Abstract
Heme, iron (Fe) protoporphyrin IX, functions as a prosthetic group in a range of hemoproteins essential to support life under aerobic conditions. The Fe contained within the prosthetic heme groups of these hemoproteins can catalyze the production of reactive oxygen species. Presumably for this reason, heme must be sequestered within those hemoproteins, thereby shielding the reactivity of its Fe-heme. However, under pathologic conditions associated with oxidative stress, some hemoproteins can release their prosthetic heme groups. While this heme is not necessarily damaging per se, it becomes highly cytotoxic in the presence of a range of inflammatory mediators such as tumor necrosis factor. This can lead to tissue damage and, as such, exacerbate the pathologic outcome of several immune-mediated inflammatory conditions. Presumably, targeting “free heme” may be used as a therapeutic intervention against these diseases.
Physiological Role of Heme
Heme acts as prosthetic group in a range of hemoproteins that play a pivotal role in many essential biological processes. These include gas transport and storage, mitochondrial electron transport chain, drug metabolism, signal transduction and regulation of gene expression (Chapman et al., 1997).
Structure and biochemistry of heme
Heme is a hydrophobic metallo-compound containing a Fe atom within its protoporphyrin ring. It is composed of four methane-bridged pyrroles (tetrapyrrole ring) bound to the central Fe atom through nitrogen atoms (Reedy and Gibney, 2004; Kumar and Bandyopadhyay, 2005; Tsiftsoglou et al., 2006). Ferrous (Fe2+) heme has a neutral chemical charge, whereas ferric (Fe3+) heme is positively charged, and can bind anions (Kumar and Bandyopadhyay, 2005). Although ferrous and ferric states are the most common within the heme structure, oxidative states of Fe-heme can vary from Fe2+ to Fe5+(Ogura et al., 1996; Karpefors et al., 2000; Dey and Ghosh, 2002).
Heme exists mainly as a, b, and c variants, which differ by subtle chemical modifications (Chapman et al., 1997; Tsiftsoglou et al., 2006; Smith et al., 2010). One of the vinyl groups of heme b, is replaced by a 17-hydroxyethylfarnesyl group in heme a. One of the methyl groups of heme b is also replaced by a formyl group in heme a (Caughey et al., 1975; Han et al., 1991). In heme c, both vinyl groups of heme b are replaced by sulfhydryl groups (Bowman and Bren, 2008). Heme b is the most common variant in mammals, present, among others, in hemoglobin (Hb) and myoglobin (Fermi et al., 1984; Evans and Brayer, 1990; Paoli et al., 1996; Park et al., 2006). Heme a and c are found in cytochrome c oxidase and cytochrome c, respectively (Bushnell et al., 1990; Tsukihara et al., 1995). Whereas heme c can bind covalently to proteins, via two thioether bonds, this is not the case for heme b and a, eventually allowing for their release from hemoproteins (Allen et al., 2003).
Hemoproteins
Hemoproteins have a diverse spectrum of biological functions. These include transport and storage of diatomic gaseous molecules such as oxygen (O2), nitric oxide (NO) and carbon monoxide (CO; Chay and Brillhart, 1974a,b; Aono, 2008; Kakar et al., 2010). They are also essential for electron transfer reactions (Makinen et al., 1983; Gray and Winkler, 1996) as well as for modulation of gene transcription (Tahara et al., 2004a,b; Youn et al., 2006; Hira et al., 2007), such as involved in the regulation of circadian rhythms (Kaasik and Lee, 2004). Prosthetic heme groups have also been implicated in the regulation of signal transduction pathways (Igarashi and Sun, 2006; Zenke-Kawasaki et al., 2007), ion-channel functions (Tang et al., 2003; Horrigan et al., 2005), micro-RNA processing (Faller et al., 2007) and the metabolism of xenobiotics and drugs (Bernhardt, 1996; Rowland et al., 2006).
Under physiologic conditions, heme is found mainly in the “heme pockets” of hemoproteins. These are characterized by a high frequency in aromatic amino acids (Table 1), such as phenylalanine (F), tyrosine (Y) or tryptophan (W) and by few or no charged amino acids (Li et al., 2011), conferring the hydrophobicity required to promote stable heme binding to hemoproteins. Five highly conserved amino acids, including histidine (H), methionine (M), cysteine (C), tyrosine (Y) and lysine (K) can act as axial heme ligands in the heme pockets of hemoproteins (Li et al., 2011). Of these, H is frequently found in hemoproteins containing heme c or b (Table 1). Moreover, C often promotes binding of heme b, whereas M binds heme c (Fufezan et al., 2008; Li et al., 2011; Table 1). Other hydrophobic amino acids such as leucine (L), isoleucine (I) and valine (V) can also create interactions with the porphyrin structure of heme, whereas arginine (R) and other positively charged amino acids can interact with the negatively charged heme propionate groups (Schneider et al., 2007).
Table 1
| Function | Representative hemoprotein | Number/type of heme | Axial ligand | References |
|---|---|---|---|---|
| Electron transport | Cytochrome c | 1/c-type | His, Met | Mirkin et al. (2008) |
| Cytochrome b5 | 1/b-type | His, His | Wirtz et al. (2000) | |
| Cytochrome c oxidase | 2/a-type | His/Vacant, His/His | Tsukihara et al. (1995) | |
| Cytochrome c reductase | 2/b-type, 1/c-type | His/His, His/His, His/Met | Xia et al. (1997); Zhang et al. (1998) | |
| Gas carriers | Hemoglobin | 4/b-type | His, Vacant | Park et al. (2006) |
| Myoglobin | 1/b-type | His, Vacant | Evans and Brayer (1988); Vojtechovsky et al. (1999) | |
| Catalytic activity – enzyme | Catalase I | 1/b-type | Tyr, Vacant | Putnam et al. (2000) |
| Cytochrome P450 | 1/b-type | Cys, Vacant or water | Rowland et al. (2006) | |
| Indoleamine 2,3-dioxygenase | 1/b-type | His, ligand | Sugimoto et al. (2006) | |
| NO synthase | 2/b-type | Cys, Vacant | Crane et al. (1997) | |
| Cystathionine β-synthase | 1/b-type | Cys/His | Meier et al. (2001) | |
| Tryptophan 2,3-dioxygenase | 4/b-type | His/Vacant | Zhang et al. (2007b) | |
| Transient heme binding | Heme oxygenase | 1/b-type | His, Water | Lad et al. (2003) |
| Hemopexin | 1/b-type | His/His | Paoli et al. (1999) | |
| Albumin | 1/b-type | Tyr/Vacant | Wardell et al. (2002) | |
| Heme-sensing/heme regulated | NPAS2 | 2/b-type | His/Cys or His/His, His/? | Dioum et al. (2002); Uchida et al. (2005) |
| Iron regulatory protein 2 (IRP2) | 1 | Cys/? | Jeong et al. (2004) | |
| Soluble guanylyl cyclase | 1/b-type | His/Vacant | Ma et al. (2007) |
Selected representatives of Mammalian hemoproteins and heme-sensing proteins.
Under physiologic conditions, heme is found mainly in the “heme pockets” of hemoproteins, characterized by a high frequency in aromatic amino acids, e.g., phenylalanine (F), tyrosine (Y) or tryptophan (W) and few or no charged amino acids (Li et al., 2011), that confer hydrophobicity, required to promote stable heme binding to hemoproteins. Five highly conserved amino acids, including histidine (H), methionine (M), cysteine (C), tyrosine (Y) and lysine (K) can act as axial heme ligands in the heme pockets of hemoproteins (Li et al., 2011). Of these, H is frequently found in hemoproteins containing heme c and heme b. Moreover, C often promotes binding of heme b, whereas M binds heme c (Fufezan et al., 2008; Li et al., 2011). Other hydrophobic amino acids such as leucine (L), isoleucine (I) and valine (V) can also create interactions with the porphyrin structure of heme, whereas arginine (R) and other positively charged amino acids interact with the negatively charged heme proprionate groups (Schneider et al., 2007).
Several heme-binding motifs have been described, namely CXXCH and CXXCK that bind primarily heme c and in which “X” refers to any amino acid (Fufezan et al., 2008; Smith et al., 2010; Li et al., 2011). The GX[HR]XC[PLAV]G heme-binding motif is associated mainly with heme b (Li et al., 2011). Another related FXXGXXCXG motif found in mammal, cytochrome P450, as well as in plant and bacterial hemoproteins binds to heme b (Li et al., 2011). Other heme-binding motifs can be formed by only two amino acids (such as the CP motif; Li et al., 2011).
In some hemoproteins, the Fe and the protoporphyrin IX ring of heme can undergo chemical and/or electronic modifications, such as demonstrated for peroxidases in which heme uses hydrogen peroxide (H2O2) to undergo several Fe oxidoreductase cycles associated with conformational alterations of its protoporphyrin ring. This can lead to the oxidation of neighboring molecules, e.g., indoles, phenols, aromatic amines, lignin, Mn2+ ions, halide ions or proteins (Badyal et al., 2006; Zederbauer et al., 2007; Battistuzzi et al., 2010). In other hemoproteins, heme does not undergo alterations in its Fe redox state or protoporphyrin conformation (Poulos, 2007). This is the case for hemoproteins that bind gaseous molecules such as guanylate cyclase, Hb, myoglobin or the transcription factor neuronal PAS domain-containing protein 2 (NPAS2), among others (Chay and Brillhart, 1974a,b; Dioum et al., 2002; Podstawka and Proniewicz, 2004; Uchida et al., 2005). Binding of NO or CO to these hemoproteins modulates their biologic activity, e.g., increased production of cGMP in the case of guanylate cyclase (Ma et al., 2007), regulation of O2 transport in the case of Hb or gene transcription in the case of CO binding to NPAS2 (Dioum et al., 2002). In other instances, biologic activity is regulated by heme binding itself, as it is the case for the transcriptional repressor Bach1 (Tahara et al., 2004a,b; Hira et al., 2007).
Heme Metabolism
Heme intracellular content must be tightly controlled to prevent heme-driven cytotoxicity while assuring that heme is available to be incorporated into nascent apo-hemoproteins. This is particularly relevant for cells with very high hemoprotein content such as erythroid or muscle cells expressing Hb and myoglobin, respectively. This regulatory mechanism is ensured largely through control of heme synthesis, transport and catabolism.
Heme synthesis
Heme de novo synthesis occurs through a sequence of eight enzymatic steps, four of which take place in the cytoplasm and four in the mitochondria (Figure 1; Heinemann et al., 2008). The first and rate-limiting step is catalyzed in the mitochondria by δ-amino levulinic acid (ALA) synthase (ALAS), an enzyme that converts glycine and succinyl-CoA into ALA. In most cells, heme synthesis is down-regulated by heme, via inhibition of ALAS mRNA expression (Hamilton et al., 1991) or ALAS translocation from the cytoplasm into the mitochondrion (Lathrop and Timko, 1993).
Figure 1
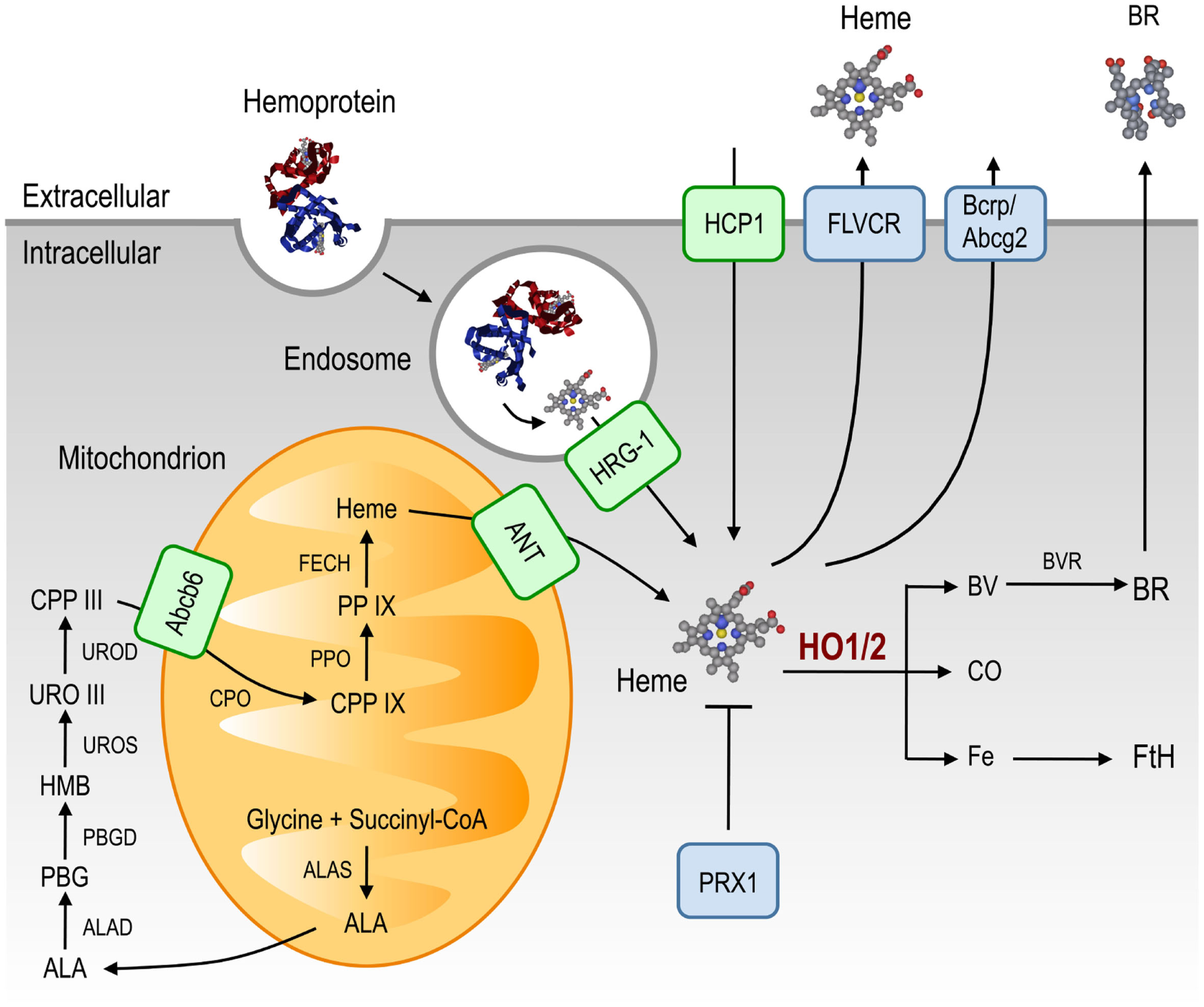
Heme metabolism. Heme is synthesized from glycine and succinyl-CoA, through eight enzymatic steps that take place in the cytoplasm and the mitochondrion. Heme and heme precursors are transported between these compartments by ANT and Abcb6, respectively. In addition to the heme produced via this pathway, extracellular heme can be imported. Hemoproteins can be taken up and denatured in endosomes, liberating heme that can be transported to the cytoplasm via HRG-1, whereas cell-free heme is taken up by the cell membrane-associated HCP1. Cytoplasmic heme can be scavenged by PRX1, which neutralizes its pro-oxidant effects. Excess heme can be exported via FLVCR and Bcrp/Abcg2 or catabolized by HO-1/2 into equimolar amounts of BV, CO and free Fe. BV can be converted into BR by BVR while labile cellular Fe induces the expression of FtH that scavenges excess cellular Fe. Abbreviations: Abcb6, ATP-binding cassette b6; ALA, δ-aminolevulinic acid; ALAD, ALA dehydratase; ALAS, ALA synthase; ANT, adenine-nucleotide translocator; Bcrp/Abcg2, breast cancer resistance protein/ATP-binding cassette g2; BR: bilirubin; BV, biliverdin; BVR, BV reductase; CO, carbon monoxide; CPP, coproporphyrinogen; CPO, CPP oxidase; Fe, iron; FECH, ferrochelatase; FLVCR, feline leukemic virus receptor; Fth, ferritin; HCP1, heme carrier protein-1; HMB, hydroxymethylbilane; HO, heme oxygenase; HRG-1, heme responsive gene-1; PBG, porphobilinogen; PBGD, PBG deaminase; PP IX, protoporphyrin IX; PPO, protoporphyrinogen oxidase; PRX1, peroxiredoxin-1; URO, uroporphyrinogen; UROD, URO decarboxylase; UROS, URO III synthase.
