less frequently than those in the RS region but can also reduce RBM20 gene-splicing activity, affecting the expression of regulated genes and leading to DCM (49, 50). Pediatric patients with DCM carrying RBM20 mutations may present with early onset heart failure and arrhythmias, particularly, an increased risk of ventricular arrhythmias (51–53).
Research has shown that most genes primarily cause DCM through autosomal-dominant inheritance patterns. Additionally, there have been a few reported cases of DCM caused by autosomal recessive, X-linked, or mitochondrial inheritance patterns (54).
Various methods have been applied to further explore new pathogenic genes and mutations. Currently, the main gene-testing strategies include: third-generation sequencing (NGS), genome-wide association studies (GWAS), whole-exome sequencing (WES), and whole-genome sequencing (WGS) (55, 56).
NGS is a targeted sequencing method that resolves genomic structural variations and complex regions. Customized DCM gene panels can cover 25–50 known DCM genes, including MYH7, MYBPC3, TTN, and LMNA (57). However, NGS cannot cover all known DCM genes and cannot detect new genes. GWAS can discover new gene loci associated with the disease by analyzing genotype and phenotype data from a large number of samples, but may miss the effects of rare variants (58). WES and WGS can rapidly identify rare pathogenic variants (59). Among these, WES is currently the preferred genetic diagnostic method for hereditary DCM. In addition, traditional methods, such as family linkage analysis, also play an important role in identifying pathogenic genes in familial DCM.
Phenotypic diversity
Pediatric DCM exhibits significant genetic heterogeneity, and its clinical manifestations also show notable heterogeneity. The main phenotypic feature of DCM is myocardial injury, with the most prominent characteristics being significant dilation of the left ventricle or both ventricles and progressive decline in cardiac function (60). Some children experience symptoms of heart failure, including shortness of breath, fatigue, and reduced exercise tolerance (61). Additionally, some children may present with various types of common arrhythmias such as atrial fibrillation and ventricular tachycardia, which can potentially exacerbate symptoms or lead to sudden events (62).
In addition to cardiac manifestations, DCM can lead to multisystem involvement. When the nervous system is affected, it can cause motor disorders and cognitive decline (62). Studies have also found that some patients may exhibit skeletal muscle involvement such as muscle fatigue, weakness, and elevated muscle enzyme levels, providing an important perspective for understanding the role of the muscle-heart axis in hereditary diseases (63). Furthermore, DCM may indirectly affect the endocrine system, particularly through fluid and electrolyte imbalances caused by heart failure (64).
The phenotypic diversity of DCM is reflected in its pathogenic mechanisms, clinical manifestations, genetic background, and treatment responses, making it a complex and variable clinical challenge. Complex regulatory mechanisms underlie this process, such as the influence of genetic modifiers and environmental factors on the phenotype (65). A deeper understanding of the molecular mechanisms underlying the phenotypic heterogeneity of DCM will facilitate the development of targeted therapeutic approaches.
Genotype-phenotype correlation studies
After reviewing a 76-year timeline in relation to studies on DCM-related gene mutations, the discovery of new DCM-related genes, gene-specific DCM outcomes, and insights into variant-environment interactions was found to have significantly advanced this field. The expansion of genomic phenotype analysis and integration of a series of prognostic factors into the variant environment are crucial (65). In Table 1, we summarized genotype and related phenotype in pediatric dilated cardiomyopathy.
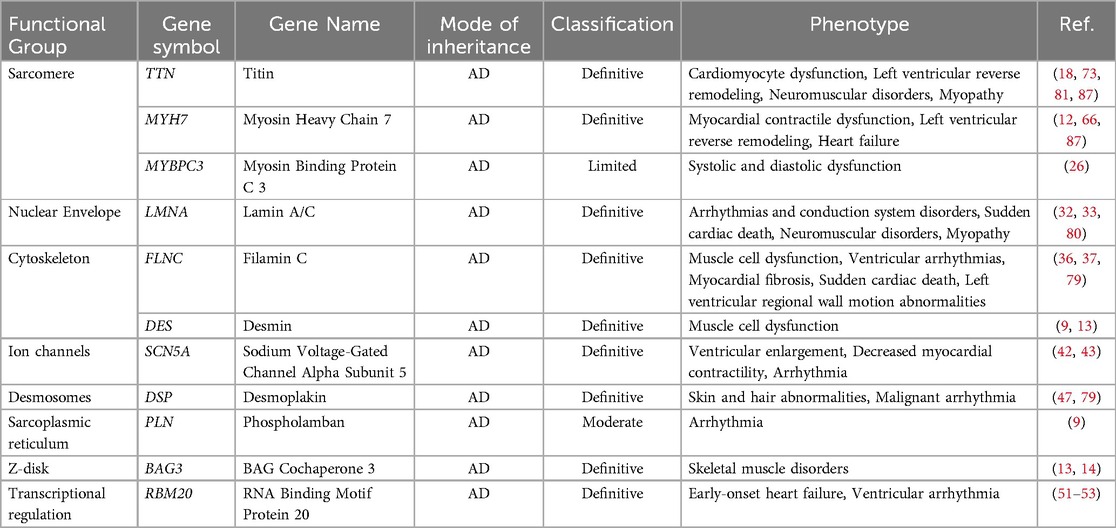
Table 1. Genotype and related phenotype in pediatric dilated cardiomyopathy.
