 Pablo J. Sáez1
Pablo J. Sáez1 Roberto Villalobos-Labra1
Roberto Villalobos-Labra1 Francisco Westermeier1,2,3
Francisco Westermeier1,2,3 Luis Sobrevia1,4,5
Luis Sobrevia1,4,5 Marcelo Farías-Jofré1*
Marcelo Farías-Jofré1*- 1Cellular and Molecular Physiology Laboratory, Division of Obstetrics and Gynecology, School of Medicine, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Facultad de Ciencia, Universidad San Sebastián, Santiago, Chile
- 3Advanced Center for Chronic Diseases, Faculty of Chemical and Pharmaceutical Sciences and Faculty of Medicine, University of Chile, Santiago, Chile
- 4University of Queensland Centre for Clinical Research, Faculty of Medicine and Biomedical Sciences, University of Queensland, Herston, QL, Australia
- 5Faculty of Pharmacy, Universidad de Sevilla, Sevilla, Spain
Adverse microenvironmental stimuli can trigger the endoplasmic reticulum (ER) stress pathway, which initiates the unfolded protein response (UPR), to restore protein-folding homeostasis. Several studies show induction of ER stress during obesity. Chronic UPR has been linked to different mechanisms of disease in obese and diabetic individuals, including insulin resistance (IR) and impaired angiogenesis. Endothelial cell (EC) migration is an initial step for angiogenesis, which is associated with remodeling of existing blood vessels. EC migration occurs according to the leader–follower model, involving coordinated processes of chemotaxis, haptotaxis, and mechanotaxis. Thus, a fine-tuning of EC migration is necessary to provide the right timing to form the required vessels during angiogenesis. ER stress modulates EC migration at different levels, usually impairing migration and angiogenesis, although different effects may be observed depending on the tissue and/or microenvironment. In the context of pregnancy, maternal obesity (MO) induces IR in the offspring. Interestingly, several proteins associated with obesity-induced IR are also involved in EC migration, providing a potential link with the ER stress-dependent alterations observed in obese individuals. Different signaling cascades that converge on cytoskeleton regulation directly impact EC migration, including the Akt and/or RhoA pathways. In addition, ER is the main intracellular reservoir for Ca2+, which plays a pivotal role during EC migration. Therefore, ER stress-related alterations in Ca2+ signaling or Ca2+ levels might also produce distorted EC migration. However, the above findings have been studied in the context of adult obesity, and no information has been reported regarding the effect of MO on fetal EC migration. Here we summarize the state of knowledge about the possible mechanisms by which ER stress and IR might impact EC migration and angiogenesis in fetal endothelium exposed to MO during pregnancy.
Introduction
Endoplasmic reticulum is the major subcellular membrane organelle, playing a pivotal role in synthesis, folding and maturation of proteins, and providing the main Ca2+ reservoir inside the cell (Berridge et al., 2003; Cnop et al., 2012; Hetz et al., 2013). Under certain conditions, the environment induces ER stress and further activation of the UPR, which triggers a cascade of signaling events to restore protein-folding homeostasis (Kozutsumi et al., 1988). This cellular condition, known as ER stress, is induced by different types of stimuli, such as accumulation of unfolded proteins, fatty acids, cytokines, redox state dysregulation, and increased intracellular Ca2+ levels (Kozutsumi et al., 1988; Hotamisligil, 2010; Cnop et al., 2012; Fu et al., 2012; Garg et al., 2012; Hetz et al., 2013). Moreover, ER stress is linked to different diseases, including cancer, type II diabetes, and obesity (Hotamisligil, 2010; Cnop et al., 2012; Hetz et al., 2013). Importantly, most if not all of these pathologies are associated with vascular pathologies such as distorted angiogenesis or endothelial dysfunction (Minamino and Kitakaze, 2010; Basha et al., 2012). By affecting EC physiology, ER stress contributes to the vascular dysfunction observed in diabetic retinopathy, cancer, obesity, atherosclerosis, and ischemia (Amin et al., 2012; Hetz et al., 2013; Zeng et al., 2013; Paridaens et al., 2014). One of the most relevant functions of EC is angiogenesis, which is the capacity to form new capillary vessels (Lamalice et al., 2007). Interestingly, ER stress affects two of the basic mechanisms that contribute to angiogenesis (Lamalice et al., 2007): VEGF signaling, and EC migration (Iwawaki et al., 2009; Ghosh et al., 2010; Pereira et al., 2010; Banerjee et al., 2011; Zeng et al., 2013; Paridaens et al., 2014). However, obesity might impact EC migration directly through ER stress and induced IR; Westermeier et al., 2014), because several of the involved proteins, such as RhoA and Akt (also called protein kinase B), also modulate EC migration (Lamalice et al., 2007).
As expected, the development of obesity in adults produces altered angiogenic responses in adipose tissue (Christiaens and Lijnen, 2010). However, in the context of pregnancy, MO not only affects the mother but also can permanently damage fetal tissues [American College of Obstetricians and Gynecologists (ACOG), 2005]. Thus, the adverse intrauterine environment in MO pregnancies could modulate offspring physiology (Bruyndonckx et al., 2013), leading to in utero development of IR (Catalano et al., 2009), which ultimately might affect EC migration and angiogenesis. In support of this notion, MO is associated with alterations in serum levels of angiogenic markers (Zera et al., 2014) and changes in VEGF receptor expression patterns in the placenta (Dubova et al., 2011; Saben et al., 2014). Interestingly, very recent studies show that MO induces ER stress in offspring in murine models (Melo et al., 2014; Wu et al., 2014), suggesting that distortions in EC migration and angiogenesis might occur. Since cell migration commands angiogenesis, our goal is to give an integrative overview of how MO-induced ER stress and IR might affect the migratory potential of EC and hence angiogenesis in the offspring, with deleterious consequences for the offspring’s development.
ER Stress and the Unfolded Protein Response
Multiple environmental stimuli are capable of triggering ER stress (Schroder and Kaufman, 2005). There are three major sensors of ER stress, all of which are ER membrane-associated proteins: ATF6 (α and β isoforms), PERK, and IRE1 (Hetz et al., 2013). While activation of both PERK and IRE1 involves dimerization and phosphorylation, ATF6 activation requires its cleavage and translocation to the nucleus (Hetz et al., 2013). These three pathways interact and produce ER-to-nucleus signaling that reduces protein translation and increases folding capacity (Hotamisligil, 2010). However, differential activation of ER sensors may occur depending on the type and timing of the ER stressor signal (Wu et al., 2007; Fu et al., 2012). The latter will produce different UPR profiles, associated with the specific stimuli triggering the ER stress, the affected cell type(s), and the microenvironment background. In addition, acute versus chronic ER stress may lead to different cellular responses (Wu et al., 2007; Fu et al., 2012).
Role of ER Stress in Cell Migration
The role of ER stress on cell migration and angiogenesis has been studied mainly in cancer cells (Hetz et al., 2013). Several ER stress-related proteins contribute to cell migration and/or angiogenesis in tumors (Paridaens et al., 2014). The three abovementioned ER stress branches usually contribute to angiogenesis in the tumoral context (Hetz et al., 2013). Interestingly, the ER chaperone immunoglobulin binding protein (BiP/GRP78), which is an early signal of ER stress activation, is required to give angiogenic potential to tumors (Dong et al., 2008), suggesting that ER stress might impact angiogenesis from the beginning of the response. However, in non-tumoral contexts, ER stress has been shown to impair cell migration and angiogenesis.
Tunicamycin is an antibiotic that inhibits synthesis of asparagine-linked glycoproteins (Takatsuki et al., 1971; Duksin and Bornstein, 1977) and is commonly used to induce ER stress in different in vitro and in vivo models. Promoting the accumulation of misfolded un-glycosylated proteins at the ER level, tunicamycin produces activation of all of the ER branches (Schroder and Kaufman, 2005). In support of the notion that ER stress impacts cell migration, early studies performed by Gipson et al. (1984) showed impaired epithelial sheet migration in the presence of tunicamycin. This study observed delayed wound healing in tunicamycin-exposed organotypic cultures of corneas (Gipson et al., 1984). Several years later, similar results were obtained in vitro with human epithelial airway cells (Dorscheid et al., 2001). In addition, recent observations in vascular smooth muscle cells show that tunicamycin activates the IRE1 and ATF6 pathways, imparing platelet-derived growth factor-induced in vitro migration (Yi et al., 2012). Similarly, neferine, an alkaloid used in cancer treatment, induces ER stress activation in an epithelial cell line, which produces concomitant inhibition of cell migration (Yoon et al., 2013).
