 Lu Cao
Lu Cao Qin Wang
Qin Wang Jianjiang Zhang
Jianjiang Zhang- Department of Pediatrics, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
This case report details a 6-year-old Han Chinese girl diagnosed with Systemic Lupus Erythematosus (SLE) associated with a frameshift variant in the SOCS1 gene. Initially presenting with fever and rash, the patient exhibited abnormal liver function, hypocomplementemia, and positive antinuclear and anti-dsDNA antibodies. Genetic testing identified a heterozygous frameshift mutation in the SOCS1 gene, inherited from her mother. The girl was treated with intravenous methylprednisolone, oral prednisolone, hydroxychloroquine, and mycophenolate mofetil, leading to significant clinical improvement. Considering the clinically relevant variant in SOCS1, the findings suggest that identifying pathogenic genes can facilitate the development of new therapeutic targets and biomarkers, with JAK inhibitors showing promise for treating SOCS1-related conditions.
Introduction
Systemic lupus erythematosus (SLE) is a chronic inflammatory condition caused by issues in immune regulation genes. Various inflammatory cytokines and genes contribute to the pathogenesis of SLE. The discovery of the Suppressor of Cytokine Signaling 1 (SOCS1) represented a significant advancement in understanding the regulation of the Janus kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathways (1). SOCS1 modulates cytokine signaling pathways and immune responses, including interferon-γ signaling, CD4T cell differentiation, influence on CD4/CD8T cell development and activation, and innate immunity (1). SOCS1 insufficiency disrupts immune homeostasis through multiple mechanisms: it impairs regulatory T cell (Treg) function, promotes hyperactivation of CD8+ T cells with a memory-like phenotype, and enhances B cell autoantibody production via elevated B-cell activating factor (BAFF) secretion (2). These abnormalities are linked to uncontrolled STAT1/STAT5 phosphorylation in response to IFN-γ and IL-2, leading to a “cytokine hypersensitivity” state.
In murine models, SOCS1 insufficiency or complete loss-of-function mutations induce lupus-like phenotypes, including autoantibody production, glomerulonephritis, and lymphoproliferation. For example, Fujimoto et al. (3) demonstrated that SOCS1+/− mice develop spontaneous autoimmunity with age, characterized by anti-dsDNA antibodies, immune complex deposition in the kidneys, and Th1/Th17 cell skewing, mirroring human SLE pathogenesis. Similarly, Hadjadj et al. (2) reported that SOCS1 insufficiency in humans leads to early-onset autoimmunity, with two patients meeting SLE criteria and exhibiting mesangial proliferative glomerulonephritis and dysregulated B cell subsets.
The JAK/STAT pathway is crucial in various physiological and pathological processes, such as immune responses, cell proliferation, differentiation, and apoptosis.
This case underscores the importance of genetic analysis in pediatric SLE, especially in young patients with severe disease, a family history of relevant diseases, or an atypical response to treatment. Such analysis can be more targeted and practical, guiding accurate diagnosis and management. Whole Exome Sequencing (WES) shows potential in this regard. The potential link between SLE and SOCS1 insufficiency lies in the latter's role in immune regulation. Further research is crucial to explore this relationship and its implications for treatment (2). In this study, we report a case of SLE in a child with a frameshift mutation in the SOCS1 gene.
