Abstract
Atrial fibrillation (AF) is the most frequently encountered clinical arrhythmia and is associated with increased morbidity and mortality. Ectopic activity and reentry are considered major arrhythmogenic mechanisms contributing to the initiation and maintenance of AF. In addition, AF is self-reinforcing through progressive electrical and structural remodeling which stabilize the arrhythmia and make it more difficult to treat. Recent research has suggested an important role for Ca2+-dysregulation in AF. Ca2+-handling abnormalities may promote ectopic activity, conduction abnormalities facilitating reentry, and AF-related remodeling. In this review article, we summarize the Ca2+-handling derangements occurring in AF and discuss their impact on fundamental arrhythmogenic mechanisms. We focus in particular on the role of the multifunctional Ca2+/calmodulin-dependent protein kinase type-II (CaMKII), which acts as a major link between Ca2+-dysregulation and arrhythmogenesis. CaMKII expression and activity are increased in AF and promote arrhythmogenesis through phosphorylation of various targets involved in cardiac electrophysiology and excitation-contraction coupling. We discuss the implications for potential novel therapeutic strategies for AF based on CaMKII and Ca2+-handling abnormalities.
Introduction
Atrial fibrillation (AF) is the most prevalent heart-rhythm disorder, estimated to affect more than 33 million people worldwide (Chugh et al., 2013). AF is associated with increased morbidity and mortality, notably as a risk factor for stroke and worsening of heart failure (Camm et al., 2012; Chugh et al., 2013). Current pharmacological treatments for rhythm-control of AF mainly include class-I and class-III antiarrhythmic drugs, which have modest efficacy, providing sinus-rhythm maintenance in only 30–70% of patients after >1 year of follow-up (Camm, 2012). In addition, these drugs are associated with substantial adverse side-effects including ventricular proarrhythmia and extra-cardiac toxicity (Zimetbaum, 2012; Heijman et al., 2013a). The AF incidence is expected to increase due to aging of the population, making the development of improved antiarrhythmic treatments of critical importance. A better understanding of AF pathophysiology is expected to foster this development (Dobrev et al., 2012). Accumulating evidence has highlighted a central role for abnormal Ca2+-handling in AF-pathophysiology (Dobrev and Nattel, 2008; Heijman et al., 2012; Nattel and Dobrev, 2012). Here, we review recent studies detailing the proarrhythmic role of AF-related Ca2+-handling abnormalities, with particular focus on the contributions of the Ca2+/calmodulin-dependent protein kinase type-II (CaMKII).
Atrial cellular electrophysiology and arrhythmogenic mechanisms
Normal atrial cellular electrophysiology and Ca2+-handling
The atrial action potential (AP) is determined by depolarizing and repolarizing ionic currents (Dobrev and Ravens, 2003). Depolarizing currents include the cardiac voltage-gated Na+-current (INa) and its persistent (“late”) component (INa,late), the L-type Ca2+-current (ICa,L) and the Na+/Ca2+-exchanger type-1 (NCX1) current (INCX), which, in its forward mode, extrudes one Ca2+-ion in exchange for 3 Na+-ions, resulting in a net depolarizing inward current. Repolarizing currents include the transient-outward K+-current (Ito), delayed-rectifier K+-currents with slow, rapid or ultra-rapid kinetics (IKs, IKr, and IKur, respectively), as well as the Na+/K+-ATPase current (INaK). In addition, AP duration (APD) and resting membrane potential are influenced by basal and acetylcholine-activated inward-rectifier K+-currents (IK1 and IK,ACh). The IKur and IK,ACh currents are predominantly expressed in the atria, thereby providing potential atrial-specific therapeutic targets.
Ca2+ entry through the L-type Ca2+-channel activates Ca2+-induced Ca2+-release from the sarcoplasmic reticulum (SR) through type-2 ryanodine receptor channels (RyR2), producing the systolic Ca2+-transient responsible for initiating contraction of atrial cardiomyocytes (Bers, 2002). In addition, inositol 1,4,5-triphosphate (IP3)-receptor-mediated Ca2+-release may contribute to Ca2+-induced Ca2+-release by activating neighboring RyR2, although direct IP3-receptor-mediated activation of NCX1 has also been described recently (Dobrev and Nattel, 2008; Roderick and Knollmann, 2013).
Structural differences between atrial and ventricular cardiomyocytes may further contribute to a unique atrial Ca2+-handling profile. Isolated atrial cardiomyocytes generally have a less well-developed T-tubular network than ventricular cardiomyocytes. However, cardiomyocytes of certain species including humans, sheep, goats, cows, and horses do have more T-tubules than those from rodents (Dibb et al., 2009; Lenaerts et al., 2009; Richards et al., 2011). At least in sheep, this T-tubular system contributes to a more uniform, ventricular-like, Ca2+-induced Ca2+-release (Dibb et al., 2009). Although a small T-tubular system is present in human atrial myocytes, it shows some variability depending on region and cardiomyocyte size (Trafford et al., 2013). Moreover, this T-tubular system can be remodeled by cardiac disease including AF (Lenaerts et al., 2009). In atrial cardiomyocytes with a less well-developed T-tubular structure, Ca2+-induced Ca2+-release starts at the plasma membrane and propagates slowly toward the cell-center (Dobrev et al., 2009; Bootman et al., 2011). Relaxation occurs when Ca2+ is extruded from the cell via NCX1 and the plasmalemmal Ca2+-ATPase (PMCA), or is taken back up into the SR by the type-2a SR Ca2+-ATPase (SERCA2a). The affinity of SERCA2a for intracellular Ca2+ is largely determined by the inhibitory proteins phospholamban (PLB) and sarcolipin. The expression of sarcolipin is atrial-specific, whereas PLB is more strongly expressed in the ventricles than in the atria (Dobrev et al., 2009).
Arrhythmogenic mechanisms in AF
AF can occur as a result of abnormalities in electrical impulse formation or impulse conduction (Nattel et al., 2008; Wakili et al., 2011; Heijman et al., 2014). Electrical impulse generation outside of the sinoatrial node, termed ectopic activity, can sustain AF as a driver, and can trigger reentry in a vulnerable substrate characterized by a slow and inhomogeneous conduction and short effective refractory periods. This vulnerable substrate can arise from genetic conditions, normal aging, or co-morbidities such as heart failure or hypertension (Wakili et al., 2011). Reentry can occur around anatomical obstacles or can be functional (i.e., occurring in the absence of anatomical obstacles). Reentry is considered the predominant mechanism for AF maintenance. When AF is maintained, atrial tachycardia-related remodeling produces electrical and structural alterations that further promote AF maintenance and stabilization, contributing to the progression toward longer-lasting AF episodes that are more difficult to treat.
At the cellular level, the effective refractory period is determined by APD and post-repolarization refractoriness. Conduction velocity is influenced by the depolarizing force through INa, and the electrical conduction between atrial cardiomyocytes is controlled by gap-junction channels as well as the structure of the atrial myocardium, notably the amount and composition of the extracellular matrix, particularly fibrosis. The cellular mechanisms of ectopic activity mainly involve early and delayed afterdepolarizations (EADs and DADs, respectively). EADs are caused primarily by recovery from inactivation of ICa,L during excessive APD-prolongation, for example due to loss of repolarizing K+-currents. DADs are likely the most common mechanism underlying ectopic (triggered) activity and result from intracellular Ca2+-handling abnormalities. Spontaneous diastolic SR Ca2+-release events resulting from SR Ca2+-overload or intrinsic RyR2-dysfunction can activate NCX1, resulting in a transient-inward current that depolarizes the membrane potential as Ca2+ is extruded from the atrial cardiomyocyte (Dobrev and Wehrens, 2010). When the threshold for excitation is reached in a sufficient number of cardiomyocytes, an ectopic impulse is generated (Wakili et al., 2011).