Heme transport
Being hydrophobic, heme was assumed to diffuse passively across cellular membranes. However, heme cellular trafficking appears to be controlled by a range of evolutionary conserved heme transporters (Figure 1; reviewed in Schultz et al., 2010; Khan and Quigley, 2011). Some of these heme transporters, e.g., mitochondrial ATP-binding cassette (ABC) family member, Abcb6, and the adenine-nucleotide translocator (ANT), control the translocation of heme as well as that of heme precursors between the cytoplasm and the mitochondria (Figure 1; Krishnamurthy et al., 2006; Azuma et al., 2008). Other heme transporters, including HRG-1 expressed in endosomes and lysosomes (Rajagopal et al., 2008) or the heme carrier protein (HCP1) expressed in the cytoplasmic membrane, control extracellular heme import (Figure 1; Shayeghi et al., 2005; Latunde-Dada et al., 2006; Figure 1). Heme can also be exported from cells via the ATP-binding cassette (Abcg2; Krishnamurthy et al., 2004; Krishnamurthy and Schuetz, 2006) and the feline leukemic virus receptor (FLVCR; Figure 1; Quigley et al., 2000, 2004; Schultz et al., 2010).
Heme catabolism
Heme degradation can be catalyzed by the enzymatic activity of heme oxygenases (HO; Tenhunen et al., 1968. There are two isoforms, i.e., HO-1 and HO-2, encoded by the HMOX1 and HMOX2 genes in humans, respectively (Figure 1; Maines et al., 1986; Trakshel et al., 1986). HO-2 is constitutively expressed by most cells (Trakshel et al., 1986) and is thought to carry out heme catabolism under homeostatic conditions. In contrast, excess intracellular heme induces the expression of the stress-responsive HO-1 isoform (see HO-Catalyzed Heme Degradation), increasing the rate of heme catabolism and, as such, preventing its putative cytotoxic effects (Gozzelino et al., 2010). HO activity catalyzes the oxidative cleavage of the heme protoporphyrin IX ring, yielding biliverdin (BV), concomitant with the release of equimolar amounts of labile Fe and carbon monoxide (CO; Figure 1; Tenhunen et al., 1968). Biliverdin is converted into bilirubin (BR) by BV reductase (Kutty and Maines, 1981; Figure 1). The physiological roles of these end products of heme catabolism, are discussed in further detail under Section “Cellular Control of Free Heme.”
Pathophysiological Effects of Free Heme
Under pathophysiological conditions, heme b can be released from hemoproteins to which it binds non-covalently (Bunn and Jandl, 1968; Hebbel et al., 1988; Pamplona et al., 2007). This produces “free heme,” that is, heme that is not bound to the heme pockets of hemoproteins. Free heme can act as a potent cytotoxic pro-oxidant (Balla et al., 1992, 1993) owing to the Fe atom contained within its protophorphyrin ring (Seixas et al., 2009; Gozzelino and Soares, 2011).
Immune-mediated inflammatory diseases can be associated with the accumulation of free heme in the circulation (Reiter et al., 2002; Pamplona et al., 2007; Seixas et al., 2009; Andrade et al., 2010a,b; Larsen et al., 2010). The observation that free heme can sensitize non-hematopoietic cells, e.g., hepatocytes, to undergo programmed cell death (PCD) in response to a variety of pro-inflammatory agonists (Seixas et al., 2009; Larsen et al., 2010; Gozzelino and Soares, 2011), suggests that free heme may participate functionally in the pathogenesis of immuno-mediated inflammatory diseases (Figure 2; Francis et al., 1997), in which the host component of inflammation would exacerbate the deleterious effects of free heme (Seixas et al., 2009; Larsen et al., 2010; Gozzelino and Soares, 2011). Taking this into account, the amount of circulating free heme, as a result of the overall heme metabolism, would dictate the outcome of these pathologic conditions. While free heme also appears to exert pro-inflammatory effects (Figueiredo et al., 2007; Porto et al., 2007; Fernandez et al., 2010) that might participate in the pathogenesis of immune-mediated inflammatory diseases these will not be covered in detail hereby.
Figure 2

Pathophysiological effects of heme. Free heme can sensitize parenchyma cells to undergo PCD in response to pro-inflammatory signals such as TNF. Under homeostatic conditions, cellular heme is degraded via HO-1 activity and the labile Fe released in this manner, is scavenged by FtH, thus preventing oxidative stress. Under a variety of inflammatory conditions, the concentration of intracellular free heme can exceed the rate of heme catabolism by HO-1. In addition, accumulation of cellular labile Fe can also overcome FtH scavenging capacity. When this occurs, free heme and/or labile Fe accumulate in cells, thereby boosting mitochondrion-driven ROS. Activation of the TNF receptor leads to further generation of ROS, resulting in sustained JNK activation which promotes FtH degradation (Antosiewicz et al., 2007), leading to further labile Fe overload and hence to further ROS generation via Fenton chemistry. Moreover, sustained JNK activation lead to the activation of effector caspases thereby inducing PCD. Abbreviations: FtH, ferritin; HO-1, heme oxygenase-1; JNK, c-Jun N-terminal kinase; ROS, reactive oxygen species; TNF, tumor necrosis factor; TNFR1, TNF receptor-1.
Presumably, the cytotoxic effect of free heme is mediated, to a large extent, via the unfettered production of free radicals leading to oxidative stress. This notion is supported by the observation that pharmacologic antioxidants can confer cytoprotection against free heme (Seixas et al., 2009; Larsen et al., 2010). Sustained free radicals production can prolong the activation of the c-Jun N-terminal Kinase (JNK) signaling transduction pathway (Ventura et al., 2004; Kamata et al., 2005), via inhibition of phosphatases controlling JNK activation in response to tumor necrosis factor (TNF; Davis, 2000; Sanchez-Perez et al., 2000; Masuda et al., 2001; Tanoue et al., 2001; Kamata et al., 2005; Figure 2). The cytotoxic effect of free heme is owed to this sustained JNK activation, as suggested by the observation that heme-driven free radical production leads to sustained JNK phosphorylation/activation (Figure 2) and that inhibition of JNK activation affords cytoprotection against free heme (Raffaella Gozzelino et al., unpublished results). The cytotoxic effect of free heme is also associated with cleavage of caspase-3, suggesting that heme sensitizes non-hematopoietic cells, e.g., hepatocytes, to undergo PCD by apoptosis. This notion is supported by the demonstration that pharmacologic caspase inhibition, including specific inhibition of caspase-3, confers cytoprotection against free heme (Figure 2; Seixas et al., 2009). More recently, free heme has also been shown to trigger necroptosis in hematopoietic cells, as demonstrated for macrophages (Fortes et al., 2012).
Physiologic Mechanisms of Protection Against Free Heme
Under pathophysiologic conditions associated with intravascular hemolysis, significant amounts of Hb can be released from red blood cells (RBCs). The resulting circulating cell-free Hb undergoes oxidation, releasing its prosthetic heme groups (Bunn and Jandl, 1968; Hebbel et al., 1988; Pamplona et al., 2007; Ferreira et al., 2008). Similarly, oxidation of other hemoproteins, e.g., myoglobin, may also contribute to the unwarranted accumulation of cytotoxic free heme, such as associated with rhabdomyolysis (Nath et al., 1992).
Protective mechanisms are in place, on systemic as well as cellular levels, to deal with this challenge, as described in this section.
Systemic control of free heme
Cell-free Hb can be scavenged by haptoglobin (Hp), an acute phase protein synthesized in the liver and present at relatively high concentrations (0.3–3 mg/mL) in plasma (Levy et al., 2010). Binding of Hb to Hp prevents Hb oxidation (Melamed-Frank et al., 2001) and thereby the release of its heme moiety (Figure 3). Hb:Hp complexes binds to CD163 (Kristiansen et al., 2001) expressed in macrophages and hepatocytes (Philippidis et al., 2004; Quaye, 2008), leading to endocytosis and degradation of the Hb:Hp complex (Figure 3). Polymorphisms in the human HPT gene can act as independent risk factors for several immune-mediated inflammatory diseases (Quaye, 2008), suggesting that Hb scavenging by Hp modulates the pathogenesis of these diseases.
Figure 3

Mechanisms of protection against free heme. A range of extracellular mechanisms confer protection against cell-free Hb and heme. Under hemolytic conditions, Hb is released from RBC into the circulation where it is scavenged by Hp, thereby preventing Hb oxidation, and allowing for the uptake of Hb:Hp complexes via the CD163 receptor. Once the scavenging capacity of Hp is exhausted, Hb becomes oxidized, releasing its heme prosthetic groups. These can be scavenged by albumin, HDL and LDL, α1-microglobulin, acting as “scavenging buffer,” and ultimately by Hx, which has the highest affinity for free heme. Hx:heme complexes are taken up via the CD91 receptor, delivering heme for catabolism by HO-1. Intracellular heme, whether originating from the extra- or intracellular compartment, is degraded via HO-1. The labile Fe released from heme catabolism is subsequently scavenged by FtH, thus conferring cytoprotection against heme-Fe. Abbreviations: Fe, iron; FtH, ferritin; Hp, haptoglobin; Hb, hemoglobin; HDL/LDL, high/low-density lipoprotein; Hx, hemopexin; ROS, reactive oxygen species.
Under pathophysiological conditions associated with more or less extensive intravascular hemolysis, such as malaria or different forms of hemolytic anemia (see Heme and the Pathogenesis of Infectious Diseases and Heme and the Pathogenesis of Non-Infectious Immune-Mediated Inflammatory Conditions), extracellular Hb can lead to the depletion of circulating Hp (Muller-Eberhard et al., 1968; Rother et al., 2005). When Hb accumulates in the circulation it becomes oxidized, releasing its heme groups (Bunn and Jandl, 1968; Hebbel et al., 1988; Pamplona et al., 2007; Ferreira et al., 2008). A number of circulating proteins can scavenge free heme in plasma (Bunn and Jandl, 1966; Figure 3), including hemopexin (Paoli et al., 1999; Tolosano et al., 2010), albumin (Fasano et al., 2007), α1-microglobulin (Allhorn et al., 2002) and high- and low-density lipoproteins (Miller and Shaklai, 1999).
Hemopexin (Hx) is an acute phase protein present at high concentrations in plasma (0.6–1.2 mg/mL), with the highest binding affinity for free heme (Kd < 10−12 in humans) of any described protein (Paoli et al., 1999; Tolosano et al., 2010). Heme:Hx complexes, which inhibits Fe-heme reactivity (Eskew et al., 1999), are recognized by the macrophage CD91 receptor (Hvidberg et al., 2005), allowing for intracellular heme catabolism (Figure 3; Alam and Smith, 1989). Hx is not degraded by macrophages, to any significant extent, being secreted back into the circulation (Figure 3; Hvidberg et al., 2005). As a consequence, Hx is less prone to complete depletion from the circulation, as compared to Hp.
Mice deficient in Hx and/or Hp (Hpx−/−Hpt−/−) develop severe renal damage when subjected to hemolysis, suggesting that Hx is part of a systemic protective mechanism against the deleterious effects of circulating free heme (Tolosano et al., 1999). The reason for which the kidney appears to be the first target of heme cytotoxicity in vivo is not clear, but is in keeping with the observation that induction of heme catabolism prevents kidney injury driven by rhabdomyolysis, where the hemoprotein, myoglobin, is released into circulation (Nath et al., 1992).
Albumin can bind heme with an affinity 104 times lower than Hx (Kd = 10−8; Little and Neilands, 1960). However, the high concentration of albumin in plasma (∼43 mg/mL) might compensate to some extent for its low affinity (Figure 3). Heme binding to albumin can neutralize the pro-oxidant effect of free heme, which may explain the protective effect of albumin infusion to individuals developing severe forms of sepsis (Delaney et al., 2011) or malaria (Maitland et al., 2005; Akech et al., 2006), the pathogenesis of which are driven to a significant extent by free heme (Pamplona et al., 2007; Ferreira et al., 2008; Seixas et al., 2009; Larsen et al., 2010; Raffaella Gozzelino et al., unpublished results).
α1-Microglobulin is a member of the lipocalin protein superfamily, that binds small lipophilic ligands (Flower et al., 2000). Cell-free Hb can cleave α1-microglobulin, into a truncated protein that binds (Kd = 10−6) and converts free heme into a yellow-brown proteinaceous chromophore, subsequently cleared by renal filtration (Allhorn et al., 2002). Binding of α1-microglobulin to heme neutralizes its pro-oxidant effect, suggesting that α1-microglobulin acts as a heme scavenger in circulation (Figure 3; Allhorn et al., 2002). Whether under pathophysiologic conditions α1-microglobulin plays a protective role against heme is unknown.
High- and low-density lipoproteins (HDL and LDL, respectively) can bind heme (Kd ∼ 10−11–10−12) with faster kinetics to those of Hx or albumin (Miller and Shaklai, 1999), thereby acting as the initial heme scavengers in circulation (Balla et al., 1991; Jeney et al., 2002). Once bound to HDL and LDL, heme can be transferred to Hx (Figure 3; Morgan et al., 1976; Hvidberg et al., 2005). In this manner, HDL and LDL may function as an initial “buffer” for circulating heme. Binding of heme to HDL or LDL oxidizes HDL and LDL that are then cleared via the CD36 and CD68 scavenger receptors expressed on macrophages (Camejo et al., 1998; Miller and Shaklai, 1999). Of note, however, oxidized HDL and LDL are cytotoxic (Jeney et al., 2002; Stocker and Perrella, 2006), suggesting that heme-mediated lipid peroxidation might be a key mechanism via which heme exerts its deleterious effects (Jeney et al., 2002; Stocker and Perrella, 2006). This also indicates that the beneficial role of HDL and LDL in circulation would be dependent on their rapid clearance that might otherwise be detrimental. Considering its high affinity for heme, Hx acts most probably as an ultimate heme scavenger in plasma, targeting heme bound to other carrier proteins or not, and allowing for heme delivery for intracellular degradation by the HO system (see HO-Catalyzed Heme Degradation).