Different genotypes have different prognoses
Mutations in different genes can lead to variations in disease progression and prognosis. In pediatric DCM, MYH7-related DCM is characterized by early onset, high phenotypic expressivity, a low incidence of left ventricular reverse remodeling, and frequent progression to end-stage heart failure (66). Truncating mutations in MYBPC3 result in loss of protein function,thereby accelerating the progression of cardiomyopathy, which may be significantly associated with disease severity and early onset (67). Patients with DCM carrying FLNC mutations may exhibit symptoms such as early onset heart failure and arrhythmia (68, 69). TTNtv mutations exhibit incomplete and age-dependent penetrance, with variable prognosis in affected children, reaching 100% penetrance by the age of 70 years; the same mutations have also been detected in unaffected relatives (70). Although TTNtv are less frequently found in pediatric cases, studies have shown a similar prevalence in adolescents and adults, suggesting that multiple clinical and genetic risk factors rather than a single TTNtv are required for its manifestation (71). Hasselberg et al. (33) found that LMNA mutation-related DCM is a highly pathogenic and age-dependent malignant disease, with affected children prone to arrhythmias and sudden death. These patients have a high incidence of adverse cardiac events and rapid disease progression, with some developing arrhythmias before left ventricular systolic dysfunction is observed. This indicates that genotype information can predict the clinical outcomes in affected children, aiding in the development of individualized treatment strategies.
Heterogeneity exists in the same genotype
There may be significant differences in the phenotypic expression even among individuals carrying the same gene mutation. This phenotypic heterogeneity can be caused by various factors including environmental factors, epigenetic regulation, and genetic background (72). For example, Tharp et al. (17) found that the phenotype and severity of DCM are related to the location of TTNtv, with TTNtv mutations frequently occurring in the A-band region. In terms of sex, males carrying TTNtv were found to be associated with more severe clinical phenotypes and adverse clinical outcomes than females. Furthermore, patients with variants in the exon 11 region (c.2721–2760) of RBM20 had a higher probability of developing DCM than those with variants in the exon 9 region (c.1881–1920) (51). Male carriers of RBM20 variants were more likely to progress to end-stage heart failure than female carriers (73). However, sex differences were not significant in pediatric and adolescent cohorts. In addition, chemotherapeutic drugs can induce or exacerbate TTNtv mutation-related DCM in children. Nonsense variants of TTN are more common in patients with DCM, whereas frameshift termination and missense variants are more common in patients with neuromuscular and myocardial skeletal disorders (73).
Genotype and electrocardiogram (ECG) abnormalities
Numerous children with DCM exhibit ECG abnormalities, such as T-wave changes, left bundle branch block, atrioventricular conduction abnormalities, and supraventricular arrhythmias (74). Children with ECG abnormalities have a higher mortality rate. Certain genetic causes of DCM result in malignant arrhythmic phenotypes. Autonomic nervous system imbalance and impaired myocardial repolarization homogeneity are two major underlying mechanisms of arrhythmia in patients with DCM (75). Laminopathies are often associated with prolonged PR intervals on ECG, which are indicators of cardiac conduction disorders. DCM associated with DSP mutations typically has a poor prognosis and may lead to malignant arrhythmias.
Patients with DCM carrying variants of LMNA, PLN, FLNC, and RBM20 have an increased risk of developing arrhythmias. Younger patients with LMNA-related DCM and RBM20-related DCM show increased expression levels of arrhythmia-related cardiomyopathy. LMNA-related DCM exhibits a highly penetrant arrhythmic phenotype accompanied by multiple muscle involvement. Taylor et al. (76) reported that patients with LMNA-related DCM simultaneously experienced muscle involvement and arrhythmias, particularly chronic arrhythmias. LMNA variants account for up to 33% of DCM cases with atrioventricular conduction block (77). Similarly, patients carrying the PLN R14del founder variant typically develop ventricular arrhythmia and end-stage heart failure at a young age (78). Studies have found that left ventricular regional wall motion abnormalities are more common in DSP/FLNC genotypes, and these genotypes cause extensive regional left ventricular damage (79).