Therefore, ER stress activation under resting or non-tumoral physiopathological conditions seems to impair collective cell migration, conversely to the tumoral context in which it seems to promote angiogenesis (Paridaens et al., 2014). This finding suggests that ER stress might play different roles in EC migration depending on the tissue environment.
Role of ER Stress in EC Migration and Angiogenesis
Collective EC migration is required as an initial event during angiogenesis. The EC migration process combines three different mechanisms: (1) chemotaxis, which is induced by soluble chemoattractants, (2) haptotaxis, which is mediated by chemoattractants bound to the substrate, and (3) mechanotaxis, which provides the mechanical forces to provide directionality (Lamalice et al., 2007). EC migrate according to the leader–follower model, in which a leader (or pioneer) cell with more protrusive and motile activity at the leading edge affects the signaling of the follower cells. Thus, the leader cell exerts mechanical pulling over the follower cells, providing the directionality of the sheet growth (Vitorino and Meyer, 2008; Rorth, 2009). At the cellular level, this process involves both actin and microtubule cytoskeleton rearrangements and changes in cell polarity (Figure 1) towards the edge of the monolayer (Etienne-Manneville, 2013).
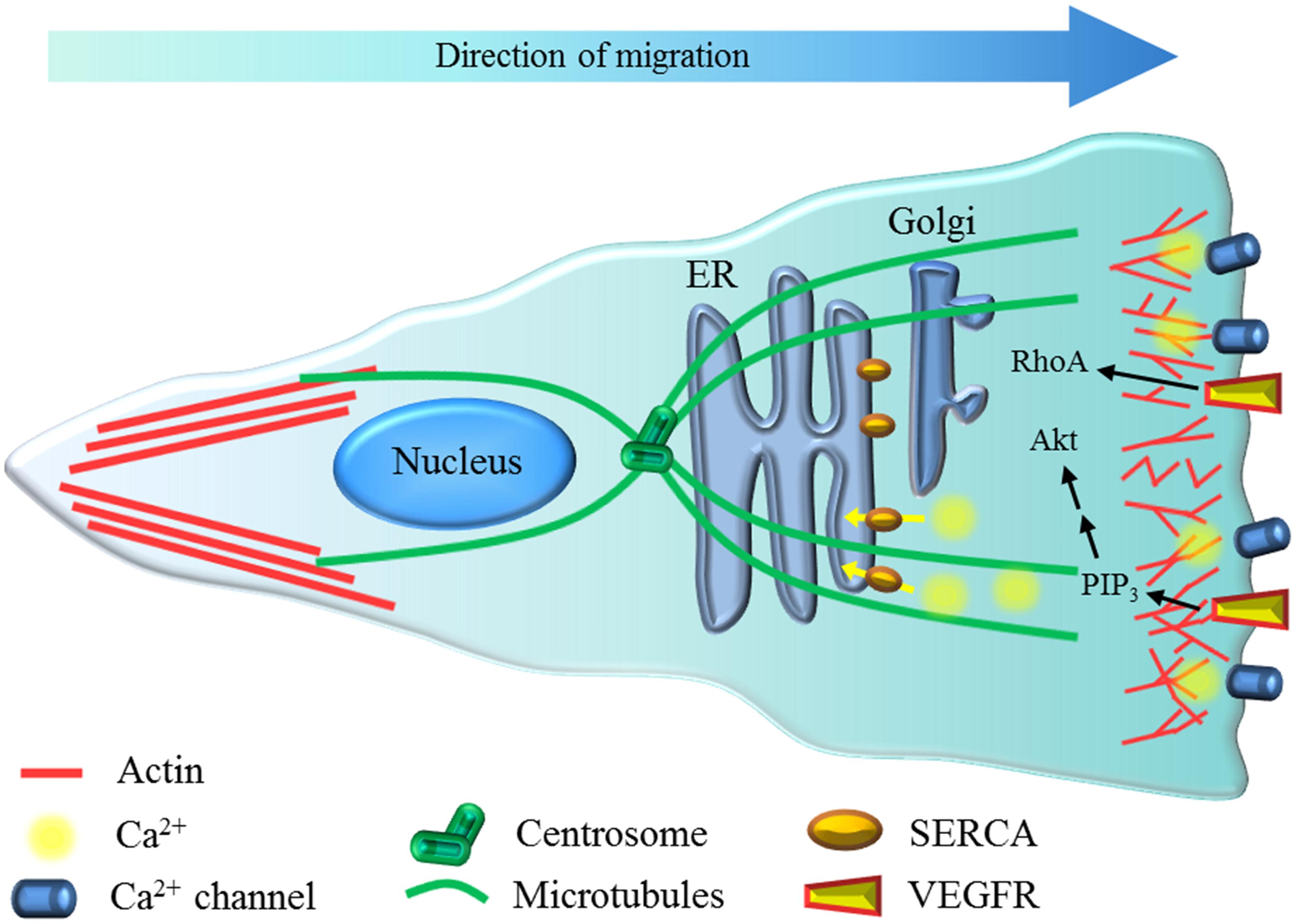
FIGURE 1. Possible role of ER stress on EC migration. Migrating EC have actin (red) dynamics at the front and also polarization of the centrosome and microtubules (green) and Golgi apparatus toward the leading edge. Ca2+ (yellow) dynamics at the front of the cell are mainly mediated by Ca2+ channels (i.e., Orai), and the sarco/endoplasmic reticulum Ca2+-ATPase pump (SERCA, orange) restores the free intracellular Ca2+ levels in the cytoplasm, allowing for sustained oscillations. At the rear of the cell, the formation of actin stress fibers is observed. Vascular endothelial growth factor receptor (VEGFR) activation induces RhoA and -through phosphatidylinositol (3,4,5)-triphosphate (PIP3) production- Akt activity, which contributes to cytoskeleton polarization and remodeling.
While wound healing is commonly used to evaluate collective EC migration, tube formation in Matrigel has been used extensively to evaluate the angiogenic potential of these cells. These and other in vitro and in vivo models are used to evaluate different pro- or anti-angiogenic compounds (Lamalice et al., 2007; Aranda and Owen, 2009). As expected, VEGF is a potent chemoattractant for EC and contributes to angiogenesis (Lamalice et al., 2007). Interestingly, signaling of VEGF and other proteins involved in EC migration and angiogenesis is affected by ER stress (Paridaens et al., 2014).
Tunicamycin exposure is associated with impairment of both spontaneous and VEGF-induced migration of capillary EC, involving inhibition of VEGF signaling (Banerjee et al., 2011). Similarly, 2-DG reduces spontaneous collective migration, showing an anti-angiogenic effect on cultures of HUVEC through activation of ER stress (Merchan et al., 2010). Moreover, this study also shows that 2-DG has an anti-angiogenic effect in vivo (Merchan et al., 2010). In addition, treatment with neferine, another ER stress inducer, inhibits in vitro angiogenesis in HUVEC (Yoon et al., 2013). Similarly, acrolein, which is an unsaturated aldehyde known as an environmental pollutant and also found in some foods, induces ER stress in EC (Haberzettl et al., 2009), affecting both migration and angiogenesis (O’Toole et al., 2014). This study shows that acrolein inhibits wound healing and tube formation in HUVEC (O’Toole et al., 2014). Importantly, the same study evaluated Akt signaling after insulin exposure, showing that acrolein impairs insulin signaling (O’Toole et al., 2014), supporting the hypothesis that IR might be linked to distorted EC migration.
In contrast, MCPIP mediates cytokine-induced angiogenesis in HUVEC by up-regulation of ER stress markers (Roy and Kolattukudy, 2012). In addition, a recent study shows that VEGF might induce PERK and ATF6 signaling, which contribute to survival and migration of EC (Karali et al., 2014). These data suggest that in EC cells, the induction of ER stress might affect both migration and angiogenesis in a stimulus-dependent manner. Moreover, in a murine model it has been shown that IRE1 activity, which contributes to proper placental development, is required for development of ER stress during pregnancy (Iwawaki et al., 2009). This study shows that lack of IRE1 reduces VEGF receptor expression and is lethal (Iwawaki et al., 2009). These data suggest that in EC, induction of ER stress might affect both migration and angiogenesis in a stimulus-dependent manner.