Structure, activation and targets of CaMKII
CaMKII is a multifunctional serine/threonine protein kinase that is abundantly expressed in various tissues including the heart (Swaminathan et al., 2012). There are four CaMKII isoforms, with CaMKIIδ being the most abundant in heart. CaMKIIδ has a hypervariable region, giving rise to multiple splice variants, including a splice variant with a nuclear localization signal (NLS; CaMKIIδB) and one without such NLS sequence (CaMKIIδC). The latter was traditionally considered cytosolic (Swaminathan et al., 2012), although this localization is not absolute (Mishra et al., 2011). CaMKII is a holoenzyme consisting of two stacked hexameric rings of subunits. Each subunit has a catalytic domain that, under resting conditions, is inhibited by regulatory domains of neighboring subunits. When intracellular Ca2+-levels periodically rise during the cellular Ca2+-transient, Ca2+ binds to calmodulin and activates CaMKII by binding to the regulatory domain (Swaminathan et al., 2012). CaMKII subunits can auto-phosphorylate Thr287 on neighboring subunits, thereby hindering the re-association of the catalytic and regulatory domains, producing sustained Ca2+-independent activation. This mechanism makes CaMKII activation strongly heart rate-dependent, with accumulating activity at faster rates. Furthermore, CaMKII can show Ca2+-independent activation following oxidation of Met281/282 by reactive oxygen species (Erickson et al., 2008), via O-linked glycosylation of Ser280 by O-linked N-acetylglucosamine (Erickson et al., 2013), and via NO-dependent nitrosylation of Cys116, Cys273, or Cys290, the exact residue being at present unknown (Gutierrez et al., 2013). In contrast, phosphorylation of Thr306/307 promotes CaMKII inactivation by reducing the binding of Ca2+/calmodulin complexes (Colbran, 1993).
CaMKII can phosphorylate multiple substrates in atrial cardiomyocytes (Figure 1). CaMKII-dependent phosphorylation of L-type Ca2+-channels produces high-activity mode-2 gating resulting in increased open probability of ICa,L, thereby augmenting the amount of Ca2+ entering the atrial cardiomyocyte. CaMKII also contributes to the increase in ICa,L following repeated depolarizing pulses (termed Ca2+-dependent ICa,L-facilitation) (Swaminathan et al., 2012). CaMKII-dependent phosphorylation of Nav1.5 slows INa inactivation and augments the non-inactivating, “late” component of INa (Wagner et al., 2011). The Kv4.3 pore-forming subunit of Ito is also regulated by CaMKII-dependent phosphorylation through the accessory protein SAP97, resulting in increased Ito that would tend to shorten APD (El-Haou et al., 2009; Wagner et al., 2009). Based on experiments involving CaMKII inhibition with an inhibitory peptide or the experimental drug KN-93, CaMKII also appears to acutely augment IK1 (Wagner et al., 2009) and IKur (Tessier et al., 1999), thereby offsetting the APD-prolonging effects of CaMKII-dependent ICa,L and INa phosphorylation. In addition, both PLB and sarcolipin can undergo CaMKII-dependent phosphorylation, causing disinhibition of SERCA2a and increasing SR Ca2+-reuptake (Dobrev and Wehrens, 2010). Finally, CaMKII-dependent hyperphosphorylation of Ser2814 on RyR2 increases channel open probability, augmenting SR Ca2+-release. Taken together, CaMKII plays a nodal role in the modulation of atrial cellular Ca2+-handling.
Figure 1
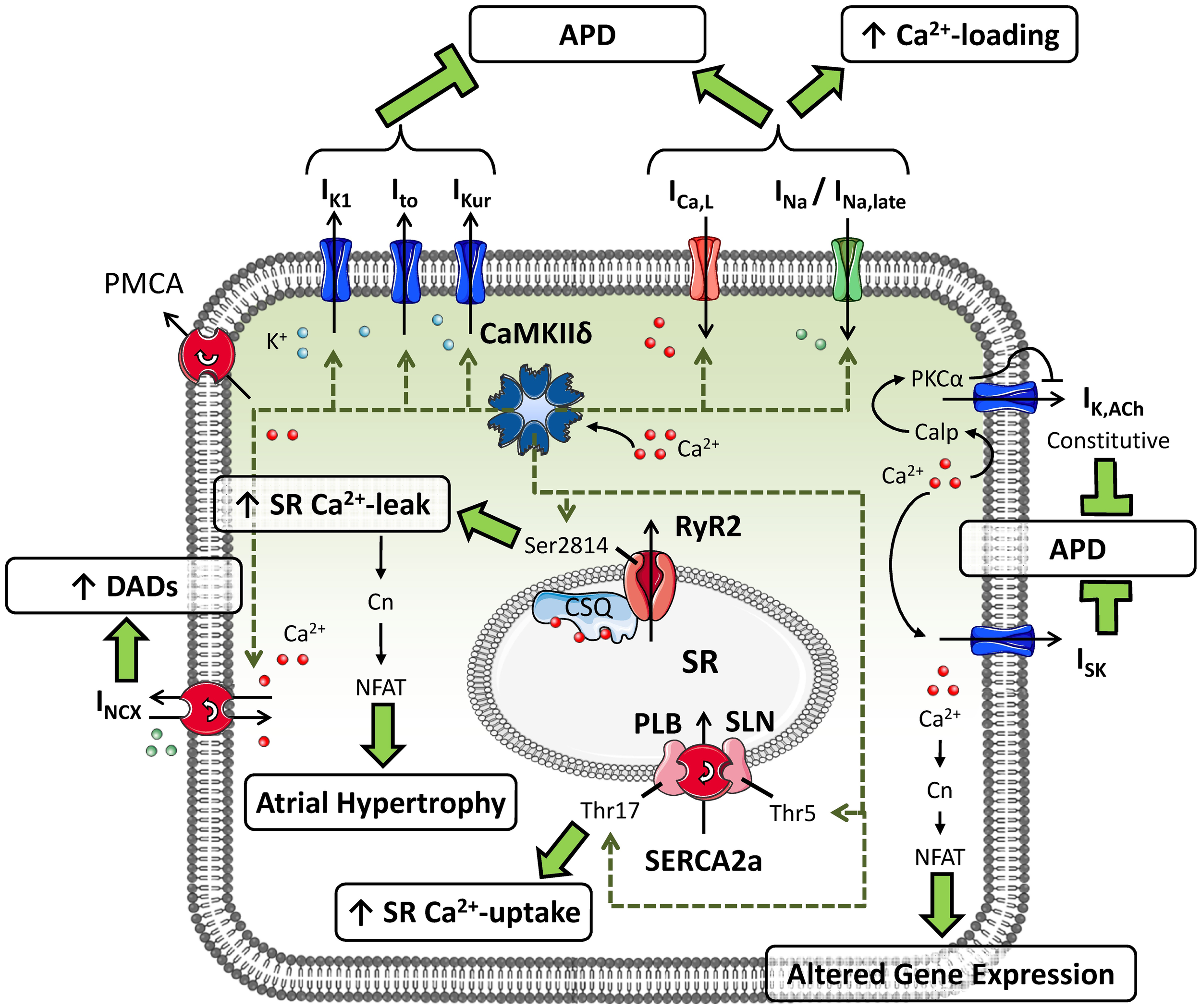
Putative substrates for CaMKII-dependent phosphorylation in atrial cardiomyocytes and their consequences for atrial cellular electrophysiology and Ca2+-handling. CaMKII can phosphorylate the transient-outward K+-current (Ito), inward-rectifier K+-current (IK1) and ultra-rapid delayed-rectifier K+-current (IKur), augmenting their functions and shortening action potential duration (APD). Phosphorylation of L-type Ca2+-current (ICa,L) and Na+-current (INa; resulting in an increased late component: INa,late) by CaMKII increases intracellular Ca2+ levels and prolongs APD. CaMKII-dependent phosphorylation of phospholamban (PLB) and sarcolipin (SLN) increases sarcoplasmic reticulum (SR) Ca2+-uptake, whereas phosphorylation of type-2 ryanodine-receptor channels (RyR2) promotes diastolic SR Ca2+-leak. CaMKII-dependent increases in expression of Na+/Ca2+-exchanger type-1 (NCX1) augment NCX-current (INCX), promoting the occurrence of delayed afterdepolarizations (DADs). In addition, Ca2+-handling abnormalities can activate small-conductance Ca2+-activated K+-currents (ISK) and agonist-independent “constitutive” IK,ACh, shortening APD, and promote altered gene expression via the Ca2+-dependent phosphatase calcineurin (Cn).