Genotyping and neuromuscular diseases
The dilated phenotype is the most common cardiomyopathic manifestation in neuromuscular diseases (1). In pediatric neuromuscular diseases (NMD), particularly in dystrophies, abnormalities in the splicing of the CUG binding protein (CUG-BP) and muscle blind-like protein (MBNL) interfere with cellular signaling, resulting in toxic effects on muscle metabolism and RNA processing. This may cause changes in the cardiac structure and function, potentially resulting in the early onset of dilated cardiomyopathy (DCM) (80). Children with DCM who carry specific genotypes are prone to developing concurrent skeletal myopathies. Relevant neuromuscular symptoms in patients or relatives are important criteria for genetic analysis, because variants of LMNA are associated with DCM related to neuromuscular diseases (limb-girdle muscular dystrophy) (81). Emery-Dreifuss muscular dystrophy (EDMD) is closely related to dilated cardiomyopathy (DCM). In a study of 53 patients, mutations in LMNA were identified as a major cause, with 12 patients exhibiting significant cardiac involvement and 41 exhibiting muscle weakness. Therefore, screening for LMNA mutations is crucial in familial and sporadic cases associated with EDMD and DCM (82).
The impact of TTNtv extends beyond the heart, with 46%–57% of children with DCM and congenital heart disease exhibiting TTN-related neuromuscular diseases of varying severity (83). Sofie et al. (84) found that TTNtv carriers had a higher skeletal muscle fat content than patients with non-TTNtv hereditary DCM. Muscle biopsies in 62% of TTNtv carriers showed characteristics of skeletal muscle involvement, manifesting as well-aligned Z-lines and T-tubules, but with uneven and discontinuous M-lines, accompanied by excessive glycogen deposition. This was surrounded by autophagosomes, lysosomes, and mitochondrial autophagy with abnormal mitochondria, suggesting that patients with DCM with monoallelic TTNtv simultaneously have mild skeletal myopathy. Phenotype overlap may also occur in genes such as DES and LMNA, where DCM may be accompanied by skeletal myopathy (85).
Treatment strategies and future research directions
Over the past few decades, while significant progress has been made in the treatment of DCM, many challenges remain. Transcription factors play a crucial role in dilated cardiomyopathy (DCM),may represent novel therapeutic targets.For instance, GATA4 and MEF2C are involved in the regulation of cardiac remodeling, and changes in their expression may lead to alterations in myocardial structure and function (86). Recent studies have also identified a correlation between mutations in the TBX5 gene and an increased risk of cardiac developmental defects and DCM. Additionally, TBX5 is associated with certain neuromuscular diseases, which often involve cardiac complications, including DCM (87, 88). Cardiovascular magnetic resonance can help identify the subclinical forms of the disease, facilitating a deeper understanding of the diverse phenotypes of DCM (89, 90) and the genes affecting pediatric DCM, with gene therapy offering a potential avenue for treating pediatric DCM (91). Cardiac function can be improved by modifying or replacing pathogenic genes in the cardiomyocytes to restore normal gene function. However, gene therapy is still in its early stages, and more clinical trials are necessary to verify the efficacy and safety of these therapeutic strategies (5).
Personalized medicine is becoming increasingly important in the treatment of pediatric DCM. Studies have indicated that patients with different phenotypes respond differently to specific treatments. For example, Verdonschot et al. (92) found differences in prognoses among different phenotype groups in DCM. Among the four phenotypic groups identified in their study, the arrhythmic group had the poorest prognosis and was highly susceptible to life-threatening malignant arrhythmias. After 12 months of regular drug therapy, 52.5% of patients experienced left ventricular reverse remodeling (LVRR), with the severe systolic dysfunction group showing the highest LVRR rate. Therefore, developing individualized treatment plans based on phenotypes and comorbidities of different patients can significantly improve treatment efficacy and patient quality of life.
Regarding future research directions, efforts should be made to expand sample sizes and establish more extensive genetic and phenotypic databases to achieve comprehensive assessments (93). At the same time, long-term follow-up of patients with DCM is crucial. Simultaneously,long-term follow-up allows for the evaluation of the effect of how different genotypes on disease progression and enables timely adjustments to treatment plans, thereby improving prognosis (94).
Conclusion
Pediatric DCM exhibits significant genetic heterogeneity, with variations in the expression of multiple genes being closely associated with its development in childhood (2). These genes affect myocardial function through different pathways and can lead to diverse clinical phenotypes (65). This variability may result from the combined effects of gene-environment interactions, epigenetic regulation, and other unknown genetic factors (Figure 2). Personalized treatment plans targeting specific genotypes are promising for improving the prognosis and quality of life of affected children. The development of technologies such as GWAS and NGS has considerably advanced understanding of the action mechanisms of pediatric DCM (95). Gene therapy has shown promising results in animal models (96). However, their safety and efficacy require further validation through clinical trials. In the future, large-sample multicenter prospective cohort studies will help deepen the understanding of the genetic mechanisms underlying pediatric DCM.