Cellular control of free heme
Heme incorporation into hemoproteins
The rate of de novo heme synthesis must correspond accurately to its rate of incorporation into newly synthesized apo-hemoproteins. This is regulated at different levels via the control of heme (see Heme Metabolism) as well as apo-hemoprotein synthesis. Free heme can induce the transcription and hence the expression of several apo-hemoproteins, as demonstrated for Hb, myoglobin and neuroglobin in erythroid, muscle and neuronal cells, respectively (Bruns and London, 1965; Tahara et al., 1978; Graber and Woodworth, 1986; Zhu et al., 2002a,b). This evolutionary conserved strategy prevents intracellular heme accumulation, presumably limiting heme cytotoxicity.
HO-catalyzed heme degradation
Whereas HO-2 is responsible for homeostatic heme catabolism (see Heme Catabolism), HO-1 can be induced by different forms of stress (Choi and Alam, 1996). Oxidative stress can lead to heme release from hemoproteins (reviewed in Gozzelino et al., 2010), requiring a cellular adaptive response maintaining intracellular free heme below a cytotoxic threshold level. This is accomplished, to a large extent, via the induction of HMOX1 transcription and HO-1 expression (Alam et al., 1994, 1995; Choi and Alam, 1996; Alam and Cook, 2007). In line with this notion, mouse cells deficient in HO-1 expression (Hmox1−/−), and thus unable to increase heme catabolism in response to different forms of stress, display exacerbated cytotoxicity in response to oxidative stress (Poss and Tonegawa, 1997a,b; Yet et al., 2003; Bishop et al., 2004). Whether this is, as speculated hereby, due to the accumulation of free heme or whether HO-1 acts in a cytoprotective manner independently of its ability to neutralize the cytotoxic effect of free heme, remains unclear.
Induction of HO-1 expression in response to infection can be essential to sustain host survival, as demonstrated for systemic polymicrobial (Larsen et al., 2010) as well as for Plasmodium infection (Pamplona et al., 2007; Seixas et al., 2009), the causative agent of malaria. Accumulation of heme in the circulation is cytotoxic, thus leading to tissue damage and, as such, exacerbating the pathologic outcome of these diseases (Pamplona et al., 2007; Seixas et al., 2009; Larsen et al., 2010). HO-1 neutralizes this cytotoxic effect and hence provides tissue damage control and host protection against infections (Pamplona et al., 2007; Larsen et al., 2010; reviewed in Gozzelino et al., 2010). This host defense strategy acts irrespectively of pathogen load, a phenomenon referred to as host tolerance to infection (Medzhitov et al., 2012).
The cytoprotective action of HO-1 (Vile et al., 1994; Soares et al., 1998) resides not only in the degradation of cytotoxic heme, but also in the production of the different end products of heme catabolism, namely CO, BV, and labile Fe (Figure 3). CO has a variety of biological effects that mitigate the pathogenesis and/or progression of immune-mediated inflammatory diseases (Otterbein et al., 1999b; Fujita et al., 2001; Sato et al., 2001). These include its ability to bind the Fe atom in the prosthetic heme groups of hemoproteins, thereby modulating their function (Kim et al., 2006; Piantadosi, 2008; Mustafa et al., 2009). This mechanism of action probably underlies the cytoprotective (Brouard et al., 2000; Silva et al., 2006) and “anti-inflammatory” effects of CO (Otterbein et al., 2000) as well as the ability of this gasotransmitter to inhibit platelet activation and aggregation (Brune and Ullrich, 1987). In addition, binding of CO to the Fe-heme prevents the oxidation of hemoproteins, thereby slowing down heme release. When exerted over cell-free Hb (Hebbel et al., 1988; Pamplona et al., 2007), CO prevents heme release from Hb, suppressing the pathogenesis of severe forms of malaria (Pamplona et al., 2007; Ferreira et al., 2008). Presumably this mechanism should contribute to prevent the pathogenesis and/or progression of other immune-mediated inflammatory diseases associated with heme release from hemoproteins (reviewed in Ferreira et al., 2008; Gozzelino et al., 2010).
Biliverdin is converted into lipid phase antioxidant BR (Stocker et al., 1987) by BV reductase (BVR; Figure 3; Singleton and Laster, 1965; Kutty and Maines, 1981). The BV/BR system affords cytoprotection against reactive oxygen species (ROS; Dore et al., 1999; Baranano et al., 2002). Both free heme and BR are lipophilic, suggesting that the antioxidant action of BR may limit lipid oxidation by free heme. Presumably, the cytoprotective effect of BV/BR contributes to their overall protective effect against a range of immune-mediated inflammatory diseases (Yamashita et al., 2004; Ollinger et al., 2005, 2007; Sarady-Andrews et al., 2005; Overhaus et al., 2006; Wang et al., 2006).
The Fe released from the protoporphyrin IX ring of heme (Figure 3) can catalyze the generation of ROS via Fenton chemistry (Fenton, 1894). Cells can neutralize this cytotoxic effect by secreting Fe via Fe efflux pumps (Ferris et al., 1999; Baranano et al., 2000) or by storing Fe inside ferritin (Figure 3; Eisenstein et al., 1991; Harrison and Arosio, 1996; Baker et al., 2003), a multimeric protein complex composed of a heavy/heart chain (FtH) and a light/liver chain (FtL) that store up to 4500 Fe molecules (Harrison and Arosio, 1996). The FtH subunit possesses ferroxidase activity that allows it to catalyze the conversion of Fe2+ into Fe3+, required for Fe storage. FtH is cytoprotective against TNF (Ferris et al., 1999; Berberat et al., 2003; Cozzi et al., 2003; Xie et al., 2005) as well as heme-driven cytotoxicity (Balla et al., 1992). This salutary effect relies on the ability of FtH to neutralize the pro-oxidant activity of labile Fe, thereby preventing sustained JNK activation (Pham et al., 2004; Raffaella Gozzelino et al., unpublished results). Presumably, the cytoprotective effect of FtH prevents the pathogenesis of immune-mediated inflammatory diseases, such as demonstrated originally for ischemia–reperfusion injury (IRI; Berberat et al., 2003) and more recently for malaria (Raffaella Gozzelino et al., unpublished results).
Heme transport and secretion
Proteins involved in heme transport or intracellular heme scavenging (see Heme Transport) should regulate the pro-oxidant effects of heme (Figure 1; reviewed in Schultz et al., 2010; Khan and Quigley, 2011). Expression of the intracellular heme-binding thioredoxin-dependent peroxidase (PRX-1) is induced transcriptionally by heme as well as by oxidative stress (Immenschuh et al., 1995, 2005). PRX1 negates the pro-oxidant effect of free heme, preventing its deleterious effects (Immenschuh et al., 1995, 2005). Expression of the ubiquitous heme exporter, FLVCR, can also provide protection against cytotoxic free heme (Quigley et al., 2004; Keel et al., 2008; reviewed in Khan and Quigley, 2011). The Abcg2 transporter can export intracellular PPIX, delivering heme to extracellular albumin (Szatmari et al., 2006) and as such may be considered, together with FLVCR, as a therapeutic target to promote cell survival against heme-mediated cytotoxicity.
Heme and the Pathogenesis of Infectious Diseases
Failure to control the deleterious effects of free heme can contribute to the pathogenesis of a number of pathologies, associated with heme release from Hb or other hemoproteins.
Severe sepsis
Severe sepsis develops from an exacerbated inflammatory response to microbial infection, leading to systemic refractory hypotension, disseminated intravascular coagulation, multiple end-stage organ failure and ultimately to death (reviewed in Cohen, 2002; Hotchkiss and Karl, 2003). The mechanisms underlying the pathogenesis of severe sepsis remains poorly understood (reviewed in Cohen, 2002; Riedemann et al., 2003; Ulloa and Tracey, 2005). We have recently identified heme as a central component in the pathogenesis of this acute inflammatory disease (Larsen et al., 2010).
Severe sepsis driven by polymicrobial infection can be induced experimentally in mice by cecal ligation and puncture (CLP; Rittirsch et al., 2009). Disease progression is associated with RBC deformation (poikilocytosis), accumulation of cell-free Hb and free heme as well as with decreased concentration of Hp and Hx in plasma (Larsen et al., 2010). Poikilocytosis (Piagnerelli et al., 2003; Bain, 2005) and decreased concentration of Hx in plasma (Larsen et al., 2010) are also observed in human sepsis. This indicates that in the absence of overt hemolysis, significant amounts of Hb can be released from RBC resulting in heme accumulation in the circulation. A number of factors can contribute to this (Kempe et al., 2007; Lang et al., 2008), including deregulated plasma osmolarity, acidosis, bacterial hemolysins (particularly Clostridium perfringens; Gutierrez et al., 1995), or free heme itself (Sil et al., 2004). The pathophysiologic relevance of these findings is supported by the observations that lethality driven by severe sepsis is associated with decreased levels of Hx in plasma and that administration of purified Hx, after the onset of lethal sepsis in mice, improves host survival (Larsen et al., 2010). This suggests that targeting free heme, such as using Hx, might be used therapeutically to treat severe forms of sepsis.
As discussed above (see Pathophysiological Effects of Free Heme) free heme sensitizes cells in parenchymal tissues to undergo PCD in response to a variety of pro-oxidant and/or inflammatory compounds (Seixas et al., 2009; Gozzelino et al., 2010; Larsen et al., 2010). This cytotoxic effect might contribute to the development of organ failure associated with severe sepsis. This may also explain why therapeutic targeting of single pro-inflammatory molecules, e.g., the cytokine TNF (Cohen and Carlet, 1996) or IL-1 (Opal et al., 1997) have failed as a treatment for severe sepsis, while targeting free heme may be a valuable therapeutic option, since heme acts as a cytoxic catalyst for these as well as other pro-inflammatory molecules.
It should be noted that the accumulation of free heme in the circulation of septic patients and mice does not have to derive exclusively from the oxidation of cell-free Hb. It is possible that heme b is released from the oxidation of other hemoproteins to which it binds non-covalently, such as myoglobin released during rhabdomyolysis associated with septic shock (Kumar et al., 2009).
Malaria
Together with sepsis, malaria remains a major cause of morbidity and mortality worldwide, in particular in subtropical countries where it affects millions of individuals every year (Murray et al., 2012). Malaria develops in response to Plasmodium infection, at a specific stage of the life cycle of this protozoan parasite associated with RBC lysis (Miller et al., 2002). This so-called “blood stage” of infection is associated with hemolysis and hence with oxidation of cell-free Hb and production of free heme (Ferreira et al., 2008).
All the clinical manifestations of malaria, from fever to respiratory distress, circulatory collapse, abnormal bleeding, jaundice, hemoglobinuria, severe anemia, convulsions, prostration, and/or impaired consciousness are associated with the blood stage of infection (Idro et al., 2005) and as such with the accumulation of cell-free Hb and heme in plasma. When an individual presents with one or more of these symptoms, with no cause of disease other than Plasmodium infection, he is defined as developing a severe form of malaria (Marsh et al., 1995; Maitland and Marsh, 2004).
That non-Hb bound heme contributes to the pathogenesis of severe forms of malaria is suggested by the following observations in mice. Severity of Plasmodium infection correlates with the accumulation of cell-free Hb and non-Hb bound free heme in plasma (Pamplona et al., 2007; Ferreira et al., 2008; Andrade et al., 2010a). The same is true in humans (Andrade et al., 2010a). Free heme is sufficient to elicit the onset of severe forms of malaria in Plasmodium infected mice (Pamplona et al., 2007; Ferreira et al., 2008, 2011; Seixas et al., 2009). Heme catabolism by HO-1 suppresses the pathogenesis of severe forms of malaria in mice (Pamplona et al., 2007; Seixas et al., 2009; Ferreira et al., 2011) and is associated with the pathologic outcome of malaria in humans as well (Andrade et al., 2010a; Sambo et al., 2010; Walther et al., 2012).
The pathologic effect of free heme is most probably driven by the Fe atom released from the protoporphyrin ring of heme, either through catabolism by HO-1 (Tenhunen et al., 1968) or via non-enzymatic oxidative degradation (Schaefer et al., 1985; Nagababu and Rifkind, 2000, 2004). The deleterious effects of labile Fe can be countered by FtH (Balla et al., 1992), probably explaining why FtH prevents the onset of severe forms of malaria in mice (Raffaella Gozzelino et al., unpublished results).
Heme and the Pathogenesis of Non-Infectious Immune-Mediated Inflammatory Conditions
Hemolytic disorders
A number of hereditary diseases associated with defects in RBC function can lead to premature RBC senescence, associated with increased hematophagocytosis as well as with the release of Hb and heme into circulation. Heme toxicity may contribute to the clinical complications of these diseases, as illustrated in this section for sickle cell anemia and G6PD deficiency.
Sickle cell disease
Sickle cell disease is a molecular disease (Pauling et al., 1949) caused by a single point mutation in the β chain of Hb (β6Glu > Val), leading to the synthesis of a modified Hb variant generally referred to as HbS (Williams et al., 2005a; Jallow et al., 2009). Other sickle mutations include HbC (β6Glu > Lys; Modiano et al., 2001) and HbE (β6Glu > Lys; Hutagalung et al., 1999).
When present in the homozygous form, the Hb β6Glu > Val sickle mutation becomes pathologic, leading under low oxygen pressure to Hb polymerization, RBC deformation (sickle shape), shortened RBC half-live and eventually to hemolysis. This is associated with Hb and heme release into circulation (Hebbel et al., 1988; Reiter et al., 2002). The disease manifests by a range of clinical outcomes that include episodes of vascular occlusion/dysfunction eventually leading to the development of stroke and/or renal failure. Sickle cell anemia is also associated with tissue Fe overload, presumably driven by chronic accumulation of free heme in the circulation (Hebbel et al., 1988; Reiter et al., 2002).
Cell-free Hb is thought to contribute to the pathogenesis of sickle cell anemia, essentially by scavenging NO (Reiter et al., 2002), thereby promoting vasoconstriction, platelet aggregation and expression of adhesion molecules associated with endothelial cell activation (Belcher et al., 2003). Interaction of NO with cell-free Hb also catalyzes the production of free radicals, promoting Hb oxidation and heme release. Further oxidation of cell-free Hb can lead to the formation of ferrylHb aggregates acting as a pro-inflammatory agonist in the vascular endothelium (Silva et al., 2009).
Expression of HO-1 or administration of exogenous CO counters the pathological outcome of sickle cell disease (Belcher et al., 2006). Presumably this protective effect is mediated via inhibition of endothelial cell activation (Soares et al., 2004; Seldon et al., 2007). In addition, this salutary effect may also be mediated via inhibition of Hb oxidation, preventing heme release from cell-free Hb. If this proves to be the case, then targeting cell-free Hb or free heme in plasma using Hp or Hx should ameliorate the pathological outcome of sickle cell disease as well.