Ca2+/CaMKII dysregulation in AF
Mechanisms promoting CaMKII dysregulation in AF
CaMKIIδ protein expression and activity are increased in dogs with pacing-induced atrial tachycardia remodeling (Wakili et al., 2010), goats with long-standing AF (Greiser et al., 2009), and patients with chronic AF (cAF); (Tessier et al., 1999; Neef et al., 2010; Voigt et al., 2012), suggesting that increased CaMKII function can be a consequence of AF. Activation of CaMKII appears to be regulated locally within the myocyte, since autophosphorylation of Thr287 was increased for CaMKIIδC but not CaMKIIδB in patients with cAF (Voigt et al., 2012). Several AF-related conditions, including sympathetic hyperactivity, oxidative stress and atrial tachycardia per se, may promote CaMKII activation (Figure 2). High atrial-rates during AF can activate CaMKII via frequency-dependent mechanisms. In addition, neuronal autonomic dysbalance can contribute to AF initiation (Park et al., 2012) and atrial tachycardia, in turn, promotes neural remodeling including heterogeneous sympathetic hyperactivity (Jayachandran et al., 2000). Increased sympathetic activity can activate CaMKII through various pathways, including protein kinase-A (PKA)-dependent augmentation of cellular Ca2+-cycling (Grimm and Brown, 2010). In addition, PKA-independent, exchange-protein activated by cAMP (Epac) can activate CaMKII following β-adrenoceptor stimulation (Mangmool et al., 2010; Pereira et al., 2013). Moreover, β1-adrenoceptor-activated Epac2 can promote SR Ca2+-leak via phosphorylation of RyR2-Ser2814 (Pereira et al., 2013). It has also been suggested that the Epac-mediated CaMKII activation involves phosphorylation of CaMKII-Thr287 by protein kinase-C type-ε (PKCε) (Oestreich et al., 2009) and the upregulation of PKCε in cAF patients (Voigt et al., 2008) might contribute to increased CaMKII activity. Since PKCε translocation to the membrane is increased in atrial myocytes following in vitro tachypacing (Makary et al., 2011), this might promote local atrial tachycardia-dependent CaMKII stimulation, although this remains to be proven in future studies. AF is also associated with oxidative stress and oxidation of CaMKII is increased in AF patients (Purohit et al., 2013). Conversely, phosphorylation of the inhibitory Thr306/307 site is decreased in cAF patients, providing another pathway of CaMKII activation in AF (Voigt et al., 2012).
Figure 2
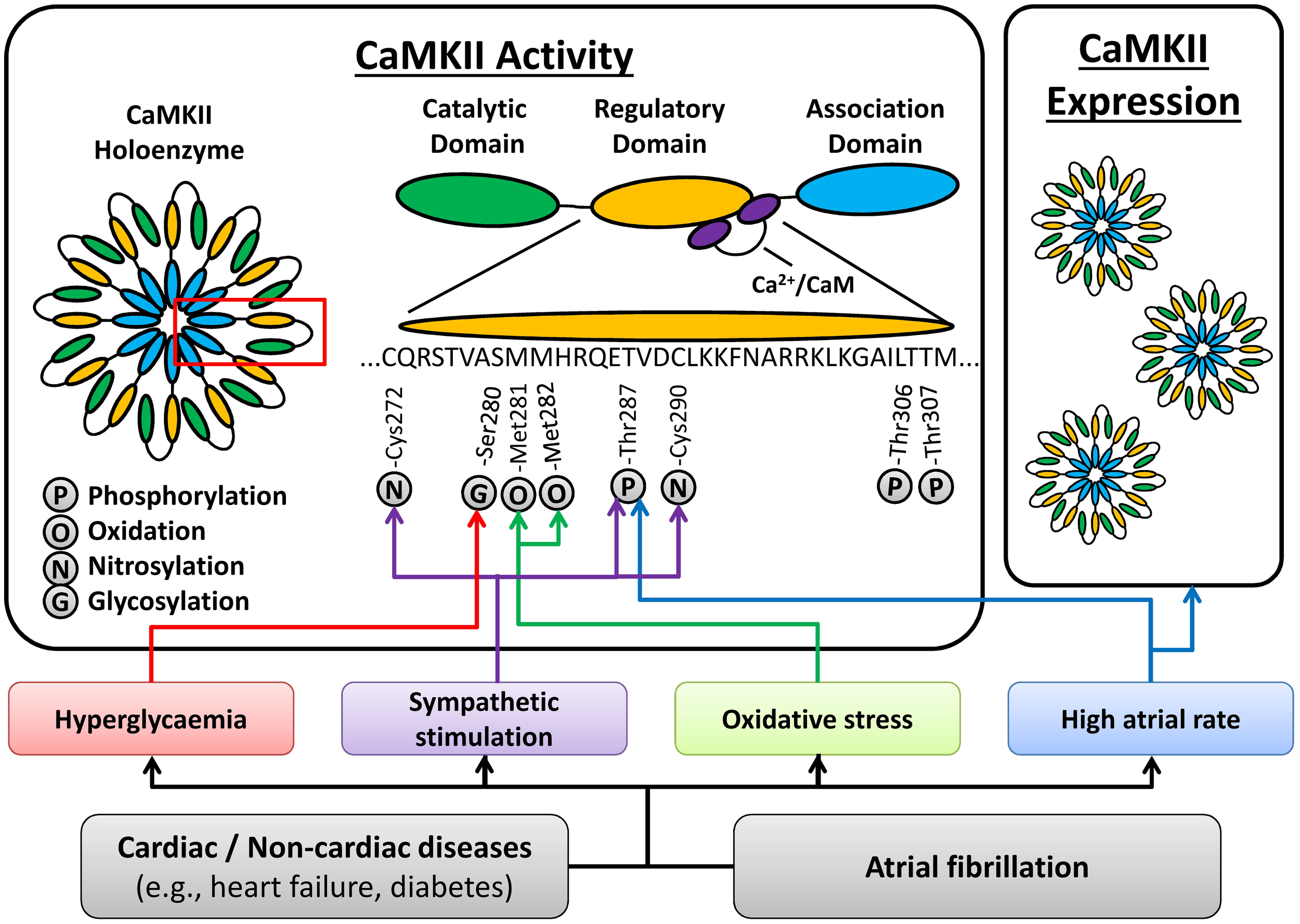
Atrial fibrillation (AF)-related mechanisms promoting CaMKII activation. AF-promoting conditions and/or AF itself can activate CaMKII via high atrial rates, oxidative stress, sympathetic hyperactivity, and hyperglycaemia, resulting in post-translational modifications (phosphorylation, oxidation, nitrosylation and glycosylation) of various residues in the regulatory domain of CaMKII. In addition, AF is associated with an increased total expression of the CaMKII holoenzyme. See text for details.
Atrial CaMKII activity is also increased in dogs with ventricular tachypacing-induced heart failure (Yeh et al., 2008), and in goats with atrial dilatation (Greiser et al., 2009), suggesting that CaMKII can be activated by AF-enabling cardiac pathologies, potentially contributing to the evolution of a vulnerable substrate for AF initiation. Similarly, increased body-mass index and diabetes are AF risk-factors (Dublin et al., 2006) that may further promote CaMKII activation via O-linked glycosylation in response to hyperglycaemia (Erickson et al., 2013). Thus, CaMKII activation is multifactorial, resulting from AF itself, as well as from AF-enabling risk factors and diseases (Figure 2).
Role of CaMKII in ectopic activity
CaMKII has been shown to promote EADs in ventricular cardiomyocytes (Qi et al., 2009), which can produce ectopic (triggered) activity. CaMKII-dependent phosphorylation of ICa,L slows ICa,L inactivation, increasing the ICa,L window current that plays a major role in the generation of EADs (Qi et al., 2009). In addition, the APD-prolonging effects of CaMKII-dependent phosphorylation of INa, increasing INa,late, could further promote the occurrence of EADs and ectopic activity (Wagner et al., 2011). However, since most forms of AF are generally associated with abbreviated APD, the relevance of such EADs may be lower in atrial compared to ventricular arrhythmogenesis. On the other hand, EADs can also arise from Ca2+-handling abnormalities that activate depolarizing NCX-current (late phase-3 EADs), which have been implicated in the initiation of AF in some animal models (Burashnikov and Antzelevitch, 2003; Patterson et al., 2006).
