 Alessandro Cannavo1,2
Alessandro Cannavo1,2 Daniela Liccardo1,2
Daniela Liccardo1,2 Klara Komici2
Klara Komici2 Graziamaria Corbi3
Graziamaria Corbi3 Claudio de Lucia1
Claudio de Lucia1 Grazia D. Femminella4
Grazia D. Femminella4 Andrea Elia5
Andrea Elia5 Leonardo Bencivenga2
Leonardo Bencivenga2 Nicola Ferrara2,5
Nicola Ferrara2,5 Walter J. Koch1
Walter J. Koch1 Nazareno Paolocci6,7
Nazareno Paolocci6,7 Giuseppe Rengo2,5*
Giuseppe Rengo2,5*- 1Lewis Katz School of Medicine, Center for Translational Medicine, Temple University, Philadelphia, PA, United States
- 2Department of Translational Medical Sciences, University of Naples Federico II, Naples, Italy
- 3Department of Medicine and Health Science, University of Molise, Campobasso, Italy
- 4Imperial College London, London, United Kingdom
- 5Istituti Clinici Scientifici Maugeri SpA Società Benefit, Telese Terme Institute (BN), Telese, Italy
- 6Division of Cardiology, Johns Hopkins University Medical Institutions, Baltimore, MD, United States
- 7Department of Experimental Medicine, University of Perugia, Perugia, Italy
The sphingosine kinases 1 and 2 (SphK1 and 2) catalyze the phosphorylation of the lipid, sphingosine, generating the signal transmitter, sphingosine 1-phosphate (S1P). The activation of such kinases and the subsequent S1P generation and secretion in the blood serum of mammals represent a major checkpoint in many cellular signaling cascades. In fact, activating the SphK/S1P system is critical for cell motility and proliferation, cytoskeletal organization, cell growth, survival, and response to stress. In the cardiovascular system, the physiological effects of S1P intervene through the binding and activation of a family of five highly selective G protein-coupled receptors, called S1PR1-5. Importantly, SphK/S1P signal is present on both vascular and myocardial cells. S1P is a well-recognized survival factor in many tissues. Therefore, it is not surprising that the last two decades have seen a flourishing of interest and investigative efforts directed to obtain additional mechanistic insights into the signaling, as well as the biological activity of this phospholipid, and of its receptors, especially in the cardiovascular system. Here, we will provide an up-to-date account on the structure and function of sphingosine kinases, discussing the generation, release, and function of S1P. Keeping the bull’s eye on the cardiovascular system, we will review the structure and signaling cascades and biological actions emanating from the stimulation of different S1P receptors. We will end this article with a summary of the most recent, experimental and clinical observations targeting S1PRs and SphKs as possible new therapeutic avenues for cardiovascular disorders, such as heart failure.
Introduction
Sphingolipids are ubiquitous components of the eukaryotic cell membrane that play important roles in the regulation of many cellular processes (Ghosh et al., 1990; Zhang et al., 1991). Among these molecules, sphingosine 1-phosphate (S1P) is a bioactive lipid with a variety of physiological roles across a broad range of organisms (Strub et al., 2010). S1P is produced by the phosphorylation of sphingosine, a reaction catalyzed by an enzyme, sphingosine kinase (SphK), present in two isoforms, SphK1 and 2. S1P degradation involves a cleavage by S1P lyase (SPL) (Imamura et al., 2001). In general, when phosphorylated, this lipid is secreted in the plasma, mainly by red blood cells, platelets, fibroblasts, and vascular endothelial cells (ECs, Tani et al., 2005; Kacimi et al., 2007; Pappu et al., 2007; Venkataraman et al., 2008; Gellings Lowe et al., 2009). In addition, extracellular SphK1 released from these cells may also contribute to S1P synthesis (Venkataraman et al., 2006).
The activation of SphK1 and 2, and the consequent S1P generation/secretion have a crucial role in many cellular signaling cascades and pathological processes. In particular, their role has been widely studied in angiogenesis, in cancer development/progression and in immune and inflammatory responses (Karliner, 2009). However, the SphK/S1P axis has received a special attention from cardiovascular scientists because implicated in the cardiovascular system development and functioning. The latter encompasses the modulation of heart rate, cardiac contractility, and vascular tone (Peters and Alewijnse, 2007). All these effects are mediated by the binding to specific G protein-coupled receptors (GPCRs), called S1PRs (Hla et al., 2001; Chun et al., 2002; Spiegel and Milstien, 2003; Usui et al., 2004). Since an increasing body of experimental evidence supports the notion that activating the SphK/S1P-S1PR system protects both the heart and the vasculature, several synthetic S1P analogs have been previously designed (Pelletier and Hafler, 2012). Of note, one of them, Fingolimod, or FTY720 (a Novartis proprietary compound) is currently approved and used in clinical practice to treat neurological degenerative disorders, such as multiple sclerosis (Pelletier and Hafler, 2012).
Given the current availability of compounds, such as Fingolimod, it is now more feasible, and should be even more attractive, to test the impact of agonists of the SphK/S1P-S1PR axis in the context of clinically relevant cardiovascular disorders, as in the recent case of a study that demonstrated an increase in myocardial salvage and a decrease in adverse post-infarction remodeling with Fingolimod in a porcine model of ischemia-reperfusion injury (Santos-Gallego et al., 2016).
Here, we will review the mechanisms by which SphKs modulate S1P generation and secretion in different cardiovascular compartments. Then, we will focus on the pathophysiological role exerted by SphK/S1P-S1PRs axis in the circulatory system. Finally, we will describe how S1PRs agonism and antagonism can improve outcome in cardiac disease states, such as post-ischemic heart failure (HF).
Structure and Function of Sphingosine Kinases
Structure of SphKs
The SphKs are members of a family of enzymes that includes the diacylglycerol (DAG) and the ceramide kinases, all of which are able to generate bioactive lipids (Wattenberg et al., 2006). Currently, two isoforms (SphK1 and 2) have been cloned and characterized, and the genes encoding for these two enzymes are localized on different chromosomes—sphk1 gene is on chromosome 17, whereas sphk2 gene is on chromosome 19—and encode for several splicing variants (Imamura et al., 2001; Alemany et al., 2007). SphK1 was originally purified from rat and show a high degree of homology with the mouse and human enzyme (Kohama et al., 1998; Nava et al., 2000). The second isoform, SphK2, was cloned and characterized from mouse and human by Spiegel and colleagues (Liu H. et al., 2000). At the structural level, SphK1, in homology with SphK2, is composed of an N-terminal (NT) and a C-terminal (CT) domain, with the catalytic one located in a cleft at the interdomain junction. Both structures appear to have an homology because they contain five conserved domains, including an adenosine triphosphate (ATP)-binding motif that allows the transfer of a γ-phosphoryl group from the ATP to the D-erythro-sphingosine, to generate the S1P (Melendez et al., 2000; Nava et al., 2000). However, while SphK2 presents a nuclear localization signal (NLS) in the NT and a nuclear exportation signal (NES) in the CT, SphK1 lacks these sequences. Importantly, their presence within the SphK2 structure increases also the number of amino acids required for the enzyme composition. In fact, SphK1 consists of 384 amino acids (42.5 kDa), whereas SphK2 presents with 618 amino acids (65.2 kDa) (Liu H. et al., 2000; Okada et al., 2005). Moreover, based on kinetic studies performed in vitro and in vivo, both SphKs cannot only phosphorylate sphingosine in a similar manner, but they can also phosphorylate the immunomodulatory drug FTY720 (Fingolimod). However, SphK2 appear to be more efficient in doing so than SphK1 (Billich et al., 2003; Paugh et al., 2003). Due to this property, it has been suggested that only SphK2 is required for metabolic activation of this drug. In fact, FTY720, that is able to cause lymphopenia, loses its effect only in mice lacking SphK2, but not in mice in which SphK1 is downregulated (Allende et al., 2004; Kharel et al., 2005). For all these very reasons, it appears crystal-clear that, based on their structure, both the kinases can have different functions and localization (please, see more below). However, as shown by previous studies, these kinases have overlapping vital functions. In this regard, although knockout mouse models for either SphK1 or SphK2 develop normally, the genetic deletion of both isoforms results in fetal death due to alterations in vasculogenesis and severe bleeding (Allende et al., 2004; Mizugishi et al., 2005; Michaud et al., 2006).
Subcellular Localization of SphKs
Concerning the localization of the two enzymes, it has been reported that SphK1 predominantly resides in the cytoplasm (Wattenberg, 2010; Pitson, 2011), and it can translocate to the plasma membrane upon cell stimulation (Pitson et al., 2003). Therefore, S1P generated by SphK1 can be exported outside the cells, and activate both proliferative and anti-apoptotic effects, in an autocrine and/or paracrine manner (Spiegel and Milstien, 2000). This phenomenon is known as “inside-out” signaling, and it has been described in cardiac myocytes too (Tao et al., 2007; Wang et al., 2012). Moreover, Ancellin et al. (2002) demonstrated that SphK1 can be released outside the cells, thus accounting for the extracellular S1P generation. Conversely, SphK2 is primarily localized at the level of the endoplasmic reticulum (ER, Maceyka et al., 2005), or it can also be associated with mitochondria (Strub et al., 2011; Chipuk et al., 2012). S1P generated in these compartments can affect cell survival (Maceyka et al., 2005; Strub et al., 2011; Chipuk et al., 2012; Maceyka et al., 2012). Moreover, since SphK2 presents an NLS and NES, it can shuttle in and out of the nucleus. S1P generated in this subcellular compartment can affect histone deacetylases activity, with a consequently enhanced transcription of genes involved in the growth arrest (Hait et al., 2009).