One additional explanation of the different effects of ER stress on angiogenesis capacity could be related to the involvement of microenvironmental factors such as inflammatory mediators. For example, diabetic and other models of retinopathy are associated with distorted retinal angiogenesis, which has been related to ER stress (Salminen et al., 2010; Wang et al., 2012). In a murine model of type I diabetes, increased levels of VEGF and TNF-α were observed in the retina, which were correlated with increased ER stress markers of the PERK and IRE1 branches (Li et al., 2009). In addition, oxygen-induced retinopathy was associated with development of ER stress, in a similar way to that induced by tunicamycin (Li et al., 2009). Importantly, resveratrol and some of its derivatives (Tabata et al., 2007), which have anti-inflammatory effects and inhibit ER stress development (Zhang and Kaufman, 2008), prevent retinal vascular degeneration induced by tunicamycin or ischemia/reperfusion (Li et al., 2012). Taken together, these data suggest that ER stress contributes to angiogenesis and neovascularization in vivo. However, the target cells are not fully elucidated, and it is possible that the effect of ER stress inhibition might first impact immune cells, which through the release of inflammatory mediators might indirectly impact EC. Interestingly, several of these inflammatory mediators show altered levels during obesity (Snyder-Cappione and Nikolajczyk, 2013).
Effects of Maternal Obesity and Insulin Resistance on Cell Migration
Altered nutritional state is becoming a relevant and growing public health issue globally [World Health Organ (WHO), 2003]. The relationship between obesity-induced chronic ER stress and IR has been well established in murine and human adipose tissues (Cnop et al., 2012; Flamment et al., 2012; Jung et al., 2013; Boden et al., 2014). Interestingly, it has been shown that the obesity-dependent induction of ER stress markers is reduced in human adipose tissue after weight loss, suggesting that body weight change constitutes an important factor that modulates the ER stress response (Gregor et al., 2009). During pregnancy, excessive gestational weight gain and MO have been associated with increased risk of maternal pathologies and detrimental long-term effects on fetal tissues, through a process known as intrauterine programming (McMillen and Robinson, 2005). Since HUVEC provides a useful model to study neonatal evidence of fetal EC programming under multiple pregnancy conditions, in this section we focus on different IR- and migration-associated proteins that might be distorted by MO.
MO-Related Fetal Programming
Obesity and overweight during pregnancy are well-recognized independent risk factors that contribute to the development of metabolic syndrome and several diet-related anomalies not only in the mother, but also in the fetus through fetal programming [American College of Obstetricians and Gynecologists (ACOG), 2005; Flenady et al., 2011; Triunfo and Lanzone, 2014]. This intrauterine programming can be observed as altered responses to physiological stimuli in HUVEC isolated from pathological pregnancies (Cheng et al., 2013; Krause et al., 2013). Indeed, it has been described that MO induces IR in fetuses in utero (Catalano et al., 2009), showing the relevance of metabolic fetal programming. Recently, it has been found that EC from obese adult subjects show ER stress (Kaplon et al., 2013), but it has not been determined whether MO induces these changes in fetal tissue. However, interesting recent evidence suggests that ER stress might be induced through fetal programming in animal models (Melo et al., 2014; Wu et al., 2014).
Using a murine model of MO, feeding dams a high-fat diet resulted in increased inflammation, ER stress markers, and IR in hypothalamic tissue of the MO offspring (post-natal day 28) compared to the control group (Melo et al., 2014). This study shows that lactation plays a major role in the development of ER stress (Melo et al., 2014). However, it was also noted that there was a significant increase in phosphorylation of eIF2α, downstream of PERK, in hypothalamic tissue at birth (day 0) of MO offspring (Melo et al., 2014), suggesting that at least the PERK ER stress branch is already activated during MO pregnancy.
In another study, using a similar model of diet-induced obesity, it was shown that MO offspring have increased ER stress and inflammatory markers compared to the control group (Wu et al., 2014). This study shows increased PERK and IRE1 activation in liver and adipose tissue of MO offspring at post-natal day 100 (Wu et al., 2014). Interestingly, treating dams during pregnancy and lactation with quercetin, which is an anti-inflammatory flavonoid (Indra et al., 2013) that inhibits ER stress (Suganya et al., 2014), prevented the development of ER stress in the offspring of MO pregnancies (Wu et al., 2014), suggesting that the development of ER in the offspring begins during pregnancy.
Altogether these data show that MO induces ER stress through fetal programming in murine models. Therefore, it is conceivable to suggest that MO in human pregnancies might produce a similar phenomenon.
How Might MO Impact HUVEC Migration and Angiogenesis?
Fetal programming is known to occur during MO pregnancies; however one remaining question is how MO might mediate fetal EC migration and angiogenesis. First, human chorionic gonadotropin has been shown to increase the proliferation of HUVEC in the presence of various adipokines, such as IL 6, leptin, adiponectin, and TNF-α (Polec et al., 2014). Moreover, there is interesting evidence that placental tissue from women with MO shows altered expression of VEGF receptors (Saben et al., 2014). In fact, a very recent study showed that increased body mass index (BMI) was associated with the presence of angiogenic markers in placental tissue (Zera et al., 2014). This work demonstrates an inverse correlation between BMI and serum levels of sFlt-1, which is associated with a pro-angiogenic profile (Zera et al., 2014). The authors propose that this might be due to excessive fetal growth, which requires a bigger placental vascular bed (Zera et al., 2014). This distorted angiogenic profile during MO is also supported by evidence showing the predominance of non-branching angiogenesis observed in placental tissue of obese women (Dubova et al., 2011).
Considering that: (1) MO results in distorted angiogenesis; (2) obesity has been associated with ER stress and IR; and (3) IR-related proteins also play a role in cell migration (RhoA, Akt), we hypothesize the possible contribution of different IR-related and others proteins to the modulation of EC migration capacity in the context of MO-dependent ER stress (Table 1).
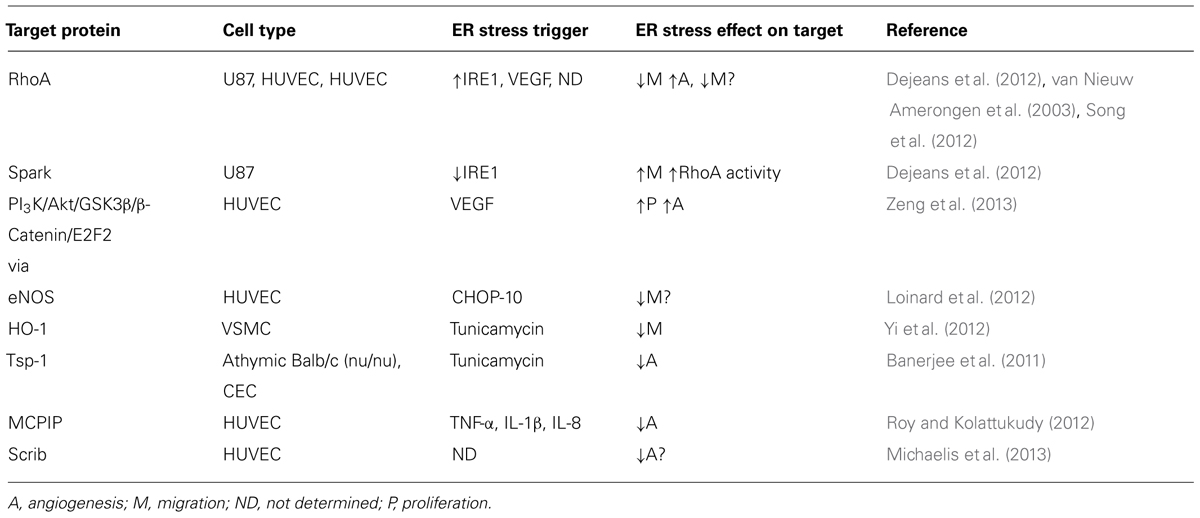
TABLE 1. Putative migration-related targets of ER stress signaling.
RhoA signaling
RhoA and its downstream signaling has been linked to IR because they have targets such as Akt and eNOS (Kanda et al., 2006; Nunes et al., 2010) but also play a relevant role in cell migration (Jaffe and Hall, 2005). As mentioned above, in 2D cultures, EC migrate according to the leader–follower model (Rorth, 2009). In HUVEC, fibroblastic growth factor-induced collective cell migration is commanded by proteins that regulate cell–cell interactions, cell density, individual cell migration, and directed cell migration (Vitorino and Meyer, 2008). In the same study, RhoA was found to contribute to collective cell migration of HUVEC, a finding corroborated later by other groups (Vitorino and Meyer, 2008; Povero et al., 2013). Moreover, the role of RhoA is also relevant in other cell types where it seems to be a typical feature of leader cells (Omelchenko et al., 2003; Jaffe and Hall, 2005; Rorth, 2009), because it contributes importantly to the mechanotaxis process (Reffay et al., 2014).