Ca2+-handling abnormalities can also cause DADs and ectopic (triggered) activity, promoting AF initiation. Genetic mouse models have revealed that intrinsic RyR2-dysfunction is sufficient to increase the susceptibility to pacing-induced AF, as reviewed in (Dobrev et al., 2011). Mice with gain-of-function RyR2 mutations causing catecholaminergic polymorphic ventricular tachycardia (CPVT), and mice lacking the RyR2-stabilizing subunit FKBP12.6, develop Ca2+-handling abnormalities including increased SR Ca2+-leak and spontaneous SR Ca2+-release events (i.e., sparks, waves). These mice also have an increased susceptibility to pacing-induced AF (Sood et al., 2008; Chelu et al., 2009; Shan et al., 2012). Rapid-pacing activates CaMKII and increases CaMKII-dependent RyR2 and PLB phosphorylation. Genetic and pharmacological CaMKII inhibition normalized the susceptibility to pacing-induced AF in mice with a CPVT mutation in RyR2 (Chelu et al., 2009). Of note, selective genetic inhibition of CaMKII-dependent RyR2-hyperphosphorylation (RyR2-Ser2814Ala) also reduced the incidence of rapid-pacing-induced AF in mice where a vulnerable substrate was created using stimulation with the muscarinic-receptor agonist carbachol, and pacing-induced AF in mice deficient of FKBP12.6 (Chelu et al., 2009; Li et al., 2012), strongly suggesting that CaMKII-dependent RyR2 hyperphosphorylation and associated Ca2+-handling abnormalities are critical AF-promoting factors (Dobrev et al., 2011). In addition, recent work has identified calmodulin as a direct regulator of RyR2 that stabilizes SR Ca2+-release (Yang et al., 2014). Although overall calmodulin levels are increased in cAF patients (Voigt et al., 2012), a reduced affinity between RyR2 and calmodulin, as observed in heart failure (Yang et al., 2014), could potentially contribute to RyR2 dysfunction in AF.
Atrial cardiomyocytes from cAF patients have unaltered RyR2 protein expression levels and SR Ca2+-load (Voigt et al., 2012). However, they exhibit CaMKII-dependent RyR2-hyperphosphorylation that increases RyR2 open probability and augments SR Ca2+-leak and spontaneous diastolic Ca2+-release events. The enhanced SR Ca2+ leak results in enhanced DADs and cellular triggered activity and can be blocked using CaMKII inhibitors, thus supporting an important proarrhythmic role for these CaMKII-dependent Ca2+-handling abnormalities in human AF (Voigt et al., 2012). In addition, cAF patients had significantly reduced levels of RyR2-stabilizing FKBP12.6 subunits (Vest et al., 2005) and larger transient-inward currents/depolarizations for a given SR Ca2+-release. The latter is in part mediated by increased NCX1 mRNA (Gaborit et al., 2005) and protein expression levels (Schotten et al., 2002; El-Armouche et al., 2006; Voigt et al., 2012) in cAF patients. There is evidence that CaMKII can upregulate NCX1 transcription following β-adrenoceptor stimulation (Mani et al., 2010), suggesting that CaMKII could also be involved in the increased NCX1 expression in AF. Although atrial cardiomyocytes from paroxysmal AF (pAF) patients also have increased SR Ca2+-leak, spontaneous SR Ca2+-release events and DADs, these effects appear to be CaMKII-independent, since CaMKII expression and Thr287 autophosphorylation were not changed in pAF patients (Voigt et al., 2014). Similarly, CaMKII-dependent PLB and RyR2 phosphorylation, as well as NCX1 expression were also unaltered in pAF patients. However, RyR2 expression and RyR2 single-channel open-probability were increased and SR Ca2+-load was larger in pAF, likely due to PKA-dependent PLB hyperphosphorylation (Voigt et al., 2014). Computational modeling showed that both increased SR Ca2+-load and RyR2 dysregulation contribute to the spontaneous diastolic SR Ca2+-release events in cardiomyocytes from pAF patients. Thus, although SR Ca2+-handling abnormalities appear a central element in experimental and human AF, the underlying molecular mechanisms are complex. In addition, it is likely that the proarrhythmic consequences of Ca2+-handling abnormalities are distinct for different types of AF (Figure 3). Whereas Ca2+-mediated triggered activity is a likely candidate for the re-initiation of AF episodes in pAF patients, its relevance for patients with long-standing persistent AF is incompletely understood. In persistent AF forms, Ca2+-dependent evolution and progression of atrial remodeling may play a prominent role in arrhythmia maintenance and stabilization (as discussed below).
Figure 3
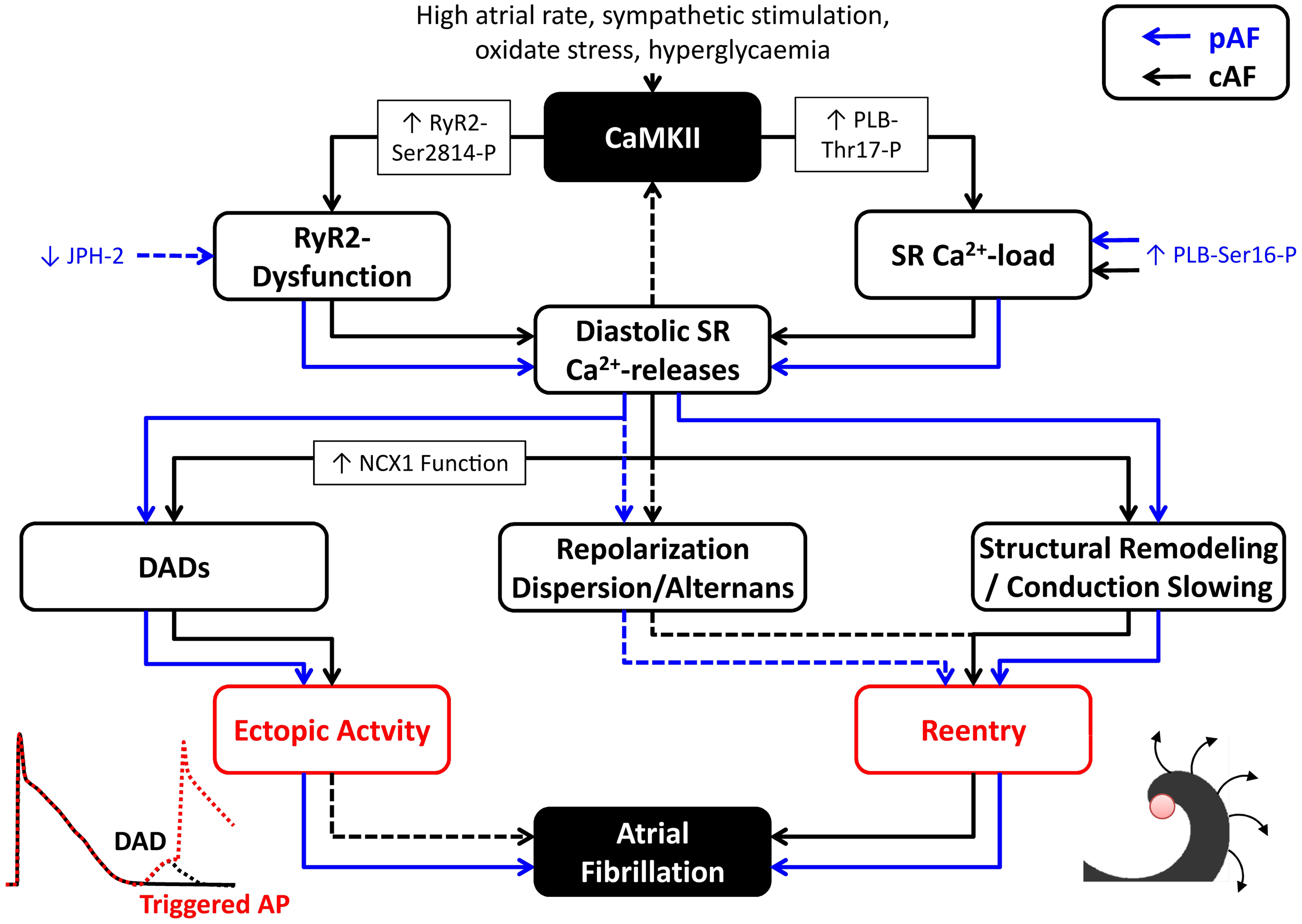
Proarrhythmic consequences of Ca2+/CaMKII dysregulation in atrial fibrillation (AF). CaMKII activation and CaMKII-dependent phosphorylation of type-2 ryanodine-receptor (RyR2) channels (RyR2-Ser2814-P) and phospholamban (PLB-Thr17-P), as well as other factors, promote spontaneous diastolic sarcoplasmic reticulum (SR) Ca2+-release events through RyR2 dysfunction and modulation of SR Ca2+-load in patients with paroxysmal AF (pAF; blue lines) and long-standing persistent, chronic AF (cAF; black lines). SR Ca2+-leak and diastolic SR Ca2+-release events can produce delayed afterdepolarizations (DADs) that contribute to ectopic activity. In addition, they can promote reentry through local repolarization abnormalities, as well as structural remodeling and conduction velocity (CV) slowing. Influences for which the proarrhythmic roles are more speculative have been indicated with dashed lines.