Case presentation
At the onset of the illness, a 6-year-old girl presented with episodic fever and rash and was admitted to a local hospital. Laboratory analysis revealed a haemoglobin level of 96 g/L, a white blood cell count of 3.42 × 109/L, an Epstein–Barr (EB) virus DNA quantification of 1.56E + 4 IU/ml, alanine aminotransferase (ALT) levels of 89 U/L, aspartate aminotransferase (AST) levels of 92 U/L, and 1 + urine protein. An upper abdominal computed tomography (CT) scan showed liver and spleen enlargement, pleural effusion on both sides, and inflammation of the lower lobe of the left lung. She was treated for infectious mononucleosis, and her temperature returned to normal, transaminases normalised, and the rash disappeared. A routine blood check-up at the local hospital revealed a white blood cell count of 3.27 × 109/L, haemoglobin of 92 g/L, erythrocyte sedimentation rate (ESR) of 97 mm/h, ALT of 262 U/L, AST of 301 U/L, and proteinuria 1+. Upon referral to our hospital due to elevated transaminase levels, the patient presented with normal results for cytomegalovirus DNA and EB virus DNA tests. Further investigation revealed hypocomplementemia, prolonged coagulation screening with an activated partial thromboplastin time (APTT) of 69 s and a prothrombin time (PT) of 18.9 s, as well as proteinuria 1+ and microscopic haematuria of 11/μl. Antiphospholipid antibodies, anti-beta-2-glycoprotein-1 antibodies, and lupus anticoagulant were positive. Coagulation factors revealed mild decreases in the activities of Factors IX, XI, and XII. Laboratory tests showed positive antinuclear antibody (1/320 titre) and positive anti-double-stranded DNA (ds-DNA) [ds-DNA: 429 IU/ml]. A bone marrow biopsy showed no concern for malignancy or dysplasia. A lymph node biopsy showed reactive lymphoid hyperplasia. Magnetic resonance imaging (MRI) of the brain revealed no abnormality. The girl's past medical and family history did not include inflammatory or autoimmune diseases. A 24-h urine protein test showed 0.3 g. Based on her history and laboratory findings, the girl was diagnosed with systemic lupus erythematosus (SLE), meeting the 2019 EULAR/ACR classification criteria (4). The girl received intravenous methylprednisolone infusion (1.6 mg/kg/day, Max dose 48 mg/day) which was subsequently changed to oral prednisolone (2 mg/kg/day). Calcium supplementation, oral hydroxychloroquine (4 mg/kg/day), and mycophenolate mofetil (30 mg/kg/day) were also prescribed, along with liver protection and plasma transfusion. Symptomatic treatment included plasma transfusion and nutritional liver support. Oral administration of Benazepril tablets at 5 mg per day for 3 consecutive months to reduce urinary protein. Given the patient's initial symptoms consistent with infectious mononucleosis, which later progressed to manifestations suggestive of systemic lupus erythematosus, and considering the child's young age of six, a comprehensive whole exome sequencing (WES) analysis was conducted to identify potential pathogenic genetic variants for targeted treatment options. WES identified a frameshift variant of the SOCS1 gene (exon 2, NM_003745.1: c.476_480dup; p.Met161AlafsTer46) inherited from her mother, who had no clinically significant systemic history (Figure 1). This variant has been previously reported in family C of a prior study (2), where a similar frameshift mutation in the SOCS1 gene was associated with early-onset autoimmunity, including Evans syndrome and lymphoproliferative manifestations. The mutation introduces a premature stop codon and a 46-residue neopeptide within the SOCS box domain, predicted to disrupt JAK-STAT pathway regulation and cause loss of function. While the mother's carrier status does not negate the pathogenic role of the SOCS1 variant, the patient's severe, early-onset phenotype—combined with the variant's presence in a known disease-associated locus (2)—strongly supports its causal involvement. The absence of other identified pathogenic variants in SLE-associated genes (e.g., TNFAIP3, TREX1) further reinforces SOCS1 insufficiency as the primary driver.