The contribution of RhoA to EC migration has been observed using a dominant-negative model and by its inhibition using ADP-ribosylation after bacterial toxin exposure. Both experimental conditions were associated with reduction of HUVEC migration (Aepfelbacher et al., 1997; Song et al., 2012) and angiogenesis (Povero et al., 2013). Previous studies suggest that RhoA mediates migration and VEGF-induced chemotaxis (van Nieuw Amerongen et al., 2003). However, interesting observations using a microfluidic device showed that RhoA contributes to HUVEC shear stress-induced mechanotaxis, although it does not affect VEGF-induced filopodia formation (Song et al., 2012). The explanation for these differential effects may also rely on the EC culture type studied; for example, RhoA contributes to VEGF-induced migration and angiogenesis of human foreskin microvascular EC (van Nieuw Amerongen et al., 2003). Thus, it is possible to suggest that RhoA contributes to migration in a stimulus- and cell type-dependent manner.
As mentioned above, ER stress is linked to cell migration. Supporting this notion, ER stress is associated with IRE1, which acts as an upstream protein of RhoA signaling (Dejeans et al., 2012). In a RhoA-dependent manner, cancer cells expressing a dominant-negative IRE1 protein show increased adhesion, impaired migration, and a reduced proliferation rate, but no change in invasive properties (Dejeans et al., 2012). As expected, RhoA inhibition restores the phenotype in IRE1 dominant-negative expressing cells (Dejeans et al., 2012). Therefore, as IRE1-lacking cells show over-activation of RhoA, it is possible to hypothesize that MO-induced ER stress, which increases IRE1 activity, might reduce RhoA activity, impairing EC migration (Figure 2).
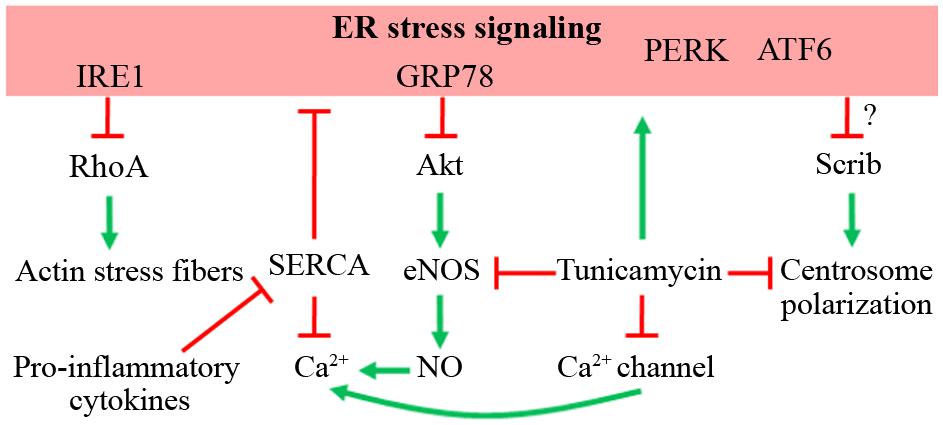
FIGURE 2. Possible role of ER stress on EC migration. The effects of ER stress signaling on different proteins that command cellular migration is depicted, as well as the effect of tunicamycin, an ER stress inducer. Red lines indicate inhibition or down-regulation of the target. Green arrows indicate activation or up-regulation of the target.
Akt signaling
The Akt gene encodes three isoforms (Akt1-3). In EC, Akt activation is related to several signaling cascades, such as the insulin pathway, eNOS activation, cell survival, and migration (Shiojima and Walsh, 2002). In EC, it has been suggested that Akt3 contributes to cell migration (Vitorino and Meyer, 2008). However, Akt involvement in cellular migration and angiogenesis depends on the tissue context and simultaneously activated signaling (Somanath et al., 2006). For instance, Akt3 has defective signaling in muscle cells from obese insulin-resistant subjects (Brozinick et al., 2003), potentially affecting EC migration response downstream of the insulin receptors (Guo, 2014). In addition, extensive evidence has shown a potential link between Akt and ER stress signaling (Appenzeller-Herzog and Hall, 2012). Interestingly, BiP/GRP78 suppresses the Ser473 phosphorylation of Akt by direct interaction, which might prevent accessibility for activating kinases (Yung et al., 2011). Moreover, this study shows that ER stress activates or inhibits Akt signaling depending on the magnitude or severity of this response (Yung et al., 2011). On the other hand, VEGF has been related to induction of the IRE1 branch of ER stress in HUVEC (Zeng et al., 2013). VEGF exposure was associated with IRE1-dependent splicing of XBP1 and activation of Akt/GSK signaling, which is required for the proliferation and angiogenesis induced by this growth factor (Zeng et al., 2013).
Downstream to Akt activation, the production of NO by eNOS has been linked in EC migration, because pharmacological inhibition of eNOS reduces in vitro migration capacity (Murohara et al., 1999; Lamalice et al., 2007). Moreover, aortic EC from eNOS-lacking mice have impaired in vitro and in vivo angiogenesis (Lee et al., 1999; Lamalice et al., 2007). Tunicamycin-induced ER stress reduces eNOS levels in mouse coronary artery EC (Galan et al., 2014). Accordingly, homocysteine-induced ER stress through C/EBP CHOP-10 signaling inhibits eNOS signaling in HUVEC (Loinard et al., 2012). As mentioned above, fetuses from pregnancies with MO develop IR (Catalano et al., 2009), which is maintained through childhood (Bruyndonckx et al., 2013). Since eNOS activation is regulated by insulin stimulation in EC, eNOS signaling could be altered in HUVEC from MO pregnancies as an outcome of fetal programming, as occurs in other maternal pathologies (Farías et al., 2006, 2010; Leiva et al., 2011; Westermeier et al., 2011). If the latter effectively occurs, HUVEC derived from MO pregnancies might have impaired Akt/eNOS signaling and migration and/or angiogenic capacity (Figure 2).
Soluble mediators and Ca2+ signaling
Pro-inflammatory mediators such as cytokines and adipokines are increased during MO (Catalano et al., 2009) and have detrimental effects on EC migration. Leptin is an adipokine with pro-angiogenic effects that induces HUVEC migration after activation of Akt and eNOS (Goetze et al., 2002). However, leptin exposure is not associated with significant effects on tube formation in vitro (Dubois et al., 2013). In contrast, adiponectin inhibits HUVEC migration in a wound-healing assay and also inhibits tube formation in vitro (Dubois et al., 2013), showing an opposite effect to that of leptin. In fact, leptin signaling is impaired by ER stress, which also contributes to leptin resistance (Hosoi et al., 2008; Ozcan et al., 2009). Conversely, adiponectin has been linked to inhibition of ER stress (Boddu et al., 2014).
Pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-8, have a pro-angiogenic effect (Dinarello, 2007). These cytokines exert this effect by up-regulation of MCPIP, which is required to induce angiogenesis in vitro by increasing ER stress (Roy and Kolattukudy, 2012), suggesting an association between cytokines and ER stress. Interestingly, most if not all cytokines regulate free intracellular Ca2+ levels in cells, providing another possible link between these soluble mediators and the development of ER stress, as we discuss next.
Ca2+ signaling is one of the most important players during cell migration (Wei et al., 2012). Ca2+ channel-dependent calcium dynamics are observed at the leading edge in migrating cells (Wei et al., 2009). Consequently, during HUVEC migration, a polarized generation of PIP3 is found at the front of the migrating cells, which further increases the Ca2+ influx, allowing cytoskeleton rearrangements required for motility (Tsai and Meyer, 2012; Tsai et al., 2014). Simultaneously, an increase in the extrusion of Ca2+ towards the extracellular milieu is observed, hence maintaining the Ca2+ dynamics at the front (Tsai et al., 2014), showing that a fine-tuning of Ca2+ signaling is required for HUVEC migration. In this context, tunicamycin links ER stress with Ca2+ signaling because this antibiotic induces distorted function of Ca2+-channels (Czyz et al., 2009). In addition, NO produced by eNOS contributes to Ca2+ dynamics (Huang et al., 2013), suggesting that deficient eNOS signaling induced by ER stress might affect Ca2+ signaling and hence cell migration. Interestingly, pro-inflammatory cytokines IL-1β and IFN-γ down-regulate the SERCA and increase ER stress markers in pancreatic β-cells (Cardozo et al., 2005). The possibility of a similar mechanism occurring in EC is interesting, because it would unveil the mechanisms by which cytokines might affect EC migration (Figure 2).