Role of CaMKII in reentry-promoting remodeling
Ca2+-handling abnormalities also play a role in AF-promoting reentry. APD-shortening is a hallmark feature of AF-related remodeling that facilitates the maintenance of reentrant circuits. It is largely mediated by a reduction in depolarizing ICa,L and an increase in several repolarizing K+-currents. Various mechanisms contribute to reduced ICa,L in AF (Dobrev et al., 2012). Cav1.2 expression is reduced in AF through a pathway involving the Ca2+-dependent phosphatase calcineurin and nuclear factor of activated T-cells (NFAT) (Qi et al., 2008) and increased activation of the Ca2+-dependent protease calpain promotes breakdown of ICa,L channels (Brundel et al., 2004). ICa,L phosphorylation is also reduced in AF, decreasing current amplitude, and could be due to either increased protein phosphatase activity or local reduction in CaMKII availability (Christ et al., 2004). IK1 is increased in cAF patients, and, together with an increase in the acetylcholine-independent “constitutive” activity of IK,ACh, results in an overall increase in inward-rectifier K+-current that contributes to APD shortening (Dobrev et al., 2005). A Ca2+-dependent NFAT-mediated reduction in the inhibitory microRNA-26 in AF results in disinhibition of Kir2.1 expression, contributing to the increase in IK1 in cAF patients (Luo et al., 2013). Increased constitutive IK,ACh may also result from Ca2+-dependent calpain-mediated reduction in inhibitory PKCα (Makary et al., 2011). Thus, the proarrhythmic increases in IK1 and constitutive IK,ACh are partially mediated by Ca2+-dependent processes, althouth the potential involvement of CaMKII needs to be specifically addressed in future studies. Finally, the Ca2+-dependent small-conductance (SK) K+-current (ISK) is upregulated in atria of cAF patients, which might contribute to APD shortening (Zhou et al., 2012), although others have reported reduced ISK in AF (Yu et al., 2012). Acute Ca2+-dependent regulation of currents such as INa, ISK or ICa,L can also contribute to beat-by-beat alterations in APD, including APD alternans and augmentation of dispersion of repolarization. These spatial and temporal repolarization heterogeneities favor unidirectional conduction block that can initiate reentry. In agreement, atrial APD alternans is emerging as a clinical index to assess the vulnerability to develop AF in patients (Lalani et al., 2013).
Ca2+-entry into atrial fibroblasts via multiple ion channels contributes to fibroblast proliferation and differentiation into collagen-secreting myofibroblasts, which promote fibrosis-induced heterogeneous conduction slowing and reentry (Yue et al., 2011). Transient-receptor potential (TRP) melastatin-related-7 (TRPM7) and canonical-3 (TRPC3) channels are major sources of Ca2+-entry into human atrial fibroblasts (Du et al., 2010; Harada et al., 2012). Atrial fibroblasts from AF-patients have larger TRPM7 currents and increased TRPC3 expression, and are more prone to differentiate into myofibroblasts. Knockdown of TRPM7 expression reduces basal differentiation of fibroblasts from cAF patients (Du et al., 2010). Furthermore, pharmacological inhibition of TRPC3 channels reduces AF substrate development and AF duration in dogs with electrically maintained AF (Harada et al., 2012). TRPM7-like channels are inhibited by CaMKII in hepatocytes, which may support hepatocellular survival during proliferation (Mishra et al., 2009). Moreover, Ca2+-influx through TRPC3 promotes CaMKII activation and NADPH-oxidase-mediated production of reactive oxygen species in a genetic mouse model (Kitajima et al., 2011). Thus, CaMKII could potentially act both upstream and downstream of TRP channels to alter fibroblast function in AF, although this requires confirmation in subsequent studies.
Ca2+-handling abnormalities can also promote reentry by reducing atrial conduction velocity through a reduction in INa or direct inhibition of gap-junction channels in atrial cardiomyocytes (Heijman et al., 2013b; King et al., 2013b). The reduction in conduction velocity observed in mice with RyR2 mutations could be reproduced in wild-type mice with acute application of caffeine to increase SR Ca2+-leak, and appears to be due to both acute Ca2+-dependent inhibition of INa, as well as downregulation of Nav1.5 subunit expression under chronic conditions (King et al., 2013a). This Ca2+-dependent reduction in INa is expected to promote reentry-mediated AF maintenance but may also reduce the likelihood of ectopic activity (Heijman et al., 2013b). At present the role of CaMKII in these reentry-promoting Ca2+-handling abnormalities is largely unknown, although it has been suggested that CaMKII-dependent phosphorylation could also reduce peak INa, particularly at fast heart rates relevant for AF (Wagner et al., 2006), which could contribute to reentry by reducing atrial conduction velocity.
Cardiac myosin-binding protein-C (cMyBPC) is a critical regulator of myofilament function (Schlossarek et al., 2011). Ser282-phosphorylation of cMyBPC is decreased in dogs with pacing-induced atrial tachycardia remodeling (Wakili et al., 2010), in dogs with ventricular tachypacing-induced heart failure (Yeh et al., 2008), goats with long-standing AF or atrial dilatation (Greiser et al., 2009), and in cAF patients (Tessier et al., 1999; Neef et al., 2010; Voigt et al., 2012). Although there is indirect evidence that this could be due to increased local dephosphorylation by phosphatases, reduced local CaMKII-dependent phosphorylation of Ser282 could also be involved. In addition, contractile dysfunction is promoted by activation of Ca2+-dependent proteases. Together, contractile dysfunction and associated atrial dilatation result in a larger vulnerable substrate, promoting reentrant arrhythmias (De Jong et al., 2011).
Accumulating evidence suggests that CaMKII-dependent RyR2-hyperphosphorylation and the related SR Ca2+-leak play an important role in AF-promoting structural remodeling. Mice with transgenic overexpression of the transcriptional repressor CREM-IbΔC-X in cardiomyocytes (CREM mice) develop age-dependent progression from spontaneous atrial ectopy to paroxysmal and long-lasting AF episodes (Li et al., 2014). The development of spontaneous AF episodes is preceded by Ca2+-handling abnormalities and atrial enlargement. Genetic inhibition of CaMKII-dependent RyR2 phosphorylation (RyR2-Ser2814Ala) in CREM mice prevents Ca2+-handling abnormalities and spontaneous AF, as well as atrial dilatation and conduction abnormalities (Li et al., 2014). Thus, CaMKII-dependent RyR2-dysregulation not only contributes to ectopic (triggered) activity, but also drives a progressive development of an AF substrate (Figure 3), promoting atrial hypertrophy and dilatation, and AF progression (Li et al., 2014). These studies suggest the interesting possibility that the progression of AF might be inhibited by targeted treatment of CaMKII or SR Ca2+-leak via RyR2. Future studies in mice and large animal models are required to confirm this concept, since the pathophysiological mechanisms and the importance of CaMKII likely vary for different species and experimental AF models, as well as for different forms of clinical AF.