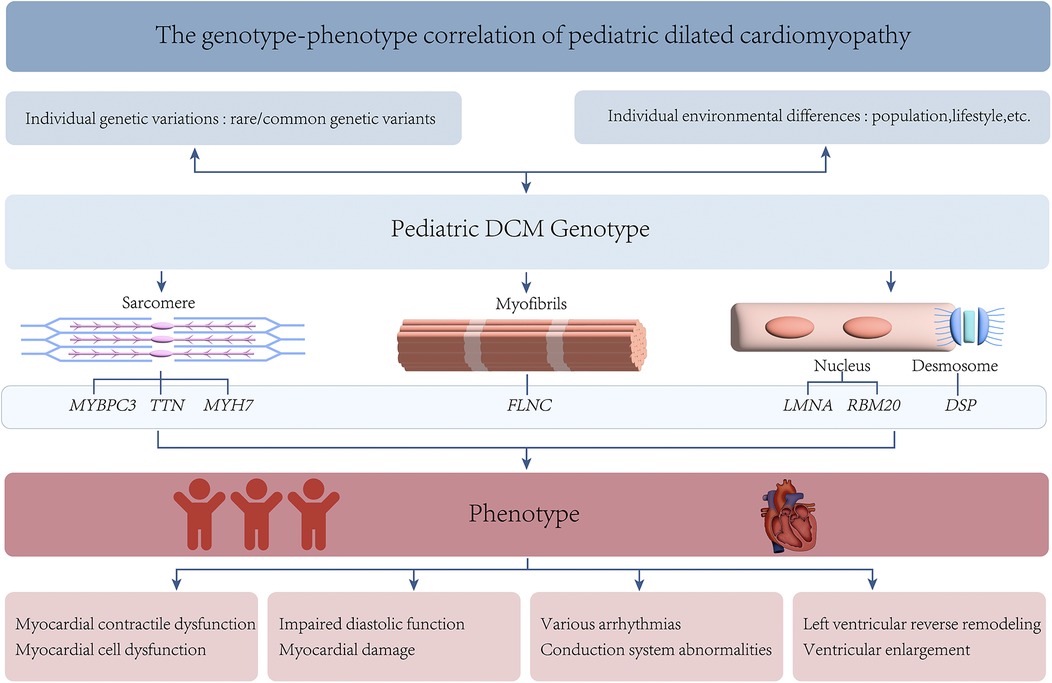
Figure 2. The genotype and phenotype of pediatric DCM.
In conclusion, research on the genotypes and phenotypes of pediatric DCM provides crucial perspectives and information for understanding its pathological mechanisms, thereby offering new hope for children with DCM. However, major challenges remain. We anticipate that as research progresses, these challenges can be addressed with the development of new therapeutic approaches for this complex disease.
Author contributions
YD: Conceptualization, Data curation, Methodology, Writing – original draft. YW: Data curation, Investigation, Writing – review & editing. YF: Conceptualization, Supervision, Writing – review & editing. BH: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China General Project (81873498), Mount Taishan Scholar Distinguished Expert (No. ts201511099), Shandong Provincial Natural Science Foundation General Project (ZR2023MH052, ZR2023MH298) and Jinan Science and Technology Plan (202134015).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lipshultz SE, Law YM, Asante-Korang A, Austin ED, Dipchand AI, Everitt MD, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American heart association. Circulation. (2019) 140(1):e9–e68. doi: 10.1161/CIR.0000000000000682
2. Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al. Pediatric cardiomyopathies. Circ Res. (2017) 121(7):855–73. doi: 10.1161/CIRCRESAHA.116.309386
3. Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation. (2021) 144(1):7–19. doi: 10.1161/CIRCULATIONAHA.120.053033
4. Harakalova M, Kummeling G, Sammani A, Linschoten M, Baas AF, van der Smagt J, et al. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific genes. Eur J Heart Fail. (2015) 17(5):484–93. doi: 10.1002/ejhf.255
5. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. (2017) 121(7):731–48. doi: 10.1161/CIRCRESAHA.116.309396
6. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. (2015) 36(18):1123–35a. doi: 10.1093/eurheartj/ehu301
7. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. (2019) 16(11):e301–e72. doi: 10.1016/j.hrthm.2019.05.007
8. Wang Y, Dobreva G. Epigenetics in LMNA-related cardiomyopathy. Cells. (2023) 12(5):783. doi: 10.3390/cells12050783
9. Eldemire R, Mestroni L, Taylor MRG. Genetics of dilated cardiomyopathy. Annu Rev Med. (2024) 75:417–26. doi: 10.1146/annurev-med-052422-020535
10. Morales A, Kinnamon DD, Jordan E, Platt J, Vatta M, Dorschner MO, et al. Variant interpretation for dilated cardiomyopathy: refinement of the American college of medical genetics and genomics/ClinGen guidelines for the DCM precision medicine study. Circ Genom Precis Med. (2020) 13(2):e002480. doi: 10.1161/CIRCGEN.119.002480
11. Burstein DS, Gaynor JW, Griffis H, Ritter A, Connor MJO, Rossano JW, et al. Genetic variant burden and adverse outcomes in pediatric cardiomyopathy. Pediatr Res. (2021) 89(6):1470–6. doi: 10.1038/s41390-020-1101-5
12. Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, et al. Genetic basis of severe childhood-onset cardiomyopathies. J Am Coll Cardiol. (2018) 72(19):2324–38. doi: 10.1016/j.jacc.2018.08.2171
13. Ware SM, Bhatnagar S, Dexheimer PJ, Wilkinson JD, Sridhar A, Fan X, et al. The genetic architecture of pediatric cardiomyopathy. Am J Hum Genet. (2022) 109(2):282–98. doi: 10.1016/j.ajhg.2021.12.006
14. Khan RS, Pahl E, Dellefave-Castillo L, Rychlik K, Ing A, Yap KL, et al. Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J Am Heart Assoc. (2022) 11(1):e022854. doi: 10.1161/JAHA.121.022854
15. Akinrinade O, Lesurf R, Genomics England Research C, Lougheed J, Mondal T, Smythe J, et al. Age and sex differences in the genetics of cardiomyopathy. J Cardiovasc Transl Res. (2023) 16(6):1287–302. doi: 10.1007/s12265-023-10411-8
16. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. (2006) 296(15):1867–76. doi: 10.1001/jama.296.15.1867
17. Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L. The giant protein Titin’s role in cardiomyopathy: genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front Physiol. (2019) 10:1436. doi: 10.3389/fphys.2019.01436
18. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. (2012) 366(7):619–28. doi: 10.1056/NEJMoa1110186
19. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. (2014) 16(8):601–8. doi: 10.1038/gim.2013.204
20. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. (2015) 7(270):270ra6. doi: 10.1126/scitranslmed.3010134
21. Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. (2017) 49(1):46–53. doi: 10.1038/ng.3719
22. Tayal U, Newsome S, Buchan R, Whiffin N, Halliday B, Lota A, et al. Phenotype and clinical outcomes of titin cardiomyopathy. J Am Coll Cardiol. (2017) 70(18):2264–74. doi: 10.1016/j.jacc.2017.08.063
23. Gao Y, Peng L, Zhao C. MYH7 In cardiomyopathy and skeletal muscle myopathy. Mol Cell Biochem. (2024) 479(2):393–417. doi: 10.1007/s11010-023-04735-x
24. Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. (2008) 358(18):1899–908. doi: 10.1056/NEJMoa075463
25. Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J Muscle Res Cell Motil. (2012) 33(1):75–80. doi: 10.1007/s10974-011-9268-3
26. Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. (2015) 573(2):188–97. doi: 10.1016/j.gene.2015.09.008
27. Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. (1997) 16(4):379–82. doi: 10.1038/ng0897-379
28. Sorrentino U, Gabbiato I, Canciani C, Calosci D, Rigon C, Zuccarello D, et al. Homozygous TNNI3 mutations and severe early onset dilated cardiomyopathy: patient report and review of the literature. Genes. (2023) 14(3):748. doi: 10.3390/genes14030748
29. Lynn ML, Tal Grinspan L, Holeman TA, Jimenez J, Strom J, Tardiff JC. The structural basis of alpha-tropomyosin linked (Asp230Asn) familial dilated cardiomyopathy. J Mol Cell Cardiol. (2017) 108:127–37. doi: 10.1016/j.yjmcc.2017.06.001
30. Captur G, Arbustini E, Bonne G, Syrris P, Mills K, Wahbi K, et al. Lamin and the heart. Heart. (2018) 104(6):468–79. doi: 10.1136/heartjnl-2017-312338
31. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. (1999) 341(23):1715–24. doi: 10.1056/NEJM199912023412302
32. Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. (2008) 52(15):1250–60. doi: 10.1016/j.jacc.2008.06.044
33. Hasselberg NE, Haland TF, Saberniak J, Brekke PH, Berge KE, Leren TP, et al. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J. (2018) 39(10):853–60. doi: 10.1093/eurheartj/ehx596
34. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. (2012) 59(5):493–500. doi: 10.1016/j.jacc.2011.08.078
35. Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. (2016) 68(22):2440–51. doi: 10.1016/j.jacc.2016.09.927
36. Ader F, De Groote P, Reant P, Rooryck-Thambo C, Dupin-Deguine D, Rambaud C, et al. FLNC Pathogenic variants in patients with cardiomyopathies: prevalence and genotype-phenotype correlations. Clin Genet. (2019) 96(4):317–29. doi: 10.1111/cge.13594
37. Verdonschot JAJ, Vanhoutte EK, Claes GRF, Helderman-van den Enden A, Hoeijmakers JGJ, Hellebrekers D, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat. (2020) 41(6):1091–111. doi: 10.1002/humu.24004
38. Reinstein E, Gutierrez-Fernandez A, Tzur S, Bormans C, Marcu S, Tayeb-Fligelman E, et al. Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. Eur J Hum Genet. (2016) 24(12):1792–6. doi: 10.1038/ejhg.2016.110
39. Baban A, Alesi V, Magliozzi M, Parlapiano G, Genovese S, Cicenia M, et al. Cardiovascular involvement in pediatric FLNC variants: a case series of fourteen patients. J Cardiovasc Dev Dis. (2022) 9(10):332. doi: 10.3390/jcdd9100332
40. Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol. (2013) 591(17):4099–116. doi: 10.1113/jphysiol.2013.256461