When present in the heterozygous form, the Hb β6Glu > Val sickle mutation is not pathogenic, conferring instead a net survival advantage to human population living in endemic areas of malaria (Williams et al., 2005b). This protective effect acts via a mechanism involving the accumulation of low (non-cytotoxic) levels of free heme in plasma that induce the expression of HO-1 via activation of the transcription factor NF-E2-related factor 2 (Nrf2; Ferreira et al., 2011). The CO produced via heme catabolism by HO-1 binds to cell-free Hb and prevents the accumulation of high levels (cytotoxic) of circulating free heme following Plasmodium infection, thus suppressing the pathogenesis of severe forms of malaria (Ferreira et al., 2011). This protective effect does not interfere with parasite load revealing that sickle Hb confers tolerance against Plasmodium infection (Ferreira et al., 2011). That is, it promotes host survival irrespectively of its pathogen load (Medzhitov et al., 2012).
Glucose-6-P dehydrogenase deficiency
RBC lack mitochondria and as such cannot generate NAPDH via oxidative phosphorylation. Glucose-6-phosphate dehydrogenase (G6PD) catalyzes the first step of the pentose phosphate pathway of glucose metabolism, and is a critical mechanism for the generation of reducing power via RBC NADPH. More than 140 G6PD mutations have been reported, leading to RBC oxidative stress and eventually to varying degrees of hemolysis (reviewed in Cappellini and Fiorelli, 2008). The clinical outcomes associated with G6PD deficiency are revealed clinically by fatigue, jaundice (i.e., hepatic damage), increased circulating unconjugated BR and lactate dehydrogenase as well as reticulocytosis (Cappellini and Fiorelli, 2008). Presumably, some of these clinical outcomes can be explained by the release of Hb and heme into circulation, associated to chronic or acute episodes of hemolysis.
G6PD deficiency is one of the most common enzyme mutation in human populations (Mason, 1996) and is particularly prevalent in geographical areas where malaria is endemic. This suggests that mutations leading to the functional silencing of the human G6PD gene were naturally selected through evolution on the basis of the survival advantage they provide against malaria, a major driving force in human evolution (Cappellini and Fiorelli, 2008). The molecular mechanism via which G6PD mutations confer protection against Plasmodium infection are not clear but, as discussed in the previous section, this is likely to be related to the mechanism involved in the protection conferred by sickle Hb against malaria.
Pathological complications of pregnancy
The early stages of pregnancy in mammals are associated with the induction of high levels of HO-1 expression by placental trophoblasts, as demonstrated in mice, rats, and humans (Ihara et al., 1998; Barber et al., 2001; Sollwedel et al., 2005; Zenclussen et al., 2011). This suggested that expression of HO-1 might be required to support early stages of pregnancy, a notion in keeping with the observation that Hmox1 deletion causes pre-natal lethality in mice (Poss and Tonegawa, 1997b; Yet et al., 1999; Tzima et al., 2009). Several studies have demonstrated that reduced HO-1 expression and/or activity can promote miscarriages in rodents as well as in humans (Zenclussen et al., 2003, 2005), which is also the case for pre-eclampsia, probably the most severe pathological complication associated to human pregnancy (Ahmed et al., 2000).
A more detailed analysis of the mechanisms involved in pre-natal lethality associated with Hmox1 deletion in mice revealed that HO-1 is required to sustain placental formation and function and hence fetal development and success of the early stages of pregnancy (Zenclussen et al., 2011). Presumably, the salutary effect of HO-1 relies on its ability to prevent the accumulation of free heme, generated in the placenta from the oxidation of cell-free Hb. This notion is supported by the observation that impairment of intrauterine fetal survival, driven by Hmox1 deletion, is associated with the accumulation of free heme in plasma, while heme administration to wild type (Hmox1+/+) mice is sufficient per se to impair fetal survival (Zenclussen et al., 2011). CO can be used therapeutically to prevent miscarriages in mice, inhibiting heme release from cell-free Hb and preventing the accumulation of circulating free heme (Zenclussen et al., 2011). This is, to the best of our knowledge, the first demonstration that heme can participate in the pathogenesis of an immune-mediated inflammatory condition that is not driven by infection.
Ischemia and reperfusion injury
Ischemia and reperfusion injury act as a major cause of pathology leading to myocardial infarction, stroke or damage associated with organ transplantation, among others. Ischemia refers to a more or less pronounced restriction of blood supply to a given organ, associated with lower than physiological supply of several essential molecules, including O2. In the event that blood flow is re-established, i.e., reperfusion, the ischemic tissue is confronted with an abrupt delivery of O2 that cannot be readily used by the mitochondrial electron transport chain. The excess O2 becomes available to oxidative enzymes, e.g., NADPH oxidase, catalyzing the generation of ROS, i.e., superoxide (O2•), hydrogen peroxide (H2O2) and hydroxyl radical (OH•). Accumulation of ROS above a certain threshold level causes oxidative stress, promoting PCD and tissue damage (Zhang et al., 2007a).
Expression of HO-1 exerts strong protective effects against IRI (reviewed in Soares and Bach, 2007), via a mechanism that is not clear but is likely to involve the catabolism of deleterious free heme, which is in keeping with the notion that hemolysis might act as an inherent component of the pathogenesis of IRI. Presumably, the cytotoxic effect of free heme contributes to promote tissue damage associated with IRI, explaining the salutary effects of HO-1 (Shen et al., 2005), CO (Fujita et al., 2001; Akamatsu et al., 2004) or FtH (Berberat et al., 2003) against this pathologic process.
Transplantation
Mouse grafts can survive indefinitely when transplanted into immunosuppressed rats (Miyatake et al., 1998). Long-term survival relies on a cytoprotective response elicited in the transplanted organ, a phenomenon termed by Fritz H. Bach as “accommodation” (Soares et al., 1999). HO-1 expression in the transplanted organ is required to promote accommodation (Soares et al., 1998, 1999), a salutary effect mediated at least partially by CO (Sato et al., 2001; reviewed in Soares and Bach, 2007). While there are probably several mechanisms that can contribute to explain the salutary effects of HO-1 and CO in the context of organ transplantation, these are likely mediated by the capacity of HO-1 to prevent the deleterious effect of free heme (see HO-Catalyzed Heme Degradation above). This remains however to be established experimentally.
Targeting Free Heme Therapeutically to Treat Immune-Mediated Inflammatory Diseases
Systemic heme targeting
Targeting heme proteins
While CO appears to be a major component of the protective action exerted by HO enzymatic activity (Otterbein et al., 1999b; Brouard et al., 2000, 2002; Fujita et al., 2001; Sato et al., 2001; Akamatsu et al., 2004; Soares et al., 2004), the mechanism underlying the salutary effects of this gasotransmitter remain unclear. CO can bind Fe2+ within the prosthetic heme groups of hemoproteins and hence modulate their biologic function (see HO-Catalyzed Heme Degradation). Presumably, this underlies the capacity of CO to act in a cytoprotective manner (Brouard et al., 2000, 2002; Silva et al., 2006) as well as to modulate macrophage activation (Otterbein et al., 2000) and to control cell proliferation (Otterbein et al., 2003).
There is another major biological output associated with CO binding to the Fe atom in the prosthetic heme groups of hemoproteins that contributes to the salutary effects of this gasotransmitter. Namely, CO can inhibit Fe-heme oxidation, preventing as well hemoprotein oxidation. This inhibits heme release from hemoproteins, as demonstrated for Hb (Pamplona et al., 2007; Figure 4), which is sufficient per se to explain how this gasotransmitter prevents the pathological outcome of malaria (Pamplona et al., 2007; Ferreira et al., 2008; Rosenthal, 2011). This suggests that CO may be used pharmacologically as a supportive therapy for the treatment of life-threatening complications of malaria.
Figure 4

Therapeutic targeting of free heme. Free heme, and its pathophysiological effects, might be targeted directly and indirectly, extra- and intracellularly, by a range of mechanisms. Extracellularly, albumin and the two naturally occurring acute phase proteins, Hp and Hx, can scavenge cell-free Hb and free heme, respectively, whereas CO can prevent the release of heme from Hb. Intracellularly, induction of HO-1 or FtH, might target excess heme and labile Fe, respectively. Antioxidants such as BR and the heme carrier protein PRX1, could prevent the participation of heme-Fe in the generation of unfettered production of free radicals (ROS). Specific heme transport between the intra- and extracellular compartments, including Abcg2, FLVCR, HCP1 and HRG-1, might be considered as potential, therapeutic targets to afford protection against heme cytotoxicity. Abbreviations: Bcrp/Abcg2, breast cancer resistance protein/ATP-binding cassette g2; BR, bilirubin; CO, carbon monoxide; Fe, iron; FLVCR, feline leukemic virus receptor; FtH, ferritin; Hb, hemoglobin; HCP1, heme carrier protein-1; Hp, haptoglobin; HO-1, heme oxygenase-1; HRG-1, heme responsive gene-1; Hx, hemopexin; ROS, reactive oxygen species.
It is likely that in a similar manner to Hb, CO may also prevent heme release from other hemoproteins such as myoglobin. Whether this would limit the pathological outcome of diseases associated with the accumulation of cell-free myoglobin, such as during rhabdomyolysis (Kumar et al., 2009), remains to be established.
While the evidence for the therapeutic application of CO in a number of diseases is solid, the clinical application of this gaseous molecule poses a number of practical challenges. Recently, the development of CO-releasing molecules (CORMs) received considerable attention as an alternative means of CO delivery (Foresti et al., 2005; Motterlini and Otterbein, 2010). A number of studies have demonstrated that the therapeutic benefits of CORMs mimic or can even excel those of CO inhalation (Pena et al., 2012).
Targeting free heme using natural heme scavengers
Under pathologic conditions associated with severe, acute, or chronic hemolysis, as occurs under certain infectious and non-infectious inherited diseases (see Heme and the Pathogenesis of Infectious Diseases and Heme and the Pathogenesis of Non-Infectious Immune-Mediated Inflammatory Conditions), the physiologic Hb and heme scavengers become saturated (Muller-Eberhard, 1970; Larsen et al., 2010). Therefore, replenishing the circulating stores of heme scavengers, thereby compensating for the loss of heme scavenging capacity in plasma, may be used as a therapeutic approach to target circulating free heme and prevent its deleterious effects.
Two clinical trials demonstrated that the administration of albumin, a heme scavenger, provides a significant survival benefit to children developing severe forms of malaria (Maitland et al., 2005; Akech et al., 2006). Similar data has recently been reported in the context of severe sepsis (Delaney et al., 2011). Whether the therapeutic benefit of albumin is due to its heme scavenging capacity remains to be established.
We have recently shown that exogenously administered Hx can prevent the lethal outcome of severe sepsis in mice (Larsen et al., 2010), suggesting that this may also be a viable therapeutic intervention against sepsis and possibly against other pathologies associated with a hemolytic component, such as malaria, sickle cell disease or other inherited hemolytic disorders (Figure 4).
Cellular heme targeting
Cellular targeting of hemoproteins or heme transporters may counter the deleterious effects of intracellular accumulation of free heme and, as such, limit tissue damage and ameliorate the pathological outcome of immune-mediated inflammatory diseases. This may be accomplished by the pharmacological use of CO that in a similar manner to other gasotransmitters can diffuse freely into cells and, as such, target intracellular hemoproteins. In keeping with this notion, the cytoprotective effect of CO (Brouard et al., 2000) may in this manner limit tissue damage associated with the pathological outcome of immune-mediated inflammatory diseases (Otterbein et al., 2000; Fujita et al., 2001; Sato et al., 2001; Akamatsu et al., 2004; Pamplona et al., 2007; Ferreira et al., 2011). Given that the cytoprotective effect of CO acts via a mechanism targeting the mitochondria (Queiroga et al., 2010; Wang et al., 2011) it is possible that CO would also target mitochondrial hemoproteins that play a central role in the execution of PCD, e.g., cythocrome c.
Targeting heme transport may also be used therapeutically to prevent heme-driven tissue damage (Figure 4). This type of approach is based on the assumption that the cytotoxic effects of free heme are mediated via a mechanism that involves its transport by dedicated heme transporters. If this proves to be the case, then targeting these transporters may provide salutary effects against immune-mediated inflammatory diseases. The general principle may be that favoring the activity of heme exporters, such as FLVCR or Abcg2 may allow cells to secrete heme and, as such, to prevent its pro-oxidant effects leading to PCD and tissue damage. In a similar manner, inhibiting cellular heme import such as by targeting HRG-1 and/or HCP1 may also be used to limit intracellular heme accumulation (Figure 4; Gozzelino et al., 2010).
Finally, intracellular heme might be targeted by the modulation of HO-1 expression and/or activity. This has been shown using recombinant virus-mediated overexpression of HO-1 in animal models of disease. These include vasospasm following subarachnoid hemorrhage in rats (Ono et al., 2002), transplantation of myogenic precursor cells in pigs (Laumonier et al., 2008), IRI following liver transplant in rats (Coito et al., 2002), hyperoxia-induced lung injury in rats (Otterbein et al., 1999a), and atherosclerotic diseases (Stocker and Perrella, 2006), as reviewed in (Abraham et al., 2007).
Alternatively, the deleterious effects of cellular heme can be neutralized by the therapeutic induction of endogenous HO-1, via the delivery of non-cytotoxic levels of heme or heme-containing proteins, e.g., by so-called preconditioning. In this manner, the administration of Hb in rats prior to challenge, was shown to improve survival in response to endotoxic shock (Otterbein et al., 1995), liver injury and kidney failure following rhabdomyolysis (Nath et al., 1992). Interestingly, a physiological example of heme preconditioning is represented by sickle cell trait, in which individuals carry the HbS mutation (see Sickle Cell Disease) in the heterozygous form (Williams et al., 2005a,b; Jallow et al., 2009). This mutation confers protection against severe forms of malaria due to the continuous release of low concentrations of heme into circulation, thereby leading to induction of endogenous HO-1 expression and protection against Plasmodium infection (Ferreira et al., 2011).
Conclusion
Although not highly cytotoxic per se, free heme sensitizes non-hematopoietic cells to undergo PCD in response to a variety of pro-inflammatory agonists. This unique pathophysiologic feature relies on the capacity of heme to promote the production of free radicals in an unfettered manner. Several protection mechanisms, regulating heme synthesis, catabolism and transport, can be considered as possible therapeutic targets to prevent the deleterious effects of free heme. In keeping with this notion, tissue damage control afforded by genes involved in heme and Fe metabolism, such as HO-1 and FtH, provide a “proof of principle” for the therapeutic benefits associated with neutralization of heme-Fe cytotoxicity in the context of immune-mediated inflammatory diseases.