Cell polarity
During cell migration, a reorientation of several cellular structures occurs in a process called polarization (Rorth, 2009). One of the intracellular features exhibited by migrating EC is the polarization of the centrosome toward the direction of movement of the endothelial sheet (Figure 1; Gotlieb et al., 1981; Rorth, 2009; Etienne-Manneville, 2013). Moreover, microtubule-binding drugs that inhibit HUVEC migration exert this blockade effect by avoiding centrosome repositioning (Hotchkiss et al., 2002; Kamath et al., 2014). Therefore, ER stress might affect cellular polarization and hence impair cell migration.
One of the candidates potentially affected by ER stress is Scrib, which mediates chemotaxis-dependent, but not spontaneous, cell migration (Figure 2) and in vitro and in vivo angiogenesis (Michaelis et al., 2013). This protein contributes to cytoskeletal rearrangements and Golgi apparatus polarization toward the leading edge in wound-healing assays (Michaelis et al., 2013). Whether a similar distortion occurs with nuclei and/or mitochondrial and/or lysosomal reorientation (Rorth, 2009; Friedl et al., 2011; Etienne-Manneville, 2013; da Silva et al., 2014) has not yet studied. Thus, cell polarity-related proteins might be affected by ER stress and thus impair proper organelle and centrosome polarization.
Concluding Remarks and Perspectives
Endothelial cell migration relies on tightly regulated signaling cascades that are activated by various stimuli. Adequate signaling events are required for proper remodeling of vessels during angiogenesis, and distorted intracellular cross-talk among the involved pathways would result in vascular dysfunction. We focus on the potential involvement of two main mechanisms of disease observed in obesity, ER stress, and IR. Interestingly, ER stress might impact EC migration and hence angiogenesis in different ways. Here, we summarize the current knowledge about how ER stress might provoke alterations in EC migration capacity and propose new targets (Figure 1). Specifically, both ER stress and IR might affect the coordination of endothelial chemotaxis, haptotaxis, mechanotaxis, Ca2+ signaling, and cell polarity modulation, which are key steps associated with EC migration. Better understanding of these processes regarding the physiopathological mechanism underlying ER stress might provide new perspectives in the design of therapeutic targets.
Different ER stress stimuli and micro-environmental contexts play major roles in regulation of EC migration, as well as the timing of stimulation signals and the magnitude of ER stress activation. For example, a physiological role of ER stress has been shown during pregnancy, where it is required for placenta development (Iwawaki et al., 2009), but it still is unknown whether its overactivation under pathological conditions remains favorable or becomes detrimental. On the other hand, intracellular cascades associated with IR development may be also associated with impaired EC migration capacity. To further address these research topics, new models of in vivo and in vitro analysis are required. An interesting approach recently validated the use of rat mesenteric EC to evaluate angiogenesis, because these cells exhibit the same behavior as HUVEC during migration and angiogenesis (Mansouri et al., 2013). Another approach proposes 3D culture of adipocytes and HUVEC in microspheres, in an attempt to mimic adipose tissue (Yao et al., 2013). The zebrafish, a well-established model to evaluate migration and angiogenesis, has been recently used to evaluate the role of Akt and ER stress pathways (Lu et al., 2014). Furthermore, the chick embryo chorioallantonic membrane assay might be used to evaluate the impact of ER stress on EC migration and angiogenesis, in a way similar to its current use in evaluating the anti-angiogenic potential of different compounds (Lange et al., 2014). Taken together, these models might provide new tools for studying EC migration and angiogenesis during obesity-induced ER stress and IR.
Considering the state of knowledge, we propose that acute and chronic ER stress might induce different effects on EC migration. In addition, as observed in tumoral versus non-tumoral environments, ER stress might promote or impair EC migration and angiogenesis, respectively. Finally, based on the hypothesis of intrauterine programming during pregnancies affected by adverse conditions and the induction of ER stress and IR in the presence of obesity, we suggest that MO might induce fetal ER stress and IR, two intracellular mechanisms associated with altered EC migration and hence distorted angiogenesis in offspring endothelium.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors thank members of the Cellular and Molecular Physiology Laboratory (CMPL) in the Division of Obstetrics and Gynecology, Faculty of Medicine, Pontificia Universidad Católica de Chile, for excellent secretarial and technical assistance, and the personnel of the Maternity Ward at Hospital Clínico of Pontificia Universidad Católica de Chile. This study was funded by Fondo Nacional de Desarrollo Científico y Tecnológico FONDECYT grants 1121145, 1130801, 1110977, 11100192, 3130583, 3140532; Programa de Investigación Interdisciplinario (PIA) from Comisión Nacional de Investigación en Ciencia y Tecnología, CONICYT Anillos ACT-73; and the International NETWORK program from CONICYT (130102).
Abbreviations
2-DG, 2-deoxy-D-glucose; ATF6, activating transcription factor 6; BiP/GRP78, immunoglobulin binding protein; BMI, body mass index; CHOP-10, C/EBP homologous protein-10; EC, endothelial cell; eIF2α, eukaryotic translational initiation factor 2α; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; GSK, Akt/glycogen synthase kinase; HUVECs, human umbilical vein endothelial cells; IFN-γ, interferon γ; IL, interleukin; IR, insulin resistance; IRE1, inositol-requiring enzyme 1α; MCPIP, monocyte chemotactic protein-induced protein; MO, maternal obesity; PERK, PKR-like eukaryotic initiation factor 2α kinase; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; RhoA, Ras homolog family member A; Scrib, scribbled planar cell polarity protein; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase pump; sFlt-1, soluble fms-like tyrosine kinase-1; TNF-α, tumor necrosis factor α; UPR, unfolded protein response; VEGF, vascular endothelial growth factor; XBP1, X-box binding protein 1.
References
Aepfelbacher, M., Essler, M., Huber, E., Sugai, M., and Weber, P. C. (1997). Bacterial toxins block endothelial wound repair. Evidence that Rho GTPases control cytoskeletal rearrangements in migrating endothelial cells. Arterioscler. Thromb. Vasc. Biol. 17, 1623–1629. doi: 10.1161/01.ATV.17.9.1623
American College of Obstetricians and Gynecologists [ACOG]. (2005). ACOG committee opinion number 315, September 2005. Obesity in pregnancy. Obstet. Gynecol. 106, 671–675.