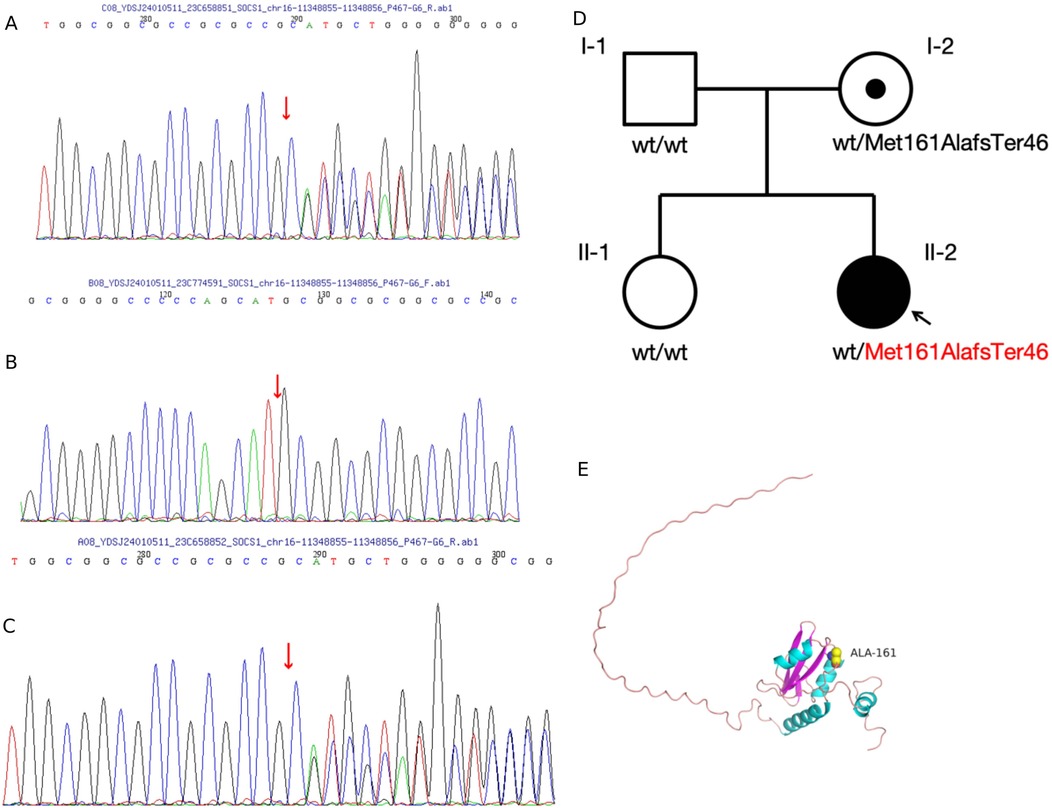
Figure 1. (A–C) sanger sequencing of identified alterations using whole exome sequencing. (D) Pedigree and variant of the SOCS1 gene in the family. (E) Amino acid change from methionine to alanine at position 161, with an extension of 44 additional amino acids before termination of expression, resulting in the loss of 6 amino acids. This alteration affects the protein conformation. The predicted structure of SOCS1, modelled using SWISS-MODEL, is shown. Structural components are highlighted as follows: alpha helices (blue), beta sheets (purple), and loop structures (pink). Hydrogen bonds are represented by stick structures, with atoms coloured as follows: yellow for carbon, grey for hydrogen, blue for nitrogen, red for oxygen, and orange for sulfur.
After two months of treatment, the patient's liver function returned to normal. Significant clinical improvement was noted during hospitalisation and at the 6-month follow-up (Table 1).
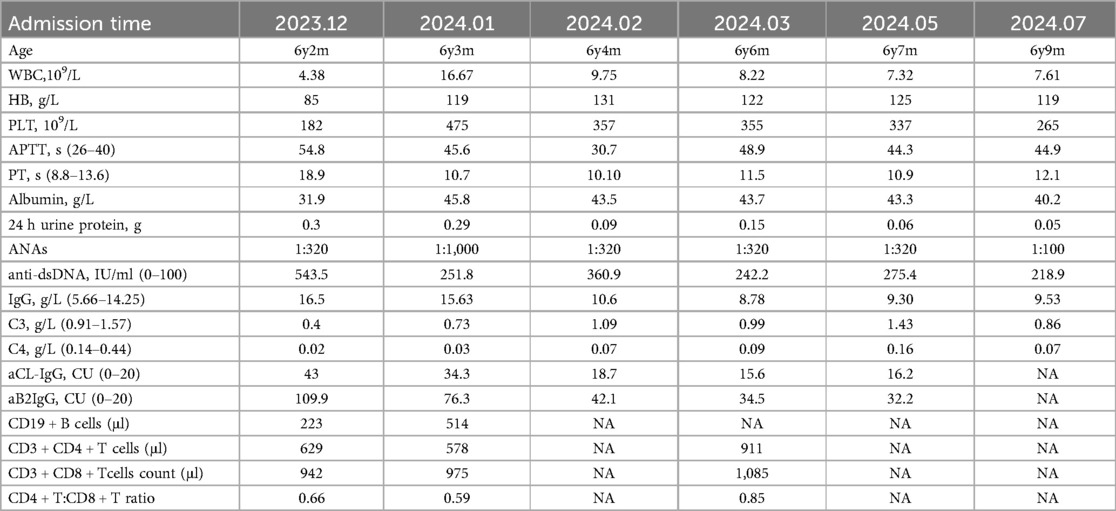
Table 1. Details of the patient's clinical dates.