Statements
Acknowledgments
The authors thank Ana Cunha (Instituto Gulbenkian de Ciência, Oeiras, Portugal) for carefully reading the manuscript and providing critical comments. This work was funded by Fundação para a Ciência e Tecnologia (FCT, Portugal) grants to RL (SFRH/BPD/25436/2005), ZG (SFRH/BD/44828/2008) and RG (SFRH/BPD/44256/2008), MPS was funded by FCT (PTDC/BIA-BCM/101311/2008, PTDC/SAU-FCF/100762/2008 and PTDC/SAU-TOX/116627/2010), the European Community, 6th Framework, Grant LSH-2005-1.2.5-1, XENOME, and The Bill & Melinda Gates Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbrahamN. G.AsijaA.DrummondG.PetersonS. (2007). Heme oxygenase -1 gene therapy: recent advances and therapeutic applications. Curr. Gene Ther.7, 89–108.10.2174/156652307780363134
2
AhmedA.RahmanM.ZhangX.AcevedoC. H.NijjarS.RushtonI.BussolatiB.St JohnJ. (2000). Induction of placental heme oxygenase-1 is protective against TNFalpha-induced cytotoxicity and promotes vessel relaxation. Mol. Med.6, 391–409.
3
AkamatsuY.HagaM.TyagiS.YamashitaK.Graca-SouzaA. V.OllingerR.CzismadiaE.MayG. A.IfedigboE.OtterbeinL. E.BachF. H.SoaresM. P. (2004). Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J.18, 771–772.
4
AkechS.GwerS.IdroR.FeganG.EziefulaA. C.NewtonC. R.LevinM.MaitlandK. (2006). Volume expansion with albumin compared to gelofusine in children with severe malaria: results of a controlled trial. PLoS Clin. Trials1, e21.10.1371/journal.pctr.0010021
5
AlamJ.CaiJ.SmithA. (1994). Isolation and characterization of the mouse heme oxygenase-1 gene. Distal 5′ sequences are required for induction by heme or heavy metals. J. Biol. Chem.269, 1001–1009.
6
AlamJ.CamhiS.ChoiA. M. (1995). Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J. Biol. Chem.270, 11977–11984.10.1074/jbc.270.20.11977
7
AlamJ.CookJ. L. (2007). How many transcription factors does it take to turn on the heme oxygenase-1 gene?Am. J. Respir. Cell. Mol. Biol.36, 166–174.10.1165/rcmb.2006-0340TR
8
AlamJ.SmithA. (1989). Receptor-mediated transport of heme by hemopexin regulates gene expression in mammalian cells. J. Biol. Chem.264, 17637–17640.
9
AllenJ. W.DaltropO.StevensJ. M.FergusonS. J. (2003). C-type cytochromes: diverse structures and biogenesis systems pose evolutionary problems. Philos. Trans. R. Soc. Lond. B Biol. Sci.358, 255–266.10.1098/rstb.2002.1192
10
AllhornM.BerggardT.NordbergJ.OlssonM. L.AkerstromB. (2002). Processing of the lipocalin alpha(1)-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood99, 1894–1901.10.1182/blood.V99.6.1894
11
AndradeB. B.Araujo-SantosT.LuzN. F.KhouriR.BozzaM. T.CamargoL. M.BarralA.BorgesV. M.Barral-NettoM. (2010a). Heme impairs prostaglandin E2 and TGF-beta production by human mononuclear cells via Cu/Zn superoxide dismutase: insight into the pathogenesis of severe malaria. J. Immunol.185, 1196–1204.10.4049/jimmunol.0904179
12
AndradeB. B.Reis-FilhoA.Souza-NetoS. M.ClarencioJ.CamargoL. M.BarralA.Barral-NettoM. (2010b). Severe Plasmodium vivax malaria exhibits marked inflammatory imbalance. Malar. J.9, 13.10.1186/1475-2875-9-S2-P13
13
AntosiewiczJ.ZiolkowskiW.KaczorJ. J.Herman-AntosiewiczA. (2007). Tumor necrosis factor-alpha-induced reactive oxygen species formation is mediated by JNK1-dependent ferritin degradation and elevation of labile iron pool. Free Radic. Biol. Med.43, 265–270.10.1016/j.freeradbiomed.2007.04.023
14
AonoS. (2008). Metal-containing sensor proteins sensing diatomic gas molecules. Dalton Trans.24, 3137–3146.10.1039/b802070c
15
AzumaM.KabeY.KuramoriC.KondoM.YamaguchiY.HandaH. (2008). Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS ONE3, e3070.10.1371/journal.pone.0003070
16
BadyalS. K.JoyceM. G.SharpK. H.SewardH. E.MewiesM.BasranJ.MacdonaldI. K.MoodyP. C.RavenE. L. (2006). Conformational mobility in the active site of a heme peroxidase. J. Biol. Chem.281, 24512–24520.10.1074/jbc.M602602200
17
BainB. J. (2005). Diagnosis from the blood smear. N. Engl. J. Med.353, 498–507.10.1056/NEJMra043442
18
BakerH. M.AndersonB. F.BakerE. N. (2003). Dealing with iron: common structural principles in proteins that transport iron and heme. Proc. Natl. Acad. Sci. U.S.A.100, 3579–3583.10.1073/pnas.0237313100
19
BallaG.JacobH. S.BallaJ.RosenbergM.NathK.AppleF.EatonJ. W.VercellottiG. M. (1992). Ferritin: a cytoprotective antioxidant strategem of endothelium. J. Biol. Chem.267, 18148–18153.
20
BallaG.JacobH. S.EatonJ. W.BelcherJ. D.VercellottiG. M. (1991). Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb.11, 1700–1711.10.1161/01.ATV.11.6.1700
21
BallaJ.JacobH. S.BallaG.NathK.EatonJ. W.VercellottiG. M. (1993). Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. U.S.A.90, 9285–9289.10.1073/pnas.90.20.9285
22
BarananoD. E.RaoM.FerrisC. D.SnyderS. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A.99, 16093–16098.10.1073/pnas.252626999
23
BarananoD. E.WoloskerH.BaeB. I.BarrowR. K.SnyderS. H.FerrisC. D. (2000). A mammalian iron ATPase induced by iron. J. Biol. Chem.275, 15166–15173.10.1074/jbc.275.20.15166
24
BarberA.RobsonS. C.MyattL.BulmerJ. N.LyallF. (2001). Heme oxygenase expression in human placenta and placental bed: reduced expression of placenta endothelial HO-2 in preeclampsia and fetal growth restriction. FASEB J.15, 1158–1168.10.1096/fj.00-0376com
25
BattistuzziG.BelleiM.BortolottiC. A.SolaM. (2010). Redox properties of heme peroxidases. Arch. Biochem. Biophys.500, 21–36.10.1016/j.abb.2010.03.002
26
BelcherJ. D.BryantC. J.NguyenJ.BowlinP. R.KielbikM. C.BischofJ. C.HebbelR. P.VercellottiG. M. (2003). Transgenic sickle mice have vascular inflammation. Blood101, 3953–3959.10.1182/blood-2002-10-3313
27
BelcherJ. D.MahasethH.WelchT. E.OtterbeinL. E.HebbelR. P.VercellottiG. M. (2006). Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J. Clin. Invest.116, 808–816.10.1172/JCI26857
28
BerberatP. O.KatoriM.KaczmarekE.AnselmoD.LassmanC.KeB.ShenX.BusuttilR. W.YamashitaK.CsizmadiaE.TyagiS.OtterbeinL. E.BrouardS.TobiaschE.BachF. H.Kupiec-WeglinskiJ. W.SoaresM. P. (2003). Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J.17, 1724–1726.
29
BernhardtR. (1996). Cytochrome P450: structure, function, and generation of reactive oxygen species. Rev. Physiol. Biochem. Pharmacol.127, 137–221.10.1007/BFb0048267
30
BishopA.YetS. F.LeeM. E.PerrellaM. A.DempleB. (2004). A key role for heme oxygenase-1 in nitric oxide resistance in murine motor neurons and glia. Biochem. Biophys. Res. Commun.325, 3–9.10.1016/j.bbrc.2004.10.010
31
BowmanS. E.BrenK. L. (2008). The chemistry and biochemistry of heme c: functional bases for covalent attachment. Nat. Prod. Rep.25, 1118–1130.10.1039/b717196j
32
BrouardS.BerberatP. O.TobiaschE.SeldonM. P.BachF. H.SoaresM. P. (2002). Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J. Biol. Chem.277, 17950–17961.10.1074/jbc.M108317200
33
BrouardS.OtterbeinL. E.AnratherJ.TobiaschE.BachF. H.ChoiA. M.SoaresM. P. (2000). Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med.192, 1015–1026.10.1084/jem.192.7.1015
34
BruneB.UllrichV. (1987). Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol. Pharmacol.32, 497–504.
35
BrunsG. P.LondonI. M. (1965). The effect of hemin on the synthesis of globin. Biochem. Biophys. Res. Commun.18, 236–242.10.1016/0006-291X(65)90746-1
36
BunnH. F.JandlJ. H. (1966). Exchange of heme among hemoglobin molecules. Proc. Natl. Acad. Sci. U.S.A.56, 974–978.10.1073/pnas.56.3.974
37
BunnH. F.JandlJ. H. (1968). Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol. Chem.243, 465–475.
38
BushnellG. W.LouieG. V.BrayerG. D. (1990). High-resolution three-dimensional structure of horse heart cytochrome c. J. Mol. Biol.214, 585–595.10.1016/0022-2836(90)90200-6
39
CamejoG.HalbergC.Manschik-LundinA.Hurt-CamejoE.RosengrenB.OlssonH.HanssonG. I.ForsbergG. B.YlhenB. (1998). Hemin binding and oxidation of lipoproteins in serum: mechanisms and effect on the interaction of LDL with human macrophages. J. Lipid Res.39, 755–766.
40
CappelliniM. D.FiorelliG. (2008). Glucose-6-phosphate dehydrogenase deficiency. Lancet371, 64–74.10.1016/S0140-6736(08)60073-2
41
CaugheyW. S.SmytheG. A.O’KeeffeD. H.MaskaskyJ. E.SmithM. I. (1975). Heme A of cytochrome c oxicase. Structure and properties: comparisons with hemes B, C, and S and derivatives. J. Biol. Chem.250, 7602–7622.
42
ChapmanS. K.DaffS.MunroA. W. (1997). Heme: the most versatile redox centre in biology?Metal Sites Proteins Models88, 39–70.10.1007/3-540-62870-3_2
43
ChayT. R.BrillhartD. (1974a). Mechanism of cooperative oxygen binding to hemoglobin: kinetic aspects. Biochemistry13, 5311–5317.10.1021/bi00719a017
44
ChayT. R.BrillhartD. K. (1974b). Mechanism of cooperative oxygen binding to hemoglobin: equilibrium aspects. Biochemistry13, 4579–4585.10.1021/bi00719a017
45
ChoiA. M.AlamJ. (1996). Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am. J. Respir. Cell Mol. Biol.15, 9–19.
46
CohenJ. (2002). The immunopathogenesis of sepsis. Nature420, 885–891.10.1038/nature01326
47
CohenJ.CarletJ. (1996). INTERSEPT: an international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International Sepsis Trial Study Group. Crit. Care Med.24, 1431–1440.10.1097/00003246-199609000-00002
48
CoitoA. J.BuelowR.ShenX. D.AmersiF.MooreC.VolkH. D.BusuttilR. W.Kupiec-WeglinskiJ. W. (2002). Heme oxygenase-1 gene transfer inhibits inducible nitric oxide synthase expression and protects genetically fat Zucker rat livers from ischemia-reperfusion injury. Transplantation74, 96–102.10.1097/00007890-200207150-00017
49
CozziA.LeviS.CorsiB.SantambrogioP.CampanellaA.GerardiG.ArosioP. (2003). Role of iron and ferritin in TNFalpha-induced apoptosis in HeLa cells. FEBS Lett.537, 187–192.10.1016/S0014-5793(03)00114-5
50
CraneB. R.ArvaiA. S.GachhuiR.WuC.GhoshD. K.GetzoffE. D.StuehrD. J.TainerJ. A. (1997). The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science278, 425–431.10.1126/science.278.5337.425
51
DavisR. J. (2000). Signal transduction by the JNK group of MAP kinases. Cell103, 239–252.10.1016/S0092-8674(00)00116-1
52
DelaneyA. P.DanA.McCaffreyJ.FinferS. (2011). The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit. Care Med.39, 386–391.10.1097/CCM.0b013e3181ffe217
53
DeyA.GhoshA. (2002). “True” iron(V) and iron(VI) porphyrins: a first theoretical exploration. J. Am. Chem. Soc.124, 3206–3207.10.1021/ja0126125
54
DioumE. M.RutterJ.TuckermanJ. R.GonzalezG.Gilles-GonzalezM. A.McKnightS. L. (2002). NPAS2: a gas-responsive transcription factor. Science298, 2385–2387.10.1126/science.1078456
55
DoreS.TakahashiM.FerrisC. D.ZakharyR.HesterL. D.GuastellaD.SnyderS. H. (1999). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U.S.A.96, 2445–2450.10.1073/pnas.96.5.2445
56
EisensteinR. S.Garcia-MayolD.PettingellW.MunroH. N. (1991). Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc. Natl. Acad. Sci. U.S.A.88, 688–692.10.1073/pnas.88.3.688
57
EskewJ. D.VanacoreR. M.SungL.MoralesP. J.SmithA. (1999). Cellular protection mechanisms against extracellular heme. heme-hemopexin, but not free heme, activates the N-terminal c-jun kinase. J. Biol. Chem.274, 638–648.10.1074/jbc.274.2.638
58
EvansS. V.BrayerG. D. (1988). Horse heart metmyoglobin. A 2.8-A resolution three-dimensional structure determination. J. Biol. Chem.263, 4263–4268.
59
EvansS. V.BrayerG. D. (1990). High-resolution study of the three-dimensional structure of horse heart metmyoglobin. J. Mol. Biol.213, 885–897.10.1016/S0022-2836(05)80270-0
60
FallerM.MatsunagaM.YinS.LooJ. A.GuoF. (2007). Heme is involved in microRNA processing. Nat. Struct. Mol. Biol.14, 23–29.10.1038/nsmb1182
61
FasanoM.FanaliG.LeboffeL.AscenziP. (2007). Heme binding to albuminoid proteins is the result of recent evolution. IUBMB Life59, 436–440.10.1080/15216540701474523
62
FentonH. J. H. (1894). Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans.65, 899–911.10.1039/ct8946500899
63
FermiG.PerutzM. F.ShaananB.FourmeR. (1984). The crystal structure of human deoxyhaemoglobin at 1.74 A resolution. J. Mol. Biol.175, 159–174.10.1016/0022-2836(84)90472-8
64
FernandezP. L.DutraF. F.AlvesL.FigueiredoR. T.Mourao-SaD.FortesG. B.BergstrandS.LonnD.CevallosR. R.PereiraR. M.LopesU. G.TravassosL. H.PaivaC. N.BozzaM. T. (2010). Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J. Biol. Chem.285, 32844–32851.10.1074/jbc.M109.064618
65
FerreiraA.BallaJ.JeneyV.BallaG.SoaresM. P. (2008). A central role for free heme in the pathogenesis of severe malaria: the missing link?J. Mol. Med.86, 1097–1111.10.1007/s00109-008-0339-x
66
FerreiraA.MargutiI.BechmannI.JeneyV.ChoraA.PalhaN. R.RebeloS.HenriA.BeuzardY.SoaresM. P. (2011). Sickle hemoglobin confers tolerance to Plasmodium infection. Cell145, 398–409.10.1016/j.cell.2011.03.049
67
FerrisC. D.JaffreyS. R.SawaA.TakahashiM.BradyS. D.BarrowR. K.TysoeS. A.WoloskerH.BarananoD. E.DoreS.PossK. D.SnyderS. H. (1999). Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat. Cell Biol.1, 152–157.10.1038/11072
68
FigueiredoR. T.FernandezP. L.Mourao-SaD. S.PortoB. N.DutraF. F.AlvesL. S.OliveiraM. F.OliveiraP. L.Graca-SouzaA. V.BozzaM. T. (2007). Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem.282, 20221–20229.10.1074/jbc.M610737200
69
FlowerD. R.NorthA. C.SansomC. E. (2000). The lipocalin protein family: structural and sequence overview. Biochim. Biophys. Acta1482, 9–24.10.1016/S0167-4838(00)00148-5
70
ForestiR.ShureyC.AnsariT.SibbonsP.MannB. E.JohnsonT. R.GreenC. J.MotterliniR. (2005). Reviewing the use of carbon monoxide-releasing molecules (CO-RMs) in biology: implications in endotoxin-mediated vascular dysfunction. Cell. Mol. Biol.51, 409–423.