Amin, A., Choi, S. K., Galan, M., Kassan, M., Partyka, M., Kadowitz, P.,et al. (2012). Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. J. Pathol. 227, 165–174. doi: 10.1002/path.3960
Appenzeller-Herzog, C., and Hall, M. N. (2012). Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 22, 274–282. doi: 10.1016/j.tcb.2012.02.006
Aranda, E., and Owen, G. I. (2009). A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol. Res. 42, 377–389. doi: 10.4067/S0716-97602009000300012
Banerjee, A., Lang, J. Y., Hung, M. C., Sengupta, K., Banerjee, S. K., Baksi, K.,et al. (2011). Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 286, 29127–29138. doi: 10.1074/jbc.M110.169771
Basha, B., Samuel, S. M., Triggle, C. R., and Ding, H. (2012). Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Exp. Diabetes Res. 2012, 481840. doi: 10.1155/2012/481840
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Boddu, N. J., Theus, S., Luo, S., Wei, J. Y., and Ranganathan, G. (2014). Is the lack of adiponectin associated with increased ER/SR stress and inflammation in the heart? Adipocyte 3, 10–18. doi: 10.4161/adip.26684
Boden, G., Cheung, P., Salehi, S., Homko, C., Loveland-Jones, C., Jayarajan, S.,et al. (2014). Insulin regulates the unfolded protein response in human adipose tissue. Diabetes 63, 912–922. doi: 10.2337/db13-0906
Brozinick, J. T. Jr., Roberts, B. R., and Dohm, G. L. (2003). Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes 52, 935–941. doi: 10.2337/diabetes.52.4.935
Bruyndonckx, L., Hoymans, V. Y., Van Craenenbroeck, A. H., Vissers, D. K., Vrints, C. J., Ramet, J.,et al. (2013). Assessment of endothelial dysfunction in childhood obesity and clinical use. Oxid. Med. Cell Longev. 2013, 174782. doi: 10.1155/2013/174782
Cardozo, A. K., Ortis, F., Storling, J., Feng, Y. M., Rasschaert, J., Tonnesen, M.,et al. (2005). Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes 54, 452–461. doi: 10.2337/diabetes.54.2.452
Catalano, P. M., Presley, L., Minium, J., and Hauguel-De Mouzon, S. (2009). Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 32, 1076–1080. doi: 10.2337/dc08-2077
Cnop, M., Foufelle, F., and Velloso, L. A. (2012). Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18, 59–68. doi: 10.1016/j.molmed.2011.07.010
Cheng, X., Chapple, S. J., Patel, B., Puszyk, W., Sugden, D., Yin, X.,et al. (2013). Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes 62, 4088–4097. doi: 10.2337/db13-0169
Christiaens, V., and Lijnen, H. R. (2010). Angiogenesis and development of adipose tissue. Mol. Cell. Endocrinol. 318, 2–9. doi: 10.1016/j.mce.2009.08.006
Czyz, A., Brutkowski, W., Fronk, J., Duszynski, J., and Zablocki, K. (2009). Tunicamycin desensitizes store-operated Ca2+ entry to ATP and mitochondrial potential. Biochem. Biophys. Res. Commun. 381, 176–180. doi: 10.1016/j.bbrc.2009.02.006
da Silva, A. F., Mariotti, F. R., Maximo, V., and Campello, S. (2014). Mitochondria dynamism: of shape, transport and cell migration. Cell. Mol. Life Sci. 71, 2313–2324. doi: 10.1007/s00018-014-1557-8
Dejeans, N., Pluquet, O., Lhomond, S., Grise, F., Bouchecareilh, M., Juin, A.,et al. (2012). Autocrine control of glioma cells adhesion and migration through IRE1alpha-mediated cleavage of SPARC mRNA. J. Cell Sci. 125, 4278–4287. doi: 10.1242/jcs.099291
Dinarello, C. A. (2007). Historical insights into cytokines. Eur. J. Immunol. 37(Suppl. 1), S34–S45. doi: 10.1002/eji.200737772
Dong, D., Ni, M., Li, J., Xiong, S., Ye, W., Virrey, J. J.,et al. (2008). Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 68, 498–505. doi: 10.1158/0008-5472.CAN-07–2950
Dorscheid, D. R., Wojcik, K. R., Yule, K., and White, S. R. (2001). Role of cell surface glycosylation in mediating repair of human airway epithelial cell monolayers. Am. J. Physiol. Lung. Cell. Mol. Physiol. 281, L982–L992.
Dubois, V., Delort, L., Billard, H., Vasson, M. P., and Caldefie-Chezet, F. (2013). Breast cancer and obesity: in vitro interferences between adipokines and proangiogenic features and/or antitumor therapies? PLoS ONE 8:e58541. doi: 10.1371/journal.pone.0058541
Dubova, E. A., Pavlov, K. A., Borovkova, E. I., Bayramova, M. A., Makarov, I. O., and Shchegolev, A. I. (2011). Vascular endothelial growth factor and its receptors in the placenta of pregnant women with obesity. Bull. Exp. Biol. Med. 151, 253–258. doi: 10.1007/s10517-011-1302-3
Duksin, D., and Bornstein, P. (1977). Changes in surface properties of normal and transformed cells caused by tunicamycin, an inhibitor of protein glycosylation. Proc. Natl. Acad. Sci. U.S.A. 74, 3433–3437. doi: 10.1073/pnas.74.8.3433
Etienne-Manneville, S. (2013). Microtubules in cell migration. Annu. Rev. Cell Dev. Biol. 29, 471–499. doi: 10.1146/annurev-cellbio-101011–155711
Farías, M., Puebla, C., Westermeier, F., Jo, M. J., Pastor-Anglada, M., Casanello, P.,et al. (2010). Nitric oxide reduces SLC29A1 promoter activity and adenosine transport involving transcription factor complex hCHOP-C/EBPalpha in human umbilical vein endothelial cells from gestational diabetes. Cardiovasc. Res. 86, 45–54. doi: 10.1093/cvr/cvp410
Farías, M., San Martin, R., Puebla, C., Pearson, J. D., Casado, J. F., Pastor-Anglada, M.,et al. (2006). Nitric oxide reduces adenosine transporter ENT1 gene (SLC29A1) promoter activity in human fetal endothelium from gestational diabetes. J. Cell. Physiol. 208, 451–460. doi: 10.1002/jcp.20680
Flamment, M., Hajduch, E., Ferre, P., and Foufelle, F. (2012). New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 23, 381–390. doi: 10.1016/j.tem.2012.06.003
Flenady, V., Koopmans, L., Middleton, P., Froen, J. F., Smith, G. C., Gibbons, K.,et al. (2011). Major risk factors for stillbirth in high-income countries: a systematic review and meta-analysis. Lancet 377, 1331–1340. doi: 10.1016/S0140-6736(10)62233-7
Friedl, P., Wolf, K., and Lammerding, J. (2011). Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 23, 55–64. doi: 10.1016/j.ceb.2010.10.015
Fu, S., Watkins, S. M., and Hotamisligil, G. S. (2012). The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 15, 623–634. doi: 10.1016/j.cmet.2012.03.007
Galan, M., Kassan, M., Kadowitz, P. J., Trebak, M., Belmadani, S., and Matrougui, K. (2014). Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim. Biophys. Acta 1843, 1063–1075. doi: 10.1016/j.bbamcr.2014.02.009
Garg, A. D., Kaczmarek, A., Krysko, O., Vandenabeele, P., Krysko, D. V., and Agostinis, P. (2012). ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol. Med. 18, 589–598. doi: 10.1016/j.molmed.2012.06.010
Ghosh, R., Lipson, K. L., Sargent, K. E., Mercurio, A. M., Hunt, J. S., Ron, D.,et al. (2010). Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 5:e9575. doi: 10.1371/journal.pone.0009575
Gipson, I. K., Kiorpes, T. C., and Brennan, S. J. (1984). Epithelial sheet movement: effects of tunicamycin on migration and glycoprotein synthesis. Dev. Biol. 101, 212–220. doi: 10.1016/0012-1606(84)90131-3
Goetze, S., Bungenstock, A., Czupalla, C., Eilers, F., Stawowy, P., Kintscher, U.,et al. (2002). Leptin induces endothelial cell migration through Akt, which is inhibited by PPARgamma-ligands. Hypertension 40, 748–754. doi: 10.1161/01.HYP.0000035522.63647.D3
Gotlieb, A. I., May, L. M., Subrahmanyan, L., and Kalnins, V. I. (1981). Distribution of microtubule organizing centers in migrating sheets of endothelial cells. J. Cell Biol. 91, 589–594. doi: 10.1083/jcb.91.2.589
Gregor, M. F., Yang, L., Fabbrini, E., Mohammed, B. S., Eagon, J. C., Hotamisligil, G. S.,et al. (2009). Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 58, 693–700. doi: 10.2337/db08-1220
Guo, S. (2014). Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J. Endocrinol. 220, T1–T23. doi: 10.1530/JOE-13-0327
Haberzettl, P., Vladykovskaya, E., Srivastava, S., and Bhatnagar, A. (2009). Role of endoplasmic reticulum stress in acrolein-induced endothelial activation. Toxicol. Appl. Pharmacol. 234, 14–24. doi: 10.1016/j.taap.2008.09.019
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
Hosoi, T., Sasaki, M., Miyahara, T., Hashimoto, C., Matsuo, S., Yoshii, M.,et al. (2008). Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 74, 1610–1619. doi: 10.1124/mol.108.050070
Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. doi: 10.1016/j.cell.2010.02.034
Hotchkiss, K. A., Ashton, A. W., Mahmood, R., Russell, R. G., Sparano, J. A., and Schwartz, E. L. (2002). Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Mol. Cancer Ther. 1, 1191–1200.
Huang, Y., Zheng, L., Yang, H., Chen, J., Wang, Y., Li, H.,et al. (2013). Calcium mobilization in HeLa cells induced by nitric oxide. Scanning doi: 10.1002/sca.21098 [Epub ahead of print].