Discussion
In this case report, a 6-year-old girl initially presented with symptoms suggestive of infectious mononucleosis and was admitted for treatment. She was later transferred to our hospital with elevated transaminases and a rapidly worsening clinical condition. Diagnostic workup revealed coagulation abnormalities, positive antiphospholipid and antinuclear antibodies, anti-dsDNA antibodies, and hypocomplementemia, all indicative of SLE. Given her young age and the aggressive nature of the disease, and considering the contraindication of renal biopsy due to bleeding risk, we opted for whole exome sequencing to identify potential pathogenic genetic variants for targeted treatment options.
SLE is characterised by tissue inflammation mediated by the activation of autoreactive lymphocytes and the subsequent production of autoantibodies, culminating in immune complex deposition and ultimately contributing to tissue and organ damage. Over 80 polygenic lupus susceptibility loci have been identified; targeted screening for these can enhance patient risk stratification and treatment optimisation (5, 6). Recent research strongly supports the involvement of various gene mutations as influential triggers for dysregulated or impaired responses in both the innate and adaptive immune systems, ultimately leading to a lupus-like syndrome (5). In SLE disease models, Fujimoto et al. (3) revealed that SOCS1-/- mice spontaneously develop a fatal disease due to aberrantly activated lymphocytes. The partial restoration of SOCS1 in lymphoid cells rescues SOCS1-/- mice from early onset fatal disease. However, in these mutant mice with partial restoration of SOCS1 expression, SOCS1 levels were insufficient to effectively downregulate its target signaling. Consequently, these mice exhibited spontaneous hyperactivation of lymphocytes, elevated anti-DNA autoantibody levels, increased serum Ig, and glomerulonephritis with IgG deposition in the glomeruli. The observations in murine models led to the hypothesis that a deficiency in SOCS1 accelerates the development of autoimmune phenotypes. Reduced SOCS1 levels may impair the negative feedback on the JAK/STAT pathway, leading to unchecked cytokine production and a heightened risk of SLE development.
In 2020, Thaventhiran et al. (7) first characterised SOCS1 haploinsufficiency in an index case, detailing recurrent bacterial infections, severe autoinflammation, and autoimmunity. Notably, Hadjadj et al. (2) subsequently reported two pediatric SLE patients (D1 and E1) with SOCS1 insufficiency, who presented with early-onset disease (9–16 years old) and autosomal dominant inheritance, distinct from the adult female-predominant pattern of polygenic SLE. Both patients developed lupus nephritis with mesangial hypercellularity and C1q deposition, associated with impaired immune complex clearance and CD21−CD38− B-cell expansion. Mechanistically, their lymphocytes showed hyperactive STAT1/STAT5 phosphorylation in response to IFN-γ and IL-2, mirroring the “cytokine hypersensitivity” observed in our patient's JAK/STAT pathway dysregulation.