Functional Role of SphK1
As we learned before, it is generally well-consolidated that SphK1 is a cell survival promoter (Karliner, 2013). Elevated cellular SphK1 levels appear to play a major role in enhanced proliferation and metastasis/invasion of several types of cancer cells (Xia et al., 2000; Johnson et al., 2005; Li et al., 2008; Shida et al., 2008; Pyne et al., 2016). In this context, more than one study has demonstrated that inhibition of SphK1 has considerable potential as an anti-cancer strategy (Shida et al., 2008; Pyne et al., 2016). Similarly, the downregulation of SphK1 has proven able to induce apoptosis and confer sensitivity to chemo- or radiation therapy of cancer cell lines (Baran et al., 2007; Shida et al., 2008; Guillermet-Guibert et al., 2009; Pyne et al., 2016). In line with these reports, ventricular cardiomyocytes and cardiac fibroblasts lacking SphK1 exhibit greater cell death when subjected to hypoxia, compared to wild-type (WT) controls (Ancellin et al., 2002; Tao et al., 2007). Interestingly, treatment of cardiomyocytes with exogenous S1P or with monoganglioside (GM-1), an acidic glycosphingolipid containing one sialic acid residue shown to elicit S1P generation (Cavallini et al., 1999), enhances the survival of both WT and SphK1 null cells (Ancellin et al., 2002; Tao et al., 2007). It is worth stressing that one of the proposed mechanism by which SphK1 controls cell death is the regulation of ceramide (Maceyka et al., 2005). In contrast to S1P, the ceramide-signaling molecule is able to exert pro-apoptotic actions. Its synthesis and accumulation are enhanced in cells lacking SphK1, while prevented in presence of high SphK1 levels (Maceyka et al., 2005). In 1996, in order to tie together the ability of S1P and ceramide to control cell fate, it has been coined the term “sphingolipid rheostat” (Cuvillier et al., 1996; Newton et al., 2015). However, although SphK1 appears to play a major role in the regulation of this “rheostat,” previous studies suggested that sphingolipid per se are able to influence the whole mechanism, thus including the “inside-out” one. In this context, Huang et al. (2014) have recently shown that S1P can activate a positive feedback amplification loop via S1PRs activation and consequent increase in SphK1 expression.
Functional Role of SphK2
Opposite to the protective role attributed to SphK1, several early studies examining SphK2’s role have documented that the overexpression of this kinase induces cell cycle arrest and apoptosis (Karliner, 2013). More in detail, SphK2 can inhibit cell growth and enhance apoptosis, in part by increasing ceramide production (Maceyka et al., 2005). Similarly, mitochondrial localization of SphK2, and specifically S1P generation at this site seems to contribute to the activation of the pro-death Bcl-2 family protein, BID, with subsequent mitochondrial membrane permeabilization and cytochrome c release (Strub et al., 2011; Chipuk et al., 2012). In keeping with this view, several reports have shown that downregulating SphK2 can effectively prevent the increase in apoptotic rates induced by the administration of either TNF-α or staurosporine (Weigert et al., 2007; Hofmann et al., 2008). Interestingly, and contrary to the dogma that, differently from Sphka1, SphK2 is a pro-death factor, recent experimental evidence now supports a key role for this kinase in promoting cell survival and proliferation, much like SphK1 does in cancer cells, or even in cardiomyocytes (Weigert et al., 2009; Gomez et al., 2011; Vessey et al., 2011). Consonant to this view, Weigert et al. (2009) have reported that the genetic ablation of SphK2 in MCF-7 breast tumor xenografts results in the inhibition of tumor growth. Moreover, in isolated murine hearts, Karliner and colleagues have shown that SphK2 is necessary for successful ischemic pre- and post-conditioning (Vessey et al., 2011). Further to this, Gomez et al. (2011) also demonstrated that SphK2-evoked cardioprotection is dependent on the ability to prevent ischemia-induced mitochondrial dysfunction. Interestingly, the apparently divergent outcome reported with SphK2 studies could be ascribed to the specific subcellular localization of the enzyme. In agreement with this eventuality, studies have suggested that, when SphK2 is localized in the nucleus, it can inhibit the synthesis of DNA, thus exerting anti-proliferative effects (Hait et al., 2009). Conversely, other contributions have shown that, in human colon carcinoma cells, S1P generated by nuclear SphK2 can inhibit the retinoic acid receptor β, attenuating the tumor suppressor effects of this receptor (Shi et al., 2017).
S1P: Generation and Function
S1P Biosynthesis
Given the variety of processes involving S1P, most cells—red blood cells, platelets, and vascular ECs, in particular—have all the enzymatic machinery necessary for S1P synthesis. Like other sphingolipids, S1P is derived from ceramide which is composed of a sphingosine base and an amide-linked acyl chain of variable length (Yu and Law, 2009). Ceramide is in turn produced from the de novo synthetic pathway initiated by serine palmitoyltransferase in the ER, or from the degradation of some sphingolipids (Dolgachev et al., 2004; Reynolds et al., 2004). The intracellular deacylation by ceramidase gives way to the formation of sphingosine and carboxylate (Park and Schuchman, 2006). Then sphingosine can be phosphorylated to produce S1P (Figure 1). However, it is worth recalling that S1P levels in the cell are regulated not only by S1P biosynthetic enzymes but also by S1P degradative pathways, such as SPLs—two S1P-specific phosphatases—and by three lipid phosphate phosphatases (Figure 1; Hannun et al., 2001).
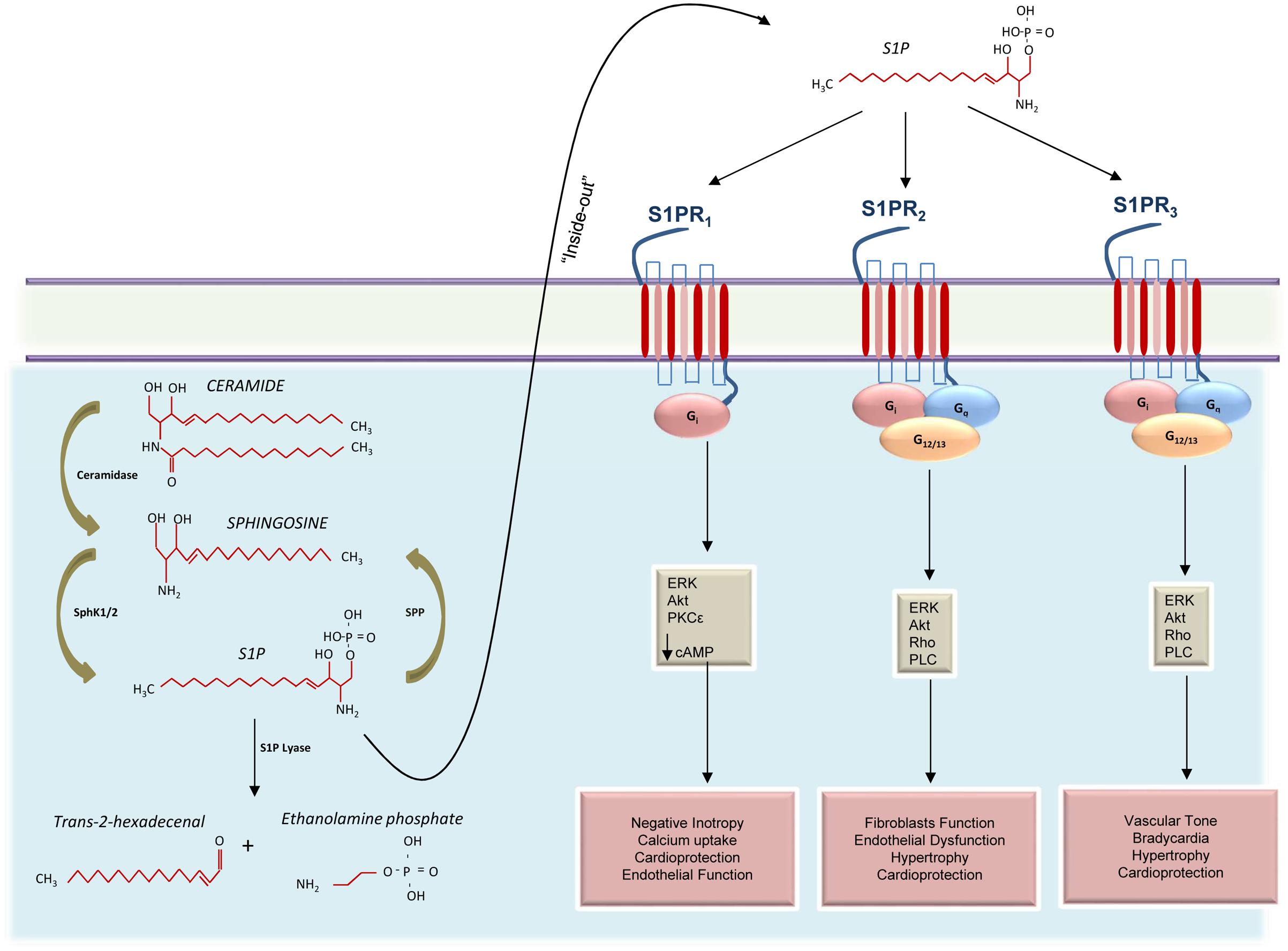
FIGURE 1. Schematic representation of S1PRs signaling activation in the cardiovascular system. Ceramide is catabolized by ceramidase to produce sphingosine that, in turn, is phosphorylated by sphingosine kinases 1 and 2 (SphK1 and 2) to generate the sphingosine 1-phosphate (S1P). Cellular S1P concentrations are regulated by the balance between its synthesis and de-phosphorylation mediated by S1P phosphatases (SPP). In the cytosol, S1P can also be irreversibly cleaved into trans-2-hexadecenal and ethanolamine phosphate or can be exported out of cells through the “inside-out” mechanism where it can bind three different receptors (S1PR1-3). Activation of S1PR1 induces negative inotropic effects via G protein (Gi) activation and decreased cAMP concentration. Moreover, S1PR1 is able to positively affect endothelial function and to confer protection to the heart, via activation of the mitogen-activated protein kinase 1 and 2 (ERK) and the protein kinase B (Akt). Both S1PR2 and S1PR3 activate Gi, Gq, and G12/13 appear to collaborate in providing cardioprotection and in regulating cardiac hypertrophy. However, while S1PR2 is involved in endothelial dysfunction and promotion of fibroblasts function, S1PR3 appears to influence the vascular tone and induce bradycardia.
S1P Functions
In general, S1P cell death-suppressing and cell survival promoting effects are opposite to those typically attributed to ceramide that, almost invariably, induces apoptosis, senescence, autophagy, and growth arrest (Cuvillier et al., 1996; Zheng et al., 2006; Pruett et al., 2008; Saddoughi and Ogretmen, 2013). Therefore, the intracellular synthesis of S1P vs. ceramide should be always under a tight control because any subtle change in this finely tuned balance, in response to environmental changes and stimuli, can direct cell in one direction or in the other (Cuvillier et al., 1996; Zheng et al., 2006; Pruett et al., 2008; Saddoughi and Ogretmen, 2013). Along with this, SPL is able to promote apoptosis under stress conditions, reducing the circulating levels of S1P (Kumar et al., 2011). Importantly, Karliner and colleagues have demonstrated that SPL activation in the myocardium following ischemia leads to reduced S1P levels and that knockout mice for this enzyme exhibit higher S1P levels and smaller infarct size (Bandhuvula et al., 2011). Moreover, mice null for SPL show significantly increased left ventricular function recovery over their WT infarcted counterparts (Bandhuvula et al., 2011). Thus, inhibition of SPL could represent a new target that can be utilized to prevent myocardium loss after ischemic injury.