Indra, M. R., Karyono, S., Ratnawati, R., and Malik, S. G. (2013). Quercetin suppresses inflammation by reducing ERK1/2 phosphorylation and NF kappa B activation in leptin-induced human umbilical vein endothelial cells (HUVECs). BMC Res. Notes 6:275. doi: 10.1186/1756-0500-6-275
Iwawaki, T., Akai, R., Yamanaka, S., and Kohno, K. (2009). Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. U.S.A. 106, 16657–16662. doi: 10.1073/pnas.0903775106
Jaffe, A. B., and Hall, A. (2005). Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269. doi: 10.1146/annurev.cellbio.21.020604.150721
Jung, D. Y., Chalasani, U., Pan, N., Friedline, R. H., Prosdocimo, D. A., Nam, M.,et al. (2013). KLF15 is a molecular link between endoplasmic reticulum stress and insulin resistance. PLoS ONE 8:e77851. doi: 10.1371/journal.pone.0077851
Kamath, K., Smiyun, G., Wilson, L., and Jordan, M. A. (2014). Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskeleton (Hoboken) 71, 46–60. doi: 10.1002/cm.21153
Kanda, T., Wakino, S., Homma, K., Yoshioka, K., Tatematsu, S., Hasegawa, K.,et al. (2006). Rho-kinase as a molecular target for insulin resistance and hypertension. FASEB J. 20, 169–171. doi: 10.1096/fj.05-4197fje
Kaplon, R. E., Chung, E., Reese, L., Cox-York, K., Seals, D. R., and Gentile, C. L. (2013). Activation of the unfolded protein response in vascular endothelial cells of nondiabetic obese adults. J. Clin. Endocrinol. Metab. 98, E1505–E1509. doi: 10.1210/jc.2013-1841
Karali, E., Bellou, S., Stellas, D., Klinakis, A., Murphy, C., and Fotsis, T. (2014). VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell. 54, 559–572. doi: 10.1016/j.molcel.2014.03.022
Kozutsumi, Y., Segal, M., Normington, K., Gething, M. J., and Sambrook, J. (1988). The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464. doi: 10.1038/332462a0
Krause, B. J., Costello, P. M., Munoz-Urrutia, E., Lillycrop, K. A., Hanson, M. A., and Casanello, P. (2013). Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 8, 944–952. doi: 10.4161/epi.25579
Lamalice, L., Le Boeuf, F., and Huot, J. (2007). Endothelial cell migration during angiogenesis. Circ. Res. 100, 782–794. doi: 10.1161/01.RES.0000259593.07661.1e
Lange, S., Gonzalez, I., Pinto, M. P., Arce, M., Valenzuela, R., Aranda, E.,et al. (2014). Independent anti-angiogenic capacities of coagulation factors X and Xa. J. Cell Physiol. doi: 10.1002/jcp.24612 [Epub ahead of print].
Lee, P. C., Salyapongse, A. N., Bragdon, G. A., Shears, L. L. II, Watkins, S. C., Edington, H. D.,et al. (1999). Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 277, H1600–H1608.
Leiva, A., Pardo, F., Ramírez, M. A., Farías, M., Casanello, P., and Sobrevia, L. (2011). Fetoplacental vascular endothelial dysfunction as an early phenomenon in the programming of human adult diseases in subjects born from gestational diabetes mellitus or obesity in pregnancy. Exp. Diabetes Res. 2011, 349286. doi: 10.1155/2011/349286
Li, C., Wang, L., Huang, K., and Zheng, L. (2012). Endoplasmic reticulum stress in retinal vascular degeneration: protective role of resveratrol. Invest. Ophthalmol. Vis. Sci. 53, 3241–3249. doi: 10.1167/iovs.11–8406
Li, J., Wang, J. J., Yu, Q., Wang, M., and Zhang, S. X. (2009). Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 583, 1521–1527. doi: 10.1016/j.febslet.2009.04.007
Loinard, C., Zouggari, Y., Rueda, P., Ramkhelawon, B., Cochain, C., Vilar, J.,et al. (2012). C/EBP homologous protein-10 (CHOP-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation 125, 1014–1026. doi: 10.1161/CIRCULATIONAHA.111.041830
Lu, J. W., Liao, C. Y., Yang, W. Y., Lin, Y. M., Jin, S. L., Wang, H. D.,et al. (2014). Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PLoS ONE 9:e85318. doi: 10.1371/journal.pone.0085318
Mansouri, K., Khodarahmi, R., and Ghadami, S. A. (2013). An in vitro model for spontaneous angiogenesis using rat mesenteric endothelial cells: possible therapeutic perspective for obesity and related disorders. Pharm. Biol. 51, 974–980. doi: 10.3109/13880209.2013.773452
McMillen, I. C., and Robinson, J. S. (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol. Rev. 85, 571–633. doi: 10.1152/physrev.00053.2003
Melo, A. M., Benatti, R. O., Ignacio-Souza, L. M., Okino, C., Torsoni, A. S., Milanski, M.,et al. (2014). Hypothalamic endoplasmic reticulum stress and insulin resistance in offspring of mice dams fed high-fat diet during pregnancy and lactation. Metabolism 63, 682–692. doi: 10.1016/j.metabol.2014.02.002
Merchan, J. R., Kovacs, K., Railsback, J. W., Kurtoglu, M., Jing, Y., Pina, Y.,et al. (2010). Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 5:e13699. doi: 10.1371/journal.pone.0013699
Michaelis, U. R., Chavakis, E., Kruse, C., Jungblut, B., Kaluza, D., Wandzioch, K.,et al. (2013). The polarity protein Scrib is essential for directed endothelial cell migration. Circ. Res. 112, 924–934. doi: 10.1161/CIRCRESAHA.112.300592
Minamino, T., and Kitakaze, M. (2010). ER stress in cardiovascular disease. J. Mol. Cell Cardiol. 48, 1105–1110. doi: 10.1016/j.yjmcc.2009.10.026
Murohara, T., Witzenbichler, B., Spyridopoulos, I., Asahara, T., Ding, B., Sullivan, A.,et al. (1999). Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler. Thromb. Vasc. Biol. 19, 1156–1161. doi: 10.1161/01.ATV.19.5.1156
Nunes, K. P., Rigsby, C. S., and Webb, R. C. (2010). RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol. Life Sci. 67, 3823–3836. doi: 10.1007/s00018-010-0460-1
Omelchenko, T., Vasiliev, J. M., Gelfand, I. M., Feder, H. H., and Bonder, E. M. (2003). Rho-imdependent formation of epithelial “leader” cells during wound healing. Proc. Natl. Acad. Sci. U.S.A. 100, 10788–10793. doi: 10.1073/pnas.1834401100
O’Toole, T. E., Abplanalp, W., Li, X., Cooper, N., Conklin, D. J., Haberzettl, P.,et al. (2014). Acrolein decreases endothelial cell migration and insulin sensitivity through induction of Let-7a. Toxicol. Sci. doi: 10.1093/toxsci/kfu087 [Epub ahead of print].