CaMKII dysregulation and Ca2+-handling abnormalities as therapeutic targets in AF
The central role of Ca2+-handling abnormalities in AF-pathophysiology suggests their potential as antiarrhythmic targets. Stabilization of RyR2 has emerged as a viable approach to normalize Ca2+-handling abnormalities. Several currently-available antiarrhythmic drugs, including the class-Ic Na+-channel blocker flecainide (Hilliard et al., 2010), the β-adrenoceptor blocker carvedilol (Zhou et al., 2011), and the antianginal drug ranolazine (Parikh et al., 2012), directly bind and inhibit RyR2 channels. Indeed, flecainide has been successfully employed in other Ca2+-dependent arrhythmias such as CPVT (Van Der Werf et al., 2011). However, flecainide also inhibits atrial K+-currents like IK,ACh (Voigt et al., 2010), which might contribute to its anti-AF efficacy. More specific RyR2 inhibitors are currently being evaluated in clinical studies (Dobrev et al., 2012).
Inhibition of CaMKII or elimination of CaMKII-dependent RyR2-phosphorylation has proven antiarrhythmic in mouse models of AF and has shown beneficial effects in atrial cardiomyocytes from cAF patients (Chelu et al., 2009; Li et al., 2012; Voigt et al., 2012). However, given the importance of CaMKII in various physiological processes, systemic CaMKII inhibition could have various undesirable side effects, including reduced fertility and impaired memory (Backs et al., 2010; Halt et al., 2012). Moreover, since CaMKII expression/autophosphorylation and CaMKII-dependent phosphorylation of RyR2 and PLB are not increased in pAF patients (Voigt et al., 2014), it is unclear whether CaMKII inhibition would be beneficial for this group of patients. Nonetheless, it appears likely that localized CaMKII inhibition could be a promising antiarrhythmic strategy for appropriately-selected AF patients. Future animal studies and clinical trials will be needed to determine which groups of AF patients are most likely to benefit from CaMKII inhibition. Local inhibition of CaMKII might be possible through inhibition of specific CaMKII-isoforms and splice variants, or by modulating different CaMKII-targeting proteins. Another potential avenue could be the modulation of microRNAs. Injection of complementary “antagomirs” to reduce the activity of certain microRNAs or overexpression of microRNAs has proven beneficial in a variety of experimental models, as reviewed in (Kumarswamy and Thum, 2013). Recent work has shown that CaMKIIδ expression is repressed by microRNA-145 (Cha et al., 2013) and microRNA-30b-5p (He et al., 2013). Increasing the levels of these microRNAs in the heart might, therefore, be an option to inhibit CaMKII.
Conclusions
Ca2+-handling abnormalities promote both focal ectopic (triggered) activity and reentry that contribute to AF initiation and maintenance. The expected increase in the incidence of AF and the limited efficacy and safety of currently available antiarrhythmic drugs, make a better understanding of these AF-modulating processes critical for the development of improved therapeutic strategies. Ca2+-handling abnormalities provide a novel set of potential antiarrhythmic targets for the treatment of AF. However, due to the multitude of etiologies and complexity of mechanisms underlying clinical AF, it is likely that tailored therapeutic strategies for specific groups of patients that target multiple pathophysiological processes will be necessary. Cardiac-specific inhibition of CaMKII could be a promising therapeutic strategy for certain groups of AF patients.
Funding
The authors' work is supported by the European–North American Atrial Fibrillation Research Alliance (07CVD03 to Dobromir Dobrev) and the Alliance for Calmodulin Kinase Signaling in Heart Disease (08CVD01, to Xander H. T. Wehrens) grants of Fondation Leducq, the European Network for Translational Research in Atrial Fibrillation (EUTRAF; 261057, to Dobromir Dobrev), the German Federal Ministry of Education and Research through the DZHK (German Center for Cardiovascular Research, to Dobromir Dobrev), the American Heart Association (13EIA14560061 to Xander H. T. Wehrens), Muscular Dystrophy Association (186530), and National Institutes of Health grants HL089598, HL091947, and HL117641 (to Xander H. T. Wehrens).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
BacksJ.SteinP.BacksT.DuncanF. E.GrueterC. E.McAnallyJ.et al. (2010). The γ isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proc. Natl. Acad. Sci. U.S.A. 107, 81–86. 10.1073/pnas.0912658106
2
BersD. M. (2002). Cardiac excitation-contraction coupling. Nature415, 198–205. 10.1038/415198a
3
BootmanM. D.SmyrniasI.ThulR.CoombesS.RoderickH. L. (2011). Atrial cardiomyocyte calcium signalling. Biochim. Biophys. Acta1813, 922–934. 10.1016/j.bbamcr.2011.01.030
4
BrundelB. J.KampingaH. H.HenningR. H. (2004). Calpain inhibition prevents pacing-induced cellular remodeling in a HL-1 myocyte model for atrial fibrillation. Cardiovasc. Res. 62, 521–528. 10.1016/j.cardiores.2004.02.007
5
BurashnikovA.AntzelevitchC. (2003). Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation107, 2355–2360. 10.1161/01.CIR.0000065578.00869.7C
6
CammA. J.LipG. Y.De CaterinaR.SavelievaI.AtarD.HohnloserS. H.et al. (2012). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. developed with the special contribution of the European Heart Rhythm Association. Eur. Heart J. 33, 2719–2747. 10.1093/eurheartj/ehs253
7
CammJ. (2012). Antiarrhythmic drugs for the maintenance of sinus rhythm: risks and benefits. Int. J. Cardiol. 155, 362–371. 10.1016/j.ijcard.2011.06.012
8
ChaM. J.JangJ. K.HamO.SongB. W.LeeS. Y.LeeC. Y.et al. (2013). MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ. Biochem. Biophys. Res. Commun. 435, 720–726. 10.1016/j.bbrc.2013.05.050
9
CheluM. G.SarmaS.SoodS.WangS.Van OortR. J.SkapuraD. G.et al. (2009). Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951. 10.1172/JCI37059
10
ChristT.BoknikP.WohrlS.WettwerE.GrafE. M.BoschR. F.et al. (2004). L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation110, 2651–2657. 10.1161/01.CIR.0000145659.80212.6A
11
ChughS. S.HavmoellerR.NarayananK.SinghD.RienstraM.BenjaminE. J.et al. (2013). Worldwide epidemiology of atrial fibrillation: a global burden of disease 2010 study. Circulation129, 837–847. 10.1161/CIRCULATIONAHA.113.005119
12
ColbranR. J. (1993). Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J. Biol. Chem. 268, 7163–7170.