S1P Levels as a Biomarker of Cardiovascular Disease
In light of the evidence discussed above, it is important to consider that blood plasma and serum typically contains, in principle, high levels of S1P (Murata et al., 2000; Deutschman et al., 2003). Importantly, changes in serum S1P may be a predictive marker for the presence and severity of cardiovascular disease, as in the case of obstructive coronary artery disease (CAD), atherosclerosis, myocardial infarction (MI) and HF in humans (Deutschman et al., 2003; Sattler et al., 2010; Argraves et al., 2011; Cannavo et al., 2013b; Egom et al., 2013; Soltau et al., 2016).
Accordingly, Knapp et al. (2013) reported a reduction in circulating S1P levels in patients with acute MI as compared to controls. Similarly, we have recently demonstrated, in mouse and rat models of post-ischemic HF, that either cardiac or circulating levels of S1P are reduced compared to non-ischemic controls (Cannavo et al., 2013b, 2017).
However, it is important to consider that a large amount (∼60–80%) of S1P in the human plasma is associated with high-density lipoprotein (HDL). The importance of HDL is mainly due to the actions exerted by this lipoprotein on S1P function. In this context, Wilkerson et al. (2012) demonstrated that HDL-S1P are able to induce a lower rate of internalization and degradation of S1PR1 than albumin-S1P (another carrier of S1P). Of note, HDLs are usually reduced in several diseases like atherosclerosis, CAD, MI, renal insufficiency, and diabetes (Sattler et al., 2010), and this could influence the levels of circulating S1P. In the same vein, Sattler et al. (2010) reported that, in patients with CAD or MI, plasma S1P levels (normalized to HDL levels) were higher than in controls. However, in the same study, the authors analyzed the levels of HDL-S1P in MI and stable CAD patients, showing that the levels of S1P conjugated with this carrier were lower than in controls. In line with the latter evidence, Argraves et al. (2011) demonstrated that circulating S1P, dihydro-S1P and c24:1-ceramide levels in HDL correlate inversely with the incidence of ischemic heart disease.
S1P Receptors and Dependent Signaling in Cardiovascular System
Many of S1P actions are mediated through specific GPCRs. Goodemote et al. (1995) demonstrated, for the first time, that S1P induced the activation of the extracellular signal-regulated kinase (ERK) 1/2 via Gi protein activation. Later on, Lee et al. (1998) identified a GPCR, originally termed endothelial differentiation gene 1 (EDG1), as the receptor of S1P. Currently, five closely related GPCRs (S1PR1-5), which differ in tissue and cell expression, have been identified for their high affinity for this bioactive lipid (Chun et al., 2002). Importantly, while S1PR1-3 are mostly expressed in the cardiovascular, central nervous system, and immune system, S1PR4 is mainly present in the lymphoid tissue, whereas S1PR5 is predominantly expressed in the central nervous system, immune system (natural killer cells), and spleen (Walzer et al., 2007; Pyne and Pyne, 2010; Jeffery et al., 2011). Importantly, the effects associated with S1PRs activation on different cell types in the cardiovascular system, either on cardiomyocytes, ECs, smooth muscle cells, or fibroblasts, are dictated by the specific G protein coupling. S1PR1 couples exclusively with the inhibitory G protein alpha subunit (Gαi), whereas S1PR2 and S1PR3 bind to Gαi, Gαq, and Gα13 and S1PR4 and S1PR5 couple to both Gαi and Gα13 (Means and Brown, 2009). Following ligand binding and subsequent activation, the α subunit of the heterotrimeric G protein is released and interacts with its downstream effectors. The main effector for Gαi is the adenylate cyclase, which is inhibited, thus leading to a reduction of the second messenger cyclic adenosine 3′,5′-monophosphate (cAMP) (Cannavo and Koch, 2017b). Moreover, Gαi is able to activate PKCα and 𝜀, thus modulating calcium uptake (Thompson et al., 2006; Marino et al., 2017). In contrast, phospholipase C (PLC) mediates the response due to Gαq activation. In turn, PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to DAG and inositol trisphosphate (IP3) (Cannavo and Koch, 2017b), whereas that for Gα13 and Gα12 is a Rho guanine nucleotide exchange factor (Rho-GEF), activating downstream low molecular Rho GTPases (Sugimoto et al., 2003).
S1PRs in Vasculature
S1P plays a key role in the development of the vasculature and its stabilization (Schuchardt et al., 2011; Cannavo et al., 2013b). Accordingly, S1P regulates the growth of ECs and vascular smooth muscle cells (VSMCs) (Skoura and Hla, 2009; Schuchardt et al., 2011; Kerage et al., 2014). ECs express S1PR1, S1PR2, and S1PR3, with the isoform 1 being the most expressed subtype (Skoura and Hla, 2009; Kerage et al., 2014). For these reasons, it is not surprising that most of the S1P-mediated responses in these cells occur via S1PR1. Importantly, S1P regulates and stimulates the migration and proliferation of ECs and alteration in S1P levels or activity are responsible for aberrant vascular maturation (Kerage et al., 2014). It has been shown, for instance, that the inability of S1P to activate the GTPase Rac, as observed in global S1PR1 knockout mice, is one of the mechanisms associated with the impaired vasculature development and embryonic lethality (Liu Y. et al., 2000). At this regard, in this study, the authors suggest that the alteration of S1PR1 activation on ECs could negatively affect VSMCs recruitment (Liu Y. et al., 2000). Importantly, in addition to S1PR1 binding in ECs, S1P also activates the S1PR3 eliciting important vascular processes, such as the formation of new vessels and stabilization of barrier integrity (Waeber et al., 2004). For example, it has been shown that S1P-mediated migration proliferation and vasculature formation require both the S1PR1 mediated activation of Gi protein and the S1PR3 coupling to Gq/G12,13 (Sugimoto et al., 2003; Kono et al., 2004; Waeber et al., 2004). The S1PR3 requirement has been further validated in studies demonstrating that a peptide derived from the second intracellular loop of the S1PR3 can induce pro-angiogenic responses (Licht et al., 2003). Conversely, the activation and upregulation of S1PR2 has been associated with impaired functions in ECs, i.e., chemotactic, wound healing, and morphogenic responses (Lu et al., 2012; Zhao et al., 2015). In particular, Lu et al. (2012) have recently demonstrated in vivo that, in aging rats, S1PR2, like others GPCRs (Vasto et al., 2010; Ferrara et al., 2014), takes part in senescence-mediated endothelial dysfunction and aging processes (Zhao et al., 2015).
Interestingly, in VSMCs the expression pattern for S1PRs significantly differs from that of ECs (Waeber et al., 2004; Kerage et al., 2014). VSMCs mainly express the S1PR2 and S1PR3 (Waeber et al., 2004; Kerage et al., 2014). While S1PR3 stimulation increases the activity of Rac with an increased VSMCs migratory capacity, the activation of S1PR2 inhibits Rac via Rho/Rho kinase pathway, thus leading to a significant reduction in VSMCs function (Waeber et al., 2004; Szczepaniak et al., 2010; Kerage et al., 2014). Importantly, S1PR3 appears to be the major mediator of S1P-induced vasoconstriction because this lipid fails to increase the vascular tone in arteries isolated from S1PR3 knockout mice (Salomone et al., 2008).
S1PRs in the Heart
Myocytes and fibroblasts represent the vast majority of the myocardial tissue. Importantly, due to the elevated activity expression of SphKs, fibroblasts appear to be the major source of cardiac S1P (Kacimi et al., 2007). Cardiac fibroblasts express predominantly S1PR3, with much lower levels of S1PR1 and S1PR2 being expressed (Landeen et al., 2008). However, most of the effects on fibroblasts function have been attributed to S1PR2 activation (Wang et al., 1997; Olivera et al., 1999; Urata et al., 2005). In particular, S1P is a positive regulator of fibroblasts function and proliferation, and these effects are associated with the activation of ERK and Rho activity downstream of S1PR2 (Wang et al., 1997; Olivera et al., 1999; Urata et al., 2005). Differently from fibroblasts, in cardiomyocytes, S1PR1 is the predominant S1P receptor subtype expressed (Means et al., 2008; Cannavo et al., 2013b). Initially, several studies evaluated the effect on S1PR1 on ion channels and contractility (Means and Brown, 2009), but decades of discoveries have later revealed that this receptor has also a prominent role in hypertrophic response and cardioprotection (Means and Brown, 2009).
S1P-dependent activation of S1PR1 in cardiomyocytes is necessary for heart development in mice (Clay et al., 2016). In fact, Clay et al. (2016) have recently shown that conditional knockout mice for S1PR1 show ventricular septal defects and perinatal lethality. Moreover, these authors reported that lacking S1PR1 is associated with decreased myofibril organization (Clay et al., 2016). Similarly, Keul et al. (2016) have shown that cardiomyocyte-restricted deletion of S1PR1 in mice results in progressive cardiomyopathy, compromised response to β-adrenergic receptor (βAR) stimulation and premature death.
The S1PR1 in cardiomyocytes is also a major regulator of contractile response (Means and Brown, 2009). Indeed, it counters the mechanical effects (positive inotropy/lusitropy, etc.) that follow cardiac β1-adrenergic receptor (β1AR)-agonism (Means and Brown, 2009; Cannavo et al., 2013b). Accordingly, S1P treatment of ventricular myocytes blocks the effects produced by the β1/β2AR agonist, isoproterenol, preventing the activation of adenylate cyclase, thus leading to a negative inotropic response (Means et al., 2008). This functional interaction between βAR and S1PR1 signaling in vivo was more recently reported also by Errami et al. (2008). In their study, these authors demonstrated that βAR-agonism in mice results in cardiac hypertrophic response via engagement of the S1PR1 signaling pathway (Errami et al., 2008). We recently proposed that this cross-talk is a major protective mechanism in response to myocardial ischemia (Cannavo et al., 2013b). In fact, we showed that isoproterenol stimulation of H9c2 cells induces the activation of S1PR1 pro-hypertrophic signaling (Cannavo et al., 2013b). In vivo, we observed that, following a cardiac ischemic attack, increased circulating levels of catecholamines lead to β1AR hyperactivation and subsequent desensitization/downregulation (Cannavo et al., 2013b), an effect coupled to increased GPCR kinase 2 (GRK2) levels that regulate both the β1AR and S1PR1 (Cannavo et al., 2013b). Importantly, the activation of such signaling pathway leads to the reciprocal downregulation of β1AR and S1PR1 in cardiac myocytes, leading to worse remodeling and progression toward HF (Cannavo et al., 2013b). This study further supported the idea that blockade of GRK2 is a valid strategy to prevent HF development and progression, but also demonstrated the cardioprotective role of S1PR1 (Cannavo et al., 2013a, 2016a,b; Cannavo and Koch, 2017a). In line with these data, we recently reported that activation of S1PR1 in the heart is also modulated by the β3AR (Cannavo et al., 2017). This receptor is the less βAR isoform expressed in the heart; however, it has important regulatory activities in cardiac hypertrophic response and in contractility (Cannavo and Koch, 2017b). In line with a previous study in adipocytes (Zhang et al., 2014), we demonstrated that selective β3AR stimulation leads to SphK1 upregulation and S1P release with a subsequent activation of S1PR1 in cardiomyocytes (Cannavo et al., 2017). Again, this mechanism appears to be relevant in a post-ischemic HF animal model. In fact, re-activation of β3AR via β1-AR blockade (Metoprolol), an indirect agonist of β3AR, is able to promote the activation of S1PR1 thus protecting the heart from failure (Cannavo et al., 2017).