71
FortesG. B.AlvesL. S.de OliveiraR.DutraF. F.RodriguesD.FernandezP. L.Souto-PadronT.De RosaM. J.KelliherM.GolenbockD.ChanF. K.BozzaM. T. (2012). Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood119, 2368–2375.10.1182/blood-2011-08-375303
72
FrancisS. E.SullivanD. J.Jr.GoldbergD. E. (1997). Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol.51, 97–123.10.1146/annurev.micro.51.1.97
73
FufezanC.ZhangJ.GunnerM. R. (2008). Ligand preference and orientation in b- and c-type heme-binding proteins. Proteins73, 690–704.10.1002/prot.22097
74
FujitaT.TodaK.KarimovaA.YanS. F.NakaY.YetS. F.PinskyD. J. (2001). Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat. Med.7, 598–604.10.1038/87929
75
GozzelinoR.JeneyV.SoaresM. P. (2010). Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol.50, 323–354.10.1146/annurev.pharmtox.010909.105600
76
GozzelinoR.SoaresM. P. (2011). Heme sensitization to TNF-mediated programmed cell death. Adv. Exp. Med. Biol.691, 211–219.10.1007/978-1-4419-6612-4_22
77
GraberS. G.WoodworthR. C. (1986). Myoglobin expression in L6 muscle cells. Role of differentiation and heme. J. Biol. Chem.261, 9150–9154.
78
GrayH. B.WinklerJ. R. (1996). Electron transfer in proteins. Annu. Rev. Biochem.65, 537–561.10.1146/annurev.bi.65.070196.002541
79
GutierrezA.FlorencioR.EzpeletaC.CisternaR.MartinezM. (1995). Fatal intravascular hemolysis in a patient with Clostridium perfringens septicemia. Clin. Infect. Dis.20, 1064–1065.10.1093/clinids/20.4.1064
80
HamiltonJ. W.BementW. J.SinclairP. R.SinclairJ. F.AlcedoJ. A.WetterhahnK. E. (1991). Heme regulates hepatic 5-aminolevulinate synthase mRNA expression by decreasing mRNA half-life and not by altering its rate of transcription. Arch. Biochem. Biophys.289, 387–392.10.1016/0003-9861(91)90428-L
81
HanS. W.ChingY. C.HammesS. L.RousseauD. L. (1991). Vibrational structure of the formyl group on heme a. Implications on the properties of cytochrome c oxidase. Biophys. J.60, 45–52.10.1016/S0006-3495(91)82029-X
82
HarrisonP. M.ArosioP. (1996). The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta1275, 161–203.10.1016/0005-2728(96)00022-9
83
HebbelR. P.MorganW. T.EatonJ. W.HedlundB. E. (1988). Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc. Natl. Acad. Sci. U.S.A.85, 237–241.10.1073/pnas.85.1.237
84
HeinemannI. U.JahnM.JahnD. (2008). The biochemistry of heme biosynthesis. Arch. Biochem. Biophys.474, 238–251.10.1016/j.abb.2008.02.015
85
HiraS.TomitaT.MatsuiT.IgarashiK.Ikeda-SaitoM. (2007). Bach1, a heme-dependent transcription factor, reveals presence of multiple heme binding sites with distinct coordination structure. IUBMB Life59, 542–551.10.1080/15216540701225941
86
HorriganF. T.HeinemannS. H.HoshiT. (2005). Heme regulates allosteric activation of the Slo1 BK channel. J. Gen. Physiol.126, 7–21.10.1085/jgp.200509262
87
HotchkissR. S.KarlI. E. (2003). The pathophysiology and treatment of sepsis. N. Engl. J. Med.348, 138–150.10.1056/NEJMra021333
88
HutagalungR.WilairatanaP.LooareesuwanS.BrittenhamG. M.AikawaM.GordeukV. R. (1999). Influence of hemoglobin E trait on the severity of Falciparum malaria. J. Infect. Dis.179, 283–286.10.1086/514287
89
HvidbergV.ManieckiM. B.JacobsenC.HojrupP.MollerH. J.MoestrupS. K. (2005). Identification of the receptor scavenging hemopexin-heme complexes. Blood106, 2572–2579.10.1182/blood-2005-03-1185
90
IdroR.JenkinsN. E.NewtonC. R. (2005). Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol.4, 827–840.10.1016/S1474-4422(05)70247-7
91
IgarashiK.SunJ. (2006). The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid. Redox Signal.8, 107–118.10.1089/ars.2006.8.107
92
IharaN.AkagiR.EjiriK.KudoT.FuruyamaK.FujitaH. (1998). Developmental changes of gene expression in heme metabolic enzymes in rat placenta. FEBS Lett.439, 163–167.10.1016/S0014-5793(98)01324-6
93
ImmenschuhS.FahimiH. D.Baumgart-VogtE. (2005). Complementary regulation of heme oxygenase-1 and peroxiredoxin I gene expression by oxidative stress in the liver. Cell. Mol. Biol.51, 471–477.
94
ImmenschuhS.IwaharaS.SatohH.NellC.KatzN.Muller-EberhardU. (1995). Expression of the mRNA of heme-binding protein 23 is coordinated with that of heme oxygenase-1 by heme and heavy metals in primary rat hepatocytes and hepatoma cells. Biochemistry34, 13407–13411.10.1021/bi00041a018
95
JallowM.TeoY. Y.SmallK. S.RockettK. A.DeloukasP.ClarkT. G.KivinenK.BojangK. A.ConwayD. J.PinderM.SirugoG.Sisay-JoofF.UsenS.AuburnS.BumpsteadS. J.CampinoS.CoffeyA.DunhamA.FryA. E.GreenA.GwilliamR.HuntS. E.InouyeM.JeffreysA. E.MendyA.PalotieA.PotterS.RagoussisJ.RogersJ.RowlandsK.SomaskantharajahE.WhittakerP.WiddenC.DonnellyP.HowieB.MarchiniJ.MorrisA.SanJoaquinM.AchidiE. A.AgbenyegaT.AllenA.AmoduO.CorranP.DjimdeA.DoloA.DoumboO. K.DrakeleyC.DunstanS.EvansJ.FarrarJ.FernandoD.HienT. T.HorstmannR. D.IbrahimM.KarunaweeraN.KokwaroG.KoramK. A.LemngeM.MakaniJ.MarshK.MichonP.ModianoD.MolyneuxM. E.MuellerI.ParkerM.PeshuN.PloweC. V.PuijalonO.ReederJ.ReyburnH.RileyE. M.SakuntabhaiA.SinghasivanonP.SirimaS.TallA.TaylorT. E.TheraM.Troye-BlombergM.WilliamsT. N.WilsonM.KwiatkowskiD. P. (2009). Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat. Genet.41, 657–665.10.1038/ng.388
96
JeneyV.BallaJ.YachieA.VargaZ.VercellottiG. M.EatonJ. W.BallaG. (2002). Pro-oxidant and cytotoxic effects of circulating heme. Blood100, 879–887.10.1182/blood.V100.3.879
97
JeongJ.RouaultT. A.LevineR. L. (2004). Identification of a heme-sensing domain in iron regulatory protein 2. J. Biol. Chem.279, 45450–45454.10.1074/jbc.M406667200
98
KaasikK.LeeC. C. (2004). Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature430, 467–471.10.1038/nature02724
99
KakarS.HoffmanF. G.StorzJ. F.FabianM.HargroveM. S. (2010). Structure and reactivity of hexacoordinate hemoglobins. Biophys. Chem.152, 1–14.10.1016/j.bpc.2010.08.008
100
KamataH.HondaS.MaedaS.ChangL.HirataH.KarinM. (2005). Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell120, 649–661.10.1016/j.cell.2004.12.041
101
KarpeforsM.AdelrothP.NamslauerA.ZhenY.BrzezinskiP. (2000). Formation of the “peroxy” intermediate in cytochrome c oxidase is associated with internal proton/hydrogen transfer. Biochemistry39, 14664–14669.10.1021/bi992719t
102
KeelS. B.DotyR. T.YangZ.QuigleyJ. G.ChenJ.KnoblaughS.KingsleyP. D.De DomenicoI.VaughnM. B.KaplanJ.PalisJ.AbkowitzJ. L. (2008). A heme export protein is required for red blood cell differentiation and iron homeostasis. Science319, 825–828.10.1126/science.1151133
103
KempeD. S.AkelA.LangP. A.HermleT.BiswasR.MuresanuJ.FriedrichB.DreischerP.WolzC.SchumacherU.PeschelA.GotzF.DoringG.WiederT.GulbinsE.LangF. (2007). Suicidal erythrocyte death in sepsis. J. Mol. Med.85, 273–281.10.1007/s00109-006-0123-8
104
KhanA. A.QuigleyJ. G. (2011). Control of intracellular heme levels: heme transporters and heme oxygenases. Biochim. Biophys. Acta1813, 668–682.10.1016/j.bbamcr.2011.01.008
105
KimH. P.RyterS. W.ChoiA. M. (2006). CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol.46, 411–449.10.1146/annurev.pharmtox.46.120604.141053
106
KrishnamurthyP.RossD. D.NakanishiT.Bailey-DellK.ZhouS.MercerK. E.SarkadiB.SorrentinoB. P.SchuetzJ. D. (2004). The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem.279, 24218–24225.10.1074/jbc.M313599200
107
KrishnamurthyP.SchuetzJ. D. (2006). Role of ABCG2/BCRP in biology and medicine. Annu. Rev. Pharmacol. Toxicol.46, 381–410.10.1146/annurev.pharmtox.46.120604.141238
108
KrishnamurthyP. C.DuG.FukudaY.SunD.SampathJ.MercerK. E.WangJ.Sosa-PinedaB.MurtiK. G.SchuetzJ. D. (2006). Identification of a mammalian mitochondrial porphyrin transporter. Nature443, 586–589.10.1038/nature05092
109
KristiansenM.GraversenJ. H.JacobsenC.SonneO.HoffmanH. J.LawS. K.MoestrupS. K. (2001). Identification of the haemoglobin scavenger receptor. Nature409, 198–201.10.1038/35051594
110
KumarA. A.BhaskarE.Palamaner Subash ShanthaG.SwaminathanP.AbrahamG. (2009). Rhabdomyolysis in community acquired bacterial sepsis–a retrospective cohort study. PLoS ONE4, e7182.10.1371/journal.pone.0007182
111
KumarS.BandyopadhyayU. (2005). Free heme toxicity and its detoxification systems in human. Toxicol. Lett.157, 175–188.10.1016/j.toxlet.2005.03.004
112
KuttyR. K.MainesM. D. (1981). Purification and characterization of biliverdin reductase from rat liver. J. Biol. Chem.256, 3956–3962.
113
LadL.SchullerD. J.ShimizuH.FriedmanJ.LiH.Ortiz de MontellanoP. R.PoulosT. L. (2003). Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J. Biol. Chem.278, 7834–7843.10.1074/jbc.M212270200
114
LangF.GulbinsE.LercheH.HuberS. M.KempeD. S.FollerM. (2008). Eryptosis, a window to systemic disease. Cell. Physiol. Biochem.22, 373–380.10.1159/000185448
115
LarsenR.GozzelinoR.JeneyV.TokajiL.BozzaF. A.JapiassuA. M.BonaparteD.CavalcanteM. M.ChoraA.FerreiraA.MargutiI.CardosoS.SepulvedaN.SmithA.SoaresM. P. (2010). A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med.2, 51ra71.10.1126/scitranslmed.3001118
116
LathropJ. T.TimkoM. P. (1993). Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science259, 522–525.10.1126/science.8424176
117
Latunde-DadaG. O.TakeuchiK.SimpsonR. J.McKieA. T. (2006). Haem carrier protein 1 (HCP1): expression and functional studies in cultured cells. FEBS Lett.580, 6865–6870.10.1016/j.febslet.2006.11.048
118
LaumonierT.YangS.KonigS.ChauveauC.AnegonI.HoffmeyerP.MenetreyJ. (2008). Lentivirus mediated HO-1 gene transfer enhances myogenic precursor cell survival after autologous transplantation in pig. Mol. Ther.16, 404–410.10.1038/sj.mt.6300354
119
LevyA. P.AslehR.BlumS.LevyN. S.Miller-LotanR.Kalet-LitmanS.AnbinderY.LacheO.NakhoulF. M.AsafR.FarbsteinD.PollakM.SoloveichikY. Z.StraussM.AlshiekJ.LivshitsA.SchwartzA.AwadH.JadK.GoldensteinH. (2010). Haptoglobin: basic and clinical aspects. Antioxid. Redox Signal.12, 293–304.10.1089/ars.2009.2793
120
LiT.BonkovskyH. L.GuoJ. T. (2011). Structural analysis of heme proteins: implications for design and prediction. BMC Struct. Biol.11, 13.10.1186/1472-6807-11-13
121
LittleH. N.NeilandsJ. B. (1960). Binding of haematin by human serum albumin. Nature188, 913–915.10.1038/188913a0
122
MaX.SayedN.BeuveA.van den AkkerF. (2007). NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J.26, 578–588.10.1038/sj.emboj.7601521
123
MainesM. D.TrakshelG. M.KuttyR. K. (1986). Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem.261, 411–419.
124
MaitlandK.MarshK. (2004). Pathophysiology of severe malaria in children. Acta Trop.90, 131–140.10.1016/j.actatropica.2003.11.010
125
MaitlandK.PambaA.EnglishM.PeshuN.MarshK.NewtonC.LevinM. (2005). Randomized trial of volume expansion with albumin or saline in children with severe malaria: preliminary evidence of albumin benefit. Clin. Infect. Dis.40, 538–545.10.1086/427505
126
MakinenM. W.SchichmanS. A.HillS. C.GrayH. B. (1983). Heme-heme orientation and electron transfer kinetic behavior of multisite oxidation-reduction enzymes. Science222, 929–931.10.1126/science.6415814
127
MarshK.ForsterD.WaruiruC.MwangiI.WinstanleyM.MarshV.NewtonC.WinstanleyP.WarnP.PeshuN.PasvolG.SnowR. (1995). Indicators of life-threatening malaria in African children. N. Engl. J. Med.332, 1399–1404.10.1056/NEJM199505253322102
128
MasonP. J. (1996). New insights into G6PD deficiency. Br. J. Haematol.94, 585–591.