13
De JongA. M.MaassA. H.Oberdorf-MaassS. U.Van VeldhuisenD. J.Van GilstW. H.Van GelderI. C. (2011). Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc. Res. 89, 754–765. 10.1093/cvr/cvq357
14
DibbK. M.ClarkeJ. D.HornM. A.RichardsM. A.GrahamH. K.EisnerD. A.et al. (2009). Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2, 482–489. 10.1161/CIRCHEARTFAILURE.109.852228
15
DobrevD.CarlssonL.NattelS. (2012). Novel molecular targets for atrial fibrillation therapy. Nat. Rev. Drug Discov. 11, 275–291. 10.1038/nrd3682
16
DobrevD.FriedrichA.VoigtN.JostN.WettwerE.ChristT.et al. (2005). The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation112, 3697–3706. 10.1161/CIRCULATIONAHA.105.575332
17
DobrevD.NattelS. (2008). Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J. Cardiovasc. Pharmacol. 52, 293–299. 10.1097/FJC.0b013e318171924d
18
DobrevD.RavensU. (2003). Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res. Cardiol. 98, 137–148. 10.1007/s00395-003-0409-8
19
DobrevD.TeosL. Y.LedererW. J. (2009). Unique atrial myocyte Ca2+ signaling. J. Mol. Cell. Cardiol. 46, 448–451. 10.1016/j.yjmcc.2008.12.004
20
DobrevD.VoigtN.WehrensX. H. (2011). The ryanodine receptor channel as a molecular motif in atrial fibrillation: pathophysiological and therapeutic implications. Cardiovasc. Res. 89, 734–743. 10.1093/cvr/cvq324
21
DobrevD.WehrensX. H. (2010). Calmodulin kinase II, sarcoplasmic reticulum Ca2+ leak, and atrial fibrillation. Trends Cardiovasc. Med. 20, 30–34. 10.1016/j.tcm.2010.03.004
22
DuJ.XieJ.ZhangZ.TsujikawaH.FuscoD.SilvermanD.et al. (2010). TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 106, 992–1003. 10.1161/CIRCRESAHA.109.206771
23
DublinS.FrenchB.GlazerN. L.WigginsK. L.LumleyT.PsatyB. M.et al. (2006). Risk of new-onset atrial fibrillation in relation to body mass index. Arch. Intern. Med. 166, 2322–2328. 10.1001/archinte.166.21.2322
24
El-ArmoucheA.BoknikP.EschenhagenT.CarrierL.KnautM.RavensU.et al. (2006). Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation114, 670–680. 10.1161/CIRCULATIONAHA.106.636845
25
El-HaouS.BalseE.NeyroudN.DilanianG.GavilletB.AbrielH.et al. (2009). Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ. Res. 104, 758–769. 10.1161/CIRCRESAHA.108.191007
26
EricksonJ. R.JoinerM. L.GuanX.KutschkeW.YangJ.OddisC. V.et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell133, 462–474. 10.1016/j.cell.2008.02.048
27
EricksonJ. R.PereiraL.WangL.HanG.FergusonA.DaoK.et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature502, 372–376. 10.1038/nature12537
28
GaboritN.SteenmanM.LamiraultG.Le MeurN.Le BouterS.LandeG.et al. (2005). Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation112, 471–481. 10.1161/CIRCULATIONAHA.104.506857
29
GreiserM.NeubergerH. R.HarksE.El-ArmoucheA.BoknikP.De HaanS.et al. (2009). Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J. Mol. Cell. Cardiol. 46, 385–394. 10.1016/j.yjmcc.2008.11.012
30
GrimmM.BrownJ. H. (2010). Beta-adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol. 48, 322–330. 10.1016/j.yjmcc.2009.10.016
31
GutierrezD. A.Fernandez-TenorioM.OgrodnikJ.NiggliE. (2013). NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 100, 392–401. 10.1093/cvr/cvt201
32
HaltA. R.DallapiazzaR. F.ZhouY.SteinI. S.QianH.JunttiS.et al. (2012). CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–1216. 10.1038/emboj.2011.482
33
HaradaM.LuoX.QiX. Y.TadevosyanA.MaguyA.OrdogB.et al. (2012). Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation126, 2051–2064. 10.1161/CIRCULATIONAHA.112.121830
34
HeJ.JiangS.LiF. L.ZhaoX. J.ChuE. F.SunM. N.et al. (2013). MicroRNA-30b-5p is involved in the regulation of cardiac hypertrophy by targeting CaMKIIδ. J. Investig. Med. 61, 604–612. 10.231/JIM.0b013e3182819ac6
35
HeijmanJ.VoigtN.DobrevD. (2013a). New directions in antiarrhythmic drug therapy for atrial fibrillation. Future Cardiol. 9, 71–88. 10.2217/fca.12.78
36
HeijmanJ.VoigtN.NattelS.DobrevD. (2012). Calcium handling and atrial fibrillation. Wien. Med. Wochenschr. 162, 287–291. 10.1007/s10354-012-0109-9
37
HeijmanJ.VoigtN.NattelS.DobrevD. (2014). Compendium: cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance and progression. Circ. Res. 10.1161/CIRCRESAHA.113.302226. [Epub ahead of print].
38
HeijmanJ.WehrensX. H.DobrevD. (2013b). Atrial arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia-is there a mechanistic link between sarcoplasmic reticulum Ca2+ leak and re-entry?Acta Physiol. (Oxf.)207, 208–211. 10.1111/apha.12038
39
HilliardF. A.SteeleD. S.LaverD.YangZ.Le MarchandS. J.ChopraN.et al. (2010). Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J. Mol. Cell. Cardiol. 48, 293–301. 10.1016/j.yjmcc.2009.10.005
40
JayachandranJ. V.SihH. J.WinkleW.ZipesD. P.HutchinsG. D.OlginJ. E. (2000). Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation101, 1185–1191. 10.1161/01.CIR.101.10.1185
41
KingJ. H.WickramarachchiC.KuaK.DuY.JeevaratnamK.MatthewsH. R.et al. (2013a). Loss of Nav1.5 expression and function in murine atria containing the RyR2-P2328S gain-of-function mutation. Cardiovasc. Res. 99, 751–759. 10.1093/cvr/cvt141
42
KingJ. H.ZhangY.LeiM.GraceA. A.HuangC. L.FraserJ. A. (2013b). Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol. (Oxf.)207, 308–323. 10.1111/apha.12006
43
KitajimaN.WatanabeK.MorimotoS.SatoY.KiyonakaS.HoshijimaM.et al. (2011). TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem. Biophys. Res. Commun. 409, 108–113. 10.1016/j.bbrc.2011.04.124
44
KumarswamyR.ThumT. (2013). Non-coding RNAs in cardiac remodeling and heart failure. Circ. Res. 113, 676–689. 10.1161/CIRCRESAHA.113.300226
45
LalaniG. G.SchrickerA. A.CloptonP.KrummenD. E.NarayanS. M. (2013). Frequency analysis of atrial action potential alternans: a sensitive clinical index of individual propensity to atrial fibrillation. Circ. Arrhythm. Electrophysiol. 6, 859–867. 10.1161/CIRCEP.113.000204
46
LenaertsI.BitoV.HeinzelF. R.DriesenR. B.HolemansP.D'hoogeJ.et al. (2009). Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ. Res. 105, 876–885. 10.1161/CIRCRESAHA.109.206276
47
LiN.ChiangD. Y.WangS.WangQ.SunL.VoigtN.et al. (2014). Ryanodine-receptor mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. [Epub ahead of print]. 10.1161/CIRCULATIONAHA.113.006611
48
LiN.WangT.WangW.CutlerM. J.WangQ.VoigtN.et al. (2012). Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ. Res. 110, 465–470. 10.1161/CIRCRESAHA.111.253229
49
LuoX.PanZ.ShanH.XiaoJ.SunX.WangN.et al. (2013). MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J. Clin. Invest. 123, 1939–1951. 10.1172/JCI62185
50
MakaryS.VoigtN.MaguyA.WakiliR.NishidaK.HaradaM.et al. (2011). Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ. Res. 109, 1031–1043. 10.1161/CIRCRESAHA.111.253120
51
MangmoolS.ShuklaA. K.RockmanH. A. (2010). β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J. Cell Biol. 189, 573–587. 10.1083/jcb.200911047
52
ManiS. K.EganE. A.AddyB. K.GrimmM.KasiganesanH.ThiyagarajanT.et al. (2010). β-Adrenergic receptor stimulated Ncx1 upregulation is mediated via a CaMKII/AP-1 signaling pathway in adult cardiomyocytes. J. Mol. Cell. Cardiol. 48, 342–351. 10.1016/j.yjmcc.2009.11.007
53
MishraR.RaoV.TaR.ShobeiriN.HillC. E. (2009). Mg2+- and MgATP-inhibited and Ca2+/calmodulin-sensitive TRPM7-like current in hepatoma and hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G687–G694. 10.1152/ajpgi.90683.2008
54
MishraS.GrayC. B.MiyamotoS.BersD. M.BrownJ. H. (2011). Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ. Res. 109, 1354–1362. 10.1161/CIRCRESAHA.111.248401
55
NattelS.BursteinB.DobrevD. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73. 10.1161/CIRCEP.107.754564
56
NattelS.DobrevD. (2012). The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur. Heart J. 33, 1870–1877. 10.1093/eurheartj/ehs079
57