Importantly, as discussed above, S1P is a cardioprotective molecule which signals in the heart via S1PRs. However, despite decades of studies from several groups, including ours, suggesting that S1PR1 is the major player in S1P-dependent cardioprotection (Cannavo et al., 2013a, 2017; Karliner, 2013; Marino et al., 2017), some reports have indicated that S1PR2 and S1PR3 can also take part in these protective molecular mechanisms activated in the myocardium in response to a specific injury (Theilmeier et al., 2006; Means et al., 2007; Means and Brown, 2009; Morel et al., 2016; Ruiz et al., 2017; Yung et al., 2017). In particular, mice lacking either S1PR2 or S1PR3, following an ischemic insult, develop infarcts equivalent to those of WT mice, whereas in S1PR2 and 3 double-knockout mice, the infarct size was increased by more than 50%, thus suggesting the potential role of these two receptors in protecting cardiomyocytes (Means et al., 2007). Mechanistically, all the beneficial effects associated with S1PR1-3, appear to be dependent mainly on the protein kinase B (Akt), that, via augmentation of eNOS expression/activity, has multiple effects, such as induction of adaptive hypertrophy, modulation of angiogenesis, and inhibition of apoptosis (Mandala et al., 2002; Means and Brown, 2009; Chaanine and Hajjar, 2011; Cannavo et al., 2013c; Keul et al., 2016). Moreover, it is also worth noting that recent reports suggest a protective role for RhoA activation in the heart (Xiang et al., 2013; Zhao et al., 2014; Yung et al., 2017) which do not involve S1PR1 (Figure 1).
Targeting S1Prs and SphKs as a Therapeutic Strategy in Cardiovascular Disorders
Currently, the majority of drugs targeting the S1P signaling are directed to the S1PRs rather than the ligand. This is due to the fact that, as anticipated above, almost all S1P actions are mediated by its receptor. Moreover, targeting a specific S1PR would render a given drug highly selective. For these reasons, many agonists/antagonists of the S1PRs have been developed and studied. For some of them, clinical data on are also available (Amiselimod, Siponimod, Ozanimod, Ceralifimod, GSK2018682, Ponesimod; O’Sullivan and Dev, 2017; Sugahara et al., 2017). One of the most tested S1PR agonists is Fingolimod or FTY720. This compound is a structural homolog of S1P, which is phosphorylated by SphK2 to form Fingolimod-phosphate (Fingolimod). It serves as a potent agonist of S1PR1 (Mandala et al., 2002; Hla and Brinkmann, 2011), or as a SphK1 inhibitor (Tonelli et al., 2010). Of relevance, this compound is a US Food and Drug Administration (FDA) approved the drug for the treatment of multiple sclerosis (Jeffery et al., 2011; Pelletier and Hafler, 2012). In fact, although the Fingolimod-phosphate initially activates S1PR1, on lymphocytes, it subsequently can induce the receptor downregulation thus preventing the egress of these cells from lymphoid tissues (Mandala et al., 2002; Jeffery et al., 2011; Pelletier and Hafler, 2012). This double mechanism of action (agonism/antagonism) of Fingolimod on S1PR1 reduces the infiltration of lymphocytes into the central nervous system blocking their noxious effect (Mandala et al., 2002; Jeffery et al., 2011; Pelletier and Hafler, 2012).
S1P and Protection Against Myocardial Ischemia
S1P is formed in the ischemic myocardium, and it is thought to be cardioprotective, mimicking the effects of ischemic preconditioning via a PKC𝜀-dependent pathway (Jin et al., 2004; Vessey et al., 2009; Marino et al., 2017). Importantly, as suggested by several studies in cardiac and non-cardiac cells, these beneficial effects are induced mainly by the selective binding to S1PR1. In line with this possibility, studies have evaluated the potential protective effect of S1PR1 agonism in cardiac cells. For instance, Wang et al. (2014) demonstrated that Fingolimod increases survival in adult murine cardiac myocytes subjected to hypoxia by inhibiting apoptosis. Following in vivo studies have demonstrated cardioprotective effects exerted by S1PR1 via Fingolimod stimulation. In particular, reports have shown that this drug is able to reduce ischemia/reperfusion (I/R) injury and to improve myocardial function in isolated mouse and rat heart preparations (Hofmann et al., 2009; Egom et al., 2010; Vessey et al., 2013). Further to this, in a mouse model of myocardial I/R, Goltz et al. (2015) have shown that, when given at reperfusion, Fingolimod provides a better hemodynamic outcome as compared to placebo-treated animals. Importantly, this effect was associated with a reduction in the number of phagocytic monocytes invading the myocardium (Goltz et al., 2015). Furthermore, in a recent report, Santos-Gallego et al. (2016) have demonstrated that Fingolimod improves myocardial function after MI in pigs. In this study, the authors showed that Fingolimod administration was associated with a reduction in the cardiac hypertrophic response and interstitial fibrosis in the remote, non-ischemic myocardium (Santos-Gallego et al., 2016). However, it is worth noting that in addition to S1PR1 activity modulation, studies have demonstrated that, albeit with minor affinity, Fingolimod can also bind and activate S1PR3 (Mandala et al., 2002; Hla and Brinkmann, 2011; Karliner, 2013). For this reason, in light of this evidence, one question would be whether or not Fingolimod induces its effects and if these are really due to the only S1PR1 modulation or if they are also related to S1PR3. Answering this intriguing would require fully dedicated studies. Anyway, other reports concerning the S1PR1 agonism protective effects against ischemic injury have explored the role of these receptors also in other organs, such as the brain and the lung (Stone et al., 2015; Brait et al., 2016). In particular, Stone et al. (2015) have demonstrated that selective stimulation of S1PR1, via Fingolimod or VPC01091, provided comparable protection from a lung injury and dysfunction after I/R. Further, in a mouse model of stroke, Brait et al. (2016) demonstrated the potential of S1PR1 agonists, such as LASW1238 or Fingolimod, in reducing the infarct size. Importantly, the authors concluded that these two drugs protect the brain only when lymphopenia is sustained for at least 24 h (Brait et al., 2016). In aggregate, these studies strongly support the overall notion that pharmacological activation of S1PR1 can reduce the detrimental effects of acute ischemia in an experimental setting, as demonstrated both in small and in large-animal models.
Use of S1PR1 Agonism in Humans: Any Alternative to Fingolimod?
Although promising, the utilization of Fingolimod in humans raises some concerns. For instance, Sanna et al. (2004) demonstrated that stimulation with a non-selective S1PR agonist reduced heart rate (bradycardia) in WT mice, but not in S1PR3 KO animals. Moreover, Fingolimod is a well-known activator of both S1PR1 and S1PR3; hence, it has been suggested that behind direct S1PR1 activation, Fingolimod can also induce bradycardia via S1PR3 (Karliner, 2013). However, data obtained in rats by Fryer et al. (2012) show that Fingolimod-induced bradycardia requires only S1PR1. In line with this evidence, Gergely et al. (2012) showed that an S1PR1 selective ligand causes transient bradycardia in humans. Taking all this into account, the FDA revised the recommendations for cardiovascular monitoring in patients with multiple sclerosis receiving Fingolimod. Most importantly, in a recent study, Racca et al. (2016) demonstrated that in multiple sclerosis patients this pharmacological agent reduces left ventricular systolic function. Therefore, based on the beneficial effects exerted by S1PR1, it is plausible that this adverse event could be related to the antagonistic effect of the Fingolimod on S1PR1 (Figure 2). Therefore, other drugs or therapeutic strategies directed to improve S1P signaling/function are currently under evaluation. For instance, Sugahara et al. (2017) have recently tested the effects and the efficacy of Amiselimod, a second-generation S1P receptor modulator that is highly selective for S1PR1 and S1PR5, with no distinct agonist activity for S1PR2 or S1PR3. Importantly, compared to Fingolimod, this compound appears to be safer because it failed to induce bradycardia (Sugahara et al., 2017). In an alternative to approaches directed to modulate S1P signaling, Duan et al. (2007) have recently demonstrated that intracardiac injection of adenoviral vectors encoding for SphK1 markedly reduces myocardial infarct size, in a rat model of I/R injury. Of note, the overexpression of SphK1 results in a significant improvement in left ventricular systolic pressure and end-diastolic pressure, and better contractility (Duan et al., 2007). Alternatively, we recently demonstrated that restoration of cardiac plasma membrane levels of S1PR1, via a recombinant adeno-associated virus serotype 6 (AAV6), produces beneficial effects, in a HF rat model (Cannavo et al., 2013b). Importantly, the overexpression of S1PR1 improves both cardiac function and enhances the re-vascularization of the ischemic cardiac tissue (Cannavo et al., 2013b).