41. Wauchop M, Rafatian N, Zhao Y, Chen W, Gagliardi M, Masse S, et al. Maturation of iPSC-derived cardiomyocytes in a heart-on-a-chip device enables modeling of dilated cardiomyopathy caused by R222Q-SCN5A mutation. Biomaterials. (2023) 301:122255. doi: 10.1016/j.biomaterials.2023.122255
42. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. (2006) 113(14):1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287
43. Remme CA. SCN5A Channelopathy: arrhythmia, cardiomyopathy, epilepsy and beyond. Philos Trans R Soc Lond B Biol Sci. (2023) 378(1879):20220164. doi: 10.1098/rstb.2022.0164
44. Proost VM, van den Berg MP, Remme CA, Wilde AAM. SCN5A-1795insD Founder variant: a unique dutch experience spanning 7 decades. Neth Heart J. (2023) 31(7-8):263–71. doi: 10.1007/s12471-023-01799-8
45. Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. (2020) 141(23):1872–84. doi: 10.1161/CIRCULATIONAHA.119.044934
46. Castelletti S, Vischer AS, Syrris P, Crotti L, Spazzolini C, Ghidoni A, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: genotype-phenotype correlation. Int J Cardiol. (2017) 249:268–73. doi: 10.1016/j.ijcard.2017.05.018
47. Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. (2000) 9(18):2761–6. doi: 10.1093/hmg/9.18.2761
48. Seidel F, Holtgrewe M, Al-Wakeel-Marquard N, Opgen-Rhein B, Dartsch J, Herbst C, et al. Pathogenic variants associated with dilated cardiomyopathy predict outcome in pediatric myocarditis. Circ Genom Precis Med. (2021) 14(4):e003250. doi: 10.1161/CIRCGEN.120.003250
49. Nishiyama T, Zhang Y, Cui M, Li H, Sanchez-Ortiz E, McAnally JR, et al. Precise genomic editing of pathogenic mutations in RBM20 rescues dilated cardiomyopathy. Sci Transl Med. (2022) 14(672):eade1633. doi: 10.1126/scitranslmed.ade1633
50. Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest. (2014) 124(8):3419–30. doi: 10.1172/JCI74523
51. Cannie DE, Protonotarios A, Bakalakos A, Syrris P, Lorenzini M, De Stavola B, et al. Risks of ventricular arrhythmia and heart failure in carriers of RBM20 variants. Circ Genom Precis Med. (2023) 16(5):434–41. doi: 10.1161/CIRCGEN.123.004059
52. Long PA, Evans JM, Olson TM. Diagnostic yield of whole exome sequencing in pediatric dilated cardiomyopathy. J Cardiovasc Dev Dis. (2017) 4(3):11. doi: 10.3390/jcdd4030011
53. Setti M, Merlo M, Gigli M, Munaretto L, Paldino A, Stolfo D, et al. Role of arrhythmic phenotype in prognostic stratification and management of dilated cardiomyopathy. Eur J Heart Fail. (2024) 26(3):581–9. doi: 10.1002/ejhf.3168
54. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American college of medical genetics and genomics (ACMG). Genet Med. (2018) 20(9):899–909. doi: 10.1038/s41436-018-0039-z
55. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. (2016) 17(6):333–51. doi: 10.1038/nrg.2016.49
56. Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. (2011) 29(10):908–14. doi: 10.1038/nbt.1975
57. Ouellette AC, Mathew J, Manickaraj AK, Manase G, Zahavich L, Wilson J, et al. Clinical genetic testing in pediatric cardiomyopathy: is bigger better? Clin Genet. (2018) 93(1):33–40. doi: 10.1111/cge.13024
58. Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. (2011) 32(9):1065–76. doi: 10.1093/eurheartj/ehr105
59. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. (2011) 12(11):745–55. doi: 10.1038/nrg3031
60. Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. (2017) 390(10092):400–14. doi: 10.1016/S0140-6736(16)31713-5
61. Puggia I, Merlo M, Barbati G, Rowland TJ, Stolfo D, Gigli M, et al. Natural history of dilated cardiomyopathy in children. J Am Heart Assoc. (2016) 5(7):e003450. doi: 10.1161/JAHA.116.003450
62. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. (2010) 375(9716):752–62. doi: 10.1016/S0140-6736(09)62023-7
63. Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. (2003) 99(1):1–19. doi: 10.1159/000068446
64. Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med. (1999) 341(8):577–85. doi: 10.1056/NEJM199908193410806
65. Njoroge JN, Mangena JC, Aribeana C, Parikh VN. Emerging genotype-phenotype associations in dilated cardiomyopathy. Curr Cardiol Rep. (2022) 24(9):1077–84. doi: 10.1007/s11886-022-01727-z
66. de Frutos F, Ochoa JP, Navarro-Penalver M, Baas A, Bjerre JV, Zorio E, et al. Natural history of MYH7-related dilated cardiomyopathy. J Am Coll Cardiol. (2022) 80(15):1447–61. doi: 10.1016/j.jacc.2022.07.023
67. Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. (2004) 44(9):1903–10. doi: 10.1016/j.jacc.2004.07.045
68. Dungu JN, Langley SG, Hardy-Wallace A, Li B, Barbagallo RM, Field D, et al. Dilated cardiomyopathy: the role of genetics, highlighted in a family with filamin C (FLNC) variant. Heart. (2022) 108(9):676–82. doi: 10.1136/heartjnl-2021-319682
69. Carruth ED, Qureshi M, Alsaid A, Kelly MA, Calkins H, Murray B, et al. Loss-of-function FLNC variants are associated with arrhythmogenic cardiomyopathy phenotypes when identified through exome sequencing of a general clinical population. Circ Genom Precis Med. (2022) 15(4):e003645. doi: 10.1161/CIRCGEN.121.003645
70. Wang Y, Han B, Fan Y, Yi Y, Lv J, Wang J, et al. Next-generation sequencing reveals novel genetic variants for dilated cardiomyopathy in pediatric Chinese patients. Pediatr Cardiol. (2022) 43(1):110–20. doi: 10.1007/s00246-021-02698-8
71. Jolfayi AG, Kohansal E, Ghasemi S, Naderi N, Hesami M, MozafaryBazargany M, et al. Exploring TTN variants as genetic insights into cardiomyopathy pathogenesis and potential emerging clues to molecular mechanisms in cardiomyopathies. Sci Rep. (2024) 14(1):5313. doi: 10.1038/s41598-024-56154-7
72. Bondue A, Arbustini E, Bianco A, Ciccarelli M, Dawson D, De Rosa M, et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: scientific update from the working group of myocardial function of the European society of cardiology. Cardiovasc Res. (2018) 114(10):1287–303. doi: 10.1093/cvr/cvy122
73. Meyer AP, Barnett CL, Myers K, Siskind CE, Moscarello T, Logan R, et al. Neuromuscular and cardiovascular phenotypes in paediatric titinopathies: a multisite retrospective study. J Med Genet. (2024) 61(4):356–62. doi: 10.1136/jmg-2023-109513
74. Ture M, Balik H, Akin A, Bilici M, Nergiz A. The relationship between electrocardiographic data and mortality in children diagnosed with dilated cardiomyopathy. Eur J Pediatr. (2020) 179(5):813–9. doi: 10.1007/s00431-020-03569-9
75. Wang M, Xu Y, Wang S, Zhao T, Cai H, Wang Y, et al. Predictive value of electrocardiographic markers in children with dilated cardiomyopathy. Front Pediatr. (2022) 10:917730. doi: 10.3389/fped.2022.917730
76. Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. (2003) 41(5):771–80. doi: 10.1016/S0735-1097(02)02954-6
77. Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. (2002) 39(6):981–90. doi: 10.1016/S0735-1097(02)01724-2
78. Serpa F, Finn CM, Tahir UA. Navigating the penetrance and phenotypic spectrum of inherited cardiomyopathies. Heart Fail Rev. (2024) 29(5):873–81. doi: 10.1007/s10741-024-10405-x
79. Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging. (2020) 21(3):326–36. doi: 10.1093/ehjci/jez188
80. Baban A, Lodato V, Parlapiano G, di Mambro C, Adorisio R, Bertini ES, et al. Myocardial and arrhythmic spectrum of neuromuscular disorders in children. Biomolecules. (2021) 11(11):1578. doi: 10.3390/biom11111578
81. Maggi L, Carboni N, Bernasconi P. Skeletal muscle laminopathies: a review of clinical and molecular features. Cells. (2016) 5(3):33. doi: 10.3390/cells5030033
82. Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D, et al. Clinical and molecular genetic spectrum of autosomal dominant emery-dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. (2000) 48(2):170–80. doi: 10.1002/1531-8249(200008)48:2%3C170::AID-ANA6%3E3.0.CO;2-J