129
MasudaK.ShimaH.WatanabeM.KikuchiK. (2001). MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J. Biol. Chem.276, 39002–39011.10.1074/jbc.M011475200
130
MedzhitovR.SchneiderD. S.SoaresM. P. (2012). Disease tolerance as a defense strategy. Science335, 936–941.10.1126/science.1214935
131
MeierM.JanosikM.KeryV.KrausJ. P.BurkhardP. (2001). Structure of human cystathionine beta-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J.20, 3910–3916.10.1093/emboj/20.15.3910
132
Melamed-FrankM.LacheO.EnavB. I.SzafranekT.LevyN. S.RicklisR. M.LevyA. P. (2001). Structure-function analysis of the antioxidant properties of haptoglobin. Blood98, 3693–3698.10.1182/blood.V98.13.3693
133
MillerL. H.BaruchD. I.MarshK.DoumboO. K. (2002). The pathogenic basis of malaria. Nature415, 673–679.10.1038/415673a
134
MillerY. I.ShaklaiN. (1999). Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim. Biophys. Acta1454, 153–164.
135
MirkinN.JaconcicJ.StojanoffV.MorenoA. (2008). High resolution X-ray crystallographic structure of bovine heart cytochrome c and its application to the design of an electron transfer biosensor. Proteins70, 83–92.10.1002/prot.21452
136
MiyatakeT.KoyamadaN.HancockW. W.SoaresM. P.BachF. H. (1998). Survival of accommodated cardiac xenografts upon retransplantation into cyclosporine-treated recipients. Transplantation65, 1563–1569.10.1097/00007890-199806270-00005
137
ModianoD.LuoniG.SirimaB. S.SimporeJ.VerraF.KonateA.RastrelliE.OlivieriA.CalissanoC.PaganottiG. M.D’UrbanoL.SanouI.SawadogoA.ModianoG.ColuzziM. (2001). Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature414, 305–308.10.1038/35104556
138
MorganW. T.LiemH. H.SutorR. P.Muller-EbergardU. (1976). Transfer of heme from heme-albumin to hemopexin. Biochim. Biophys. Acta444, 435–445.10.1016/0304-4165(76)90387-1
139
MotterliniR.OtterbeinL. E. (2010). The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov.9, 728–743.10.1038/nrd3228
140
Muller-EberhardU. (1970). Hemopexin. N. Engl. J. Med.283, 1090–1094.10.1056/NEJM197011122832007
141
Muller-EberhardU.JavidJ.LiemH. H.HansteinA.HannaM. (1968). Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood32, 811–815.
142
MurrayC. J.RosenfeldL. C.LimS. S.AndrewsK. G.ForemanK. J.HaringD.FullmanN.NaghaviM.LozanoR.LopezA. D. (2012). Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet379, 413–431.10.1016/S0140-6736(12)60034-8
143
MustafaA. K.GadallaM. M.SnyderS. H. (2009). Signaling by gasotransmitters. Sci. Signal.2, re2.10.1126/scisignal.2000464
144
NagababuE.RifkindJ. M. (2000). Heme degradation during autoxidation of oxyhemoglobin. Biochem. Biophys. Res. Commun.273, 839–845.10.1006/bbrc.2000.3025
145
NagababuE.RifkindJ. M. (2004). Heme degradation by reactive oxygen species. Antioxid. Redox Signal.6, 967–978.10.1089/1523086042259823
146
NathK. A.BallaG.VercellottiG. M.BallaJ.JacobH. S.LevittM. D.RosenbergM. E. (1992). Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Invest.90, 267–270.10.1172/JCI115847
147
OguraT.HirotaS.ProshlyakovD. A.ShinzawaItohK.YoshikawaS.KitagawaT. (1996). Time-resolved resonance Raman evidence for tight coupling between electron transfer and proton pumping of cytochrome c oxidase upon the change from the Fe-V oxidation level to the Fe-IV oxidation level. J. Am. Chem. Soc.118, 5443–5449.10.1021/ja951922i
148
OllingerR.BilbanM.EratA.FroioA.McDaidJ.TyagiS.CsizmadiaE.Graca-SouzaA. V.LiloiaA.SoaresM. P.OtterbeinL. E.UshevaA.YamashitaK.BachF. H. (2005). Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation112, 1030–1039.10.1161/CIRCULATIONAHA.104.528802
149
OllingerR.WangH.YamashitaK.WegielB.ThomasM.MargreiterR.BachF. H. (2007). Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid. Redox Signal.9, 2175–2185.10.1089/ars.2007.1807
150
OnoS.KomuroT.MacdonaldR. L. (2002). Heme oxygenase-1 gene therapy for prevention of vasospasm in rats. J. Neurosurg.96, 1094–1102.10.3171/jns.2002.96.6.1094
151
OpalS. M.FisherC. J.Jr.DhainautJ. F.VincentJ. L.BraseR.LowryS. F.SadoffJ. C.SlotmanG. J.LevyH.BalkR. A.ShellyM. P.PribbleJ. P.LaBrecqueJ. F.LookabaughJ.DonovanH.DubinH.BaughmanR.NormanJ.DeMariaE.MatzelK.AbrahamE.SeneffM. (1997). Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med.25, 1115–1124.10.1097/00003246-199707000-00010
152
OtterbeinL.SylvesterS. L.ChoiA. M. (1995). Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am. J. Respir. Cell Mol. Biol.13, 595–601.
153
OtterbeinL. E.BachF. H.AlamJ.SoaresM.Tao LuH.WyskM.DavisR. J.FlavellR. A.ChoiA. M. (2000). Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med.6, 422–428.10.1038/74680
154
OtterbeinL. E.KollsJ. K.MantellL. L.CookJ. L.AlamJ.ChoiA. M. (1999a). Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J. Clin. Invest.103, 1047–1054.10.1172/JCI5342
155
OtterbeinL. E.MantellL. L.ChoiA. M. (1999b). Carbon monoxide provides protection against hyperoxic lung injury. Am. J. Physiol.276(Pt 1), L688–L694.
156
OtterbeinL. E.ZuckerbraunB. S.HagaM.LiuF.SongR.UshevaA.StachulakC.BodyakN.SmithR. N.CsizmadiaE.TyagiS.AkamatsuY.FlavellR. J.BilliarT. R.TzengE.BachF. H.ChoiA. M.SoaresM. P. (2003). Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat. Med.9, 183–190.10.1038/nm817
157
OverhausM.MooreB. A.BarbatoJ. E.BehrendtF. F.DoeringJ. G.BauerA. J. (2006). Biliverdin protects against polymicrobial sepsis by modulating inflammatory mediators. Am. J. Physiol. Gastrointest. Liver Physiol.290, G695–G703.10.1152/ajpgi.00152.2005
158
PamplonaA.FerreiraA.BallaJ.JeneyV.BallaG.EpiphanioS.ChoraA.RodriguesC. D.GregoireI. P.Cunha-RodriguesM.PortugalS.SoaresM. P.MotaM. M. (2007). Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med.13, 703–710.10.1038/nm1586
159
PaoliM.AndersonB. F.BakerH. M.MorganW. T.SmithA.BakerE. N. (1999). Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat. Struct. Biol.6, 926–931.10.1038/13294
160
PaoliM.LiddingtonR.TameJ.WilkinsonA.DodsonG. (1996). Crystal structure of T state haemoglobin with oxygen bound at all four haems. J. Mol. Biol.256, 775–792.10.1006/jmbi.1996.0124
161
ParkS. Y.YokoyamaT.ShibayamaN.ShiroY.TameJ. R. (2006). 1.25 A resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J. Mol. Biol.360, 690–701.10.1016/j.jmb.2006.05.036
162
PaulingL.ItanoH. A.SingerS. J.WellsI. C. (1949). Sickle cell anemia a molecular disease. Science110, 543–548.10.1126/science.110.2865.543
163
PenaA. C.PenachoN.Mancio-SilvaL.NeresR.SeixasJ. D.FernandesA. C.RomaoC. C.MotaM. M.BernardesG. J.PamplonaA. (2012). A novel carbon monoxide-releasing molecule (CO-RM) fully protects mice from severe malaria. Antimicrob. Agents Chemother.56, 1281–1290.10.1128/AAC.05991-11
164
PhamC. G.BubiciC.ZazzeroniF.PapaS.JonesJ.AlvarezK.JayawardenaS.De SmaeleE.CongR.BeaumontC.TortiF. M.TortiS. V.FranzosoG. (2004). Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell119, 529–542.10.1016/j.cell.2004.10.017
165
PhilippidisP.MasonJ. C.EvansB. J.NadraI.TaylorK. M.HaskardD. O.LandisR. C. (2004). Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ. Res.94, 119–126.10.1161/01.RES.0000109414.78907.F9
166
PiagnerelliM.BoudjeltiaK. Z.VanhaeverbeekM.VincentJ. L. (2003). Red blood cell rheology in sepsis. Intensive Care Med.29, 1052–1061.10.1007/s00134-003-1783-2
167
PiantadosiC. A. (2008). Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med.45, 562–569.10.1016/j.freeradbiomed.2008.05.013
168
PodstawkaE.ProniewiczL. M. (2004). Resonance Raman study of deoxy and ligated (O2 and CO) mesoheme IX-reconstituted myoglobin, hemoglobin and its alpha and beta subunits. J. Inorg. Biochem.98, 1502–1512.10.1016/j.jinorgbio.2004.06.001
169
PortoB. N.AlvesL. S.FernandezP. L.DutraT. P.FigueiredoR. T.Graca-SouzaA. V.BozzaM. T. (2007). Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem.282, 24430–24436.10.1074/jbc.M703570200
170
PossK. D.TonegawaS. (1997a). Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. U.S.A.94, 10919–10924.10.1073/pnas.94.20.10919
171
PossK. D.TonegawaS. (1997b). Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. U.S.A.94, 10925–10930.10.1073/pnas.94.20.10919
172
PoulosT. L. (2007). The Janus nature of heme. Nat. Prod. Rep.24, 504–510.10.1039/b604195g
173
PutnamC. D.ArvaiA. S.BourneY.TainerJ. A. (2000). Active and inhibited human catalase structures: ligand and NADPH binding and catalytic mechanism. J. Mol. Biol.296, 295–309.10.1006/jmbi.1999.3458
174
QuayeI. K. (2008). Haptoglobin, inflammation and disease. Trans. R. Soc. Trop. Med. Hyg.102, 735–742.10.1016/j.trstmh.2008.04.010
175
QueirogaC. S.AlmeidaA. S.MartelC.BrennerC.AlvesP. M.VieiraH. L. (2010). Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem.285, 17077–17088.10.1074/jbc.M109.065052
176
QuigleyJ. G.BurnsC. C.AndersonM. M.LynchE. D.SaboK. M.OverbaughJ.AbkowitzJ. L. (2000). Cloning of the cellular receptor for feline leukemia virus subgroup C (FeLV-C), a retrovirus that induces red cell aplasia. Blood95, 1093–1099.
177
QuigleyJ. G.YangZ.WorthingtonM. T.PhillipsJ. D.SaboK. M.SabathD. E.BergC. L.SassaS.WoodB. L.AbkowitzJ. L. (2004). Identification of a human heme exporter that is essential for erythropoiesis. Cell118, 757–766.10.1016/j.cell.2004.08.014
178
RajagopalA.RaoA. U.AmigoJ.TianM.UpadhyayS. K.HallC.UhmS.MathewM. K.FlemingM. D.PawB. H.KrauseM.HamzaI. (2008). Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature453, 1127–1131.10.1038/nature06934
179
ReedyC. J.GibneyB. R. (2004). Heme protein assemblies. Chem. Rev.104, 617–649.10.1021/cr0206115
180
ReiterC. D.WangX.Tanus-SantosJ. E.HoggN.CannonR. O.IIISchechterA. N.GladwinM. T. (2002). Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med.8, 1383–1389.10.1038/nm1202-799
181
RiedemannN. C.GuoR. F.WardP. A. (2003). Novel strategies for the treatment of sepsis. Nat. Med.9, 517–524.10.1038/nm0503-517
182
RittirschD.Huber-LangM. S.FlierlM. A.WardP. A. (2009). Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc.4, 31–36.10.1038/nprot.2008.214
183
RosenthalP. J. (2011). Lessons from sickle cell disease in the treatment and control of malaria. N. Engl. J. Med.364, 2549–2551.10.1056/NEJMp1104428
184
RotherR. P.BellL.HillmenP.GladwinM. T. (2005). The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA293, 1653–1662.10.1001/jama.293.13.1653
185
RowlandP.BlaneyF. E.SmythM. G.JonesJ. J.LeydonV. R.OxbrowA. K.LewisC. J.TennantM. G.ModiS.EgglestonD. S.CheneryR. J.BridgesA. M. (2006). Crystal structure of human cytochrome P450 2D6. J. Biol. Chem.281, 7614–7622.10.1074/jbc.M511232200
186
SamboM. R.TrovoadaM. J.BenchimolC.QuinhentosV.GoncalvesL.VelosaR.MarquesM. I.SepulvedaN.ClarkT. G.MustafaS.WagnerO.CoutinhoA.Penha-GoncalvesC. (2010). Transforming growth factor beta 2 and heme oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS ONE5, e11141.10.1371/journal.pone.0011141
187
Sanchez-PerezI.Martinez-GomarizM.WilliamsD.KeyseS. M.PeronaR. (2000). CL100/MKP-1 modulates JNK activation and apoptosis in response to cisplatin. Oncogene19, 5142–5152.10.1038/sj.onc.1203887
188
Sarady-AndrewsJ. K.LiuF.GalloD.NakaoA.OverhausM.OllingerR.ChoiA. M.OtterbeinL. E. (2005). Biliverdin administration protects against endotoxin-induced acute lung injury in rats. Am. J. Physiol. Lung Cell Mol. Physiol.289, L1131–L1137.10.1152/ajplung.00458.2004
189
SatoK.BallaJ.OtterbeinL.SmithR. N.BrouardS.LinY.CsizmadiaE.SevignyJ.RobsonS. C.VercellottiG.ChoiA. M.BachF. H.SoaresM. P. (2001). Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J. Immunol.166, 4185–4194.