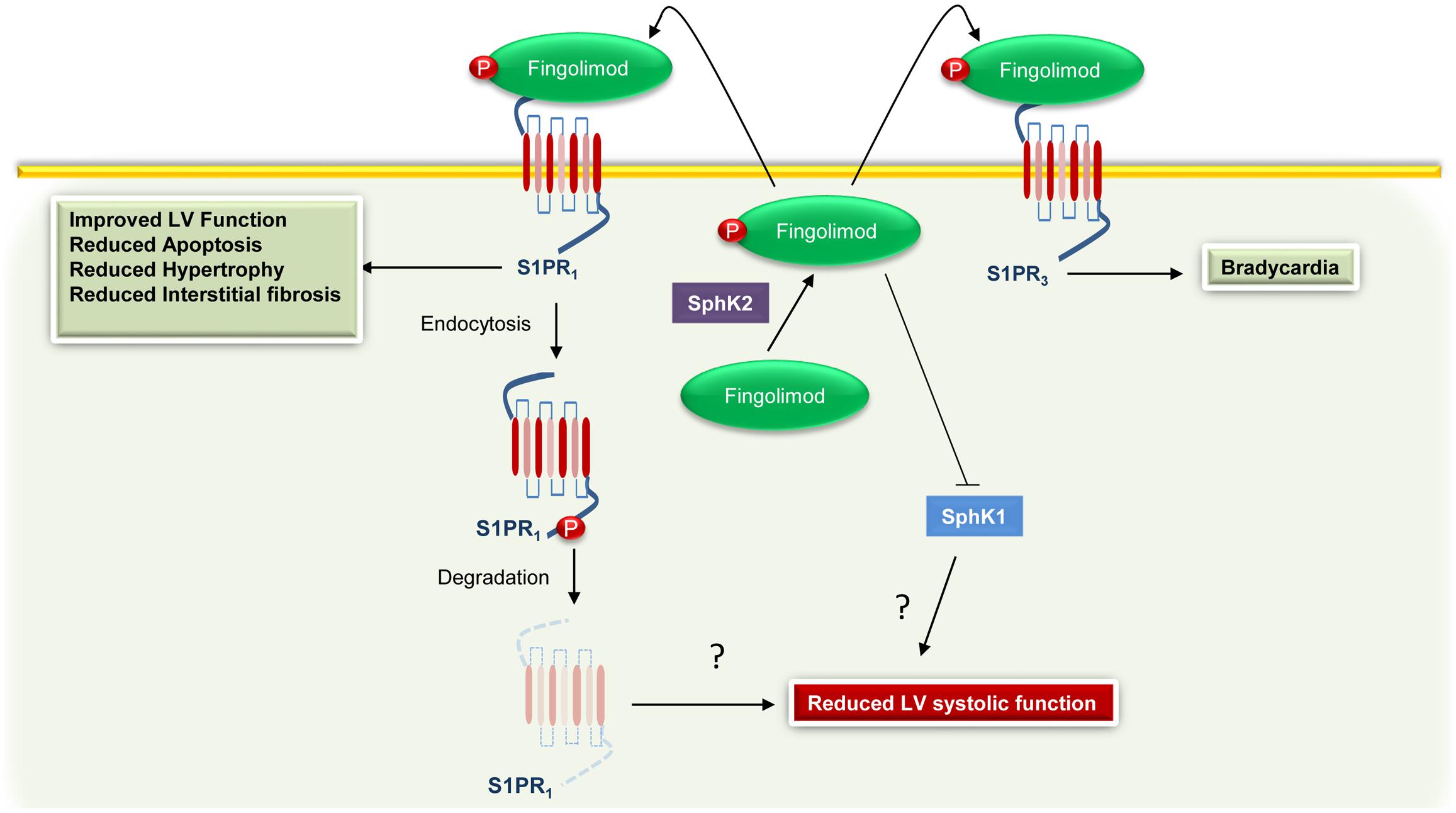
FIGURE 2. Cardiac effects associated with Fingolimod-mediated activation of S1PR1 and S1PR3. Fingolimod is phosphorylated by sphingosine kinase 2 (SphK2) to generate Fingolimod-phosphate (Fingolimod) that in turn binds to S1PR1. However, the sustained stimulation leads to S1PR1 internalization and degradation and to sphingosine kinase 1 (SphK1) inhibition. These effects are probably related to an impairment of the function of the left ventricle (LV). Moreover, via binding and activation of S1PR3, Fingolimod can induce bradycardia.
Conclusion
The GPCRs are cell surface receptors that mediate fundamental processes in all cell types of the cardiovascular system. Therefore, it is not surprising that these receptors are currently the largest family of targets for drugs in clinical use. Accordingly, approximately 20% of medicaments used to treat cardiovascular disorders have GPCR-binding properties. Importantly, the discovery that S1P signals via GPCRs, influencing the entire mammal physiology—from the immune to the nervous system, from the circulation to the skeletal muscle apparatus—have significantly advanced the field of cardiovascular pharmacology. Several drug candidates, targeting both S1PRs and downstream molecules, are currently undergoing clinical trials, and novel compounds of diverse pharmacodynamics have been identified in the attempt to optimize the benefits afforded by S1PR1 stimulation during the course of acute and chronic cardiac diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the American Heart Association-16POST30980005 (to AC); Italian Ministry of Health-GR-2011-02346878 and San Paolo Bank of Naples and University of Naples Federico II - STAR program 2016 (both to GR); AHA Grant-in-Aid 17070027 (to NP) and RO1 HL063030 (to NP, co-PI).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alemany, R., van Koppen, C. J., Dannebeg, K., ter Braak, M., and Meyer zu Heringdorf, D. (2007). Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch. Pharmacol. 374, 413–428. doi: 10.1007/s00210-007-0132-3
Allende, M. L., Sasaki, T., Kawai, H., Olivera, A., Mi, Y., Echten-Deckert, G., et al. (2004). Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 279, 52487–52492. doi: 10.1074/jbc.M406512200
Ancellin, N., Colmont, C., Su, J., Li, Q., Mittereder, N., Chae, S. S., et al. (2002). Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem. 277, 6667–6675. doi: 10.1074/jbc.M102841200
Argraves, K. M., Sethi, A. A., Gazzolo, P. J., Wilkerson, B. A., Remaley, A. T., Tybjaerg-Hansen, A., et al. (2011). S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease. Lipids Health Dis. 10:70. doi: 10.1186/1476-511X-10-70
Bandhuvula, P., Honbo, N., Wang, G. Y., Jin, Z. Q., Fyrst, H., Zhang, M., et al. (2011). S1P lyase: a novel therapeutic target for ischemia-reperfusion injury of the heart. Am. J. Physiol. Heart Circ. Physiol. 300, H1753–H1761. doi: 10.1152/ajpheart.00946.2010
Baran, Y., Salas, A., Senkal, C. E., Gunduz, U., Bielawski, J., Obeid, L. M., et al. (2007). Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J. Biol. Chem. 282, 10922–10934. doi: 10.1074/jbc.M610157200
Billich, A., Bornancin, F., Devay, P., Mechtcheriakova, D., Urtz, N., and Baumruker, T. (2003). Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J. Biol. Chem. 278, 47408–47415. doi: 10.1074/jbc.M307687200
Brait, V. H., Tarrasón, G., Gavaldà, A., Godessart, N., and Planas, A. M. (2016). Selective sphingosine 1-phosphate receptor 1 agonist is protective against ischemia/reperfusion in mice. Stroke 47, 3053–3056. doi: 10.1161/STROKEAHA.116.015371
Cannavo, A., and Koch, W. J. (2017a). GRK2 as negative modulator of NO bioavailability: implications for cardiovascular disease. Cell. Signal. doi: 10.1016/j.cellsig.2017.01.014 [Epub ahead of print].
Cannavo, A., and Koch, W. J. (2017b). Targeting β3-adrenergic receptors in the heart: selective agonism and β-blockade. J. Cardiovasc. Pharmacol. 69, 71–78. doi: 10.1097/FJC.0000000000000444
Cannavo, A., Liccardo, D., Eguchi, A., Elliott, K. J., Traynham, C. J., Ibetti, J., et al. (2016a). Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat. Commun. 7:10877. doi: 10.1038/ncomms10877
Cannavo, A., Liccardo, D., and Koch, W. J. (2013a). Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 4:264. doi: 10.3389/fphys.2013.00264
Cannavo, A., Liccardo, D., Lymperopoulos, A., Gambino, G., D’Amico, M. L., Rengo, F., et al. (2016b). β adrenergic receptor kinase C-terminal peptide gene-therapy improves β2-adrenergic receptor-dependent neoangiogenesis after hindlimb ischemia. J. Pharmacol. Exp. Ther. 356, 503–513. doi: 10.1124/jpet.115.228411
Cannavo, A., Rengo, G., Liccardo, D., Pagano, G., Zincarelli, C., De Angelis, M. C., et al. (2013b). β1-adrenergic receptor and sphingosine-1-phosphate receptor 1 (S1PR1) reciprocal downregulation influences cardiac hypertrophic response and progression to heart failure: protective role of S1PR1 cardiac gene therapy. Circulation 128, 1612–1622. doi: 10.1161/CIRCULATIONAHA.113.002659
Cannavo, A., Rengo, G., Liccardo, D., Pironti, G., Scimia, M. C., Scudiero, L., et al. (2013c). Prothymosin alpha protects cardiomyocytes against ischemia-induced apoptosis via preservation of Akt activation. Apoptosis 18, 1252–1261. doi: 10.1007/s10495-013-0876-9
Cannavo, A., Rengo, G., Liccardo, D., Pun, A., Gao, E., George, A. J., et al. (2017). β1-blockade prevents post-ischemic myocardial decompensation via β3AR-dependent protective sphingosine-1 phosphate signaling. J. Am. Coll. Cardiol. 70, 182–192. doi: 10.1016/j.jacc.2017.05.020
Cavallini, L., Venerando, R., Miotto, G., and Alexandre, A. (1999). Ganglioside GM1 protection from apoptosis of rat heart fibroblasts. Arch. Biochem. Biophys. 370, 156–162. doi: 10.1006/abbi.1999.1378
Chaanine, A. H., and Hajjar, R. J. (2011). AKT signalling in the failing heart. Eur. J. Heart Fail. 13, 825–829. doi: 10.1093/eurjhf/hfr080
Chipuk, J. E., McStay, G. P., Bharti, A., Kuwana, T., Clarke, C. J., Siskind, L. J., et al. (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000. doi: 10.1016/j.cell.2012.01.038
Chun, J., Goetzl, E. J., Hla, T., Igarashi, Y., Lynch, K. R., Moolenaar, W., et al. (2002). International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 54, 265–269. doi: 10.1124/pr.54.2.265
Clay, H., Wilsbacher, L. D., Wilson, S. J., Duong, D. N., McDonald, M., Lam, I., et al. (2016). Sphingosine 1-phosphate receptor-1 in cardiomyocytes is required for normal cardiac development. Dev. Biol. 418, 157–165. doi: 10.1016/j.ydbio.2016.06.024
Cuvillier, O., Pirianov, G., Keluser, B., Vanek, P. G., Coso, A., Gutkind, S., et al. (1996). Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381, 800–803. doi: 10.1038/381800a0
Deutschman, D. H., Carstens, J. S., Klepper, R. L., Smith, W. S., Page, M. T., Young, T. R., et al. (2003). Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J. 146, 62–68. doi: 10.1016/S0002-8703(03)00118-2
Dolgachev, V., Farooqui, M. S., Kulaeva, O. I., Tainsky, M. A., Nagy, B., Hanada, K., et al. (2004). De novo ceramide accumulation due to inhibition of its conversion to complex sphingolipids in apoptotic photosensitized cells. J. Biol. Chem. 279, 23238–23249. doi: 10.1074/jbc.M311974200
Duan, H. F., Wang, H., Yi, J., Liu, H. J., Zhang, Q. W., Li, L. B., et al. (2007). Adenoviral gene transfer of sphingosine kinase 1 protects heart against ischemia/reperfusion-induced injury and attenuates its postischemic failure. Hum. Gene Ther. 18, 1119–1128. doi: 10.1089/hum.2007.036
Egom, E. E., Ke, Y., Musa, H., Mohamed, T. M., Wang, T., Cartwright, E., et al. (2010). FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J. Mol. Cell. Cardiol. 48, 406–414. doi: 10.1016/j.yjmcc.2009.10.009
Egom, E. E., Mamas, M. A., Chacko, S., Stringer, S. E., Charlton-Menys, V., El-Omar, M., et al. (2013). Serum sphingolipids level as a novel potential marker for early detection of human myocardial ischaemic injury. Front. Physiol. 4:130. doi: 10.3389/fphys.2013.00130
Errami, M., Galindo, C. L., Tassa, A. T., Dimaio, J. M., Hill, J. A., and Garner, H. R. (2008). Doxycycline attenuates isoproterenol- and transverse aortic banding-induced cardiac hypertrophy in mice. J. Pharmacol. Exp. Ther. 324, 1196–1203. doi: 10.1124/jpet.107.133975
Ferrara, N., Komici, K., Corbi, G., Pagano, G., Furgi, G., Rengo, C., et al. (2014). β-adrenergic receptor responsiveness in aging heart and clinical implications. Front. Physiol. 4:396. doi: 10.3389/fphys.2013.00396
Fryer, R. M., Muthukumarana, A., Harrison, P. C., Nodop Mazurek, S., Chen, R. R., Harrington, K. E., et al. (2012). The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P1) and hypertension (S1P3) in rat. PLoS ONE 7:e52985. doi: 10.1371/journal.pone.0052985
Gellings Lowe, N., Swaney, J. S., Moreno, K. M., and Sabbadini, R. A. (2009). Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 82, 303–312. doi: 10.1093/cvr/cvp056
Gergely, P., Nuesslein-Hildesheim, B., Guerini, D., Brinkmann, V., Traebert, M., Bruns, C., et al. (2012). The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 167, 1035–1047. doi: 10.1111/j.1476-5381.2012.02061.x