Subsequently, a second case and her father, both with a similar clinical presentation of severe infections, atopy, and autoimmunity, were identified; the father also exhibited jaundice and unexplained liver disease. Lee et al. (8) reported on two additional patients with autoimmune cytopenias in the context of acute infections and one with paediatric multisystem inflammatory syndrome. They revealed heightened type I and II interferon responses, underscoring SOCS1's pivotal role as a negative regulator of interferon pathways. Michniacki et al. (9) reported a pediatric patient with SOCS1 insufficiency resulting from a complete gene deletion. Notably, the patient exhibited a remarkable therapeutic response to tofacitinib, a Janus kinase inhibitor known for its capacity to dampen interferon signaling pathways. Our patient's antiphospholipid antibody positivity and early liver involvement represent unique clinical features not explicitly reported in Hadjadj's cohort, highlighting the phenotypic variability of SOCS1 insufficiency. Nevertheless, the shared themes of JAK-STAT hyperactivation and B-cell dysregulation across cases support SOCS1 mutations as a monogenic driver of “cytokine-driven” SLE subsets. The response to glucocorticoids and mycophenolate mofetil in our patient, combined with the documented efficacy of JAK inhibitors in SOCS1-deficient models, reinforces the rationale for personalized therapy targeting this pathway. Type I interferonopathies are systemic disorders characterized by autoimmune and inflammatory traits. These conditions frequently manifest in early childhood, with more than 80% of cases commencing prior to the age of three (10). The heterogeneity of their clinical manifestations poses a significant challenge to early diagnosis. In the case of our patient, WES was instrumental in identifying the SOCS1 frameshift variant, which was inherited from her mother. This genetic finding provided critical insights into the pathogenesis of the patient's SLE and paved the way for a more personalized treatment approach.
During the initial six-month treatment period, glucocorticoids, hydroxychloroquine, and immunosuppressive agents formed the cornerstone of anti-inflammatory and immunosuppressive therapy for this SLE patient. The patient's condition gradually improved. Complement levels returned to normal, autoantibodies became negative, and proteinuria decreased. Fresh frozen plasma infusions kept her abnormal coagulation function under control. At present, the child is still under follow-up, with urine protein remaining negative. Her parents refused a renal pathological biopsy. However, due to the physical examination report indicating decreased serum complements C3 and C4, positive lupus anticoagulant, and persistently positive dsDNA, the parents consented to treatment with tofacitinib (weight >15–25 kg, 7.5 mg/day). A detailed description of the treatment conditions can be found in Figure 2. We chose tofacitinib over other JAK inhibitors for several reasons. First, the patient's clinical and laboratory findings indicated significant JAK-STAT pathway activation, and tofacitinib has shown efficacy in inhibiting this pathway in previous studies. Second, considering the patient's young age, tofacitinib was deemed a suitable option due to its relatively mild yet effective profile. Third, tofacitinib has been approved by the FDA for juvenile idiopathic arthritis, supporting its use in pediatric patients. Additionally, since its introduction in Chinese hospitals, tofacitinib has become more affordable and accessible, facilitating convenient access for our patient.
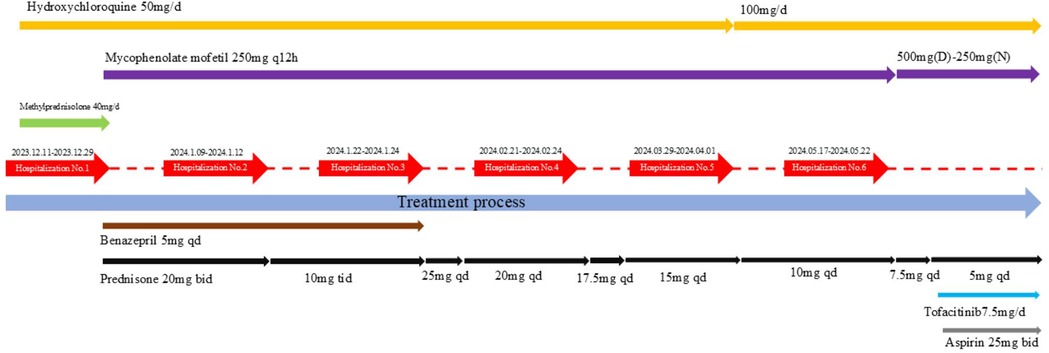
Figure 2. A detailed description of the treatment conditions.