NeefS.DybkovaN.SossallaS.OrtK. R.FluschnikN.NeumannK.et al. (2010). CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 106, 1134–1144. 10.1161/CIRCRESAHA.109.203836
58
OestreichE. A.MalikS.GoonasekeraS. A.BlaxallB. C.KelleyG. G.DirksenR. T.et al. (2009). Epac and phospholipase Cε regulate Ca2+ release in the heart by activation of protein kinase Cε and calcium-calmodulin kinase II. J. Biol. Chem. 284, 1514–1522. 10.1074/jbc.M806994200
59
ParikhA.MantravadiR.KozhevnikovD.RocheM. A.YeY.OwenL. J.et al. (2012). Ranolazine stabilizes cardiac ryanodine receptors: a novel mechanism for the suppression of early afterdepolarization and torsades de pointes in long QT type 2. Heart Rhythm9, 953–960. 10.1016/j.hrthm.2012.01.010
60
ParkH. W.ShenM. J.LinS. F.FishbeinM. C.ChenL. S.ChenP. S. (2012). Neural mechanisms of atrial fibrillation. Curr. Opin. Cardiol. 27, 24–28. 10.1097/HCO.0b013e32834dc4e8
61
PattersonE.LazzaraR.SzaboB.LiuH.TangD.LiY. H.et al. (2006). Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J. Am. Coll. Cardiol. 47, 1196–1206. 10.1016/j.jacc.2005.12.023
62
PereiraL.ChengH.LaoD. H.NaL.Van OortR. J.BrownJ. H.et al. (2013). Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation127, 913–922. 10.1161/CIRCULATIONAHA.12.148619
63
PurohitA.RokitaA. G.GuanX.ChenB.KovalO. M.VoigtN.et al. (2013). Oxidized Ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation128, 1748–1757. 10.1161/CIRCULATIONAHA.113.003313
64
QiX.YehY. H.ChartierD.XiaoL.TsujiY.BrundelB. J.et al. (2009). The calcium/calmodulin/kinase system and arrhythmogenic afterdepolarizations in bradycardia-related acquired long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2, 295–304. 10.1161/CIRCEP.108.815654
65
QiX. Y.YehY. H.XiaoL.BursteinB.MaguyA.ChartierD.et al. (2008). Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ. Res. 103, 845–854. 10.1161/CIRCRESAHA.108.175463
66
RichardsM. A.ClarkeJ. D.SaravananP.VoigtN.DobrevD.EisnerD. A.et al. (2011). Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Heart Circ. Physiol. 301, H1996–H2005. 10.1152/ajpheart.00284.2011
67
RoderickH. L.KnollmannB. C. (2013). Inositol 1,4,5-trisphosphate receptors: “exciting” players in cardiac excitation-contraction coupling?Circulation128, 1273–1275. 10.1161/CIRCULATIONAHA.113.005157
68
SchlossarekS.MeariniG.CarrierL. (2011). Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 50, 613–620. 10.1016/j.yjmcc.2011.01.014
69
SchottenU.GreiserM.BenkeD.BuerkelK.EhrenteidtB.StellbrinkC.et al. (2002). Atrial fibrillation-induced atrial contractile dysfunction: a tachycardiomyopathy of a different sort. Cardiovasc. Res. 53, 192–201. 10.1016/S0008-6363(01)00453-9
70
ShanJ.XieW.BetzenhauserM.ReikenS.ChenB. X.WronskaA.et al. (2012). Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 111, 708–717. 10.1161/CIRCRESAHA.112.273342
71
SoodS.CheluM. G.Van OortR. J.SkapuraD.SantonastasiM.DobrevD.et al. (2008). Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm5, 1047–1054. 10.1016/j.hrthm.2008.03.030
72
SwaminathanP. D.PurohitA.HundT. J.AndersonM. E. (2012). Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ. Res. 110, 1661–1677. 10.1161/CIRCRESAHA.111.243956
73
TessierS.KarczewskiP.KrauseE. G.PansardY.AcarC.Lang-LazdunskiM.et al. (1999). Regulation of the transient outward K+ current by Ca2+/calmodulin-dependent protein kinases II in human atrial myocytes. Circ. Res. 85, 810–819. 10.1161/01.RES.85.9.810
74
TraffordA. W.ClarkeJ. D.RichardsM. A.EisnerD. A.DibbK. M. (2013). Calcium signalling microdomains and the t-tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovasc. Res. 98, 192–203. 10.1093/cvr/cvt018
75
Van Der WerfC.KannankerilP. J.SacherF.KrahnA. D.ViskinS.LeenhardtA.et al. (2011). Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 57, 2244–2254. 10.1016/j.jacc.2011.01.026
76
VestJ. A.WehrensX. H.ReikenS. R.LehnartS. E.DobrevD.ChandraP.et al. (2005). Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation111, 2025–2032. 10.1161/01.CIR.0000162461.67140.4C
77
VoigtN.HeijmanJ.WangQ.ChiangD. Y.LiN.KarckM.et al. (2014). Cellular and Molecular Mechanisms of Atrial Arrhythmogenesis in Patients With Paroxysmal Atrial Fibrillation. Circulation129, 145–156. 10.1161/CIRCULATIONAHA.113.006641
78
VoigtN.LiN.WangQ.WangW.TraffordA. W.Abu-TahaI.et al. (2012). Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation125, 2059–2070. 10.1161/CIRCULATIONAHA.111.067306
79
VoigtN.MaguyA.YehY. H.QiX.RavensU.DobrevD.et al. (2008). Changes in IK,ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc. Res. 77, 35–43. 10.1093/cvr/cvm051
80
VoigtN.RozmaritsaN.TrauschA.ZimniakT.ChristT.WettwerE.et al. (2010). Inhibition of IK,ACh current may contribute to clinical efficacy of class I and class III antiarrhythmic drugs in patients with atrial fibrillation. Naunyn Schmiedebergs Arch. Pharmacol. 381, 251–259. 10.1007/s00210-009-0452-6
81
WagnerS.DybkovaN.RasenackE. C.JacobshagenC.FabritzL.KirchhofP.et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. 10.1172/JCI26620
82
WagnerS.HackerE.GrandiE.WeberS. L.DybkovaN.SossallaS.et al. (2009). Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2, 285–294. 10.1161/CIRCEP.108.842799
83
WagnerS.RuffH. M.WeberS. L.BellmannS.SowaT.SchulteT.et al. (2011). Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late INa augmentation leading to cellular Na and Ca overload. Circ. Res. 108, 555–565. 10.1161/CIRCRESAHA.110.221911
84
WakiliR.VoigtN.KaabS.DobrevD.NattelS. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968. 10.1172/JCI46315
85
WakiliR.YehY. H.Yan QiX.GreiserM.ChartierD.NishidaK.et al. (2010). Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ. Arrhythm. Electrophysiol. 3, 530–541. 10.1161/CIRCEP.109.933036
86
YangY.GuoT.OdaT.ChakrabortyA.ChenL.UchinoumiH.et al. (2014). Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 114, 295–306. 10.1161/CIRCRESAHA.114.302857
87
YehY. H.WakiliR.QiX. Y.ChartierD.BoknikP.KaabS.et al. (2008). Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol. 1, 93–102. 10.1161/CIRCEP.107.754788
88
YuT.DengC.WuR.GuoH.ZhengS.YuX.et al. (2012). Decreased expression of small-conductance Ca2+-activated K+ channels SK1 and SK2 in human chronic atrial fibrillation. Life Sci. 90, 219–227. 10.1016/j.lfs.2011.11.008
89
YueL.XieJ.NattelS. (2011). Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 89, 744–753. 10.1093/cvr/cvq329
90
ZhouQ.XiaoJ.JiangD.WangR.VembaiyanK.WangA.et al. (2011). Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med. 17, 1003–1009. 10.1038/nm.2406
91
ZhouX.-B.VoigtN.WielandT.DobrevD. (2012). Enhanced frequency-dependent retograde trafficking of small conductance Ca2+-activated K+ channels may contribute to electrical remodeling in human atrial fibrillation. Heart Rhythm9, S319.
92
ZimetbaumP. (2012). Antiarrhythmic drug therapy for atrial fibrillation. Circulation125, 381–389. 10.1161/CIRCULATIONAHA.111.019927
Summary
Keywords
atrial fibrillation, calcium, CaMKII, ectopic activity, reentry
Citation
Heijman J, Voigt N, Wehrens XHT and Dobrev D (2014) Calcium dysregulation in atrial fibrillation: the role of CaMKII. Front. Pharmacol. 5:30. doi: 10.3389/fphar.2014.00030
Received
21 January 2014
Accepted
15 February 2014
Published
04 March 2014
Volume
5 - 2014
Edited by
Andrew G. Edwards, Oslo University Hospital, Norway
Reviewed by
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USA; Sandeep Pandit, University of Michigan, USA; Eleonora Grandi, University of California Davis, USA
Copyright
© 2014 Heijman, Voigt, Wehrens and Dobrev.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dobromir Dobrev, Institute of Pharmacology, Faculty of Medicine, University Duisburg-Essen, Hufelandstrasse 55, 45122 Essen, Germany e-mail: dobromir.dobrev@uk-essen.de
This article was submitted to Pharmacology of Ion Channels and Channelopathies, a section of the journal Frontiers in Pharmacology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.