Ghosh, T. K., Bian, J., and Gill, D. L. (1990). Intracellular calcium release mediated by sphingosine derivatives generated in cells. Science 248, 1653–1656. doi: 10.1126/science.2163543
Goltz, D., Huss, S., Ramadori, E., Büttner, R., Diehl, L., and Meyer, R. (2015). Immunomodulation by splenectomy or by FTY720 protects the heart against ischemia reperfusion injury. Clin. Exp. Pharmacol. Physiol. 42, 1168–1177. doi: 10.1111/1440-1681.12465
Gomez, L., Paillard, M., Price, M., Chen, Q., Teixeira, G., Spiegel, S., et al. (2011). A novel role for mitochondrial sphingosine-1-phosphate produced by sphingosine kinase-2 in PTP-mediated cell survival during cardioprotection. Basic Res. Cardiol. 106, 1341–1353. doi: 10.1007/s00395-011-0223-7
Goodemote, K. A., Mattie, M. E., Berger, A., and Spiegel, S. (1995). Involvement of a pertussis toxin-sensitive G protein in the mitogenic signaling pathways of sphingosine 1-phosphate. J. Biol. Chem. 270, 10272–10277. doi: 10.1074/jbc.270.17.10272
Guillermet-Guibert, J., Davenne, L., Pchejetski, D., Saint-Laurent, N., Brizuela, L., Guilbeau-Frugier, C., et al. (2009). Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol. Cancer Ther. 8, 809–820. doi: 10.1158/1535-7163.MCT-08-1096
Hait, N. C., Allegood, J., Maceyka, M., Strub, G. M., Harikumar, K. B., Singh, S. K., et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. doi: 10.1126/science.1176709
Hannun, Y. A., Luberto, C., and Argraves, K. M. (2001). Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry 40, 4893–4903. doi: 10.1021/bi002836k
Hla, T., and Brinkmann, V. (2011). Sphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology 76(8 Suppl. 3), S3–S8. doi: 10.1212/WNL.0b013e31820d5ec1
Hla, T., Lee, M. J., Ancellin, N., Paik, J. H., and Kluk, M. J. (2001). Lysophospholipid-receptor revelations. Science 294, 1875–1878. doi: 10.1126/science.1065323
Hofmann, L. P., Ren, S., Schwalm, S., Pfeilschifter, J., and Huwiler, A. (2008). Sphingosine kinase 1 and 2 regulate the capacity of mesangial cells to resist apoptotic stimuli in an opposing manner. Biol. Chem. 389, 1399–1407. doi: 10.1515/BC.2008.160
Hofmann, U., Burkard, N., Vogt, C., Thoma, A., Frantz, S., Ertl, G., et al. (2009). Protective effects of sphingosine-1-phosphate receptor agonist treatment after myocardial ischaemia-reperfusion. Cardiovasc. Res. 83, 285–293. doi: 10.1093/cvr/cvp137
Huang, K., Huang, J., Chen, C., Hao, J., Wang, S., Huang, J., et al. (2014). AP-1 regulates sphingosine kinase 1 expression in a positive feedback manner in glomerular mesangial cells exposed to high glucose. Cell. Signal. 26, 629–638. doi: 10.1016/j.cellsig.2013.12.002
Imamura, T., Ohgane, J., Ito, S., Ogawa, T., Hattori, N., Tanaka, S., et al. (2001). CpG Island of rat sphingosine kinase-1 gene: tissue-dependent DNA methylation status and multiple alternative first exons. Genomics 76, 117–125. doi: 10.1006/geno.2001.6607
Jeffery, D. R., Markowitz, C. E., Reder, A. T., Weinstock-Guttman, B., and Tobias, K. (2011). Fingolimod for the treatment of relapsing multiple sclerosis. Expert Rev. Neurother. 11, 165–183. doi: 10.1586/ern.10.193
Jin, Z. Q., Goetzl, E. J., and Karliner, J. S. (2004). Sphingosine kinase activation mediates ischemic preconditioning in murine heart. Circulation 110, 1980–1989. doi: 10.1161/01.CIR.0000143632.06471.93
Johnson, K. R., Johnson, K. Y., Crellin, H. G., Ogretmen, B., Boylan, A. M., Harley, R. A., et al. (2005). Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J. Histochem. Cytochem. 53, 1159–1166. doi: 10.1369/jhc.4A6606.2005
Kacimi, R., Vessey, D. A., Honbo, N., and Karliner, J. S. (2007). Adult cardiac fibroblasts null for sphingosine kinase-1 exhibit growth dysregulation and an enhanced proinflammatory response. J. Mol. Cell. Cardiol. 43, 85–91. doi: 10.1016/j.yjmcc.2007.04.007
Karliner, J. S. (2009). Sphingosine kinase and sphingosine 1-phosphate in cardioprotection. J. Cardiovasc. Pharmacol. 53, 189–197. doi: 10.1097/FJC.0b013e3181926706
Karliner, J. S. (2013). Sphingosine kinase and sphingosine 1-phosphate in the heart: a decade of progress. Biochim. Biophys. Acta 1831, 203–212. doi: 10.1016/j.bbalip.2012.06.006
Kerage, D., Brindley, D. N., and Hemmings, D. G. (2014). Review: novel insights into the regulation of vascular tone by sphingosine 1-phosphate. Placenta 35(Suppl.), S86–S92. doi: 10.1016/j.placenta.2013.12.006
Keul, P., van Borren, M. M., Ghanem, A., Müller, F. U., Baartscheer, A., Verkerk, A. O., et al. (2016). Sphingosine-1-phosphate receptor 1 regulates cardiac function by modulating Ca2+ sensitivity and Na+/H+ exchange and mediates protection by ischemic preconditioning. J. Am. Heart Assoc. 5:e003393. doi: 10.1161/JAHA.116.003393
Kharel, Y., Lee, S., Snyder, A. H., Sheasley-O’Neill, S. L., Morris, M. A., Setiady, Y., et al. (2005). Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J. Biol. Chem. 280, 36865–36872. doi: 10.1074/jbc.M506293200
Knapp, M., Lisowska, A., Zabielski, P., Musiał, W., and Baranowski, M. (2013). Sustained decrease in plasma sphingosine-1-phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediat. 106, 53–61. doi: 10.1016/j.prostaglandins.2013.10.001
Kohama, T., Olivera, A., Edsall, L., Nagiec, M. M., Dickson, R., and Spiegel, S. (1998). Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 273, 23722–23728. doi: 10.1074/jbc.273.37.23722
Kono, M., Mi, Y., Liu, Y., Sasaki, T., Allende, M. L., Wu, Y. P., et al. (2004). The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J. Biol. Chem. 279, 29367–29373. doi: 10.1074/jbc.M403937200
Kumar, A., Byun, H. S., Bittman, R., and Saba, J. D. (2011). The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell. Signal. 23, 1144–1152. doi: 10.1016/j.cellsig.2011.02.009
Landeen, L. K., Dederko, D. A., Kondo, C. S., Hu, B. S., Aroonsakool, N., Haga, J. H., et al. (2008). Mechanisms of the negative inotropic effects of sphingosine-1-phosphate on adult mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H736–H749. doi: 10.1152/ajpheart.00316.2007
Lee, M. J., Thangada, S., Liu, C. H., Thompson, B. D., and Hla, T. (1998). Lysophosphatidic acid stimulates the G-protein-coupled receptor EDG-1 as a low affinity agonist. J. Biol. Chem. 273, 22105–22112. doi: 10.1074/jbc.273.34.22105
Li, J., Guan, H. Y., Gong, L. Y., Song, L. B., Zhang, N., Wu, J., et al. (2008). Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin. Cancer Res. 14, 6996–7003. doi: 10.1158/1078-0432.CCR-08-0754
Licht, T., Tsirulnikov, L., Reuveni, H., Yarnitzky, T., and Ben-Sasson, S. A. (2003). Induction of pro-angiogenic signaling by a synthetic peptide derived from the second intracellular loop of S1P3 (EDG3). Blood 102, 2099–2107. doi: 10.1182/blood-2002-12-3634
Liu, H., Sugiura, M., Nava, V. E., Edsal, L. C., Kono, K., Poulton, S., et al. (2000). Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275, 19513–19520. doi: 10.1074/jbc.M002759200
Liu, Y., Wada, R., Yamashita, T., Mi, Y., Deng, C. X., Hobson, J. P., et al. (2000). Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Invest. 106, 951–961. doi: 10.1172/JCI10905
Lu, H., Yuan, H., Chen, S., Huang, L., Xiang, H., Yang, G., et al. (2012). Senescent endothelial dysfunction is attributed to the up-regulation of sphingosine-1-phosphate receptor-2 in aged rats. Mol. Cell. Biochem. 363, 217–224. doi: 10.1007/s11010-011-1173-y
Maceyka, M., Harikumar, K. B., Milstien, S., and Spiegel, S. (2012). Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 22, 50–60. doi: 10.1016/j.tcb.2011.09.003
Maceyka, M., Sankkala, H., Hait, N. C., Le Stunff, H., Liu, H., Toman, R., et al. (2005). SphK1 and Sphk2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 280, 37118–37129. doi: 10.1074/jbc.M502207200
Mandala, S., Hajdu, R., Bergstrom, J., Quackenbush, E., Xie, J., Milligan, J., et al. (2002). Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296, 346–349. doi: 10.1126/science.1070238
Marino, A., Sakamoto, T., Robador, P. A., Tomita, K., and Levi, R. (2017). S1P1-mediated anti-RAS cardioprotection: pivotal role of mast cell ALDH2. J. Pharmacol. Exp. Ther. 362, 230–242. doi: 10.1124/jpet.117.241976
Means, C. K., and Brown, J. H. (2009). Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc. Res. 82, 193–200. doi: 10.1093/cvr/cvp086
Means, C. K., Miyamoto, S., Chun, J., and Brown, J. H. (2008). S1P1 receptor localization confers selectivity for Gi-mediated cAMP and contractile responses. J. Biol. Chem. 283, 11954–11963. doi: 10.1074/jbc.M707422200
Means, C. K., Xiao, C. Y., Li, Z., Zhang, T., Omens, J. H., Ishii, I., et al. (2007). Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 292, H2944–H2951. doi: 10.1152/ajpheart.01331.2006
Melendez, A. H., Carlos-Dias, E., Gosink, M., Allen, J. M., and Takacs, L. (2000). Human sphingosine kinase: molecular cloning, functional characterization, and tissue distribution. Gene 251, 19–26. doi: 10.1016/S0378-1119(00)00205-5
Michaud, J., Kohno, M., Proia, R. L., and Hla, T. (2006). Normal acute and chronic inflammatory responses in sphingosine kinase 1 knockout mice. FEBS Lett. 580, 4607–4612. doi: 10.1016/j.febslet.2006.07.035
Mizugishi, K., Yamashita, T., Olivera, A., Miller, G. F., Spiegel, S., and Proia, R. L. (2005). Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 25, 11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005