190
SchaeferW. H.HarrisT. M.GuengerichF. P. (1985). Characterization of the enzymatic and nonenzymatic peroxidative degradation of iron porphyrins and cytochrome P-450 heme. Biochemistry24, 3254–3263.10.1021/bi00334a027
191
SchneiderS.Marles-WrightJ.SharpK. H.PaoliM. (2007). Diversity and conservation of interactions for binding heme in b-type heme proteins. Nat. Prod. Rep.24, 621–630.10.1039/b604186h
192
SchultzI. J.ChenC.PawB. H.HamzaI. (2010). Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem.285, 26753–26759.10.1074/jbc.R110.119503
193
SeixasE.GozzelinoR.ChoraA.FerreiraA.SilvaG.LarsenR.RebeloS.PenidoC.SmithN. R.CoutinhoA.SoaresM. P. (2009). Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. U.S.A.106, 15837–15842.10.1073/pnas.0903419106
194
SeldonM. P.SilvaG.PejanovicN.LarsenR.GregoireI. P.FilipeJ.AnratherJ.SoaresM. P. (2007). Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. J. Immunol.179, 7840–7851.
195
ShayeghiM.Latunde-DadaG. O.OakhillJ. S.LaftahA. H.TakeuchiK.HallidayN.KhanY.WarleyA.McCannF. E.HiderR. C.FrazerD. M.AndersonG. J.VulpeC. D.SimpsonR. J.McKieA. T. (2005). Identification of an intestinal heme transporter. Cell122, 789–801.10.1016/j.cell.2005.06.025
196
ShenX. D.KeB.ZhaiY.GaoF.BusuttilR. W.ChengG.Kupiec-WeglinskiJ. W. (2005). Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am. J. Transplant.5, 1793–1800.10.1111/j.1600-6143.2005.00932.x
197
SilS.BoseT.RoyD.ChakrabortiA. S. (2004). Protoporphyrin IX-induced structural and functional changes in human red blood cells, haemoglobin and myoglobin. J. Biosci.29, 281–291.10.1007/BF02702610
198
SilvaG.CunhaA.GregoireI. P.SeldonM. P.SoaresM. P. (2006). The antiapoptotic effect of heme oxygenase-1 in endothelial cells involves the degradation of p38 alpha MAPK isoform. J. Immunol.177, 1894–1903.
199
SilvaG.JeneyV.ChoraA.LarsenR.BallaJ.SoaresM. P. (2009). Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem.284, 29582–29595.10.1074/jbc.M109.016410
200
SingletonJ. W.LasterL. (1965). Biliverdin reductase of guinea pig liver. J. Biol. Chem.240, 4780–4789.
201
SmithL. J.KahramanA.ThorntonJ. M. (2010). Heme proteins–diversity in structural characteristics, function, and folding. Proteins78, 2349–2368.10.1002/prot.22747
202
SoaresM. P.BachF. H. (2007). Heme oxygenase-1 in organ transplantation. Front. Biosci.12, 4932–4945.10.2741/2439
203
SoaresM. P.LinY.AnratherJ.CsizmadiaE.TakigamiK.SatoK.GreyS. T.ColvinR. B.ChoiA. M.PossK. D.BachF. H. (1998). Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat. Med.4, 1073–1077.10.1038/2063
204
SoaresM. P.LinY.SatoK.StuhlmeierK. M.BachF. H. (1999). Accommodation. Immunol. Today20, 434–437.10.1016/S0167-5699(99)01530-3
205
SoaresM. P.SeldonM. P.GregoireI. P.VassilevskaiaT.BerberatP. O.YuJ.TsuiT. Y.BachF. H. (2004). Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol.172, 3553–3563.
206
SollwedelA.BertojaA. Z.ZenclussenM. L.GerlofK.LisewskiU.WafulaP.SawitzkiB.WoiciechowskyC.VolkH. D.ZenclussenA. C. (2005). Protection from abortion by heme oxygenase-1 up-regulation is associated with increased levels of Bag-1 and neuropilin-1 at the fetal-maternal interface. J. Immunol.175, 4875–4885.
207
StockerR.PerrellaM. A. (2006). Heme oxygenase-1: a novel drug target for atherosclerotic diseases?Circulation114, 2178–2189.10.1161/CIRCULATIONAHA.105.598698
208
StockerR.YamamotoY.McDonaghA. F.GlazerA. N.AmesB. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science235, 1043–1046.10.1126/science.3029864
209
SugimotoH.OdaS.OtsukiT.HinoT.YoshidaT.ShiroY. (2006). Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl. Acad. Sci. U.S.A.103, 2611–2616.10.1073/pnas.0508996103
210
SzatmariI.VamosiG.BrazdaP.BalintB. L.BenkoS.SzelesL.JeneyV.Ozvegy-LaczkaC.SzantoA.BartaE.BallaJ.SarkadiB.NagyL. (2006). Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J. Biol. Chem.281, 23812–23823.10.1074/jbc.M604890200
211
TaharaS. M.TraughJ. A.SharpS. B.LundakT. S.SaferB.MerrickW. C. (1978). Effect of hemin on site-specific phosphorylation of eukaryotic initiation factor 2. Proc. Natl. Acad. Sci. U.S.A.75, 789–793.10.1073/pnas.75.2.789
212
TaharaT.SunJ.IgarashiK.TaketaniS. (2004a). Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem. Biophys. Res. Commun.324, 77–85.10.1016/j.bbrc.2004.09.022
213
TaharaT.SunJ.NakanishiK.YamamotoM.MoriH.SaitoT.FujitaH.IgarashiK.TaketaniS. (2004b). Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J. Biol. Chem.279, 5480–5487.10.1074/jbc.M302733200
214
TangX. D.XuR.ReynoldsM. F.GarciaM. L.HeinemannS. H.HoshiT. (2003). Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature425, 531–535.10.1038/nature02003
215
TanoueT.YamamotoT.MaedaR.NishidaE. (2001). A Novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38 alpha and beta MAPKs. J. Biol. Chem.276, 26629–26639.10.1074/jbc.M101981200
216
TenhunenR.MarverH. S.SchmidR. (1968). The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U.S.A.61, 748–755.10.1073/pnas.61.2.748
217
TolosanoE.FagooneeS.MorelloN.VinchiF.FioritoV. (2010). Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal.12, 305–320.10.1089/ars.2009.2787
218
TolosanoE.HirschE.PatruccoE.CamaschellaC.NavoneR.SilengoL.AltrudaF. (1999). Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood94, 3906–3914.
219
TrakshelG. M.KuttyR. K.MainesM. D. (1986). Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J. Biol. Chem.261, 11131–11137.
220
TsiftsoglouA. S.TsamadouA. I.PapadopoulouL. C. (2006). Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol. Ther.111, 327–345.10.1016/j.pharmthera.2005.10.017
221
TsukiharaT.AoyamaH.YamashitaE.TomizakiT.YamaguchiH.Shinzawa-ItohK.NakashimaR.YaonoR.YoshikawaS. (1995). Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science269, 1069–1074.10.1126/science.7652554
222
TzimaS.VictoratosP.KranidiotiK.AlexiouM.KolliasG. (2009). Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J. Exp. Med.206, 1167–1179.10.1084/jem.20081582
223
UchidaT.SatoE.SatoA.SagamiI.ShimizuT.KitagawaT. (2005). CO-dependent activity-controlling mechanism of heme-containing CO-sensor protein, neuronal PAS domain protein 2. J. Biol. Chem.280, 21358–21368.10.1074/jbc.M412350200
224
UlloaL.TraceyK. J. (2005). The “cytokine profile”: a code for sepsis. Trends. Mol. Med.11, 56–63.10.1016/j.molmed.2004.12.007
225
VenturaJ. J.CogswellP.FlavellR. A.BaldwinA. S.Jr.DavisR. J. (2004). JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev.18, 2905–2915.10.1101/gad.1223004
226
VileG. F.Basu-ModakS.WaltnerC.TyrrellR. M. (1994). Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl. Acad. Sci. U.S.A.91, 2607–2610.10.1073/pnas.91.7.2607
227
VojtechovskyJ.ChuK.BerendzenJ.SweetR. M.SchlichtingI. (1999). Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys. J.77, 2153–2174.10.1016/S0006-3495(99)77056-6
228
WaltherM.De CaulA.AkaP.NjieM.Amambua-NgwaA.WaltherB.PredazziI. M.CunningtonA.DeiningerS.TakemE. N.EbonyiA.WeisS.WaltonR.Rowland-JonesS.SirugoG.WilliamsS. M.ConwayD. J. (2012). HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog.8, e1002579.10.1371/journal.ppat.1002579
229
WangH.LeeS. S.Dell’AgnelloC.TchipashviliV.d’AvilaJ. C.CzismadiaE.ChinB. Y.BachF. H. (2006). Bilirubin can induce tolerance to islet allografts. Endocrinology147, 762–768.10.1210/en.2005-1485
230
WangX.WangY.LeeS. J.KimH. P.ChoiA. M.RyterS. W. (2011). Carbon monoxide inhibits Fas activating antibody-induced apoptosis in endothelial cells. Med. Gas Res.1, 8.10.1186/2045-9912-1-8
231
WardellM.WangZ.HoJ. X.RobertJ.RukerF.RubleJ.CarterD. C. (2002). The atomic structure of human methemalbumin at 1.9 A. Biochem. Biophys. Res. Commun.291, 813–819.10.1006/bbrc.2002.6540
232
WilliamsT. N.MwangiT. W.RobertsD. J.AlexanderN. D.WeatherallD. J.WambuaS.KortokM.SnowR. W.MarshK. (2005a). An immune basis for malaria protection by the sickle cell trait. PLoS Med.2, e128.10.1371/journal.pmed.0020128
233
WilliamsT. N.MwangiT. W.WambuaS.AlexanderN. D.KortokM.SnowR. W.MarshK. (2005b). Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J. Infect. Dis.192, 178–186.10.1086/430744
234
WirtzM.OganesyanV.ZhangX.StuderJ.RiveraM. (2000). Modulation of redox potential in electron transfer proteins: effects of complex formation on the active site microenvironment of cytochrome b5. Faraday Discuss.221–234; discussion 257–268.10.1039/b001520m
235
XiaD.YuC. A.KimH.XiaJ. Z.KachurinA. M.ZhangL.YuL.DeisenhoferJ. (1997). Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science277, 60–66.10.1126/science.277.5322.60
236
XieC.ZhangN.ZhouH.LiJ.LiQ.ZarubinT.LinS. C.HanJ. (2005). Distinct roles of basal steady-state and induced H-ferritin in tumor necrosis factor-induced death in L929 cells. Mol. Cell. Biol.25, 6673–6681.10.1128/MCB.25.15.6673-6681.2005
237
YamashitaK.McDaidJ.OllingerR.TsuiT. Y.BerberatP. O.UshevaA.CsizmadiaE.SmithR. N.SoaresM. P.BachF. H. (2004). Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J.18, 765–767.
238
YetS. F.LayneM. D.LiuX.ChenY. H.IthB.SibingaN. E.PerrellaM. A. (2003). Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J.17, 1759–1761.
239
YetS. F.PerrellaM. A.LayneM. D.HsiehC. M.MaemuraK.KobzikL.WieselP.ChristouH.KourembanasS.LeeM. E. (1999). Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Invest.103, R23–R29.10.1172/JCI6163
240
YounH.ConradM.ChungS. Y.RobertsG. P. (2006). Roles of the heme and heme ligands in the activation of CooA, the CO-sensing transcriptional activator. Biochem. Biophys. Res. Commun.348, 345–350.10.1016/j.bbrc.2006.06.200
241
ZederbauerM.FurtmullerP. G.BrogioniS.JakopitschC.SmulevichG.ObingerC. (2007). Heme to protein linkages in mammalian peroxidases: impact on spectroscopic, redox and catalytic properties. Nat. Prod. Rep.24, 571–584.10.1039/b604178g
242
ZenclussenA. C.LimE.KnoellerS.KnackstedtM.HertwigK.HagenE.KlappB. F.ArckP. C. (2003). Heme oxygenases in pregnancy II: HO-2 is downregulated in human pathologic pregnancies. Am. J. Reprod. Immunol.50, 66–76.10.1034/j.1600-0897.2003.00047.x
243
ZenclussenA. C.SollwedelA.BertojaA. Z.GerlofK.ZenclussenM. L.WoiciechowskyC.VolkH. D. (2005). Heme oxygenase as a therapeutic target in immunological pregnancy complications. Int. Immunopharmacol.5, 41–51.10.1016/j.intimp.2004.09.011
244
ZenclussenM. L.CasalisP. A.El-MouslehT.RebeloS.LangwischS.LinzkeN.VolkH. D.FestS.SoaresM. P.ZenclussenA. C. (2011). Haem oxygenase-1 dictates intrauterine fetal survival in mice via carbon monoxide. J. Pathol.225, 293–304.10.1002/path.2946
245
Zenke-KawasakiY.DohiY.KatohY.IkuraT.IkuraM.AsaharaT.TokunagaF.IwaiK.IgarashiK. (2007). Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol.27, 6962–6971.10.1128/MCB.02415-06
246
ZhangW.WangM.XieH. Y.ZhouL.MengX. Q.ShiJ.ZhengS. (2007a). Role of reactive oxygen species in mediating hepatic ischemia-reperfusion injury and its therapeutic applications in liver transplantation. Transplant. Proc.39, 1332–1337.10.1016/j.transproceed.2007.06.092
247
ZhangY.KangS. A.MukherjeeT.BaleS.CraneB. R.BegleyT. P.EalickS. E. (2007b). Crystal structure and mechanism of tryptophan 2,3-dioxygenase, a heme enzyme involved in tryptophan catabolism and in quinolinate biosynthesis. Biochemistry46, 145–155.10.1021/bi700672v
248
ZhangZ.HuangL.ShulmeisterV. M.ChiY. I.KimK. K.HungL. W.CroftsA. R.BerryE. A.KimS. H. (1998). Electron transfer by domain movement in cytochrome bc1. Nature392, 677–684.10.1038/33612
249
ZhuY.LeeH. C.ZhangL. (2002a). An examination of heme action in gene expression: heme and heme deficiency affect the expression of diverse genes in erythroid k562 and neuronal PC12 cells. DNA Cell Biol.21, 333–346.10.1089/104454902753759744
250
ZhuY.SunY.JinK.GreenbergD. A. (2002b). Hemin induces neuroglobin expression in neural cells. Blood100, 2494–2498.10.1182/blood-2002-01-0280
Summary
Keywords
heme, heme oxygenase, cytotoxicity, programmed cell death, immune-mediated inflammatory diseases
Citation
Larsen R, Gouveia Z, Soares MP and Gozzelino R (2012) Heme Cytotoxicity and the Pathogenesis of Immune-Mediated Inflammatory Diseases. Front. Pharmacol. 3:77. doi: 10.3389/fphar.2012.00077
Received
10 February 2012
Accepted
11 April 2012
Published
04 May 2012
Volume
3 - 2012
Edited by
Jaime Kapitulnik, The Hebrew University of Jerusalem, Israel
Reviewed by
Stefan W. Ryter, Harvard Medical School, USA; Stephan Immenschuh, Hannover Medical School, Germany
Copyright
© 2012 Larsen, Gouveia, Soares and Gozzelino.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Raffaella Gozzelino, Instituto Gulbenkian de Ciência, Rua da Quinta Grande, 6, 2780-156 Oeiras, Portugal. e-mail: rgozzelino@igc.gulbenkian.pt
This article was submitted to Frontiers in Drug Metabolism and Transport, a specialty of Frontiers in Pharmacology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.