Ozcan, L., Ergin, A. S., Lu, A., Chung, J., Sarkar, S., Nie, D.,et al. (2009). Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51. doi: 10.1016/j.cmet.2008.12.004
Paridaens, A., Laukens, D., Vandewynckel, Y. P., Coulon, S., Van Vlierberghe, H., Geerts, A.,et al. (2014). Endoplasmic reticulum stress and angiogenesis: is there an interaction between them? Liver Int. 34, e10–e18. doi: 10.1111/liv.12457
Pereira, E. R., Liao, N., Neale, G. A., and Hendershot, L. M. (2010). Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS ONE 5:e12521. doi: 10.1371/journal.pone.0012521
Polec, A., Fedorcsak, P., Eskild, A., and Tanbo, T. G. (2014). The interplay of human chorionic gonadotropin (hCG) with basic fibroblast growth factor and adipokines on angiogenesis in vitro. Placenta 35, 249–253. doi: 10.1016/j.placenta.2014.02.002
Povero, D., Eguchi, A., Niesman, I. R., Andronikou, N., De Mollerat Du Jeu, X., Mulya, A.,et al. (2013). Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci. Signal. 6, ra88. doi: 10.1126/scisignal.2004512
Reffay, M., Parrini, M. C., Cochet-Escartin, O., Ladoux, B., Buguin, A., Coscoy, S.,et al. (2014). Interplay of RhoA and mechanical forces in collective cell migration driven by leader cells. Nat. Cell Biol. 16, 217–223. doi: 10.1038/ncb2917
Rorth, P. (2009). Collective cell migration. Annu. Rev. Cell Dev. Biol. 25, 407–429. doi: 10.1146/annurev.cellbio.042308.113231
Roy, A., and Kolattukudy, P. E. (2012). Monocyte chemotactic protein-induced protein (MCPIP) promotes inflammatory angiogenesis via sequential induction of oxidative stress, endoplasmic reticulum stress and autophagy. Cell. Signal. 24, 2123–2131. doi: 10.1016/j.cellsig.2012.07.014
Saben, J., Lindsey, F., Zhong, Y., Thakali, K., Badger, T. M., Andres, A.,et al. (2014). Maternal obesity is associated with a lipotoxic placental environment. Placenta 35, 171–177. doi: 10.1016/j.placenta.2014.01.003
Salminen, A., Kauppinen, A., Hyttinen, J. M., Toropainen, E., and Kaarniranta, K. (2010). Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol. Med. 16, 535–542. doi: 10.2119/molmed.2010.00070
Schroder, M., and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789. doi: 10.1146/annurev.biochem.73.011303.074134
Shiojima, I., and Walsh, K. (2002). Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 90, 1243–1250. doi: 10.1161/01.RES.0000022200.71892.9F
Snyder-Cappione, J. E., and Nikolajczyk, B. S. (2013). When diet and exercise are not enough, think immunomodulation. Mol. Aspects Med. 34, 30–38. doi: 10.1016/j.mam.2012.10.003
Somanath, P. R., Razorenova, O. V., Chen, J., and Byzova, T. V. (2006). Akt1 in endothelial cell and angiogenesis. Cell Cycle 5, 512–518. doi: 10.4161/cc.5.5.2538
Song, J. W., Daubriac, J., Tse, J. M., Bazou, D., and Munn, L. L. (2012). RhoA mediates flow-induced endothelial sprouting in a 3-D tissue analogue of angiogenesis. Lab Chip 12, 5000–5006. doi: 10.1039/c2lc40389g
Suganya, N., Bhakkiyalakshmi, E., Suriyanarayanan, S., Paulmurugan, R., and Ramkumar, K. M. (2014). Quercetin ameliorates tunicamycin-induced endoplasmic reticulum stress in endothelial cells. Cell Prolif. 47, 231–240. doi: 10.1111/cpr.12102
Tabata, Y., Takano, K., Ito, T., Iinuma, M., Yoshimoto, T., Miura, H.,et al. (2007). Vaticanol B, a resveratrol tetramer, regulates endoplasmic reticulum stress and inflammation. Am. J. Physiol. Cell Physiol. 293, C411–C418. doi: 10.1152/ajpcell.00095.2007
Takatsuki, A., Arima, K., and Tamura, G. (1971). Tunicamycin, a new antibiotic. I. Isolation and characterization of tunicamycin. J. Antibiot. (Tokyo) 24, 215–223. doi: 10.7164/antibiotics.24.215
Triunfo, S., and Lanzone, A. (2014). Impact of overweight and obesity on obstetric outcomes. J. Endocrinol. Invest. doi: 10.1007/s40618-014-0058-9 [Epub ahead of print].
Tsai, F. C., and Meyer, T. (2012). Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 22, 837–842. doi: 10.1016/j.cub.2012.03.037
Tsai, F. C., Seki, A., Yang, H. W., Hayer, A., Carrasco, S., Malmersjo, S.,et al. (2014). A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 16, 133–144. doi: 10.1038/ncb2906
van Nieuw Amerongen, G. P., Koolwijk, P., Versteilen, A., and Van Hinsbergh, V. W. (2003). Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol. 23, 211–217. doi: 10.1161/01.ATV.0000054198.68894.88
Vitorino, P., and Meyer, T. (2008). Modular control of endothelial sheet migration. Genes Dev. 22, 3268–3281. doi: 10.1101/gad.1725808
Wang, S., Park, J. K., and Duh, E. J. (2012). Novel targets against retinal angiogenesis in diabetic retinopathy. Curr. Diab. Rep. 12, 355–363. doi: 10.1007/s11892-012-0289-0
Wei, C., Wang, X., Chen, M., Ouyang, K., Song, L. S., and Cheng, H. (2009). Calcium flickers steer cell migration. Nature 457, 901–905. doi: 10.1038/nature07577
Wei, C., Wang, X., Zheng, M., and Cheng, H. (2012). Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 24, 254–261. doi: 10.1016/j.ceb.2011.12.002
Westermeier, F., Sáez, P. J., Villalobos-Labra, R., Sobrevia, L., and Farías-Jofré, M. (2014). Programming of fetal insulin resistance in pregnancies with maternal obesity by ER stress and inflammation. BioMed Res. Int. 2014:917672. doi: 10.1155/2014/917672
Westermeier, F., Salomón, C., Gonzalez, M., Puebla, C., Guzmán-Gutierrez, E., Cifuentes, F.,et al. (2011). Insulin restores gestational diabetes mellitus-reduced adenosine transport involving differential expression of insulin receptor isoforms in human umbilical vein endothelium. Diabetes 60, 1677–1687. doi: 10.2337/db11-0155
World Health Organ [WHO]. (2003). Diet, nutrition and the prevention of chronic diseases. World Health Organ Tech. Rep. Ser. 916(i–viii), 1–149.
Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders, T., Wang, J.,et al. (2007). ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364. doi: 10.1016/j.devcel.2007.07.005
Wu, Z., Zhao, J., Xu, H., Lyv, Y., Feng, X., Fang, Y.,et al. (2014). Maternal quercetin administration during gestation and lactation decrease endoplasmic reticulum stress and related inflammation in the adult offspring of obese female rats. Eur. J. Nutr. doi: 10.1007/s00394-014-0673i4 [Epub ahead of print].
Yao, R., Du, Y., Zhang, R., Lin, F., and Luan, J. (2013). A biomimetic physiological model for human adipose tissue by adipocytes and endothelial cell cocultures with spatially controlled distribution. Biomed. Mater. 8, 045005. doi: 10.1088/1748-6041/8/4/045005
Yi, N., Chen, S. Y., Ma, A., Chen, P. S., Yao, B., Liang, T. M.,et al. (2012). Tunicamycin inhibits PDGF-BB-induced proliferation and migration of vascular smooth muscle cells through induction of HO-1. Anat. Rec. (Hoboken) 295, 1462–1472. doi: 10.1002/ar.22539
Yoon, J. S., Kim, H. M., Yadunandam, A. K., Kim, N. H., Jung, H. A., Choi, J. S.,et al. (2013). Neferine isolated from Nelumbo nucifera enhances anti-cancer activities in Hep3B cells: molecular mechanisms of cell cycle arrest, ER stress induced apoptosis and anti-angiogenic response. Phytomedicine 20, 1013–1022. doi: 10.1016/j.phymed.2013.03.024
Yung, H. W., Charnock-Jones, D. S., and Burton, G. J. (2011). Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 6:e17894. doi: 10.1371/journal.pone.0017894
Zeng, L., Xiao, Q., Chen, M., Margariti, A., Martin, D., Ivetic, A.,et al. (2013). Vascular endothelial cell growth-activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation 127, 1712–1722. doi: 10.1161/CIRCULATIONAHA.112.001337
Zera, C. A., Seely, E. W., Wilkins-Haug, L. E., Lim, K. H., Parry, S. I., and Mcelrath, T. F. (2014). The association of body mass index with serum angiogenic markers in normal and abnormal pregnancies. Am. J. Obstet. Gynecol. doi: 10.1016/j.ajog.2014.03.020 [Epub ahead of print].
Keywords: mesenchymal migration, unfolded protein response, RhoA, Akt, Scrib, polarization, cytoskeleton
Citation: Sáez PJ, Villalobos-Labra R, Westermeier F, Sobrevia L and Farías-Jofré M (2014) Modulation of endothelial cell migration by ER stress and insulin resistance: a role during maternal obesity? Front. Pharmacol. 5:189. doi: 10.3389/fphar.2014.00189
Received: 30 April 2014; Accepted: 24 July 2014;
Published online: 19 August 2014.
Edited by:
Martin Michel, Boehringer Ingelheim Pharma GmbH & Co KG, GermanyReviewed by:
Martin Michel, Boehringer Ingelheim Pharma GmbH & Co KG, GermanyTereza Cindrova-Davies, University of Cambridge, UK
Copyright © 2014 Sáez, Villalobos-Labra, Westermeier, Sobrevia and Farías-Jofré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo Farías-Jofré, Cellular and Molecular Physiology Laboratory, Division of Obstetrics and Gynecology, School of Medicine, Faculty of Medicine, Pontificia Universidad Católica de Chile, Marcoleta 391, P. O. Box 114-D, Santiago 8330024, Chile e-mail:bWZhcmlhc0BtZWQucHVjLmNs