Morel, S., Christoffersen, C., Axelsen, L. N., Montecucco, F., Rochemont, V., and Frias, M. A. (2016). Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc. Res. 109, 385–396. doi: 10.1093/cvr/cvw004
Murata, N., Sato, K., Kon, J., Tomura, H., Yanagita, M., Kuwabara, A., et al. (2000). Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem. J. 352, 809–815. doi: 10.1042/bj3520809
Nava, V. E., Lacana, E., Poulton, S., Liu, H., Sugiura, M., Kono, K., et al. (2000). Functional characterization of human sphingosine kinase-1. FEBS Lett. 473, 81–84. doi: 10.1016/S0014-5793(00)01510-6
Newton, J., Lima, S., Maceyka, M., and Spiegel, S. (2015). Revisiting the sphingolipid rheostat: evolving concepts in cancer therapy. Exp. Cell Res. 333, 195–200. doi: 10.1016/j.yexcr.2015.02.025
Okada, T., Ding, G., Sonoda, H., Kajimoto, T., Haga, Y., Khosrowbeygi, A., et al. (2005). Involvement of N-terminal-extended form of sphingosine kinase 2 in serum-dependent regulation of cell proliferation and apoptosis. J. Biol. Chem. 280, 36318–36325. doi: 10.1074/jbc.M504507200
Olivera, A., Kohama, T., Edsall, L., Nava, V., Cuvillier, O., Poulton, S., et al. (1999). Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Cell Biol. 147, 545–548. doi: 10.1083/jcb.147.3.545
O’Sullivan, S., and Dev, K. K. (2017). Sphingosine-1-phosphate receptor therapies: advances in clinical trials for CNS-related diseases. Neuropharmacology 113, 597–607. doi: 10.1016/j.neuropharm.2016.11.006
Pappu, R., Schwab, S. R., Cornelissen, I., Pereira, J. P., Regard, J. B., Xu, Y., et al. (2007). Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316, 295–298. doi: 10.1126/science.1139221
Park, J. H., and Schuchman, E. H. (2006). Acid ceramidase and human disease. Biochim. Biophys. Acta. 1758, 2133–2138. doi: 10.1016/j.bbamem.2006.08.019
Paugh, S. W., Payne, S. G., Barbour, S. E., Milstien, S., and Spiegel, S. (2003). The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 554, 189–193. doi: 10.1016/S0014-5793(03)01168-2
Pelletier, D., and Hafler, D. A. (2012). Fingolimod for multiple sclerosis. New Engl. J. Med. 366, 339–347. doi: 10.1056/NEJMct1101691
Peters, S. L. M., and Alewijnse, A. E. (2007). Sphingosine-1-phosphate signaling in the cardiovascular system. Curr. Opin. Pharmacol. 7, 186–192. doi: 10.1016/j.coph.2006.09.008
Pitson, S. M. (2011). Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 36, 97–107. doi: 10.1016/j.tibs.2010.08.001
Pitson, S. M., Morretti, P. A., Zebol, J. R., Lynn, H. E., Xia, P., Vadas, M. A., et al. (2003). Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 22, 5491–5500. doi: 10.1093/emboj/cdg540
Pruett, S. T., Bushnev, A., Hagedorn, K., Adiga, M., Haynes, C. A., Sullards, M. C., et al. (2008). Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. J. Lipid Res. 49, 1621–1639. doi: 10.1194/jlr.R800012-JLR200
Pyne, N. J., and Pyne, S. (2010). Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 10, 489–503. doi: 10.1038/nrc2875
Pyne, S., Adams, D. R., and Pyne, N. J. (2016). Sphingosine 1-phosphate and sphingosine kinases in health and disease: recent advances. Prog. Lipid Res. 62, 93–106. doi: 10.1016/j.plipres.2016.03.001
Racca, V., Di Rienzo, M., Cavarretta, R., Toccafondi, A., Vaini, E., Ferratini, M., et al. (2016). Fingolimod effects on left ventricular function in multiple sclerosis. Mult. Scler. 22, 201–211. doi: 10.1177/1352458515587753
Reynolds, C. P., Maurer, B. J., and Kolesnick, R. N. (2004). Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett. 206, 169–180. doi: 10.1016/j.canlet.2003.08.034
Ruiz, M., Okada, H., and Dahlbäck, B. (2017). HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Health Dis. 16:36. doi: 10.1186/s12944-017-0429-2
Saddoughi, S. A., and Ogretmen, B. (2013). Diverse functions of ceramide in cancer cell death and proliferation. Adv. Cancer Res. 117, 37–58. doi: 10.1016/B978-0-12-394274-6.00002-9
Salomone, S., Potts, E. M., Tyndall, S., Ip, P. C., Chun, J., Brinkmann, V., et al. (2008). Analysis of sphingosine 1-phosphate receptors involved in constriction of isolated cerebral arteries with receptor null mice and pharmacological tools. Br. J. Pharmacol. 153, 140–147. doi: 10.1038/sj.bjp.0707581
Sanna, M. G., Liao, J., Jo, E., Alfonso, C., Ahn, M. Y., Peterson, M. S., et al. (2004). Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 279, 13839–13848. doi: 10.1074/jbc.M311743200
Santos-Gallego, C. G., Vahl, T. P., Goliasch, G., Picatoste, B., Arias, T., Ishikawa, K., et al. (2016). Sphingosine-1-phosphate receptor agonist fingolimod increases myocardial salvage and decreases adverse postinfarction left ventricular remodeling in a porcine model of ischemia/reperfusion. Circulation 133, 954–966. doi: 10.1161/CIRCULATIONAHA.115.012427
Sattler, K. J., Elbasan, S., Keul, P., Elter-Schulz, M., Bode, C., and Gräler, M. H. (2010). Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res. Cardiol. 105, 821–832. doi: 10.1007/s00395-010-0112-5
Schuchardt, M., Tölle, M., Prüfer, J., and van der Giet, M. (2011). Pharmacological relevance and potential of sphingosine 1-phosphate in the vascular system. Br. J. Pharmacol. 163, 1140–1162. doi: 10.1111/j.1476-5381.2011.01260.x
Shi, W. N., Cui, S. X., Song, Z. Y., Wang, S. Q., Sun, S. Y., Yu, X. F., et al. (2017). Overexpression of SphK2 contributes to ATRA resistance in colon cancer through rapid degradation of cytoplasmic RXRα by K48/K63-linked polyubiquitination. Oncotarget 8, 39605–39617. doi: 10.18632/oncotarget.17174
Shida, D., Takabe, K., Kapitonov, D., Milstien, S., and Spiegel, S. (2008). Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 9, 662–673. doi: 10.2174/138945008785132402
Skoura, A., and Hla, T. (2009). Lysophospholipid receptors in vertebrate development, physiology, and pathology. J. Lipid Res. 50(Suppl.), S293–S298. doi: 10.1194/jlr.R800047-JLR200
Soltau, I., Mudersbach, E., Geissen, M., Schwedhelm, E., Winkler, M. S., Geffken, M., et al. (2016). Serum-sphingosine-1-phosphate concentrations are inversely associated with atherosclerotic diseases in humans. PLoS ONE 11:e0168302. doi: 10.1371/journal.pone.0168302
Spiegel, S., and Milstien, S. (2000). Sphingosine-1-phosphate: signaling inside and out. FEBS Lett. 476, 55–57. doi: 10.1016/S0014-5793(00)01670-7
Spiegel, S., and Milstien, S. (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407. doi: 10.1038/nrm1103
Stone, M. L., Sharma, A. K., Zhao, Y., Charles, E. J., Huerter, M. E., Johnston, W. F., et al. (2015). Sphingosine-1-phosphate receptor 1 agonism attenuates lung ischemia-reperfusion injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L1245–L1252. doi: 10.1152/ajplung.00302.2014
Strub, G. M., Maceyka, M., Hait, N. C., Milstien, S., and Spiegel, S. (2010). Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 688, 141–155. doi: 10.1007/978-1-4419-6741-1_10
Strub, G. M., Paillard, M., Liang, J., Gomez, L., Allegood, J. C., Hait, N. C., et al. (2011). Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612. doi: 10.1096/fj.10-167502
Sugahara, K., Maeda, Y., Shimano, K., Mogami, A., Kataoka, H., Ogawa, K., et al. (2017). Amiselimod, a novel sphingosine 1-phosphate receptor-1 modulator, has potent therapeutic efficacy for autoimmune diseases, with low bradycardia risk. Br. J. Pharmacol. 174, 15–27. doi: 10.1111/bph.13641
Sugimoto, N., Takuwa, N., Okamoto, H., Sakurada, S., and Takuwa, Y. (2003). Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell. Biol. 23, 1534–1545. doi: 10.1128/MCB.23.5.1534-1545.2003
Szczepaniak, W. S., Pitt, B. R., and McVerry, B. J. (2010). S1P2 receptor-dependent Rho-kinase activation mediates vasoconstriction in the murine pulmonary circulation induced by sphingosine 1-phosphate. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, L137–L145. doi: 10.1152/ajplung.00233.2009
Tani, M., Sano, T., Ito, M., and Igarashi, Y. (2005). Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J. Lipid Res. 46, 2458–2467. doi: 10.1194/jlr.M500268-JLR200
Tao, R., Zhang, J., Vessey, D. A., Honbo, N., and Karliner, J. S. (2007). Deletion of the sphingosine kinase-1 gene influences cell fate during hypoxia and glucose deprivation in adult mouse cardiomyocytes. Cardiovasc. Res. 74, 56–63. doi: 10.1016/j.cardiores.2007.01.015
Theilmeier, G., Schmidt, C., Herrmann, J., Keul, P., Schäfers, M., Herrgott, I., et al. (2006). High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114, 1403–1409. doi: 10.1161/CIRCULATIONAHA.105.607135
Thompson, B., Ancellin, N., Fernandez, S. M., Hla, T., and Sha’afi, R. I. (2006). Protein kinase C-alpha and sphingosine 1-phosphate-dependent signaling in endothelial cell. Prostaglandins Other Lipid Mediat. 80, 15–27. doi: 10.1016/j.prostaglandins.2006.03.002
Tonelli, F., Lim, K. G., Loveridge, C., Long, J., Pitson, S. M., Tigyi, G., et al. (2010). FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell. Signal. 22, 1536–1542. doi: 10.1016/j.cellsig.2010.05.022
Urata, Y., Nishimura, Y., Hirase, T., and Yokoyama, M. (2005). Sphingosine 1-phosphate induces alpha-smooth muscle actin expression in lung fibroblasts via Rho-kinase. Kobe J. Med. Sci. 51, 17–27.