In conclusion, this case highlights the importance of genetic analysis, particularly in children with severe or atypical symptoms, a family history of relevant diseases, and individuals from consanguineous families. Incorporating Whole Exome Sequencing into the diagnostic workflow for paediatric-onset lupus cases enables a more accurate and timely aetiologic diagnosis, facilitating improved clinical management. Identifying pathogenic genes will aid in the discovery of novel therapeutic targets and the development of effective biomarkers.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this Case Report.
Author contributions
LC: Data curation, Conceptualization, Writing – review & editing, Formal analysis, Writing – original draft. QW: Writing – original draft, Data curation, Conceptualization, Writing – review & editing. HZ: Writing – original draft, Formal analysis, Conceptualization, Writing – review & editing. JZ: Writing – original draft, Formal analysis, Data curation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Körholz J, Chen LS, Strauss T, Schuetz C, Dalpke AH. One gene to rule them all—clinical perspectives of a potent suppressor of cytokine signaling—SOCS1. Front Immunol. (2024) 15:1385190. doi: 10.3389/fimmu.2024.1385190
2. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. (2020) 11(1):5341. doi: 10.1038/s41467-020-18925-4
3. Fujimoto M, Tsutsui H, Xinshou O, Tokumoto M, Watanabe D, Shima Y, et al. Inadequate induction of suppressor of cytokine signaling-1 causes systemic autoimmune diseases. Int Immunol. (2004) 16(2):303–14. doi: 10.1093/intimm/dxh030
4. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European league against rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis & Rheumatol. (2019) 71(9):1400–12. doi: 10.1002/art.40930
5. Zhang C, Han X, Sun L, Yang S, Peng J, Chen Y, et al. Novel loss-of-function mutations in Tnfaip3 gene in patients with lupus nephritis. Clin Kidney J. (2022) 15(11):2027–38. doi: 10.1093/ckj/sfac130
6. Rossano M, Conti EA, Bocca P, Volpi S, Mastrangelo A, Cavalli R, et al. Novel heterozygous Trex1 mutation in a juvenile systemic lupus erythematosus patient with severe cutaneous involvement treated successfully with JAK-inhibitors: a case report. Front Immunol. (2023) 14:1288675. doi: 10.3389/fimmu.2023.1288675
7. Thaventhiran JED, Lango Allen H, Burren O, S RW, Greene D, Staples E, et al. Whole-Genome sequencing of a sporadic primary immunodeficiency cohort. Nature. (2020) 583(7814):90–5. doi: 10.1038/s41586-020-2265-1
8. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (mis-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. (2020) 146(5):1194–200.e1. doi: 10.1016/j.jaci.2020.07.033
9. Michniacki TF, Walkovich K, DeMeyer L, Saad N, Hannibal M, Basiaga ML, et al. SOCS1 haploinsufficiency presenting as severe enthesitis, bone marrow hypocellularity, and refractory thrombocytopenia in a pediatric patient with subsequent response to JAK inhibition. J Clin Immunol. (2022) 42(8):1766–77. doi: 10.1007/s10875-022-01346-x
Keywords: systemic lupus erythematosus, SOCS1 insufficiency, child, phospholipid antibody, case report
Citation: Cao L, Wang Q, Zhang H and Zhang J (2025) SOCS1 insufficiency in systemic lupus erythematosus in a child: a case report. Front. Pediatr. 13:1618483. doi: 10.3389/fped.2025.1618483
Received: 26 April 2025; Accepted: 4 July 2025;
Published: 18 July 2025.
Edited by:
Klaus Tenbrock, RWTH Aachen University, GermanyReviewed by:
Dragana Lazarevic, University Clinical Center Nis, SerbiaJulia Körholz, University Hospital Carl Gustav Carus, Germany
Martina Rossano, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright: © 2025 Cao, Wang, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lu Cao, bWFpbHRvamVzc2llQDEyNi5jb20=