83. Rees M, Nikoopour R, Fukuzawa A, Kho AL, Fernandez-Garcia MA, Wraige E, et al. Making sense of missense variants in TTN-related congenital myopathies. Acta Neuropathol. (2021) 141(3):431–53. doi: 10.1007/s00401-020-02257-0
84. Skriver SV, Krett B, Poulsen NS, Krag T, Walas HR, Christensen AH, et al. Skeletal muscle involvement in patients with truncations of titin and familial dilated cardiomyopathy. JACC Heart Fail. (2024) 12(4):740–53. doi: 10.1016/j.jchf.2023.10.010
85. Johnson R, Otway R, Chin E, Horvat C, Ohanian M, Wilcox JAL, et al. DMD-associated dilated cardiomyopathy: genotypes, phenotypes, and phenocopies. Circ Genom Precis Med. (2023) 16(5):421–30. doi: 10.1161/CIRCGEN.123.004221
86. Mikšiūnas R, Labeit S, Bironaite D. Class I and II histone deacetylase inhibitors as therapeutic modulators of dilated cardiac tissue-derived mesenchymal stem/stromal cells. Int J Mol Sci. (2024) 25(12):6758. doi: 10.3390/ijms25126758
87. Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM, Li RG, et al. TBX5 loss-of-function mutation contributes to familial dilated cardiomyopathy. Biochem Biophys Res Commun. (2015) 459(1):166–71. doi: 10.1016/j.bbrc.2015.02.094
88. Patterson J, Coats C, McGowan R. Familial dilated cardiomyopathy associated with pathogenic TBX5 variants: expanding the cardiac phenotype associated with Holt-Oram syndrome. Am J Med Genet A. (2020) 182(7):1725–34. doi: 10.1002/ajmg.a.61635
89. Antonopoulos AS, Xintarakou A, Protonotarios A, Lazaros G, Miliou A, Tsioufis K, et al. Imagenetics for precision medicine in dilated cardiomyopathy. Circ Genom Precis Med. (2024) 17(2):e004301. doi: 10.1161/CIRCGEN.123.004301
90. de Frutos F, Ochoa JP, Fernandez AI, Gallego-Delgado M, Navarro-Penalver M, Casas G, et al. Late gadolinium enhancement distribution patterns in non-ischaemic dilated cardiomyopathy: genotype-phenotype correlation. Eur Heart J Cardiovasc Imaging. (2023) 25(1):75–85. doi: 10.1093/ehjci/jead184
91. Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. (2015) 349(6251):982–6. doi: 10.1126/science.aaa5458
92. Verdonschot JAJ, Merlo M, Dominguez F, Wang P, Henkens M, Adriaens ME, et al. Phenotypic clustering of dilated cardiomyopathy patients highlights important pathophysiological differences. Eur Heart J. (2021) 42(2):162–74. doi: 10.1093/eurheartj/ehaa841
93. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy-A heart failure society of America practice guideline. J Card Fail. (2018) 24(5):281–302. doi: 10.1016/j.cardfail.2018.03.004
94. Taylor DO, Edwards LB, Boucek MM, Trulock EP, Deng MC, Keck BM, et al. Registry of the international society for heart and lung transplantation: twenty-second official adult heart transplant report–2005. J Heart Lung Transplant. (2005) 24(8):945–55. doi: 10.1016/j.healun.2005.05.018
95. Mizusawa Y. Recent advances in genetic testing and counseling for inherited arrhythmias. J Arrhythm. (2016) 32(5):389–97. doi: 10.1016/j.joa.2015.12.009
Keywords: children, dilated cardiomyopathy, genetics, inherited cardiomyopathy, genotype-phenotype correlation
Citation: Dai Y, Wang Y, Fan Y and Han B (2025) Genotype-phenotype insights of pediatric dilated cardiomyopathy. Front. Pediatr. 13:1505830. doi: 10.3389/fped.2025.1505830
Received: 3 October 2024; Accepted: 21 January 2025;
Published: 31 January 2025.
Edited by:
Nathalie Jeanne M. Bravo-valenzuela, Federal University of Rio de Janeiro, BrazilReviewed by:
Cecilia Lazea, University of Medicine and Pharmacy Iuliu Hatieganu, RomaniaYangpo Cao, Southern University of Science and Technology, China
Copyright: © 2025 Dai, Wang, Fan and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Han, aGFuYm8zNUAxNjMuY29t; Youfei Fan, ZmFueW91ZmVpMDcyMEAxMjYuY29t