Usui, S., Sugimoto, N., Takuwa, N., Sakagami, S., Takata, S., Kaneko, S., et al. (2004). Blood lipid mediator sphingosine 1-phosphate potently stimulates platelet-derived growth factor-A and -B chain expression through S1P1-Gi-Ras-MAPK-dependent induction of Kruppel-like factor 5. J. Biol. Chem. 279, 12300–12311. doi: 10.1074/jbc.M305025200
Vasto, S., Scapagnini, G., Bulati, M., Candore, G., Castiglia, L., Colonna-Romano, G., et al. (2010). Biomarkes of aging. Front. Biosci. (Schol Ed) 2:392–402.
Venkataraman, K., Lee, Y. M., Michaud, J., Thangada, S., Ai, Y., Bonkovsky, H. L., et al. (2008). Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 102, 669–676. doi: 10.1161/CIRCRESAHA.107.165845
Venkataraman, K., Thangada, S., Michaud, J., Oo, M. L., Ai, Y., Lee, Y. M., et al. (2006). Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 397, 461–471. doi: 10.1042/BJ20060251
Vessey, D. A., Kelley, M., Li, L., and Huang, Y. (2009). Sphingosine protects aging hearts from ischemia/reperfusion injury: superiority to sphingosine 1-phosphate and ischemic pre- and post-conditioning. Oxid Med. Cell. Longev. 2, 146–151. doi: 10.4161/oxim.2.3.8622
Vessey, D. A., Li, L., Imhof, I., Honbo, N., and Karliner, J. S. (2013). FTY720 postconditions isolated perfused heart by a mechanism independent of sphingosine kinase 2 and different from S1P or ischemic postconditioning. Med. Sci. Monit. Basic Res. 19, 126–132. doi: 10.12659/MSMBR.883877
Vessey, D. A., Li, L., Jin, Z. Q., Kelley, M., Honbo, N., Zhang, J., et al. (2011). A sphingosine kinase form 2 knockout sensitizes mouse myocardium to ischemia/reoxygenation injury and diminishes responsiveness to ischemic preconditioning. Oxid. Med. Cell. Longev. 2011:961059. doi: 10.1155/2011/961059
Waeber, C., Blondeau, N., and Salomone, S. (2004). Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect. 17, 365–382.
Walzer, T., Jaeger, S., Chaix, J., and Vivier, E. (2007). Natural killer cells: from CD3(-) NKp46(+) to post-genomics meta-analyses. Curr. Opin. Immunol. 19, 365–372. doi: 10.1016/j.coi.2007.04.004
Wang, F., Nobes, C. D., Hall, A., and Spiegel, S. (1997). Sphingosine 1-phosphate stimulates Rho-mediated tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 fibroblasts. Biochem. J. 324, 481–488. doi: 10.1042/bj3240481
Wang, M., Lu, L., Liu, Y., Gu, G., and Tao, R. (2014). FTY720 attenuates hypoxia-reoxygenation-induced apoptosis in cardiomyocytes. Exp. Mol. Pathol. 97, 218–224. doi: 10.1016/j.yexmp.2014.07.008
Wang, S., Lin, X., Wang, L. Y., Ruan, K. F., Feng, Y., and Li, X. Y. (2012). A polysaccharides MDG-1 augments survival in the ischemic heart by inducing S1P release and S1P1 expression. Int. J. Biol. Macromol. 50, 734–740. doi: 10.1016/j.ijbiomac.2011.12.005
Wattenberg, B. W. (2010). Role of sphingosine kinase localization in sphingolipid signaling. World J. Biol. Chem. 26, 362–368. doi: 10.4331/wjbc.v1.i12.362
Wattenberg, B. W., Pitson, S. M., and Raben, D. M. (2006). The sphingosine and diacylglycerol kinase superfamily of signaling kinases: localization as a key to signaling function. J. Lipid Res. 47, 1128–1139. doi: 10.1194/jlr.R600003-JLR200
Weigert, A., Schiffmann, S., Sekar, D., Ley, S., Menrad, H., Werno, C., et al. (2009). Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int. J. Cancer 125, 2114–2121. doi: 10.1002/ijc.24594
Weigert, A., Tzieply, N., von Knethen, A., Johann, A. M., Schmidt, H., Geisslinger, G., et al. (2007). Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol. Biol. Cell 18, 3810–3819. doi: 10.1091/mbc.E06-12-1096
Wilkerson, B. A., Grass, G. D., Wing, S. B., Argraves, W. S., and Argraves, K. M. (2012). Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: high density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem. 283, 44645–44653. doi: 10.1074/jbc.M112.423426
Xia, P., Gamble, J. R., Wang, L., Pitson, S. M., Moretti, P. A., Wattenberg, B. W., et al. (2000). An oncogenic role of sphingosine kinase. Curr. Biol. 10, 1527–1530. doi: 10.1016/S0960-9822(00)00834-4
Xiang, S. Y., Ouyang, K., Yung, B. S., Miyamoto, S., Smrcka, A. V., Chen, J., et al. (2013). PLC𝜀, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 6:ra108. doi: 10.1126/scisignal.2004405
Yu, R. K., and Law, J. H. (2009). Herbert Edmund Carter 1910–2007 – A Biographical Memoir. Washington, DC: National Academy of Sciences.
Yung, B. S., Brand, C. S., Xiang, S. Y., Gray, C. B., Means, C. K., Rosen, H., et al. (2017). Selective coupling of the S1P3 receptor subtype to S1P-mediated RhoA activation and cardioprotection. J. Mol. Cell. Cardiol. 103, 1–10. doi: 10.1016/j.yjmcc.2016.12.008
Zhang, H., Desai, N. N., Olivera, A., Seki, T., Brooker, G., and Spiegel, S. (1991). Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell. Biol. 114, 155–167. doi: 10.1083/jcb.114.1.155
Zhang, W., Mottillo, E. P., Zhao, J., Gartung, A., VanHecke, G. C., Lee, J. F., et al. (2014). Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. J. Biol. Chem. 289, 32178–32185. doi: 10.1074/jbc.M114.601096
Zhao, J., Garcia, D., Gartung, A., and Lee, M. J. (2015). Sphingosine-1-phosphate receptor subtype 2 signaling in endothelial senescence-associated functional impairments and inflammation. Curr. Atheroscler. Rep. 17:504. doi: 10.1007/s11883-015-0504-y
Zhao, X., Ding, E. Y., Yu, O. M., Xiang, S. Y., Tan-Sah, V. P., Yung, B. S., et al. (2014). Induction of the matricellular protein CCN1 through RhoA and MRTF-A contributes to ischemic cardioprotection. J. Mol. Cell. Cardiol. 75, 152–161. doi: 10.1016/j.yjmcc.2014.07.017
Zheng, W., Kollmeyer, J., Symolon, H., Momin, A., Munter, E., Wang, E., et al. (2006). Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 1758, 1864–1884. doi: 10.1016/j.bbamem.2006.08.009
Keywords: sphingosine 1-phosphate, G protein-coupled receptors, sphingosine kinase, fingolimod, cardiovascular, heart failure, gene-therapy
Citation: Cannavo A, Liccardo D, Komici K, Corbi G, de Lucia C, Femminella GD, Elia A, Bencivenga L, Ferrara N, Koch WJ, Paolocci N and Rengo G (2017) Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 8:556. doi: 10.3389/fphar.2017.00556
Received: 11 May 2017; Accepted: 07 August 2017;
Published: 23 August 2017.
Edited by:
Nicolau Beckmann, Novartis Institutes for BioMedical Research, United StatesReviewed by:
Roberto Levi, Weill Cornell Medical College, United StatesMiguel A. Frias, Geneva University Hospitals (HUG), Switzerland
Joan Heller Brown, University of California, San Diego, United States
Copyright © 2017 Cannavo, Liccardo, Komici, Corbi, de Lucia, Femminella, Elia, Bencivenga, Ferrara, Koch, Paolocci and Rengo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Rengo, Z2l1c2VwcGUucmVuZ29AdW5pbmEuaXQ=